
Method Article
Decifrare l'organizzazione della cromatina 3D ad alta risoluzione tramite Capture Hi-C
In questo articolo
Riepilogo
Questo protocollo descrive il metodo Capture Hi-C utilizzato per caratterizzare l'organizzazione 3D di regioni genomiche mirate di dimensioni megabased ad alta risoluzione, compresi i confini dei domini topologicamente associati (TAD) e le interazioni cromatiniche a lungo raggio tra elementi regolatori e altri elementi di sequenza del DNA.
Abstract
L'organizzazione spaziale del genoma contribuisce alla sua funzione e regolazione in molti contesti, tra cui la trascrizione, la replicazione, la ricombinazione e la riparazione. Comprendere l'esatta causalità tra topologia e funzione del genoma è quindi cruciale e sempre più oggetto di un'intensa ricerca. Le tecnologie di cattura della conformazione cromosomica (3C) consentono di dedurre la struttura 3D della cromatina misurando la frequenza delle interazioni tra qualsiasi regione del genoma. Qui descriviamo un protocollo semplice e veloce per eseguire Capture Hi-C, un metodo di arricchimento del bersaglio basato su 3C che caratterizza l'organizzazione 3D specifica dell'allele di bersagli genomici di dimensioni megabased ad alta risoluzione. In Capture Hi-C, le regioni target vengono catturate da una serie di sonde biotinilate prima del sequenziamento ad alta produttività a valle. Pertanto, si ottengono una risoluzione più elevata e una specificità allele, migliorando al contempo l'efficacia temporale e l'accessibilità della tecnologia. Per dimostrare i suoi punti di forza, il protocollo Capture Hi-C è stato applicato al centro di inattivazione X del topo ( Xic), il principale luogo regolatore dell'inattivazione del cromosoma X (XCI).
Introduzione
Il genoma lineare contiene tutte le informazioni necessarie affinché un organismo subisca uno sviluppo embrionale e sopravviva per tutta l'età adulta. Tuttavia, istruire cellule geneticamente identiche a svolgere funzioni diverse è fondamentale per controllare con precisione quali informazioni vengono utilizzate in contesti specifici, inclusi diversi tessuti e / o fasi di sviluppo. Si ritiene che l'organizzazione tridimensionale del genoma partecipi a questa accurata regolazione spazio-temporale dell'attività genica facilitando o impedendo l'interazione fisica tra elementi regolatori che possono essere separati da diverse centinaia di kilobasi nel genoma lineare (per le recensioni 1,2,3). Negli ultimi 20 anni, la nostra comprensione dell'interazione tra ripiegamento del genoma e attività è rapidamente aumentata, in gran parte a causa dello sviluppo di tecnologie di cattura della conformazione cromosomica (3C) (per la revisione 4,5,6,7). Questi metodi misurano la frequenza delle interazioni tra qualsiasi regione del genoma e si basano sulla legatura delle sequenze di DNA che si trovano in stretta vicinanza 3D all'interno del nucleo. I protocolli 3C più comuni iniziano con la fissazione delle popolazioni cellulari con un agente reticolante come la formaldeide. La cromatina reticolata viene quindi digerita con un enzima di restrizione, sebbene sia stata utilizzata anche la digestione della MNasi 8,9. Dopo la digestione, le estremità del DNA libero in stretta vicinanza spaziale vengono ri-legate e la reticolazione viene invertita. Questo passaggio dà origine alla "libreria" o "modello" 3C, un pool misto di frammenti ibridi in cui le sequenze che erano in prossimità 3D del nucleo hanno maggiori probabilità di essere legate nello stesso frammento di DNA. La quantificazione a valle di questi frammenti ibridi consente di dedurre la conformazione 3D di regioni genomiche che si trovano a migliaia di coppie di basi distanti nel genoma lineare, ma potrebbero interagire nello spazio 3D.
Sono stati sviluppati molti approcci diversi per caratterizzare la libreria 3C, che differiscono sia in termini di quali sottoinsiemi di frammenti di legatura vengono analizzati sia in quale tecnologia viene utilizzata per la loro quantificazione a valle. Il protocollo originale 3C si basava sulla selezione di due regioni di interesse e sulla quantificazione della loro frequenza di interazione "uno contro uno" mediante PCR10,11. L'approccio 4C (cattura della conformazione cromosomica circolare) misura le interazioni tra un singolo locus di interesse (cioè il "view-point") e il resto del genoma ("uno contro tutti")12,13,14. In 4C, la libreria 3C subisce un secondo ciclo di digestione e re-legazione per generare piccole molecole di DNA circolari che vengono amplificate con PCR da primer specifici del punto di vista15. 5C (chromosome conformation capture carbon copy) consente la caratterizzazione delle interazioni 3D in regioni di interesse più ampie, fornendo approfondimenti sul ripiegamento della cromatina di ordine superiore all'interno di quella regione ("molti contro molti")16. In 5C, la libreria 3C è ibridata in un pool di oligonucleotidi sovrapposti a siti di restrizione che possono essere successivamente amplificati mediante multiplex PCR con primer universali15. Sia in 4C che in 5C, i frammenti informativi di DNA sono stati inizialmente quantificati da microarray e successivamente da sequenziamento di nuova generazione (NGS)17,18,19. Queste strategie caratterizzano regioni di interesse mirate, ma non possono essere applicate per mappare le interazioni a livello di genoma. Quest'ultimo obiettivo è raggiunto con Hi-C, una strategia ad alto rendimento basata su 3C in cui il sequenziamento massivamente parallelo del modello 3C consente la caratterizzazione imparziale del ripiegamento della cromatina a livello dell'intero genoma ("tutti contro tutti")20. Il protocollo Hi-C prevede l'incorporazione di un residuo biotinilato alle estremità dei frammenti digeriti, seguito dal pull-down dei frammenti di legatura con perle di streptavidina per aumentare il recupero dei frammenti legati20.
Hi-C ha rivelato che i genomi dei mammiferi sono strutturalmente organizzati su più scale nel nucleo 3D. Alla scala megabase, il genoma è diviso in regioni di cromatina attiva e inattiva, i compartimenti A e B, rispettivamente20,21. L'esistenza di ulteriori sottocompartimenti rappresentati da diversi stati di cromatina e attività è stata successivamente mostrata22. A una risoluzione più elevata, il genoma viene ulteriormente suddiviso in domini auto-interagenti sub-megabase chiamati domini topologicamente associati (TAD), rivelati per la prima volta dall'analisi Hi-C e 5C dei genomi umani e murini23,24. A differenza dei compartimenti che variano in modo tessuto-specifico, i TAD tendono ad essere costanti (anche se ci sono molte eccezioni). È importante sottolineare che i confini TAD sono conservati tra le specie25. Nelle cellule di mammifero, i TAD comprendono frequentemente geni che condividono lo stesso panorama regolatorio e hanno dimostrato di rappresentare un quadro strutturale che facilita la co-regolazione genica limitando le interazioni con i domini regolatori vicini (per la revisione 3,26,27,28). Inoltre, all'interno dei TAD, le interazioni dovute ai siti CTCF alla base dei loop estrusi di coesina possono aumentare la probabilità di interazioni promotore-potenziatore o enhancer-enhancer (per la revisione29).
