
Method Article
Decifrando a organização da cromatina 3D de alta resolução via captura Hi-C
Neste Artigo
Resumo
Este protocolo descreve o método Capture Hi-C usado para caracterizar a organização 3D de regiões genômicas alvo de tamanho megabaseado em alta resolução, incluindo limites de domínios topologicamente associados (TADs) e interações de cromatina de longo alcance entre elementos regulatórios e outros elementos de sequência de DNA.
Resumo
A organização espacial do genoma contribui para sua função e regulação em muitos contextos, incluindo transcrição, replicação, recombinação e reparo. A compreensão da causalidade exata entre a topologia e a função do genoma é, portanto, crucial e cada vez mais objeto de pesquisa intensiva. As tecnologias de captura da conformação cromossômica (3C) permitem inferir a estrutura 3D da cromatina medindo a frequência de interações entre qualquer região do genoma. Aqui descrevemos um protocolo rápido e simples para realizar o Capture Hi-C, um método de enriquecimento de alvos baseado em 3C que caracteriza a organização 3D alelo-específica de alvos genômicos de tamanho megabaseado em alta resolução. No Capture Hi-C, as regiões-alvo são capturadas por uma matriz de sondas biotiniladas antes do sequenciamento de alto rendimento downstream. Assim, maior resolução e especificidade alélica são alcançadas, melhorando a eficácia do tempo e a acessibilidade da tecnologia. Para demonstrar seus pontos fortes, o protocolo Capture Hi-C foi aplicado ao centro de inativação X de camundongos ( Xic), o locus regulador mestre da inativação do cromossomo X (XCI).
Introdução
O genoma linear contém todas as informações necessárias para que um organismo passe pelo desenvolvimento embrionário e sobreviva ao longo da vida adulta. No entanto, instruir células geneticamente idênticas a desempenhar diferentes funções é fundamental para controlar com precisão quais informações são usadas em contextos específicos, incluindo diferentes tecidos e/ou estágios de desenvolvimento. Acredita-se que a organização tridimensional do genoma participe dessa regulação espaço-temporal precisa da atividade gênica, facilitando ou impedindo a interação física entre elementos regulatórios que podem ser separados por várias centenas de quilobases no genoma linear (para revisões 1,2,3). Nos últimos 20 anos, nossa compreensão da interação entre dobramento e atividade do genoma aumentou rapidamente, em grande parte devido ao desenvolvimento de tecnologias de captura de conformação cromossômica (3C) (para revisão 4,5,6,7). Esses métodos medem a frequência de interações entre quaisquer regiões do genoma e dependem da ligadura de sequências de DNA que estão em estreita proximidade 3D dentro do núcleo. Os protocolos 3C mais comuns começam com a fixação das populações celulares com um agente de reticulação como o formaldeído. A cromatina reticulada é então digerida com uma enzima de restrição, embora a digestão com MNase também tenha sido utilizada 8,9. Após a digestão, as extremidades livres de DNA em estreita proximidade espacial são religadas, e as ligações cruzadas são invertidas. Essa etapa dá origem à 'biblioteca' ou 'modelo' 3C, um pool misto de fragmentos híbridos em que sequências que estavam em 3D próximas ao núcleo têm maiores chances de serem ligadas no mesmo fragmento de DNA. A quantificação a jusante desses fragmentos híbridos permite inferir a conformação 3D de regiões genômicas que estão localizadas a milhares de pares de bases no genoma linear, mas que podem interagir no espaço 3D.
Muitas abordagens diferentes foram desenvolvidas para caracterizar a biblioteca 3C, diferindo tanto em termos de quais subconjuntos de fragmentos de ligadura são analisados quanto qual tecnologia é usada para sua quantificação a jusante. O protocolo 3C original baseava-se na seleção de duas regiões de interesse e na quantificação de sua frequência de interação "um contra um" por PCR10,11. A abordagem 4C (captura circular da conformação cromossômica) mede as interações entre um único locus de interesse (i.e., o "ponto de vista") e o resto do genoma ("um contra todos")12,13,14. Em 4C, a biblioteca 3C passa por uma segunda rodada de digestão e religadura para gerar pequenas moléculas circulares de DNA que são amplificadas por PCR por primers específicos do ponto de visão15. O 5C (chromosome conformation capture carbon copy) permite a caracterização de interações 3D em regiões maiores de interesse, fornecendo informações sobre o dobramento de cromatina de ordem superior dentro dessa região ('muitos versus muitos')16. Em 5C, a biblioteca 3C é hibridizada a um pool de oligonucleotídeos sobrepostos em sítios de restrição que podem ser posteriormente amplificados por PCR multiplex com primers universais15. Tanto no 4C quanto no 5C, os fragmentos informativos de DNA foram inicialmente quantificados por microarrays e, posteriormente, por sequenciamento de última geração (NGS)17,18,19. Essas estratégias caracterizam regiões-alvo de interesse, mas não podem ser aplicadas para mapear interações genômicas amplas. Este último objetivo é alcançado com Hi-C, uma estratégia de alto rendimento baseada em 3C na qual o sequenciamento massivamente paralelo do molde 3C permite a caracterização imparcial do dobramento da cromatina no nível do genoma amplo ('todos versus todos')20. O protocolo Hi-C inclui a incorporação de um resíduo biotinilado nas extremidades dos fragmentos digeridos, que é seguido pelo pull-down dos fragmentos de ligadura com esferas de estreptavidina, para aumentar a recuperação dos fragmentosligados20.
Hi-C revelou que genomas de mamíferos são estruturalmente organizados em múltiplas escalas no núcleo 3D. Na escala de megabases, o genoma é dividido em regiões de cromatina ativa e inativa, compartimentos A e B, respectivamente20,21. A existência de outros subcompartimentos representados por diferentes estados de cromatina e atividade também foi posteriormente demonstrada22. Em maior resolução, o genoma é posteriormente particionado em domínios de auto-interação sub-megabase chamados domínios topologicamente associados (TADs), revelados pela primeira vez pela análise Hi-C e 5C dos genomas humano e de camundongos23,24. Ao contrário dos compartimentos que variam de maneira específica do tecido, os TADs tendem a ser constantes (embora haja muitas exceções). É importante ressaltar que os limites do TAD são conservados ao longo da espécie25. Em células de mamíferos, as DATs frequentemente englobam genes que compartilham a mesma paisagem regulatória e têm demonstrado representar uma estrutura estrutural que facilita a co-regulação gênica enquanto limita as interações com domínios regulatórios vizinhos (para revisão 3,26,27,28). Além disso, dentro de DATs, interações devidas a sítios CTCF na base de alças extrudadas por coesina podem aumentar a probabilidade de interações promotor-potenciador ou potenciador-potenciador (para revisão29).
Em Hi-C, compartimentos e DATs podem ser detectados com resolução de 1 Mb a 40 kb, mas maior resolução pode ser alcançada para caracterizar contatos de menor escala, como interações em looping entre elementos distais na escala de 5-10 kb. No entanto, aumentar a resolução para ser capaz de detectar tais loops de forma eficiente por HiC requer um aumento significativo na profundidade de sequenciamento e, portanto, nos custos de sequenciamento. Isso é exacerbado se a análise precisar ser alelo-específica. De fato, um aumento de resolução X requer um aumento X2 na profundidade de sequenciamento, o que significa que abordagens genômicas de alta resolução e alelo-específicas podem ser proibitivamente caras30.
Para melhorar a relação custo-benefício e a acessibilidade, mantendo a alta resolução, as regiões-alvo de interesse podem ser fisicamente retiradas de bibliotecas 3C ou Hi-C de todo o genoma após sua hibridização com sondas complementares de oligonucleotídeos marcados com biotina antes do sequenciamento a jusante. Essas estratégias de enriquecimento de alvos são chamadas de métodos de Captura-C e permitem a interrogação de interações de centenas de loci alvo espalhados pelo genoma (i.e., Promoter Capture (PC) Hi-C; Captura de Próxima Geração (NG)-C; Captura de Baixa Entrada (LI)-C; Captura Nuclear Titulada-C; Tri-C)31,32,33,34,35,36,37,38,39,40, ou entre regiões abrangendo até várias megabases (i.e., Capture HiC; Captura Hi-C (Hi-C2); Telha-C)41,42,43. Dois aspectos podem variar nos métodos baseados em captura: (1) a natureza e o planejamento de oligonucleotídeos biotinilados (isto é, RNA ou DNA, oligos únicos capturando alvos genômicos dispersos ou oligos múltiplos mosaicando uma região de interesse); e (2) o modelo que é usado para puxar para baixo alvos que podem ser a biblioteca 3C ou Hi-C, esta última consistindo em fragmentos de restrição biotinilados retirados da biblioteca 3C.
Aqui, um protocolo Capture Hi-C baseado no enriquecimento de contatos de destino da biblioteca 3C é descrito. O protocolo baseia-se no projeto de uma matriz de mosaico personalizada de sondas de RNA biotinilado e pode ser realizado em 1 semana desde a preparação da biblioteca 3C até o sequenciamento NGS. O protocolo é rápido, simples e permite caracterizar a organização 3D de ordem superior de regiões de interesse do tamanho de megabases com resolução de 5 kb, melhorando a eficácia do tempo e a acessibilidade em comparação com outros métodos 3C. O protocolo Capture Hi-C foi aplicado ao locus regulador mestre da inativação do cromossomo X (XCI), o centro de inativação X (Xic), que hospeda o RNA não-codificante Xist. O Xic já foi objeto de extensas análises estruturais e funcionais (para revisão44,45). Em mamíferos, XCI compensa a dosagem de genes ligados ao X entre fêmeas (XX) e machos (XY) e envolve o silenciamento transcricional de quase a totalidade de um dos dois cromossomos X em células femininas. O Xic tem representado um poderoso locus padrão-ouro para estudos em topologia genômica 3D e a interação com a regulação gênica44. A análise 5C do Xic em células-tronco embrionárias de camundongos (mESCs) levou à descoberta e nomeação de DATs, fornecendo os primeiros insights sobre a relevância funcional da partição topológica e da co-regulação gênica24. A organização topológica do Xic foi subsequentemente mostrada para ser criticamente envolvida no momento apropriado do desenvolvimento de Xist upregulation e XCI 46, e elementos cis-regulatórios insuspeitos que podem influenciar a atividade gênica dentro e entre TADs também foram recentemente descobertos dentro do Xic47,48,49. A aplicação do Capture Hi-C a 3 Mb do cromossomo X do mouse que abrange o Xic demonstra o poder dessa abordagem em dissecar o dobramento de cromatina em larga escala em alta resolução. Um protocolo detalhado e fácil de seguir é fornecido, desde o projeto da matriz de sondas biotiniladas em cada local de restrição de DpnII dentro da região de interesse até a geração da biblioteca 3C de todo o genoma, a hibridização e captura de contatos alvo e análise de dados a jusante. Uma visão geral dos controles de qualidade apropriados e dos resultados esperados também é incluída, e tanto os pontos fortes quanto as limitações da abordagem são discutidos à luz de métodos similares existentes.