In Hi-C, compartimenti e TAD possono essere rilevati con una risoluzione da 1 Mb a 40 kb, ma è possibile ottenere una risoluzione più elevata per caratterizzare contatti su scala più piccola come le interazioni di looping tra elementi distali su scala di 5-10 kb. Tuttavia, aumentare la risoluzione per essere in grado di rilevare tali loop in modo efficiente da HiC richiede un aumento significativo della profondità di sequenziamento e, quindi, dei costi di sequenziamento. Ciò è esacerbato se l'analisi deve essere allele-specifica. In effetti, un aumento X volte della risoluzione richiede un aumento di X2 della profondità di sequenziamento, il che significa che gli approcci ad alta risoluzione e allele-specifici dell'intero genoma possono essere proibitivamente costosi30.
Per migliorare l'economicità e l'accessibilità mantenendo al contempo un'alta risoluzione, le regioni target di interesse possono essere fisicamente estratte dalle librerie 3C o Hi-C a livello di genoma dopo la loro ibridazione con sonde oligonucleotidiche complementari marcate con biotina prima del sequenziamento a valle. Queste strategie di arricchimento del bersaglio sono indicate come metodi Capture-C e consentono l'interrogazione delle interazioni di centinaia di loci bersaglio sparsi nel genoma (ad esempio, Promoter Capture (PC) Hi-C; Next Generation (NG) Capture-C; Low Input (LI) Capture-C; Cattura nucleare titolata (NuTi); Tri-C)31,32,33,34,35,36,37,38,39,40, o attraverso regioni che si estendono fino a diverse megabasi (ad esempio, Capture HiC; HYbrid Capture Hi-C (Hi-C2); Piastrellato-C)41,42,43. Due aspetti possono variare nei metodi basati sulla cattura: (1) la natura e la progettazione degli oligonucleotidi biotinilati (cioè RNA o DNA, singoli oligo che catturano bersagli genomici dispersi o oligo multipli che piastrellano una regione di interesse); e (2) il modello utilizzato per abbattere i bersagli che possono essere la libreria 3C o Hi-C, quest'ultima costituita da frammenti di restrizione biotinilati estratti dalla libreria 3C.
Qui viene descritto un protocollo Capture Hi-C basato sull'arricchimento dei contatti target dalla libreria 3C. Il protocollo si basa sulla progettazione di una serie personalizzata di sonde di RNA biotinilato e può essere eseguito in 1 settimana dalla preparazione della libreria 3C al sequenziamento NGS. Il protocollo è veloce, semplice e consente di caratterizzare l'organizzazione 3D di ordine superiore di regioni di interesse di dimensioni megabase con una risoluzione di 5 kb, migliorando al contempo l'efficacia temporale e l'accessibilità rispetto ad altri metodi 3C. Il protocollo Capture Hi-C è stato applicato al locus regolatore principale dell'inattivazione del cromosoma X (XCI), il centro di inattivazione dell'X (Xic), che ospita l'RNA non codificante Xist. L'Xic è stato precedentemente oggetto di ampie analisi strutturali e funzionali (per la revisione44,45). Nei mammiferi, XCI compensa il dosaggio dei geni legati all'X tra femmine (XX) e maschi (XY) e comporta il silenziamento trascrizionale di quasi la totalità di uno dei due cromosomi X nelle cellule femminili. L'Xic ha rappresentato un potente locus gold standard per gli studi sulla topologia del genoma 3D e l'interazione con la regolazione genica44. L'analisi 5C dello Xic nelle cellule staminali embrionali di topo (mESC) ha portato alla scoperta e alla denominazione dei TAD, fornendo le prime intuizioni sulla rilevanza funzionale del partizionamento topologico e della co-regolazione genica24. L'organizzazione topologica dello Xic ha successivamente dimostrato di essere criticamente coinvolta nella tempistica di sviluppo appropriata della upregolazione di Xist e XCI 46, e sono stati recentemente scoperti anche elementi cis-regolatori insospettati che possono influenzare l'attività genica all'interno e tra i TAD all'interno dello Xic47,48,49. L'applicazione di Capture Hi-C a 3 Mb del cromosoma X del topo che attraversa l'Xic dimostra la potenza di questo approccio nel sezionare il ripiegamento della cromatina su larga scala ad alta risoluzione. Viene fornito un protocollo dettagliato e facile da seguire, a partire dalla progettazione della serie di sonde biotinilate in ogni sito di restrizione DpnII all'interno della regione di interesse fino alla generazione della libreria 3C genome-wide, all'ibridazione e cattura dei contatti target e all'analisi dei dati a valle. È inclusa anche una panoramica dei controlli di qualità appropriati e dei risultati attesi, e sia i punti di forza che i limiti dell'approccio sono discussi alla luce di metodi simili esistenti.
Protocollo
Le cellule staminali embrionali di topo (mESC) utilizzate in questo studio sono state derivate da un incrocio di una femmina TX/TX R26rtTA/rtTA 50 con un maschio di Mus musculus castaneus secondo le linee guida per la cura degli animali dell'Institut Curie (Parigi)51.
1. Design della sonda
- Progettare una serie di sonde biotinilate (oligonucleotidi RNA 120-mer) che coprano la regione target di interesse.
- Affiancare la regione di interesse con oligonucleotidi sovrapposti in modo che in media ogni sequenza all'interno del bersaglio sia coperta da due sonde uniche (copertura 2x) (Figura 1).
- Escludere le sequenze ripetitive dalla copertura della sonda per evitare l'arricchimento di interazioni non specifiche.
NOTA: Per massimizzare l'arricchimento dei frammenti di legatura informativa, sono state definite regioni che coprono 300 bp a monte e a valle di ciascun sito di restrizione DpnII attraverso il target (ChrX: 102.475.000-105.475.000) e 28.913 sonde biotinilate sono state progettate secondo la tecnologia di arricchimento del target SureSelect DNA attraverso la piattaforma Sure Design52. Secondo questa strategia, sono consentite fino a un massimo di 40 basi di sequenze ripetitive in ciascun oligonucleotide per ridurre al minimo l'arricchimento di interazioni non specifiche. L'array di sonde è stato sintetizzato da Agilent. Qui, DpnII è usato come enzima di restrizione per due motivi: (1) è un quattro taglieri usato abitualmente in diversi metodi basati su 3C53; e (2) massimizza le possibilità di catturare polimorfismi informativi a singolo nucleotide (SNP) in prossimità dei siti di taglio rispetto ad altri enzimi di restrizione che sono stati testati in silico in linee ibride F1 utilizzate in questo studio (C57BL / 6J x CASTEi / J).