Protocolo
As células-tronco embrionárias de camundongos (mESCs) utilizadas neste estudo foram derivadas do cruzamento de uma fêmea TX/TX R26rtTA/rtTA 50 com um macho Mus musculus castaneus de acordo com as diretrizes de cuidados com animais do Institut Curie (Paris)51.
1. Projeto da sonda
- Projetar uma matriz de sondas biotiniladas (oligonucleotídeos de RNA de 120 meros) cobrindo a região alvo de interesse.
- Mosaicar a região de interesse com oligonucleotídeos sobrepostos de modo que, em média, cada sequência dentro do alvo seja coberta por duas sondas únicas (cobertura 2x) (Figura 1).
- Exclua sequências repetitivas da cobertura da sonda para evitar o enriquecimento de interações inespecíficas.
NOTA: Para maximizar o enriquecimento de fragmentos informativos de ligadura, regiões abrangendo 300 pb a montante e a jusante de cada sítio de restrição DpnII em todo o alvo foram definidas (ChrX: 102.475.000-105.475.000), e 28.913 sondas biotiniladas foram projetadas de acordo com a tecnologia de enriquecimento de alvo de DNA SureSelect através da plataforma Sure Design52. De acordo com essa estratégia, até um máximo de 40 bases de sequências repetitivas são permitidas em cada oligonucleotídeo para minimizar o enriquecimento de interações inespecíficas. A matriz de sonda foi sintetizada por Agilent. Aqui, a DpnII é usada como enzima de restrição por duas razões: (1) é uma máquina de quatro cortes rotineiramente usada em vários métodos baseados em 3C53; e (2) maximiza as chances de captura de polimorfismos informativos de nucleotídeo único (SNPs) na proximidade dos locais de corte em comparação com outras enzimas de restrição que foram testadas in silico em linhagens híbridas F1 utilizadas neste estudo (C57BL/6J x CASTEi/J).
2. Procedimento experimental
- Preparação celular
- Semeando o número adequado de células em uma ou várias placas de cultura celular para atingir um número total de células de ≥ 5 x 107 células no dia da fixação.
NOTA: Células-tronco embrionárias de camundongo (mESCs) foram utilizadas neste estudo. As mESCs são plaqueadas em placas de cultura celular gelatinizadas (0,1% de gelatina em 1x PBS - o/n a 37 °C, 5% de incubadora de CO2) em meio mESCs contendo 2i + LIF e soro fetal de bezerro testado em lote (DMEM, 15% FBS, 0,1 mM β-mercaptoetanol, 1.000 U/mL−1 fator inibidor de leucemia (LIF), CHIR99021 (3 μM) e PD0325901 (1 μM)). Para este tipo celular, uma placa de 10 cm confluente 80% contém aproximadamente 2 x 107 células. - Prepare uma placa de cultura celular adicional para contagem celular.
NOTA: Uma placa de cultura de células menor pode ser usada para reduzir o uso de mídia. Nesse caso, o número de células a serem semeadas na placa menor precisa ser ajustado de acordo (por exemplo, 3x menos células em uma placa de 10 cm em comparação com uma placa de 15 cm).
- Semeando o número adequado de células em uma ou várias placas de cultura celular para atingir um número total de células de ≥ 5 x 107 células no dia da fixação.
- Fixação de formaldeído
- Estimar o número total de células a serem cruzadas.
- Antes de iniciar a reação de reticulação, tripsinize e conte as células da placa de controle preparada especificamente para contagem de células usando um contador de células automatizado de acordo com as instruções do fabricante.
- Incluir uma coloração de viabilidade (por exemplo, azul de Trypan) para determinar a porcentagem de células viáveis54. A partir dessa contagem de células, estimar o número total de células na(s) placa(s) preparada(s) para reticulação.
- Retirar o meio de cultura das placas preparadas para reticulação e substituí-lo pela quantidade adequada de solução de fixação (formaldeído a 2% em meio de cultura celular). Use 10 mL em uma placa de 10 cm (por exemplo, ~20 mL para uma placa de 15 cm).
NOTA: Adicione um volume exato de solução de fixação. Se não for possível a fixação de células aderentes, esta etapa pode ser adaptada às células tripsinizadas e realizada em 30 mL de solução de fixação em tubos centrífugos cônicos de 50 mL. O formaldeído não deve ter mais de 1 ano de idade. É preferível usar frascos descartáveis. A solução de fixação deve ser levada à temperatura ambiente (TR) antes da utilização.
CUIDADO: O formaldeído é perigoso e precisa ser manuseado de acordo com as normas de saúde e segurança adequadas. - Fixe por 10 minutos no RT sob mistura suave em uma coqueteleira.
- Atenuar a reação de fixação pela adição de 2,5 M glicina-1x PBS a uma concentração final de 0,125 M. Adicionar 530 μL de glicina-1x PBS 2,5 M a 10 mL em uma placa de 10 cm (por exemplo, 1060 μL a 20 mL em uma placa de 15 cm).
NOTA: Se as células foram fixadas em solução, extinguir a reação de fixação com 1590 μL de glicina-1x PBS 2,5 M. - Incubar durante 5 minutos em RT, misturando suavemente numa coqueteleira.
- Transfira as placas para o gelo e incube por mais 15 minutos no gelo enquanto mistura suavemente em uma coqueteleira.
NOTA: A partir de agora, as células devem ser mantidas no gelo, e os buffers devem ser pré-resfriados para evitar novas ligações cruzadas. Mude para uma câmara fria se muitas placas precisarem ser processadas. - Retire a solução de fixação das células, despejando-a em um copo para garantir um manuseio rápido.
NOTA: Certifique-se de descartar os resíduos líquidos contendo formaldeído de acordo com as normas de saúde e segurança apropriadas. - Enxaguar a placa de 10 cm rapidamente duas vezes com 5 mL de glicina-1x PBS 0,125 M fria (8 mL para uma placa de 15 cm) para lavar os detritos e as células mortas. Retire o líquido da placa despejando-o em um copo para garantir um manuseio rápido.
- Adicione 5 mL de glicina-1x PBS 0,125 M fria à placa de 10 cm (10 mL para uma placa de 15 cm) e raspe rapidamente as células da placa usando um raspador de células de plástico.
- Transfira a suspensão celular para um tubo de centrífuga cônica de 50 mL pré-resfriado usando uma pipeta sorológica.
- Enxaguar a placa duas vezes com 5 mL de glicina-1x PBS 0,125 M fria e adicionar a suspensão celular ao tubo cônico da centrífuga.
- Gire para baixo a 480 x g por 10 min a 4 °C.
NOTA: Se as células foram fixadas em solução, transfira a célula para um tubo de centrífuga cónica pré-refrigerado e gire para baixo a 480 x g durante 10 minutos a 4 °C. Retirar a solução de fixação despejando-a em um copo e lavar três vezes em 10 mL de glicina-1x PBS fria 0,125 M. Certifique-se de ressuspender as células em cada etapa de lavagem. - Remova o sobrenadante aspirando com um sistema de aspiração de bancada. Ressuspenda as células em 500 μL de 1x PBS por 1 x 107 células, pipetando cuidadosamente para cima e para baixo com uma pipeta P1000. Para ressuspender células no volume exato, consulte a estimativa do número total de células obtida em 2.2.1.
- Alíquota 500 μL da suspensão celular no número calculado de tubos de microcentrífuga de 1,5 mL (1 x 107 células/tubo).
- Gire para baixo a 480 x g por 10 min a 4 °C.
- Remova o sobrenadante com um sistema de aspiração de bancada e congele rapidamente os pellets de células em nitrogênio líquido. Conservar os pellets de células secas a -80 °C.
NOTA: As amostras podem ser armazenadas por pelo menos 1 ano.
- Estimar o número total de células a serem cruzadas.
- Lise celular
- Descongele o(s) pellet(s) congelado(s) no gelo.
- Preparar 1,5 ml de tampão de lise em H 2 0 por amostra: Adicionar 10 mM Tris-HCl, pH 8,0, 10 mM NaCl e0,2% NP40.
- Adicionar 600 μL do tampão de lise a frio e ressuspender bem sobre gelo.
- Incubar no gelo por 15 min para deixar as células incharem.
- Gire para baixo a 2655 x g durante 5 minutos a 4 °C e remova o sobrenadante utilizando um sistema de aspiração de bancada.
- Para remover os detritos, ressuspenda o pellet em 1 mL do tampão de lise a frio, gire para baixo a 2655 x g por 5 min a 4 °C e remova o sobrenadante.
- Gire novamente brevemente a 2655 x g e 4 °C e remova o máximo possível do sobrenadante restante usando um sistema de aspiração de bancada equipado com uma ponta P200.
- Ressuspender em 100 μL de SDS a 0,5% (vol/vol).
- Incubar num termomisturador a 62 °C, rodopiando a 1400 rpm durante 10 min.
- Adicione 290 μL de H2O + 50 μL de 10% TritonX-100 e misture bem, evitando bolhas de ar.
- Incubar num termomisturador a 37 °C, rodopiando a 1400 rpm durante 15 min.
- Adicione 50 μL de tampão 10x Dpnll e inverta o tubo para misturar.
- Tome 50 μL de DNA não digerido para controle de qualidade em um tubo separado. Não se esqueça de tomar a amostra de controle não digerida.
- Digestão DpnII
- Adicionar 10 μL de alta concentração de Dpnll (total de 500 U) e misturar invertendo.
- Incubar as amostras e o controle não digerido em um termomisturador a 37 °C, rodopiando a 1400 rpm por >4 h.
- Adicionar 10 μL de alta concentração de Dpnll (500 U total) no final do dia.
- Incubar as amostras e o controle não digerido a 37 °C, rodopiando a 1400 rpm durante a noite.
- Adicionar 10 μL de alta concentração de Dpnll (500 U total) no início do dia seguinte às amostras.
- Incubar as amostras e o controle não digerido em um termomisturador a 37 °C, rodopiando a 1400 rpm por 4 h.
- Ligadura e reversão de ligações cruzadas
- Incubar os tubos a 65 °C durante 20 min a 1400 rpm.
Observação : não adicione SDS neste ponto. A ideia é preservar a integridade nuclear, por isso a ligadura é realizada dentro dos núcleos, driblando a necessidade de diluição extrema. - Resfriar as amostras no gelo por no máximo 5-10 min. Para evitar a precipitação de SDS, não deixe as amostras no gelo por mais tempo do que isso.
- Tomar 50 μL do ADN digerido não ligado para controlo de qualidade num tubo separado. Conservar os controlos não digeridos e não ligados a -20 °C.
NOTA: Não se esqueça de colher a amostra de controle não ligada. - Adicionar 800 μL de coquetel de ligadura: 122 μL de tampão ligase 10x, 8 μL de T4 ligase (30 U/μL) e 670 μL de H20.
- Incubar a 16 °C, rodopiando a 1000 rpm durante a noite.
- Adicionar 7,5 μL de Proteinase K (20 mg/mL) às amostras e 2 μL aos controles.
- Incubar a 65 °C durante 4 h a 1000 rpm.
- Incubar os tubos a 65 °C durante 20 min a 1400 rpm.
- Purificação do DNA
- Transferir as amostras em gelo para tubos de centrífuga cônica de 15 mL pré-resfriados e adicionar 2 mL de água, 10,5 mL de EtOH gelado e 583 μL de NaAC 3 M.
NOTA: A água adicional visa evitar o transporte de TDT para o pellet. - Adicionar 200 μL de EtOH gelado, 10,8 μL de NaAC e 1 μL do coprecipitante aos controles de qualidade não digeridos e não ligados.
- Incubar a -80 °C durante pelo menos 4 h até à noite.
- Girar os tubos de 15 mL a 2200 x g a 4 °C por 45 min.
- Gire os tubos de controle de 1,5 mL a 20.500 x g a 4 °C por 30 min.
- Lavar uma vez com 3 mL (amostras) e 1 mL (controles) de EtOH 70% gelado.
- Girar a 2200 x g (amostras) ou 20.500 x g (controles) a 4 °C por 10 min.
- Retire cuidadosamente o EtOH e seque ao ar em RT por 10-15 min; Não seque demais.
- Ressuspender as amostras e controles em 100 μL e 20 μL de H20, respectivamente.
- Adicionar 1 μL de RNAseA e incubar a 37 °C, rodopiando a 1400 rpm durante 30 min.
- Transferir as amostras em gelo para tubos de centrífuga cônica de 15 mL pré-resfriados e adicionar 2 mL de água, 10,5 mL de EtOH gelado e 583 μL de NaAC 3 M.
- Controle de qualidade da preparação do molde 3C
- Quantifique cada amostra e controle usando um kit de fluorômetro para medições de concentração de DNA de alta sensibilidade.
- Carregar 100-200 ng de cada amostra e de cada controle em um gel de agarose/1x TBE a 1%.
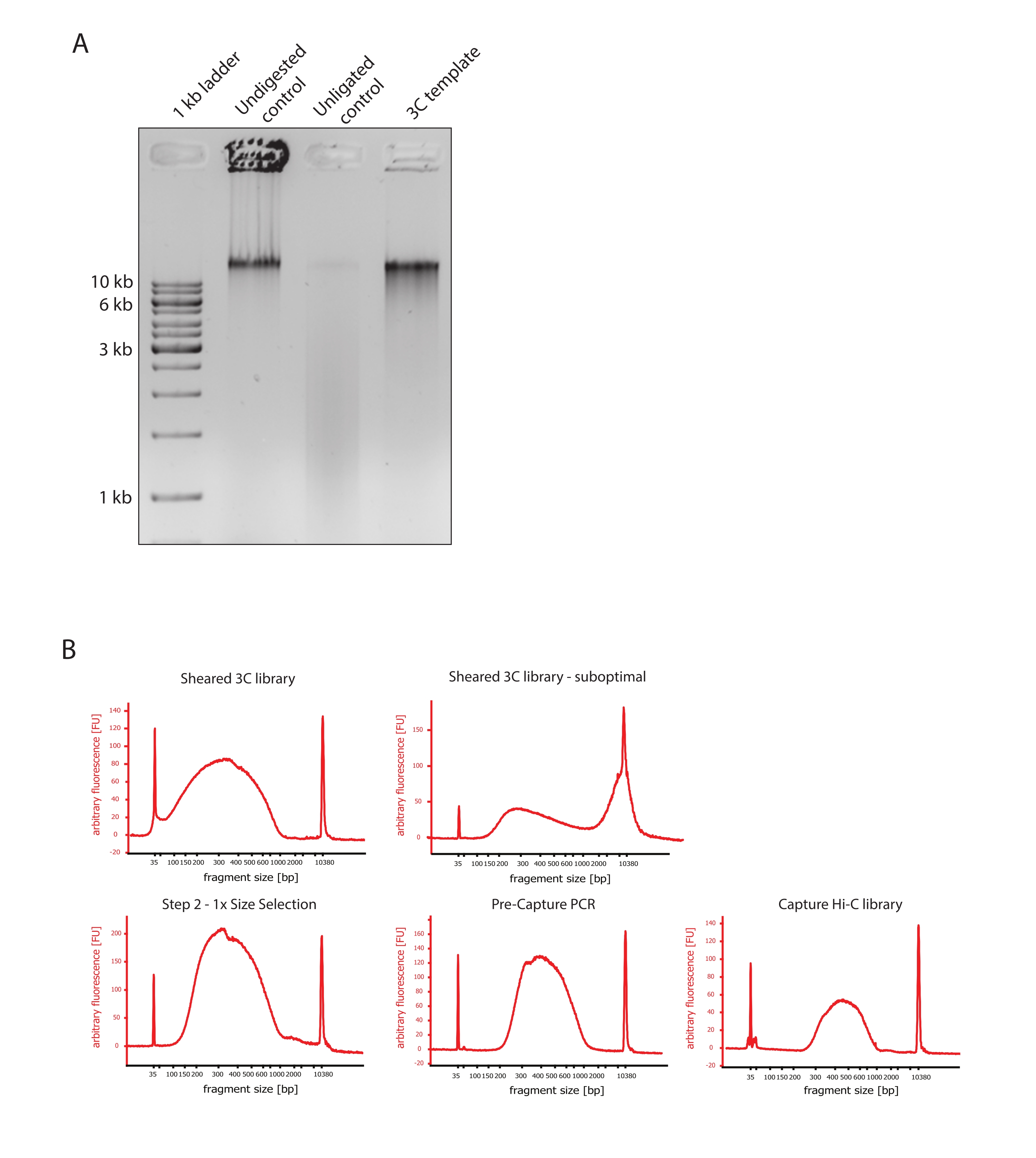
- Verifique se a imagem em gel mostra o resultado esperado, comparando as diferenças no tamanho dos fragmentos de DNA dos controles e do molde 3C, como mostrado na Figura 2A.
- Conservar as amostras e os controlos a -20 °C.
- Hibridização, captura e processamento de amostras para sequenciamento multiplexado
- Para hibridizar a matriz de sondas de RNA biotinilado para o molde 3C, capturar os fragmentos de ligadura alvo e preparar as amostras para sequenciamento multiplexado de acordo com o sistema de enriquecimento alvo usado neste estudo para sequenciamento multiplexado de extremidade pareada (ver Tabela de Materiais). Siga o protocolo de acordo com as instruções do fabricante, introduzindo as seguintes pequenas modificações:
- Secção 2 do protocolo do fabricante: Preparação da amostra
- Siga as instruções para o enriquecimento do alvo a partir de 3 μg de entrada de gDNA.
- Cisalhe o DNA em um sonicador usando as seguintes especificações: ciclo de trabalho de 10%, intensidade de 4, 200 cyc/burst e 130 s. Comece com 4 μg de molde 3C ressuspenso em 130 μL de água para cada reação de captura para garantir material suficiente para continuar a preparação da amostra com 3 μg do DNA cisalhado.
- Avaliar a qualidade do DNA cisalhado. Execute 1 μL do DNA cortado em um bioanalisador de DNA de acordo com o protocolo de alta sensibilidade. Espere uma distribuição de tamanho de fragmento entre 150-700 pb (Figura 2).
- Purificar a amostra usando esferas de imobilização reversível de fase sólida (SPRI). Adicionar 124 μL de contas SPRI a 124 μL da amostra de DNA para realizar uma seleção de tamanho do lado esquerdo de 1:1 de acordo com as instruções do fabricante e eluir em 25 μL de água livre de nucleases. Esta etapa de purificação removerá fragmentos mais curtos para enriquecer fragmentos de cerca de 300 pb (Figura 2).
NOTA: A quantidade de amostras e contas SPRI utilizadas nesta etapa leva em conta a perda de volume ocorrida durante a transferência das amostras para novos tubos e execução dos controles de qualidade no Bioanalyzer. Todas as etapas subsequentes de seleção de tamanho são realizadas de acordo com as proporções recomendadas pelo protocolo do fabricante. A eluição de DNA das esferas de SPRI é realizada em RT durante todo o protocolo. - Avaliar a qualidade do DNA cisalhado selecionado por tamanho. Executar 1 μL do DNA cortado no bioanalisador de DNA de acordo com o protocolo de alta sensibilidade (HS). Espera-se uma distribuição de tamanhos de fragmentos com o maior enriquecimento em 300 pb (Figura 2). Vá em frente com a quantificação do DNA cortado se a tosquia foi bem-sucedida.
- Quantifique o DNA cisalhado com um kit de fluorômetro para medições de concentração de DNA HS.
NOTA: Se o cisalhamento do ADN resultar num rendimento de ADN de <3 μg, efectue uma segunda ronda de cisalhamento do ADN com outros 4 μg de ADN e combine as amostras de ADN cortadas após a primeira etapa de purificação do grânulo SPRI para atingir um total de 3 μg de ADN cisalhado. - Adicionar água livre de nuclease à amostra de DNA limpa selecionada por tamanho (3 μg no total) a um volume final de 48 μL e prosseguir com a reação de reparo final de acordo com o protocolo do fabricante.
- Após a ligadura dos adaptadores pareados, amplie a biblioteca realizando cinco ciclos de PCR pré-captura de acordo com as instruções do fabricante (as condições para PCR e os primers são fornecidas no kit).
- Seção 4 do protocolo do fabricante: Hibridização e captura
- Para hibridizar as amostras de DNA preparadas para as sondas de RNA alvo-específicas, diluir 750 ng de amostras de DNA em um volume final de 3,4 μL, resultando em uma concentração inicial de 221 ng/μL. Para amostras de DNA diluídas em volumes maiores, use um concentrador de vácuo de velocidade para reduzir ao volume final. Uma concentração de velocidade-vácuo (250 x g; ≤45 °C) durante 15-20 minutos é normalmente suficiente para as amostras ressuspensas em 10 μL. Certifique-se de ter o mesmo volume de entrada para cada amostra antes de iniciar o concentrador de vácuo de velocidade.
- Incubar a mistura de hibridização durante 16-18 h a 65 °C com uma tampa aquecida a 105 °C de acordo com as instruções do fabricante.
- Secção 5 do protocolo do fabricante: Indexação e processamento de amostras para sequenciação multiplexada
- Para amplificar as bibliotecas capturadas com primers de indexação, realize 12 ciclos de PCR pós-captura de acordo com as instruções do fabricante (as condições para PCR e os primers são fornecidas no kit).