2. Procedura sperimentale
- Preparazione cellulare
- Seminare il numero appropriato di cellule su una o più piastre di coltura cellulare per ottenere un numero totale di cellule di ≥ 5 x 107 cellule il giorno della fissazione.
NOTA: In questo studio sono state utilizzate cellule staminali embrionali di topo (mESC). Le mESC sono placcate su piastre di coltura cellulare gelatinizzate (0,1% gelatina in 1x PBS - o/n a 37 °C, incubatore a CO2 al 5%) in terreno di coltura mESCs contenente 2i + LIF e siero fetale di vitello testato in batch (DMEM, 15% FBS, 0,1 mM β-mercaptoetanolo, fattore inibitorio della leucemia 1.000 U/mL−1 (LIF), CHIR99021 (3 μM) e PD0325901 (1 μM)). Per questo tipo di cellula, una piastra confluente all'80% da 10 cm contiene circa 2 x 107 celle. - Preparare una piastra di coltura cellulare aggiuntiva per il conteggio delle cellule.
NOTA: è possibile utilizzare una piastra di coltura cellulare più piccola per ridurre l'utilizzo dei supporti. In questo caso, il numero di cellule da seminare sulla piastra più piccola deve essere regolato di conseguenza (ad esempio, 3 volte meno celle su una piastra di 10 cm rispetto a una piastra di 15 cm).
- Seminare il numero appropriato di cellule su una o più piastre di coltura cellulare per ottenere un numero totale di cellule di ≥ 5 x 107 cellule il giorno della fissazione.
- Fissazione della formaldeide
- Stimare il numero totale di celle da reticolare.
- Prima di iniziare la reazione di reticolazione, tripsinizzare e contare le cellule dalla piastra di controllo preparata specificamente per il conteggio delle cellule utilizzando un contatore automatico di celle secondo le istruzioni del produttore.
- Includere una colorazione di vitalità (ad esempio, Trypan Blue) per determinare la percentuale di cellule vitali54. Da questo conteggio delle cellule, stimare il numero totale di cellule nelle piastre preparate per la reticolazione.
- Rimuovere il terreno di coltura dalle piastre preparate per la reticolazione e sostituirlo con la quantità appropriata di soluzione di fissazione (2% di formaldeide nel terreno di coltura cellulare). Utilizzare 10 ml su una piastra da 10 cm (ad esempio, ~ 20 ml per una piastra da 15 cm).
NOTA: aggiungere un volume esatto della soluzione di fissazione. Se il fissaggio delle cellule aderenti non è possibile, questa fase può essere adattata alle cellule tripsinizzate ed eseguita in 30 ml di soluzione di fissazione in provette coniche da centrifuga da 50 ml. La formaldeide non deve avere più di 1 anno. Si preferisce utilizzare fiale monouso. La soluzione di fissaggio deve essere portata a temperatura ambiente (RT) prima dell'uso.
ATTENZIONE: La formaldeide è pericolosa e deve essere maneggiata secondo le norme di salute e sicurezza appropriate. - Fissare per 10 minuti a RT mescolando delicatamente su uno shaker.
- Estinguere la reazione di fissazione aggiungendo 2,5 M glicina-1x PBS ad una concentrazione finale di 0,125 M. Aggiungere 530 μL di 2,5 M glicina-1x PBS a 10 ml su una piastra da 10 cm (ad esempio, da 1060 μL a 20 ml su una piastra da 15 cm).
NOTA: Se le cellule sono state fissate in soluzione, spegnere la reazione di fissazione con 1590 μL di 2,5 M glicina-1x PBS. - Incubare per 5 minuti a RT, mescolando delicatamente su uno shaker.
- Trasferire le piastre sul ghiaccio e incubare per altri 15 minuti sul ghiaccio mescolando delicatamente su uno shaker.
NOTA: D'ora in poi, le celle devono essere mantenute sul ghiaccio e i tamponi devono essere pre-raffreddati per evitare ulteriori reticoli. Passare a una cella frigorifera se è necessario elaborare molte piastre. - Rimuovere la soluzione di fissaggio dalle celle versandola in un becher per garantire una manipolazione rapida.
NOTA: Assicurarsi di smaltire i rifiuti liquidi contenenti formaldeide secondo le norme di salute e sicurezza appropriate. - Risciacquare rapidamente la piastra da 10 cm due volte con 5 ml di glicina-1x PBS fredda 0,125 M (8 ml per una piastra da 15 cm) per lavare via i detriti e le cellule morte. Rimuovere il liquido dalla piastra versandolo in un becher per garantire una rapida manipolazione.
- Aggiungere 5 ml di 0,125 M glicina-1x PBS freddo alla piastra da 10 cm (10 ml per una piastra da 15 cm) e raschiare rapidamente le cellule dalla piastra usando un raschietto per celle di plastica.
- Trasferire la sospensione cellulare in una provetta conica da centrifuga da 50 mL preraffreddata utilizzando una pipetta sierologica.
- Risciacquare la piastra due volte con 5 ml di glicina-1x PBS freddo 0,125 M e aggiungere la sospensione cellulare al tubo conico della centrifuga.
- Centrifugare a 480 x g per 10 minuti a 4 °C.
NOTA: Se le celle sono state fissate in soluzione, trasferire la cella in una provetta conica da centrifuga pre-raffreddata e centrifugare a 480 x g per 10 minuti a 4 °C. Rimuovere la soluzione di fissazione versandola in un becher e lavare tre volte in 10 ml di glicina-1x PBS freddo 0,125 M. Assicurati di risospendere le celle ad ogni fase di lavaggio. - Rimuovere il surnatante aspirando con un sistema di aspirazione da banco. Risospendere le celle in 500 μL di 1x PBS per 1 x 107 celle mediante pipettaggio accurato su e giù con una pipetta P1000. Per risospendere le celle nel volume esatto, fare riferimento alla stima del numero totale di celle ottenuta in 2.2.1.
- Aliquot 500 μL della sospensione cellulare nel numero calcolato di provette da microcentrifuga da 1,5 mL (1 x 107 celle/provetta).
- Centrifugare a 480 x g per 10 minuti a 4 °C.
- Rimuovere il surnatante con un sistema di aspirazione da banco e congelare a scatto i pellet cellulari in azoto liquido. Conservare il pellet a secco a -80 °C.
NOTA: I campioni possono essere conservati per almeno 1 anno.
- Stimare il numero totale di celle da reticolare.
- Lisi cellulare
- Scongelare il pellet congelato sul ghiaccio.
- Preparare 1,5 mL di tampone di lisi in H 2 0 per campione: aggiungere 10 mM Tris-HCl, pH 8,0, 10 mM NaCl e0,2% NP40.
- Aggiungere 600 μL del tampone di lisi fredda e risospendere bene sul ghiaccio.
- Incubare sul ghiaccio per 15 minuti per far gonfiare le cellule.
- Girare a 2655 x g per 5 minuti a 4 °C e rimuovere il surnatante utilizzando un sistema di aspirazione da banco.
- Per rimuovere i detriti, risospendere il pellet in 1 mL di tampone di lisi a freddo, centrifugare a 2655 x g per 5 minuti a 4 °C e rimuovere il surnatante.
- Ruotare nuovamente brevemente a 2655 x g e 4 °C e rimuovere il più possibile il surnatante rimanente utilizzando un sistema di aspirazione da banco dotato di punta P200.
- Risospendere in 100 μL di SDS allo 0,5% (vol/vol).
- Incubare in un termomiscelatore a 62 °C, roteare a 1400 giri/min per 10 min.
- Aggiungere 290 μL di H2O + 50 μL di TritonX-100 al 10% e mescolare bene, evitando bolle d'aria.
- Incubare in un termomiscelatore a 37 °C, roteare a 1400 giri/min per 15 min.
- Aggiungere 50 μL di tampone 10x Dpnll e capovolgere il tubo per miscelare.
- Prelevare 50 μL di DNA non digerito per il controllo qualità in una provetta separata. Non dimenticare di prendere il campione di controllo non digerito.
- Digestione DpnII
- Aggiungere 10 μL di Dpnll ad alta concentrazione (500 U totali) e mescolare invertendo.
- Incubare i campioni e il controllo non digerito in un termomiscelatore a 37 °C, roteando a 1400 rpm per >4 h.
- Aggiungere 10 μL di Dpnll ad alta concentrazione (500 U totali) alla fine della giornata.
- Incubare i campioni e il controllo non digerito a 37 °C, roteando a 1400 giri/min durante la notte.
- Aggiungere 10 μL di Dpnll ad alta concentrazione (500 U totali) all'inizio del giorno successivo ai campioni.
- Incubare i campioni e il controllo non digerito in un termomiscelatore a 37 °C, roteando a 1400 giri/min per 4 ore.
- Legatura e inversione del reticolazione
- Incubare i tubi a 65 °C per 20 minuti a 1400 giri/min.
NOTA: non aggiungere SDS a questo punto. L'idea è quella di preservare l'integrità nucleare, quindi la legatura viene effettuata all'interno dei nuclei, aggirando la necessità di una diluizione estrema. - Raffreddare i campioni sul ghiaccio per un massimo di 5-10 minuti. Per evitare precipitazioni SDS, non lasciare i campioni sul ghiaccio più a lungo di questo.
- Prendere 50 μL di DNA digerito non legato per il controllo di qualità in una provetta separata. Conservare i controlli non digeriti e non ligati a -20 °C.
NOTA: non dimenticare di prelevare il campione di controllo non legato. - Aggiungere 800 μL di cocktail di legatura: 122 μL di tampone 10x ligasi, 8 μL di T4 ligasi (30 U/μL) e 670 μL di H20.
- Incubare a 16 °C, turbinare a 1000 giri/min durante la notte.
- Aggiungere 7,5 μL di proteinasi K (20 mg/ml) ai campioni e 2 μL ai controlli.
- Incubare a 65 °C per 4 h a 1000 giri/min.
- Incubare i tubi a 65 °C per 20 minuti a 1400 giri/min.
- Purificazione del DNA
- Trasferire i campioni su ghiaccio in provette coniche da centrifuga da 15 ml preraffreddate e aggiungere 2 ml di acqua, 10,5 ml di EtOH ghiacciato e 583 μL di 3 M NaAC.
NOTA: l'acqua supplementare ha lo scopo di prevenire il trascinamento di DTT nel pellet. - Aggiungere 200 μL di EtOH ghiacciato, 10,8 μL di NaAC e 1 μL di coprecipitante ai controlli di qualità non digeriti e non legati.
- Incubare a -80 °C per almeno 4 ore fino a tutta la notte.
- Ruotare i tubi da 15 mL a 2200 x g a 4 °C per 45 minuti.
- Ruotare i tubi di controllo da 1,5 mL a 20.500 x g a 4 °C per 30 minuti.
- Lavare una volta con 3 ml (campioni) e 1 ml (controlli) di EtOH ghiacciato al 70%.
- Centrifugare a 2200 x g (campioni) o 20.500 x g (controlli) a 4 °C per 10 min.
- Rimuovere con attenzione EtOH e asciugare all'aria a RT per 10-15 minuti; Non asciugare troppo.
- Risospendere i campioni e i controlli rispettivamente in 100 μL e 20 μL di H20.
- Aggiungere 1 μL di RNAseA e incubare a 37 °C, ruotando a 1400 giri/min per 30 minuti.
- Trasferire i campioni su ghiaccio in provette coniche da centrifuga da 15 ml preraffreddate e aggiungere 2 ml di acqua, 10,5 ml di EtOH ghiacciato e 583 μL di 3 M NaAC.
- Controllo qualità della preparazione del modello 3C
- Quantificare ogni campione e controllare utilizzando un kit fluorometrico per misurazioni della concentrazione di DNA ad alta sensibilità.
- Caricare 100-200 ng di ciascun campione e ciascun controllo su un gel di agarosio all'1% / 1x TBE.