- Secção 2 do protocolo do fabricante: Preparação da amostra
- Para hibridizar a matriz de sondas de RNA biotinilado para o molde 3C, capturar os fragmentos de ligadura alvo e preparar as amostras para sequenciamento multiplexado de acordo com o sistema de enriquecimento alvo usado neste estudo para sequenciamento multiplexado de extremidade pareada (ver Tabela de Materiais). Siga o protocolo de acordo com as instruções do fabricante, introduzindo as seguintes pequenas modificações:
- Sequenciamento de última geração
- Para executar várias bibliotecas Hi-C de captura na mesma célula de fluxo, prepare uma mistura equimolar das bibliotecas de captura e sequencie leituras de 100-120 M por biblioteca.
- Se a análise alelo-específica for necessária, sequencie 150 pb emparelhado para garantir cobertura SNP suficiente.
3. Análise dos dados
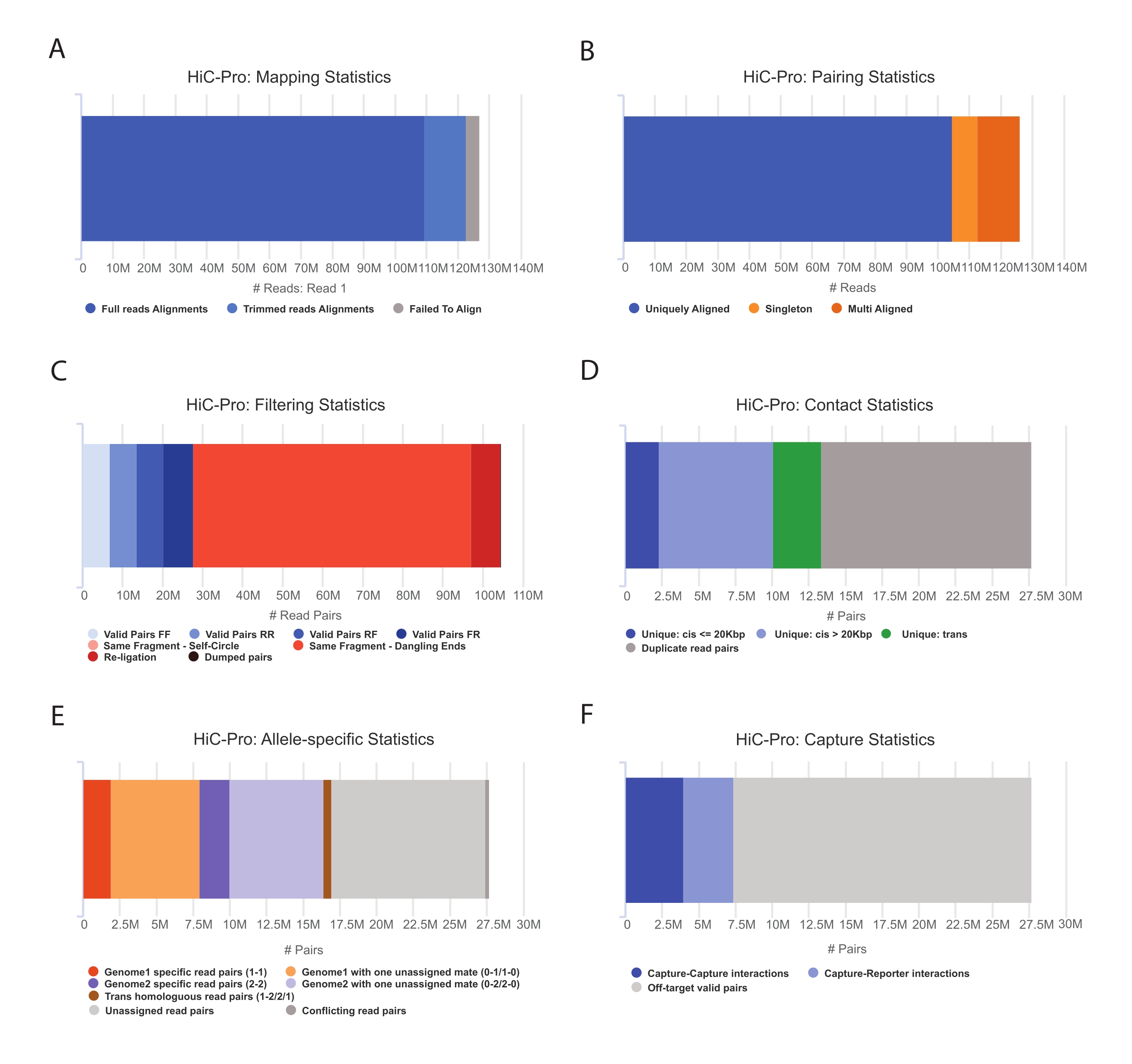
- Aplique o pipeline HiC-Pro para executar a análise de dados do Capture Hi-C55. O HiC-Pro fornece controles de qualidade em cada etapa do processamento, inclusive (Figura 3):
(i) A taxa de alinhamento no genoma de referência especificando a fração de leituras que abrange um sítio de ligadura, bem como o número de pares e singletons.
(ii) A fração de produtos de ligadura válidos e pares de leitura não informativos (ponta pendurada, auto-ligadura, etc).
(iii) A fração de contatos de curto/longo alcance e intra/intercromossômicos.
(iv) A fração de contatos no destino para o Capture Hi-C.
(v) A fração de leituras alelo-específicas se especificada.
NOTA: HiC-Pro suporta uma ampla gama de protocolos, incluindo in situ Hi-C e Capture Hi-C. Neste último caso, o usuário simplesmente precisa especificar a região de destino (formato BED) no arquivo de configuração. Uma vez que os dados são processados, as saídas HiC-Pro podem ser facilmente convertidas em um objeto mais frio para análise downstream56. Nessa etapa, os mapas de contato em várias resoluções são normalizados usando o método ICE descrito anteriormente por Imakaev e colaboradores57. Várias análises podem então ser executadas para chamar compartimentos cromossômicos, TADs ou laços de cromatina (para revisão58). O fluxo de trabalho do protocolo é mostrado na Figura 4. Aqui, o conjunto 'cooltools' é aplicado para calcular a pontuação de isolamento e os limites de TADs, conforme ilustrado na Figura 5 e na Figura 659.
Resultados
O protocolo Capture Hi-C descrito é baseado na preparação do modelo 3C do genoma usando um cortador de quatro bases (DpnII). O subsequente enriquecimento de fragmentos de ligadura através da região genômica de interesse é obtido pela hibridização de um arranjo de sondas de RNA em mosaico e sua captura baseada em estreptavidina de acordo com o sistema de enriquecimento alvo utilizado neste estudo (Figura 1). Sondas de RNA biotinilado foram selecionadas por apresentarem afinidade de ligação mais estreita a seus alvos em comparação com sondas de DNA52,60. As bibliotecas capturadas são então indexadas e agrupadas para sequenciamento multiplexado de alta taxa de transferência. Os dados Hi-C de captura podem ser visualizados como mapas de interação Hi-C de alta resolução, mas também como mapas de contato de ponto de vista único semelhantes a 4C para visualizar especificamente as interações de sequências menores, como promotores ou intensificadores em toda a região capturada. O fluxo de trabalho do protocolo é mostrado na Figura 4. Os controles de qualidade pré-seqüenciamento são mostrados na Figura 2 e incluem a avaliação da digestão e religadura adequadas do molde 3C e sua eficiente cisalhamento e purificação nas diferentes etapas do protocolo. Espera-se que o DNA do molde 3C cortado funcione entre 150 e 700 pb, e nenhum enriquecimento de fragmentos >2 kb deve ser detectado. Durante as etapas seguintes, várias etapas de limpeza de DNA baseadas em contas e seleção de tamanho são realizadas, primeiro após o cisalhamento, depois após as PCRs pré-captura e pós-captura. O tamanho médio dos fragmentos aumenta ao longo da preparação da biblioteca devido à ligadura dos adaptadores, sequenciamento e iniciadores de indexação. Os controles de qualidade pós-seqüenciamento são obtidos via Hi-C Pro e mostrados na Figura 3. Muitos softwares de bioinformática têm sido propostos para processamento e análise de dados do tipo 3C. Entre elas, o pipeline HiC-Pro é uma das soluções mais populares, permitindo o processamento de dados brutos de sequenciamento até os mapas de contato finais em diversas resoluções55. O HiC-Pro usa uma estratégia de mapeamento em duas etapas para alinhar as leituras de sequenciamento no genoma de referência. Os produtos 3C são então reconstruídos e filtrados para remover pares de contato não informativos e gerar os mapas de contato. Além disso, é capaz de usar uma lista de polimorfismos conhecidos para realizar análises alelo-específicas e separar os contatos provenientes dos dois alelos parentais em mapas de contatos distintos. Mais recentemente, o HiC-Pro foi incluído e estendido na estrutura nf-core (nf-core-hic), fornecendo um pipeline altamente escalável e reprodutível orientado pela comunidade61,62.