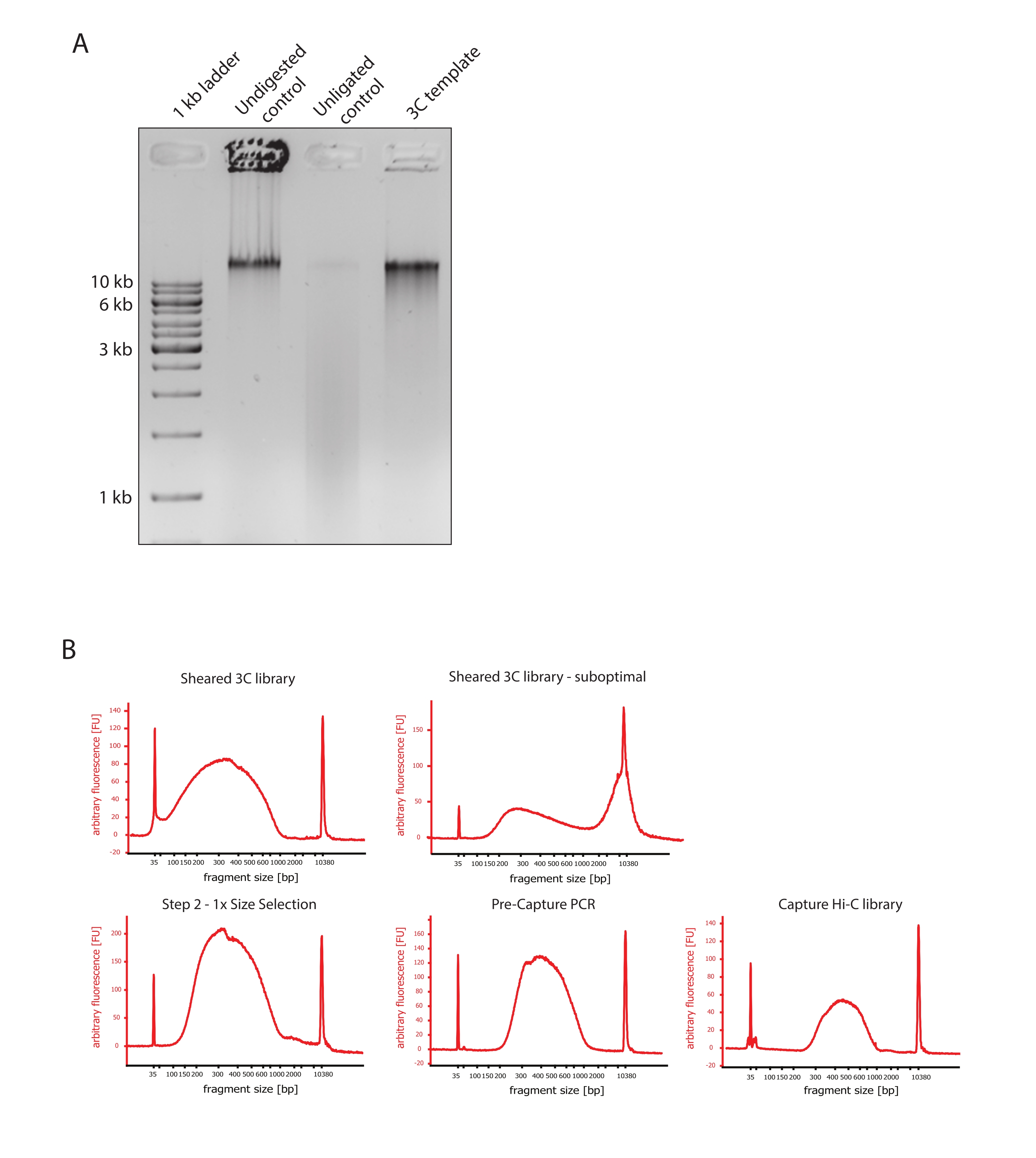
- Verificare che l'immagine del gel mostri il risultato atteso confrontando le differenze nelle dimensioni dei frammenti di DNA dei controlli e il modello 3C come mostrato nella Figura 2A.
- Conservare i campioni e i controlli a -20 °C.
- Ibridazione, acquisizione ed elaborazione dei campioni per il sequenziamento multiplex
- Per ibridare la serie di sonde di RNA biotinilato al modello 3C, catturare i frammenti di legatura mirati e preparare i campioni per il sequenziamento multiplexato secondo il sistema di arricchimento target utilizzato in questo studio per il sequenziamento multiplexato paired-end (vedere Tabella dei materiali). Seguire il protocollo secondo le istruzioni del produttore introducendo le seguenti modifiche minori:
- Sezione 2 del protocollo del fabbricante: preparazione del campione
- Seguire le istruzioni per l'arricchimento del target a partire da 3 μg di gDNA in ingresso.
- Tagliare il DNA in un sonicatore utilizzando le seguenti specifiche: ciclo di lavoro al 10%, intensità 4, 200 cyc/burst e 130 s. Iniziare con 4 μg di modello 3C risospeso in 130 μL di acqua per ogni reazione di cattura per garantire materiale sufficiente per continuare la preparazione del campione con 3 μg di DNA tranciato.
- Valutare la qualità del DNA tranciato. Eseguire 1 μL del DNA tranciato su un bioanalizzatore di DNA secondo il protocollo ad alta sensibilità. Aspettatevi una distribuzione della dimensione del frammento tra 150-700 bp (Figura 2).
- Purificare il campione utilizzando sfere di immobilizzazione reversibile in fase solida (SPRI). Aggiungere 124 μL di sfere SPRI a 124 μL del campione di DNA per eseguire una selezione 1:1 delle dimensioni del lato sinistro secondo le istruzioni del produttore ed eluire in 25 μL di acqua priva di nucleasi. Questa fase di purificazione rimuoverà i frammenti più corti per arricchire frammenti di circa 300 bp (Figura 2).
NOTA: la quantità di campioni e sfere SPRI utilizzate in questa fase tiene conto della perdita di volume che si è verificata durante il trasferimento dei campioni in nuove provette e l'esecuzione dei controlli di qualità presso il Bioanalyzer. Tutte le successive fasi di selezione delle dimensioni vengono eseguite secondo i rapporti raccomandati dal protocollo del produttore. L'eluizione del DNA dalle perle SPRI viene eseguita a RT durante tutto il protocollo. - Valutare la qualità del DNA tranciato selezionato per dimensioni. Eseguire 1 μL del DNA tranciato sul bioanalizzatore del DNA secondo il protocollo ad alta sensibilità (HS). Aspettatevi una distribuzione delle dimensioni dei frammenti con il massimo arricchimento a 300 bp (Figura 2). Vai avanti con la quantificazione del DNA tranciato se la tosatura ha avuto successo.
- Quantificare il DNA tranciato con un kit fluorometrico per le misurazioni della concentrazione di DNA HS.
NOTA: Se la cesoiatura del DNA determina una resa del DNA di <3 μg, eseguire un secondo ciclo di taglio del DNA con altri 4 μg di DNA e combinare i campioni di DNA tranciati dopo la prima fase di purificazione delle sfere SPRI per ottenere un totale di 3 μg di DNA tranciato. - Aggiungere acqua priva di nucleasi al campione di DNA pulito selezionato (3 μg totali) a un volume finale di 48 μL e procedere con la reazione di riparazione finale secondo il protocollo del produttore.
- Dopo la legatura degli adattatori accoppiati, amplificare la libreria eseguendo cinque cicli di PCR pre-acquisizione secondo le istruzioni del produttore (le condizioni per la PCR e i primer sono fornite nel kit).
- Sezione 4 del protocollo del produttore: ibridazione e cattura
- Per ibridare i campioni di DNA preparati alle sonde di RNA specifiche del bersaglio, diluire 750 ng di campioni di DNA in un volume finale di 3,4 μL, ottenendo una concentrazione iniziale di 221 ng / μL. Per i campioni di DNA diluiti in volumi maggiori, utilizzare un concentratore di velocità-vuoto per ridurre al volume finale. Una concentrazione di velocità-vuoto (250 x g; ≤45 °C) per 15-20 minuti è normalmente sufficiente per i campioni risospesi in 10 μL. Assicurarsi di avere lo stesso volume di ingresso per ciascun campione prima di avviare il concentratore di velocità-vuoto.
- Incubare la miscela di ibridazione per 16-18 h a 65 °C con coperchio riscaldato a 105 °C secondo le istruzioni del produttore.
- Sezione 5 del protocollo del produttore: indicizzazione ed elaborazione dei campioni per il sequenziamento multiplex
- Per amplificare le librerie acquisite con primer indicizzati, eseguire 12 cicli di PCR post-acquisizione secondo le istruzioni del produttore (le condizioni per PCR e primer sono fornite nel kit).
- Sezione 2 del protocollo del fabbricante: preparazione del campione
- Per ibridare la serie di sonde di RNA biotinilato al modello 3C, catturare i frammenti di legatura mirati e preparare i campioni per il sequenziamento multiplexato secondo il sistema di arricchimento target utilizzato in questo studio per il sequenziamento multiplexato paired-end (vedere Tabella dei materiali). Seguire il protocollo secondo le istruzioni del produttore introducendo le seguenti modifiche minori:
- Sequenziamento di nuova generazione
- Per eseguire più librerie Hi-C di acquisizione sulla stessa cella di flusso, preparare una miscela equimolare delle librerie di acquisizione e sequenziare 100-120 M letture per libreria.
- Se è necessaria l'analisi specifica dell'allele, sequenziare 150 bp paired-end per garantire una copertura SNP sufficiente.
3. Analisi dei dati
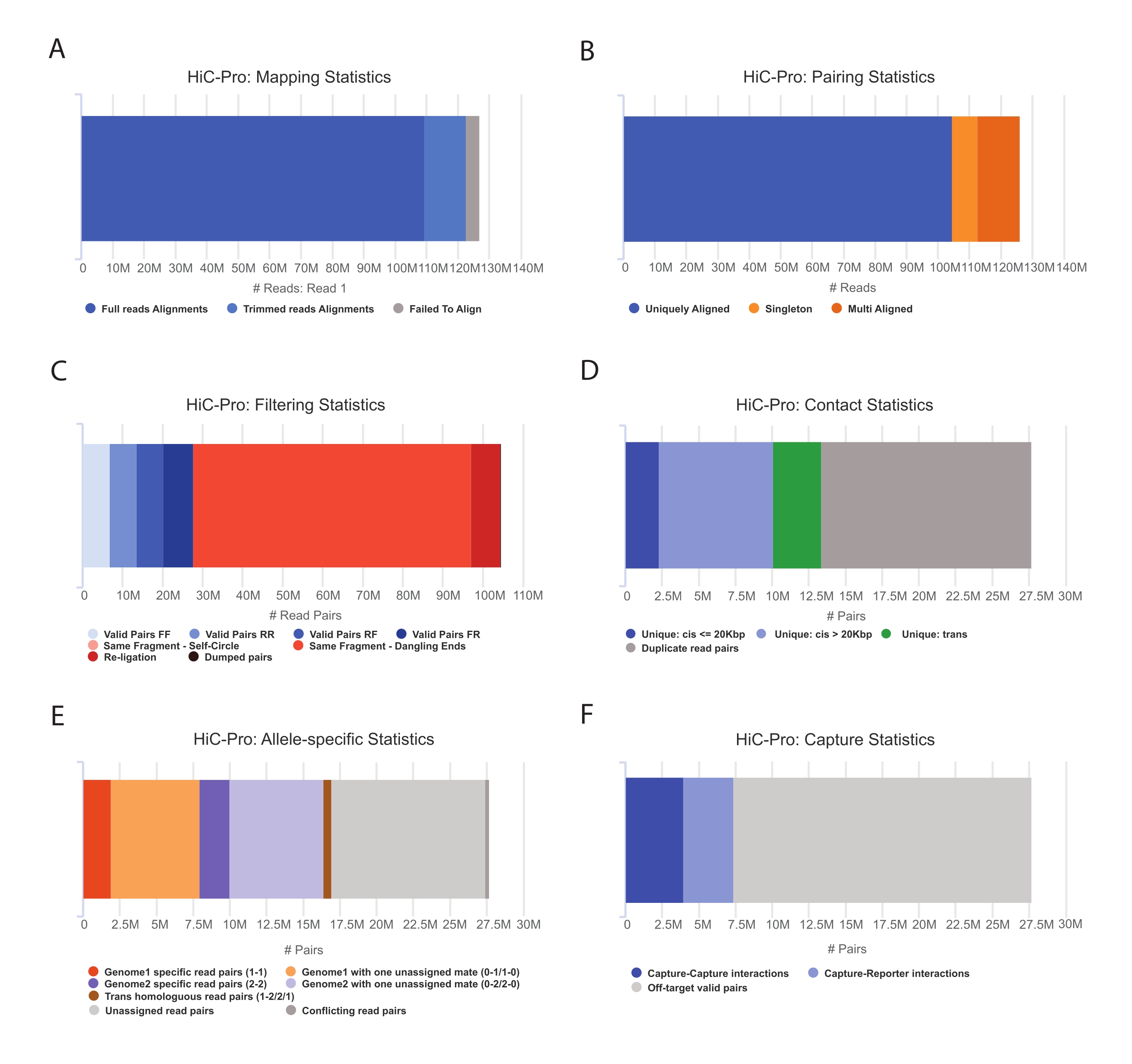
- Applicare la pipeline HiC-Pro per eseguire l'analisi dei dati Capture Hi-C55. HiC-Pro fornisce controlli di qualità in ogni fase della lavorazione, tra cui (Figura 3):
(i) Il tasso di allineamento sul genoma di riferimento specifica la frazione di letture che attraversano un sito di legatura, nonché il numero di coppie e singleton.
(ii) La frazione di prodotti di legatura validi e coppie di lettura non informative (dangling-end, self-ligation, ecc.).
(iii) La frazione di contatti a corto/lungo raggio e intra/intercromosomici.
iv) La frazione di contatti di destinazione per Capture Hi-C.
(v) La frazione di allele-specifica legge se specificata.
NOTA: HiC-Pro supporta un'ampia gamma di protocolli, tra cui Hi-C in situ e Capture Hi-C. In quest'ultimo caso, l'utente deve semplicemente specificare la regione di destinazione (formato BED) nel file di configurazione. Una volta elaborati i dati, le uscite HiC-Pro possono essere facilmente convertite in un oggetto più freddo per l'analisi a valle56. In questa fase, le mappe dei contatti a varie risoluzioni vengono normalizzate utilizzando il metodo ICE precedentemente descritto da Imakaev e colleghi57. Diverse analisi possono quindi essere eseguite per chiamare compartimenti cromosomici, TAD o anelli di cromatina (per la revisione58). Il flusso di lavoro del protocollo è illustrato nella Figura 4. Qui, la suite 'cooltools' viene applicata per calcolare il punteggio di isolamento e i limiti TADs, come illustrato nella Figura 5 e nella Figura 659.