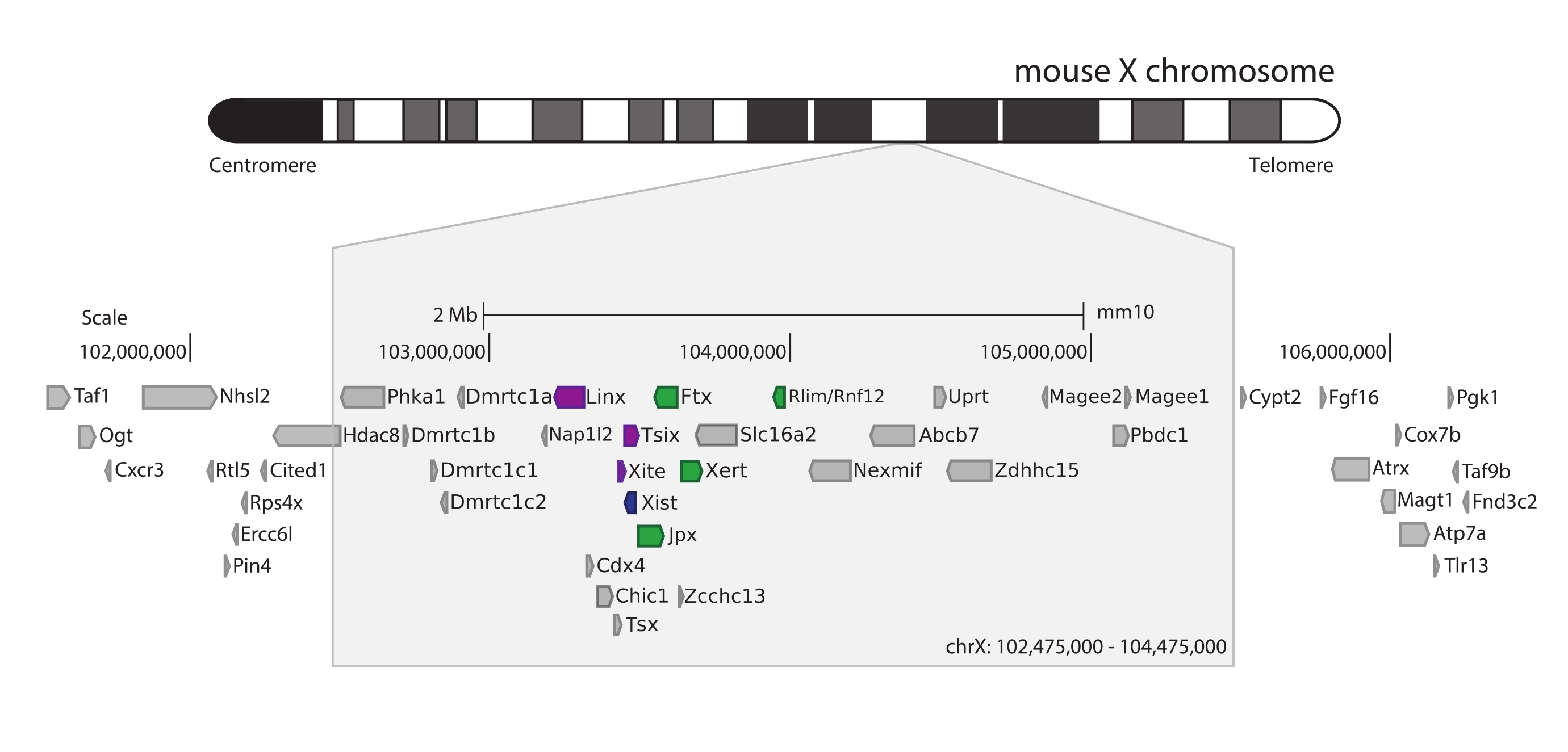
Para capturar o camundongo Xic, um conjunto de 28.913 sondas de RNA til 3 Mb do cromossomo X foi projetado. Essa região inclui o jogador-chave no XCI, o longo gene não codificante Xist, e seu conhecido cenário regulatório de ~800 kb (Figura 5). Esta região de ~800 kb é particionada em dois TADs: um incluindo o promotor Xist e seus reguladores positivos conhecidos (ou seja, os transcritos não-codificantes Ftx, Jpx e Xert e o gene codificador de proteínas Rnf12), e o TAD vizinho englobando os cis-reguladores negativos de Xist (ou seja, seu transcrito antisenso Tsix, o elemento intensificador Xite, e o transcrito não-codificante Linx) (para a revisão 44,45).
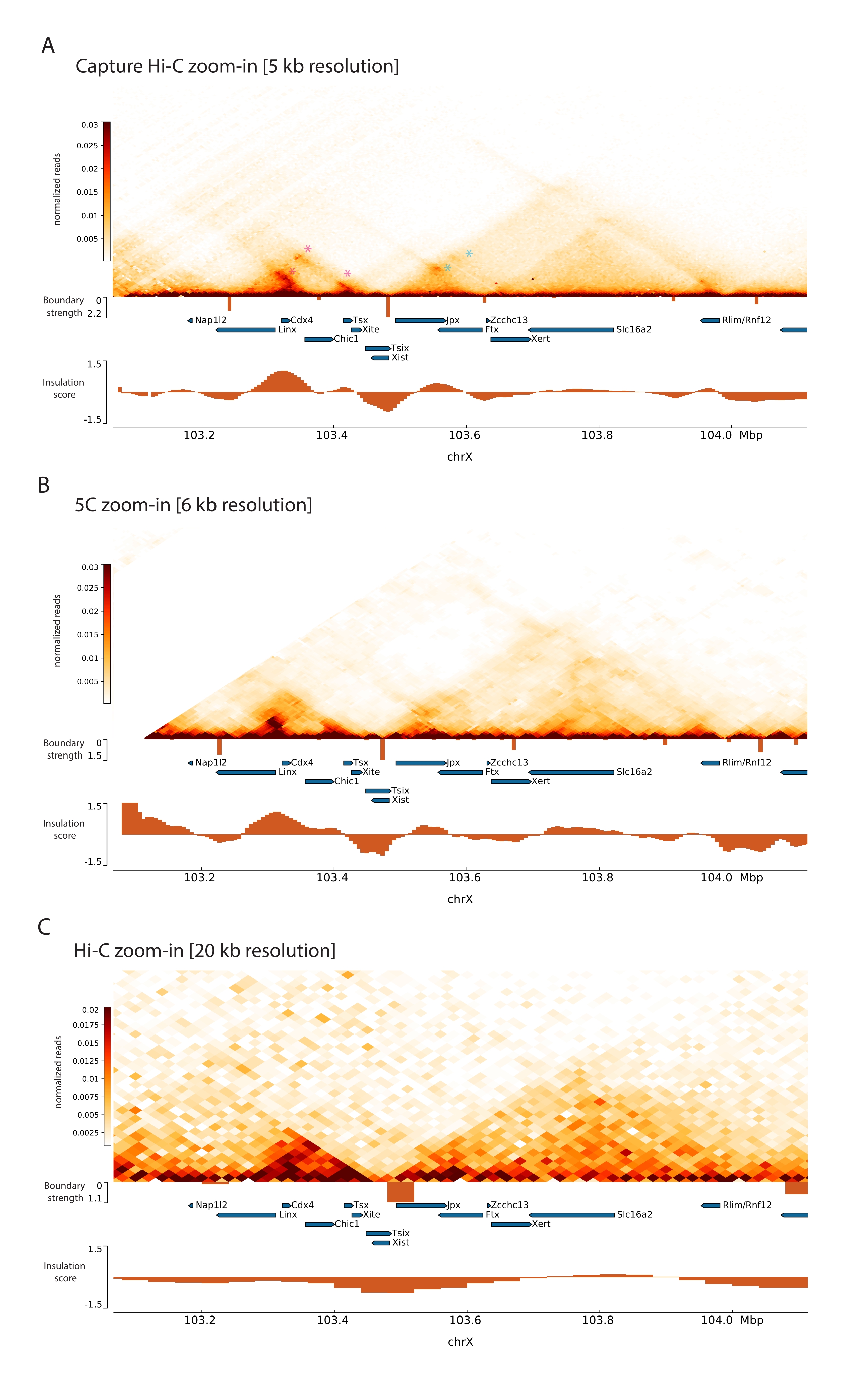
Aplicando-se o protocolo Capture Hi-C descrito ao Xic, obteve-se a organização topológica deste locus em resolução inédita (Figura 6 e Figura 7). Isso fica particularmente claro quando se compara o perfil Capture Hi-C com o 5C47 publicado anteriormente (Figura 6 e Figura 7; Quadro Complementar 1) e Hi-C61 (Figura 6 e Figura 7; Quadro Complementar 1) Perfis. Por exemplo, as estruturas sub-TAD são mais evidentes — o TAD contendo o promotor Xist ( Xist-TAD ) é claramente subdividido em dois domínios menores (Figura 6A, ponta de seta azul). Anteriormente, isso só podia ser visualmente "adivinhado" a partir do perfil 5C (Figura 6B), embora a detecção de um limite nessa região usasse o algoritmo de escore de isolamento. Da mesma forma, a resolução do perfil Capture Hi-C permite a identificação de dois domínios menores no TAD vizinho (Figura 6A, B), que contém o promotor do locus Tsix ( Tsix-TAD ); isso não foi obtido anteriormente com 5C (Figura 6B). É importante notar que os limites topológicos determinados pela pontuação de isolamento dos dados Capture Hi-C e 5C são geralmente detectados em locais ligeiramente diferentes e com diferentes forças relativas.
Além disso, outras estruturas sub-TAD, como loops de contato, são claramente visíveis a partir dos dados do Capture Hi-C, como o loop entre Xist e Ftx (Figura 7A), previamente identificado com o Capture-C63, e o loop entre Xist e Xert (Figura 7B), recentemente identificado usando um protocolo semelhante para o Capture Hi-C48. Outros contatos também podem ser mapeados com mais precisão devido ao aumento da resolução dos perfis Capture Hi-C, como aqueles que formam os hotspots de contato conhecidos dentro do Tsix-TAD entre os loci Linx, Chic1 e Xite (Figura 7A).
Em comparação com os dados Hi-C mostrados na Figura 7, o Capture Hi-C permitiu um aumento de quatro vezes na resolução, mas exigiu apenas um quarto da profundidade de sequenciamento (ou seja, 126 M leituras versus 571 M) (Tabela Suplementar 1). Esse aumento na resolução permite a detecção de subTADs e interações de looping que não puderam ser detectadas pelo Hi-C na profundidade de sequenciamento mostrada na Figura 6 e na Figura 7. O protocolo descrito para Capture Hi-C permite, portanto, uma caracterização muito mais detalhada e de alta resolução de uma grande região genômica de interesse, quando comparado a abordagens anteriores.

Figura 1: Projeto da sonda. Representação esquemática da estratégia utilizada para o projeto da sonda. Regiões de 300 pb a montante e a jusante de cada sítio de restrição de DpnII ao longo da região alvo de 3 Mb foram selecionadas e agrupadas com sondas de RNA biotinilado sobrepostas. Uma dessas regiões selecionadas é mostrada, chrX: 102.474.805-102.475.500. Não são permitidas mais de 40 bases de sequências repetitivas em cada sonda. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Captura de controles de qualidade de pré-seqüenciamento Hi-C. (A) Exemplo representativo de controles de qualidade de modelo 3C. 200 ng de DNA foram carregados em gel de agarose a 1%. Faixa 1: escada de 1 kb. Faixa 2: Cromatina não digerida, reticulada e intacta funciona como uma banda afiada a >10 kb. Faixa 3: A cromatina reticulada digerida por DpnII funciona como um esfregaço entre 1 kb e 3 kb de tamanho. Faixa 4: Biblioteca ou modelo 3C final; extremidades livres de fragmentos de DNA reticulados digeridos são religadas. O esfregaço de DNA de menor tamanho molecular é quase indetectável, e o produto da ligadura é detectado como uma banda de >10 kb. (B) Exemplos representativos de perfis de DNA de bioanalisadores de alta sensibilidade. Canto superior esquerdo: biblioteca 3C cortada com sucesso mostrando uma distribuição de tamanho de fragmento entre 150 pb e 700 pb. Canto superior direito: biblioteca 3C cortada insatisfatória. DNA não cisalhado é detectado como amplo enriquecimento de fragmentos >2 kb. (C) Parte inferior esquerda: amostra de DNA cisalhada após uma seleção de tamanho do lado esquerdo de 1:1 usando contas SPRI. Fragmentos de ~300 pb são enriquecidos. Meio inferior: Perfil de PCR pré-captura após ligadura de adaptadores de extremidade pareada de acordo com o protocolo do fabricante. Canto inferior direito: biblioteca final do Capture Hi-C, incluindo adaptadores, sequenciamento e primers de indexação para sequenciamento multiplexado. Abreviações: bp = pares de bases, FU = unidade de fluorescência arbitrária. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Captura de controles de qualidade pós-seqüenciamento Hi-C com HiC-Pro . (A) Exemplo de taxa de mapeamento no genoma de referência para o primeiro parceiro dos pares de sequenciamento. A fração azul claro representa as leituras alinhadas por HiC-Pro e abrangendo uma junção de ligadura. Essa métrica pode, portanto, ser utilizada para validar a etapa experimental de ligadura. (B) Uma vez que os parceiros de sequenciamento estejam alinhados no genoma, apenas pares de leitura alinhados exclusivamente são mantidos para análise. (C) Pares não válidos (em vermelho), como pendurados, autocírculos ou religaduras, são descartados da análise. A fração de pares válidos é um bom indicador da eficiência da ligadura e do pull-down. (D) Os pares válidos podem ser divididos em contatos intra/intercromossômicos e de curto/longo alcance. Pares de leitura duplicados que provavelmente representam artefatos de PCR são descartados da análise. (E) Para análise alelo-específica, HiC-Pro relata o número de leituras alélicas suportadas por um ou dois parceiros para cada genoma parental (ou seja, C57BL/6J x CASTEi/J). A mesma fração de leituras atribuídas ao alelo materno e paterno é esperada. (F) Finalmente, apenas pares válidos sobrepostos à região de captura são selecionados para construir os mapas de contato. Os pares captura-captura representam contatos dentro da região de destino, enquanto os pares captura-repórter envolvem interação entre a região de destino e uma fora do alvo. Clique aqui para ver uma versão maior desta figura.
{kind=link}

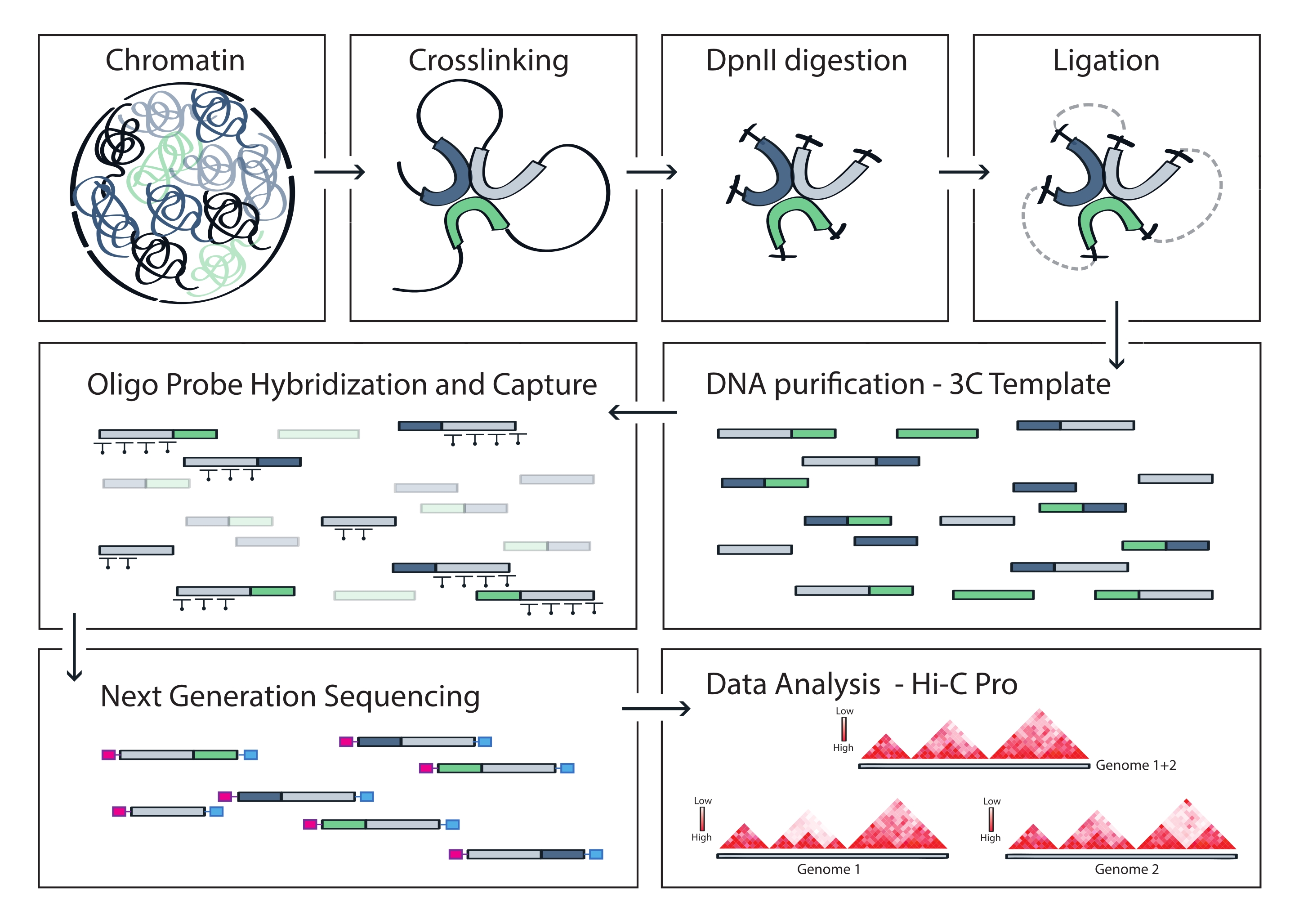
Figura 4: Fluxo de trabalho do protocolo Capture Hi-C. Representação esquemática de diferentes etapas do protocolo. Para gerar o modelo 3C do genoma, a cromatina é primeiro reticulada com formaldeído e, em seguida, digerida com a enzima de restrição DpnII. As extremidades livres do DNA são então religadas, as ligações cruzadas são revertidas e o DNA é purificado. Para enriquecer fragmentos que abrangem a região alvo, uma matriz de sondas de RNA biotinilado é hibridizada ao molde 3C e capturada por pull-down mediado por estreptavidina. Bibliotecas de captura são processadas para sequenciamento multiplexado, e fragmentos de ligadura válidos são quantificados para inferir a frequência de contatos de cromatina através do alvo, que são visualizados como mapas de interação de alta resolução. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Visão geral da região que engloba o Xic no cromossomo X do camundongo. Representação esquemática do cromossomo X do mouse e zoom in da região capturada de 3 Mb (ChrX: 102.475.000-105.475.000). A região alvo inclui ~800 kb de DNA correspondente ao Xic, o locus regulador mestre do XCI. O Xic inclui os longos genes não codificantes, Xist, um jogador-chave do XCI, e seu cenário regulatório. Os reguladores positivos do Xist são mostrados em verde e os reguladores negativos em roxo. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: Capture mapas de interação Hi-C, 5C e Hi-C na região capturada de 3 Mb. (A) Capturar mapa de interação Hi-C do alvo de 3 Mb englobando o mouse Xic com resolução de 10 kb (este estudo). (B) Mapa de interação 5C da mesma região alvo que em A com resolução de 6 kb (dados reprocessados a partir de47). As regiões repetitivas não incluídas nas análises são mascaradas em branco. Os dados 5C requerem o seu próprio processamento de bioinformática (ver47). Após a limpeza e alinhamento, os mapas 5C na resolução do primer são agrupados usando uma mediana de corrida (janela = 30 kb, passo = 5) para atingir uma resolução final de 6 kb. (C) Mapa de interação Hi-C da mesma região genômica de A e B com resolução de 40 kb (dados reprocessados a partir de64). Todos os mapas de interação foram gerados a partir de CTEs de camundongos. O escore de isolamento foi calculado usando cooltools e é representado como histogramas com mínimos de isolamento nos limites do TAD. Os limites do TAD são mostrados como linhas verticais abaixo do mapa. A altura de cada linha indica a força de contorno. Os genes são mostrados como setas apontando na direção da transcrição. Os limites de sub-TAD que são detectados exclusivamente ou mais precisamente em mapas Capture Hi-C são indicados por pontas de seta magenta e azul para sub-TADs nos TADs Tsix e Xist, respectivamente. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 7: Capture mapas de interação Hi-C, 5C e Hi-C em 1 Mb dentro da região capturada. (A) Capturar mapa de interação Hi-C da região genômica de 1 Mb englobando o camundongo Xic com resolução de 5 kb (este estudo). (B) Mapa de interação 5C da mesma região genômica de A. Com resolução de 6 kb (dados reprocessados a partir de47). As regiões repetitivas não incluídas nas análises são mascaradas em branco. É importante notar que os dados 5C requerem o seu próprio processamento de bioinformática (ver47). Após a limpeza e alinhamento, os mapas 5C na resolução do primer são agrupados usando uma mediana de corrida (janela = 30 kb, passo = 5) para atingir uma resolução final de 6 kb. (C) Mapa de interação Hi-C da mesma região genômica de A e B de Hi-C com resolução de 20 kb (dados reprocessados a partir de64). Todos os mapas de interação foram gerados a partir de mESCs. O escore de isolamento foi calculado usando cooltools e é representado como histogramas com mínimos de isolamento nos limites do TAD. Os limites do TAD são mostrados como linhas verticais abaixo do mapa. A altura de cada linha indica a força de contorno. Os genes são mostrados como setas apontando para a direção da transcrição. Os loops de contato que são detectados exclusivamente ou mais precisamente no Capture Hi-C são indicados por asteriscos magenta e azul para loops nos TADs Tsix e Xist, respectivamente. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Tabela Suplementar 1: Estatísticas pós-seqüenciamento para os conjuntos de dados utilizados neste manuscrito: Capture Hi-C (este estudo), Hi-C64 e 5C47. Clique aqui para baixar este arquivo.
Discussão
Aqui descrevemos um protocolo Capture Hi-C relativamente rápido e fácil para caracterizar a organização de ordem superior de regiões genômicas do tamanho de megabases com resolução de 5-10 kb. O Capture Hi-C pertence à família de tecnologias Capture-C que são projetadas para enriquecer interações de cromatina direcionadas a partir de modelos 3C ou Hi-C em todo o genoma. Até o momento, a grande maioria das aplicações de Capture-C tem sido explorada para mapear contatos de cromatina de elementos regulatórios relativamente pequenos espalhados por todo o genoma. No primeiro protocolo Capture-C, múltiplas sondas biotiniladas de RNA sobrepostas foram usadas para capturar >400 promotores pré-selecionados em bibliotecas 3C preparadas a partir de células eritroides31. A mesma estratégia foi posteriormente aprimorada em Next Generation (NG) e Nuclear Titrated (NuTi) Capture-C para alcançar perfis de interação de alta resolução de >8.000 promotores usando iscas únicas de DNA de 120 pb abrangendo sítios de restrição única e duas rodadas sequenciais de captura para maximizar o enriquecimento de fragmentos informativos de ligadura32,40. Essas estratégias levaram à dissecção funcional de elementos de ação cis em diversos contextos, incluindo desenvolvimento embrionário de camundongos, diferenciação celular, inativação do cromossomo X e desregulação gênica em condições patológicas 46,63,65,66,67,68,69,70,71.