Risultati
Il protocollo Capture Hi-C descritto si basa sulla preparazione del modello 3C genome-wide utilizzando un cutter a quattro basi (DpnII). Il successivo arricchimento dei frammenti di legatura attraverso la regione genomica di interesse è ottenuto mediante ibridazione di una serie di sonde di RNA piastrellate e la loro cattura basata sulla streptavidina secondo il sistema di arricchimento target utilizzato in questo studio (Figura 1). Le sonde di RNA biotinilato sono state selezionate in quanto mostrano un'affinità di legame più stretta ai loro bersagli rispetto alle sonde di DNA52,60. Le librerie acquisite vengono quindi indicizzate e raggruppate per la sequenziazione multiplex a throughput elevato. I dati Hi-C di acquisizione possono essere visualizzati come mappe di interazione Hi-C ad alta risoluzione, ma anche come mappe di contatto a punto di vista singolo simili a 4C per visualizzare in modo specifico le interazioni di sequenze più piccole come promotori o potenziatori all'interno dell'intera regione acquisita. Il flusso di lavoro del protocollo è illustrato nella Figura 4. I controlli di qualità pre-sequenziamento sono mostrati nella Figura 2 e includono la valutazione della corretta digestione e re-ligazione del modello 3C e la sua efficiente suddivisione e purificazione nelle diverse fasi del protocollo. Il DNA modello 3C tranciato dovrebbe funzionare tra 150 e 700 bp e non dovrebbe essere rilevato alcun arricchimento di frammenti >2 kb. Durante le fasi successive, vengono eseguite diverse fasi di pulizia del DNA basate su perline e selezione delle dimensioni, prima dopo la tosatura, poi dopo le PCR pre-cattura e post-cattura. Le librerie pulite mostrano un profilo di arricchimento dei frammenti distinto come visualizzato su un bioanalizzatore di DNA ad alta sensibilità (Figura 2). La dimensione media dei frammenti aumenta nel corso della preparazione della libreria a causa della legatura degli adattatori, del sequenziamento e dei primer di indicizzazione. I controlli di qualità post-sequenziamento sono ottenuti tramite Hi-C Pro e mostrati nella Figura 3. Sono state proposte molte diverse applicazioni software bioinformatiche per l'elaborazione e l'analisi dei dati simili a 3C. Tra questi, la pipeline HiC-Pro è una delle soluzioni più popolari, consentendo l'elaborazione di dati di sequenziamento grezzi alle mappe di contatto finali a varie risoluzioni55. HiC-Pro utilizza una strategia di mappatura in due fasi per allineare le letture di sequenziamento sul genoma di riferimento. I prodotti 3C vengono quindi ricostruiti e filtrati per rimuovere coppie di contatti non informative e generare le mappe dei contatti. Inoltre, è in grado di utilizzare un elenco di polimorfismi noti per eseguire analisi allele-specifiche e per separare i contatti provenienti dai due alleli parentali in mappe di contatto distinte. Più recentemente, HiC-Pro è stato incluso ed esteso nel framework nf-core (nf-core-hic), fornendo una pipeline altamente scalabile e riproducibile guidata dalla comunità61,62.
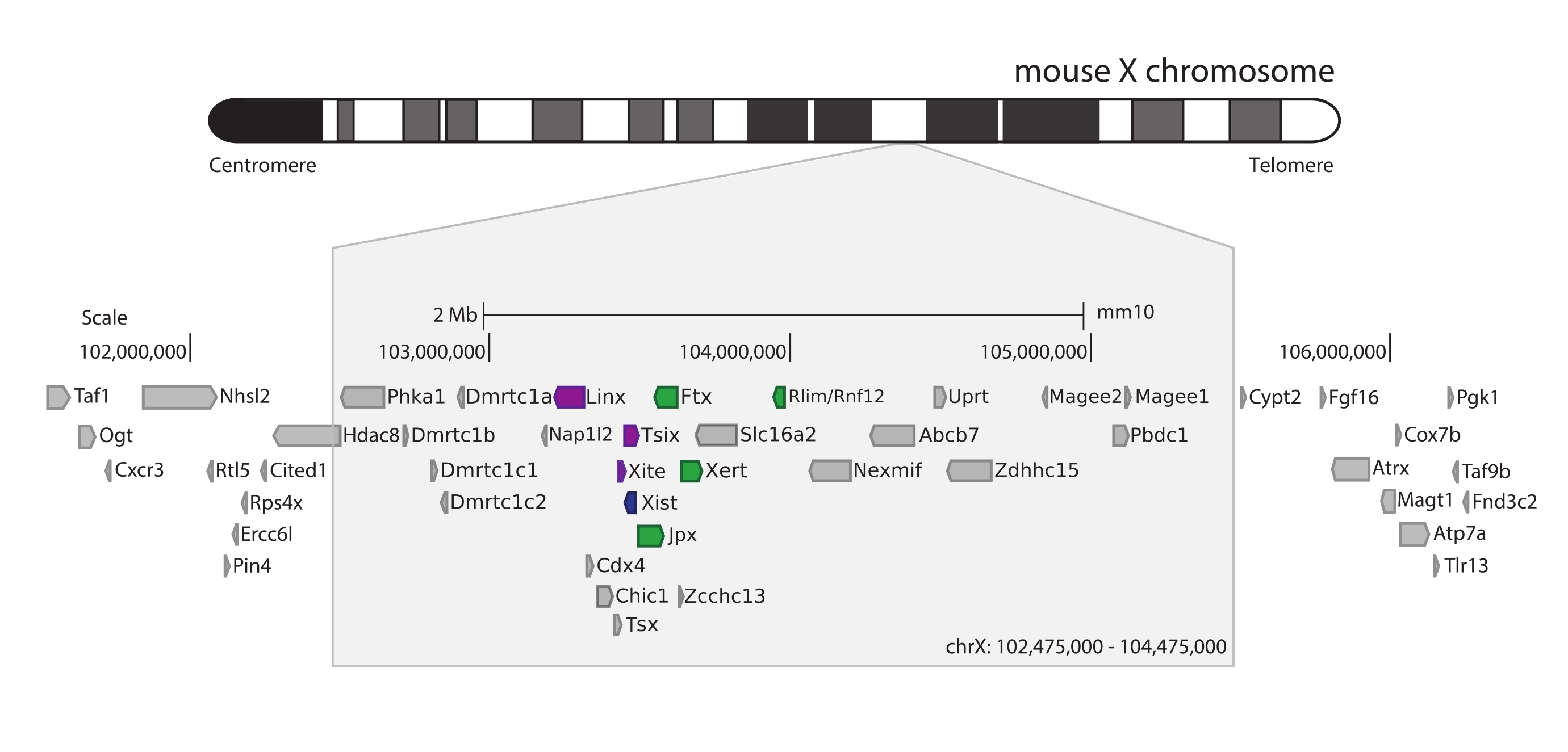
Per catturare il topo Xic, è stato progettato un array di 28.913 sonde di RNA che piastrellano 3 Mb del cromosoma X. Questa regione include l'attore chiave in XCI, il gene lungo non codificante Xist, e il suo noto panorama normativo di ~ 800 kb (Figura 5). Questa regione ~800 kb è partizionata in due TAD: uno che include il promotore Xist e i suoi regolatori positivi noti (cioè i trascritti non codificanti Ftx, Jpx e Xert e il gene codificante la proteina Rnf12), e il TAD vicino che comprende i regolatori cis negativi di Xist (cioè il suo trascritto antisenso Tsix, l'elemento enhancer Xite e il trascritto non codificante Linx) (per la revisione44, 45).