No Promoter Capture Hi-C (PCHi-C), >22.000 promotores anotados contendo fragmentos de restrição foram retirados das bibliotecas Hi-C por hibridização de sondas biotiniladas de RNA 120 mers em uma ou ambas as extremidades do fragmento de restrição34,72. Esse método permitiu a dissecção do interatoma de milhares de promotores em um número rapidamente crescente de tipos celulares, incluindo células-tronco embrionárias de camundongo, células hepáticas fetais e adipócitos 34,35,72,73, mas também linhagens linfoblastoides humanas, progenitores hematopoéticos, queratinócitos epidérmicos e células pluripotentes37,74,75,76,77.
Em comparação com essas tecnologias de enriquecimento de alvo, o Capture Hi-C tem como alvo regiões genômicas contíguas até a escala de megabase, abrangendo assim um ou mais TADs e abrangendo cenários regulatórios de genes. Toda a região de interesse deve ser coberta por uma matriz de sondas biotiniladas abrangendo cada sítio de restrição DpnII dentro do alvo. A hibridização da matriz biotinilada para o molde 3C, sua subsequente captura baseada em estreptavidina e processamento para sequenciamento multiplexado é realizada usando um sistema de enriquecimento de alvo para sequenciamento multiplexado Illumina Paired-End. Todo o protocolo é rápido, pois pode ser realizado em 1 semana, desde a preparação da biblioteca 3C até o sequenciamento NGS, e requer apenas pequenas adaptações e/ou solução de problemas personalizada.
O protocolo também oferece vantagens em comparação com outros métodos baseados em 3C. Para obter mapas de interação com resolução de 5-10 kb, sequenciamos leituras pareadas de 100-120 M. Como comparação, usamos aqui um conjunto de dados Hi-C de 571 M leituras para atingir uma resolução de 20 kb64 (GSM2053973), e pelo menos 1 bilhão de leituras seriam necessárias para atingir uma resolução de 5 kb com Hi-C22 em todo o cromossomo.
A captura Hi-C utilizada no presente estudo atinge uma resolução muito maior do que a 5C publicada anteriormente, baseada em uma enzima de restrição de corte de 6 pb47 (Tabela Suplementar 1). É importante ressaltar que a estratégia projetada para enriquecer e amplificar interações direcionadas em 5C não permite a análise alelo-específica das interações da cromatina. Pelo contrário, os dados do Capture Hi-C podem ser mapeados alelo-especificamente, permitindo a dissecção das paisagens estruturais 3D de pares de cromossomos homólogos, por exemplo, em células humanas ou em linhagens celulares híbridas F1 derivadas do cruzamento de linhagens de camundongos geneticamente diferentes78. Para gerar mapas de interação Capture Hi-C específicos para alelos com resolução de 5 kb, sequenciamos leituras de extremidade emparelhada de 150 pb para aumentar a cobertura SNP. Abordagens alelo-específicas semelhantes podem ser aplicadas a linhagens celulares humanas, para as quais a anotação de SNPs está disponível22.
É importante ressaltar que, embora o Capture Hi-C geralmente garanta alta resolução enquanto melhora a acessibilidade dos custos de sequenciamento, a produção de oligonucleotídeos biotinilados personalizados tem um impacto no custo geral desse método. Portanto, a escolha do método 3C mais adequado será diferente para diferentes aplicações, e dependerá da questão biológica que está sendo abordada e da resolução necessária, bem como do tamanho da região de interesse. Outros protocolos Capture Hi-C desenvolvidos compartilham os principais recursos com o protocolo descrito aqui. Por exemplo, uma estratégia de Captura Hi-C foi aplicada para caracterizar regiões genômicas de ~50 kb a 1 Mb abrangendo variantes não codificantes associadas ao risco de câncer de mama e colorretal; neste protocolo, as regiões-alvo foram retiradas das bibliotecas Hi-C hibridizando iscas de RNA de 120 meros que ladeiam as regiões-alvo a uma coberturade 3x 33,38,79. Da mesma forma, o HYbrid Capture Hi-C (Hi-C 2) foi usado para direcionar interações dentro de regiões de interesse de até2 Mb80. Em ambos os protocolos, o uso de um molde Hi-C enriquecido para fragmentos de ligadura puxados para baixo de biotina aumentou a porcentagem de leituras informativas totais em comparação com nosso protocolo. Por exemplo, no conjunto de dados Hi-C que usamos aqui para comparação64 (GSM2053973), a porcentagem de pares válidos após a remoção de duplicatas é 4,8 vezes maior do que os pares válidos obtidos no Capture Hi-C, conforme descrito na Figura 3 e na Tabela Suplementar 1. No entanto, o pull-down consecutivo de fragmentos ligados biotinilados e sondas hibridizadas torna o protocolo significativamente mais complexo e demorado, ao mesmo tempo em que possivelmente diminui a complexidade da região capturada.
Outro método disponível para enriquecer modelos 3C com sondas de mosaico é o Tiled-C, que foi aplicado para estudar a arquitetura da cromatina em alta resolução espacial e temporal durante a diferenciação eritroide de camundongos43. Em Tiled-C, um painel de sondas biotiniladas de 70 pb é usado para enriquecer contatos dentro de regiões de grande escala em duas rodadas consecutivas de captura para gerar mapas de altíssima resolução de interações direcionadas43,81. O enriquecimento de captura dupla também torna o protocolo mais longo e complexo quando comparado ao Capture Hi-C. No entanto, diferentemente das estratégias de Captura-C direcionadas a locais de restrição única, no Tiled-C a segunda rodada de captura não parece aumentar significativamente a eficiência de captura e, portanto, provavelmente pode ser omitida43. Finalmente, uma abordagem similar baseada na mesma estratégia de enriquecimento de alvos usada neste estudo foi aplicada à dissecção de paisagens regulatórias englobando variantes estruturais descritas em pacientes com malformações congênitas e redesenhadas em camundongos transgênicos41,42. Nesse caso, a matriz de sondas lado a lado foi projetada em todo o destino, em vez de na proximidade dos locais de restrição DpnII41. No entanto, este trabalho foi seminal ao destacar a sensibilidade e o poder dessa estratégia para alcançar a caracterização de alta resolução de grandes regiões genômicas em diferentes contextos41,42,48.
Em conclusão, o protocolo aqui descrito representa uma estratégia fácil, robusta e poderosa para a caracterização 3D de alta resolução de quaisquer regiões genômicas de interesse. A aplicação dessa abordagem a diferentes sistemas modelo, tipos celulares, paisagens de cromatina reguladas pelo desenvolvimento e regulação gênica em condições saudáveis e patológicas provavelmente facilitará nossa compreensão da interação e causalidade entre topologia genômica e regulação gênica, uma das questões fundamentais em aberto no campo da epigenética. Além disso, a aplicação do Capture Hi-C para mapear interações de longo alcance e dobramento de cromatina de ordem superior de variantes de risco identificadas por estudos GWAS tem o potencial de revelar a relevância funcional de loci genômicos não codificantes associados a doenças humanas em diferentes contextos, fornecendo assim novos insights sobre os processos potencialmente subjacentes à patogênese.
Divulgações
Kai Hauschulz é Cientista de Aplicação de Campo na Agilent Technologies - Grupo de Diagnóstico e Genômica. Todos os outros autores declaram não haver interesses concorrentes.
Agradecimentos
O trabalho no laboratório Heard foi apoiado por um prémio de Investigador Avançado do Conselho Europeu de Investigação (XPRESS - AdG671027). A.L. é apoiada por uma bolsa individual Marie Skłodowska-Curie Actions da União Europeia (IF-838408). A A.H. é apoiada pela ITN Innovative and Interdisciplinary Network ChromDesign, ao abrigo do acordo de subvenção Marie Skłodowska-Curie 813327. Os autores agradecem a Daniel Ibrahim (MPI for Molecular Genetics, Berlim) pelo aconselhamento técnico útil, à plataforma NGS no Institut Curie (Paris), e a Vladimir Benes e ao Genomics Core Facility na EMBL (Heidelberg), pelo apoio e assistência.
Materiais
| Name | Company | Catalog Number | Comments |
| 10x PBS pH 7.4 | Gibco | 10010-023 | |
| 37% (vol/vol) paraformaldehyde solution | Electron Microscopy Sciences | 15686 | single use glass-vials; do not reuse |
| 50 mL PP conical tube | Falcon | 352070 | |
| Agarose | Sigma | A9539-500g | |
| Bioanalyzer | Agilent | G2939BA | |
| Cell Scrapers - 25 cm Handle and 3.0 cm Blade | Falcon | 353089 | |
| CHIR99021 | Axon Medchem BV | Axon 1386 | |
| cOmplete Mini, Protease inhibitor cocktail (EDTA-free) | Merck | 11836170001 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | |
| Countess II FL | Invitrogen | ZGEXSCCOUNTESS2FL | Automated cell counter |
| Covaris S2 | Covaris | 500217 | Sonicator |
| DNA LoBind tube, 1.5 mL | Eppendorf | 30108051 | |
| DpnII (50000 units/mL) | New England Biolabs | R0543M | |
| Dulbecco's Modified Eagle Medium (DMEM) | Merck | D6429 | |
| Ethanol (100%) | Merck | 1.00983.2500 | |
| Fetal Bovine Serum (FBS) | Thermo Scientific | 10270106 | |
| gelatine from porcine skin | Sigma | G1890 | |
| GeneRuler 1 kb Plus DNA Ladder | Thermo Scientific | SM0313 | |
| GlycoBlue | Thermo Scientific | AM9516 | Coprecipitant |
| High-Sensitivity Bioanlayzer chips | Agilent | 5067-4626 | |
| Large Cooling Centrifuge 5920 R | Eppendorf | 5948000018 | |
| leukaemia inhibitory factor (LIF) | Merck | ESG1107 | |
| Liquiport | KNF | NF300 | Benchtop aspiration system |
| Low-binding filter tips | Biozym | VT0260U, VT0240, VT0220, VT0200U | |
| Molecular biology grade water | Merck | W3500-6x500ML | |
| Next Seq 500 | Illumina | SY-415-1001 | |
| Next Seq 500 High Output v2 Kit (300 cycles) | Illumina | FC-404-2004 | |
| Nonidet P40 Substitute (NP40) | Merck | 11332473001 | |
| PD0325901 | Axon Medchem BV | Axon 1408 | |
| Protease inhibitor cocktail (EDTA-free) | Merck | 11873580001 | |
| Proteinase K - recombinant, PCR-grade (20 mg/mL) | Thermo Scientific | EO0491 | |
| Qubit 2.0 | Thermo Scientific | Q32871 | |
| Qubit assay tubes | Thermo Scientific | Q32856 | |
| Qubit dsDNA High Sensitivity kit | Thermo Scientific | Q32851 | |
| RNase A (10 mg/mL) | Thermo Scientific | EN0531 | |
| Sodium acetate pH 5.2 (3M) | Merck | S7899 | |
| speed vacuum concentrator | Eppendorf | EP5305000100-1EA | |
| Agencourt AMPureXP | Beckman Coulter | A63881 | SPRI beads |
| SureSelect Target Enrichment Box 1 | Agilent | 5190-8645 | |
| SureSelect Target Enrichment Kit ILM Indexing Hyb Module Box 2 | Agilent | 5190-4455 | |
| SureSelect XT Library Prep Kit ILM | Agilent | 5500-0132 | |
| T4 ligase (30 units/µL) | Thermo Scientific | EL0013 | |
| table-top Centrifuge 5427 R | Eppendorf | 5409000012 | |
| Triton-X-100 (500 mL) | Merck | X100-500ML | |
| Trypan Blue | Invitrogen | T10282 | |
| Trypsine | Thermo Scientific | 25300054 | |
| UltraPure Glycine | Thermo Scientific | 15527013 | |
| β-mercaptoethanol | Thermo Scientific | 31350010 |
Referências
- Ibrahim, D. M., Mundlos, S. The role of 3D chromatin domains in gene regulation: a multi-facetted view on genome organization. Current Opinion in Genetics & Development. 61, 1-8 (2020).