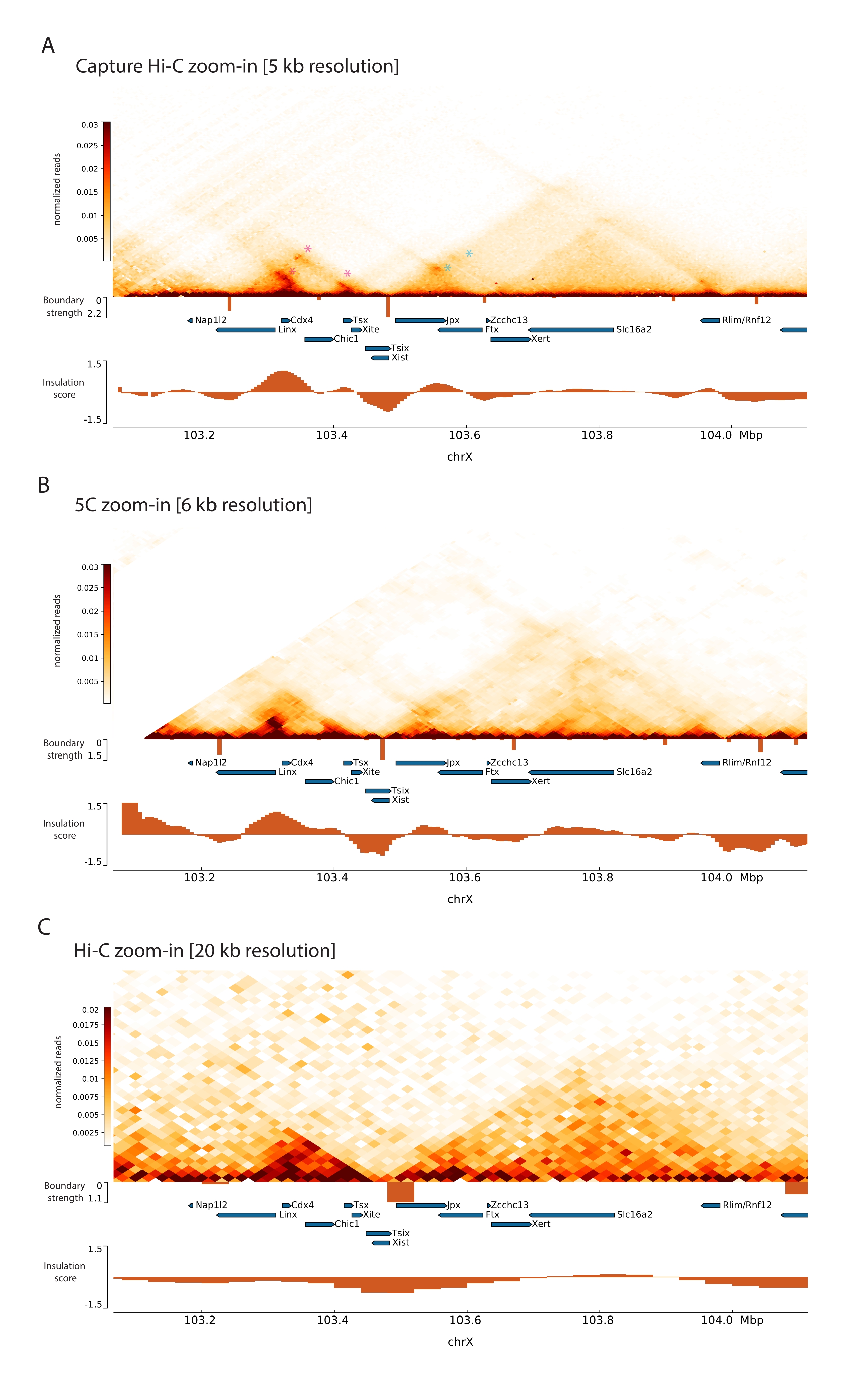
Applicando il protocollo Capture Hi-C descritto allo Xic, l'organizzazione topologica di questo locus è stata ottenuta con una risoluzione senza precedenti (Figura 6 e Figura 7). Ciò è particolarmente evidente quando si confronta il profilo Capture Hi-C con 5C47 pubblicato in precedenza (Figura 6 e Figura 7; Tabella supplementare 1) e Hi-C61 (Figura 6 e Figura 7; Tabella supplementare 1) Profili. Ad esempio, le strutture sub-TAD sono più evidenti: il TAD contenente il promotore Xist ( Xist-TAD ) è chiaramente suddiviso in due domini più piccoli (Figura 6A, punta di freccia blu). In precedenza, questo poteva essere solo visivamente "indovinato" dal profilo 5C (Figura 6B), sebbene il rilevamento di un confine in questa regione utilizzando l'algoritmo del punteggio di isolamento. Allo stesso modo, la risoluzione del profilo Capture Hi-C consente l'identificazione di due domini più piccoli nel TAD vicino (Figura 6A, B), che contiene il promotore del locus Tsix ( Tsix-TAD ); questo non è stato precedentemente raggiunto con 5C (Figura 6B). Da notare che i confini topologici determinati dal punteggio di isolamento dai dati Capture Hi-C e 5C vengono generalmente rilevati in posizioni leggermente diverse e con diversi punti di forza relativi.
Inoltre, altre strutture sub-TAD come i loop di contatto sono chiaramente visibili dai dati di Capture Hi-C, come il loop tra Xist e Ftx (Figura 7A), precedentemente identificato con Capture-C63, e il loop tra Xist e Xert (Figura 7B), recentemente identificato utilizzando un protocollo simile per Capture Hi-C48. Altri contatti possono anche essere mappati in modo più preciso grazie alla maggiore risoluzione dei profili Capture Hi-C, come quelli che formano gli hotspot di contatto noti all'interno del Tsix-TAD tra i loci Linx, Chic1 e Xite (Figura 7A).
Rispetto ai dati Hi-C mostrati nella Figura 7, Capture Hi-C ha permesso un aumento di quattro volte