- Bolt, C. C., Duboule, D. The regulatory landscapes of developmental genes. Development. 147 (3), (2020).
- Glaser, J., Mundlos, S. 3D or not 3D: Shaping the genome during development. Cold Spring Harbor Perspectives in Biology. 14 (5), 040188(2021).
- Denker, A., De Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Kempfer, R., Pombo, A. Methods for mapping 3D chromosome architecture. Nature Reviews Genetics. 21 (4), 207-226 (2020).
- McCord, R. P., Kaplan, N., Giorgetti, L. Chromosome conformation capture and beyond: Toward an integrative view of chromosome structure and function. Molecular Cell. 77 (4), 688-708 (2020).
- Jerkovic, I., Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nature ReviewsMolecular Cell Biology. 22 (8), 511-528 (2021).
- Hsieh, T. -H. S., et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 162 (1), 108-119 (2015).
- Krietenstein, N., et al. Ultrastructural details of mammalian chromosome architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Naumova, N., Smith, E. M., Zhan, Y., Dekker, J. Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods. 58 (3), 192-203 (2012).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Zhao, Z., et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra-and interchromosomal interactions. Nature Genetics. 38 (11), 1341-1347 (2006).
- Würtele, H., Chartrand, P. Genome-wide scanning of HoxB1-associated loci in mouse ES cells using an open-ended Chromosome Conformation Capture methodology. Chromosome Research. 14 (5), 477-495 (2006).
- De Wit, E., De Laat, W. A decade of 3C technologies: insights into nuclear organization. Genes & Development. 26 (1), 11-24 (2012).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Splinter, E., et al. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes & Development. 25 (13), 1371-1383 (2011).
- Ferraiuolo, M. A., Sanyal, A., Naumova, N., Dekker, J., Dostie, J. From cells to chromatin: capturing snapshots of genome organization with 5C technology. Methods. 58 (3), 255-267 (2012).
- Kim, J. H., et al. 5C-ID: Increased resolution Chromosome-Conformation-Capture-Carbon-Copy with in situ 3C and double alternating primer design. Methods. 142, 39-46 (2018).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Zhang, Y., et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 148 (5), 908-921 (2012).
- Rao, S. S. P., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Dixon, J. R., et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 485 (7398), 376-380 (2012).
- Nora, E. P., et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 485 (7398), 381-385 (2012).
- Krefting, J., Andrade-Navarro, M. A., Ibn-Salem, J. Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biology. 16 (1), 87(2018).
- Galupa, R., Heard, E. Topologically associating domains in chromosome architecture and gene regulatory landscapes during development, disease, and evolution. Cold Spring Harbor Symposia on Quantitative Biology. 82, 267-278 (2017).
- Tena, J. J., Santos-Pereira, J. M. Topologically associating domains and regulatory landscapes in development, evolution and disease. Frontiers in Cell and Developmental Biology. 9, 702787(2021).
- Lupiáñez, D. G., Spielmann, M., Mundlos, S. Breaking TADs: How alterations of chromatin domains result in disease. Trends in Genetics. 32 (4), 225-237 (2016).
- Davidson, I. F., Peters, J. -M. Genome folding through loop extrusion by SMC complexes. Nature Reviews Molecular Cell Biology. 22 (7), 445-464 (2021).
- Schmitt, A. D., Hu, M., Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nature Reviews Molecular Cell Biology. 17 (12), 743-755 (2016).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Davies, J. O. J., et al. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nature Methods. 13 (1), 74-80 (2016).
- Jäger, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178(2015).
- Schoenfelder, S., et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Research. 25 (4), 582-597 (2015).
- Sahlén, P., et al. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology. 16, 156(2015).
- Joshi, O., et al. Dynamic reorganization of extremely long-range promoter-promoter interactions between two states of pluripotency. Cell Stem Cell. 17 (6), 748-757 (2015).
- Mifsud, B., et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics. 47 (6), 598-606 (2015).
- Dryden, N. H., et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research. 24 (11), 1854-1868 (2014).
- Oudelaar, A. M., Davies, J. O. J., Downes, D. J., Higgs, D. R., Hughes, J. R. Robust detection of chromosomal interactions from small numbers of cells using low-input Capture-C. Nucleic Acids Research. 45 (22), 184(2017).
- Oudelaar, A. M., et al. Single-allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nature Genetics. 50 (12), 1744-1751 (2018).
- Franke, M., et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 538 (7624), 265-269 (2016).
- Despang, A., et al. Functional dissection of the Sox9-Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nature Genetics. 51 (8), 1263-1271 (2019).
- Oudelaar, A. M., et al. Dynamics of the 4D genome during in vivo lineage specification and differentiation. Nature Communications. 11 (1), 1-12 (2020).
- Galupa, R., Heard, E. X-chromosome inactivation: A crossroads between chromosome architecture and gene regulation. Annual Review of Genetics. 52, 535-566 (2018).
- Loda, A., Collombet, S., Heard, E. Gene regulation in time and space during X-chromosome inactivation. Nature Reviews. Molecular Cell Biology. 23 (4), 231-249 (2022).
- van Bemmel, J. G., et al. The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nature Genetics. 51 (6), 1024-1034 (2019).
- Galupa, R., et al. A conserved noncoding locus regulates random monoallelic Xist expression across a topological boundary. Molecular Cell. 77 (2), 352-367 (2020).
- Gjaltema, R. A. F., et al. Distal and proximal cis-regulatory elements sense X chromosome dosage and developmental state at the Xist locus. Molecular Cell. 82 (1), 190-208 (2022).
- Galupa, R., et al. Inversion of a topological domain leads to restricted changes in its gene expression and affects inter-domain communication. Development. 149 (9), (2022).
- Savarese, F., Flahndorfer, K., Jaenisch, R., Busslinger, M., Wutz, A. Hematopoietic precursor cells transiently reestablish permissiveness for X inactivation. Molecular and Cellular Biology. 26 (19), 7167-7177 (2006).
- Schulz, E. G., et al. The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell. 14 (2), 203-216 (2014).
- Gnirke, A., et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology. 27 (2), 182-189 (2009).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Piccinini, F., Tesei, A., Arienti, C., Bevilacqua, A. Cell counting and viability assessment of 2D and 3D Cell cultures: Expected reliability of the trypan blue assay. Biological Procedures Online. 19 (1), 8(2017).
- Servant, N., et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology. 16, 259(2015).
- Abdennur, N., Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 36 (1), 311-316 (2020).
- Imakaev, M., et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nature Methods. 9 (10), 999-1003 (2012).
- Forcato, M., et al. Comparison of computational methods for Hi-C data analysis. Nature Methods. 14 (7), 679-685 (2017).
- Venev, S., et al. open2c/cooltools: v0.4.1. , (2021).
- Wages, J. M. NUCLEIC ACIDS | Immunoassays. Encyclopedia of Analytical Science. , Elsevier. 408-417 (2005).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Servant, N., Peltzer, A. nf-core/hic: Initial release of nf-core/hic. Zenodo. , (2019).
- Furlan, G., et al. The Ftx noncoding locus controls X chromosome inactivation independently of its RNA products. Molecular Cell. 70 (3), 462-472 (2018).
- Giorgetti, L., et al. Structural organization of the inactive X chromosome in the mouse. Nature. 535 (7613), 575-579 (2016).
- Simon, C. S., et al. Functional characterisation of cis-regulatory elements governing dynamic Eomes expression in the early mouse embryo. Development. 144 (7), 1249-1260 (2017).
- Williams, R. M., et al. Reconstruction of the global neural crest gene regulatory network in vivo. Developmental Cell. 51 (2), 255-276 (2019).
- Godfrey, L., et al. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nature Communications. 10 (1), 2803(2019).
- Hanssen, L. L. P., et al. Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nature Cell Biology. 19 (8), 952-961 (2017).
- Larke, M. S. C., et al. Enhancers predominantly regulate gene expression during differentiation via transcription initiation. Molecular Cell. 81 (5), 983-997 (2021).
- Oudelaar, A. M., et al. A revised model for promoter competition based on multi-way chromatin interactions at the α-globin locus. Nature Communications. 10 (1), 5412(2019).
- Long, H. K., et al. Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell. 27 (5), 765-783 (2020).
- Schoenfelder, S., Javierre, B. -M., Furlan-Magaril, M., Wingett, S. W., Fraser, P. Promoter Capture Hi-C: High-resolution, genome-wide profiling of promoter interactions. Journal of Visualized Experiments. (136), e57320(2018).
- Siersbæk, R., et al. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Molecular Cell. 66 (3), 420-435 (2017).
- Rubin, A. J., et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nature Genetics. 49 (10), 1522-1528 (2017).
- Freire-Pritchett, P., et al. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. eLife. 6, 21926(2017).
- Javierre, B. M., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 167 (5), 1369-1384 (2016).
- Miguel-Escalada, I., et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nature Genetics. 51 (7), 1137-1148 (2019).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Baxter, J. S., et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nature Communications. 9 (1), 1028(2018).
- Sanborn, A. L., et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proceedings of the National Academy of Sciences. 112 (47), 6456-6465 (2015).
- Owens, D. D. G., et al. Dynamic Runx1 chromatin boundaries affect gene expression in hematopoietic development. Nature Communications. 13 (1), 773(2022).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados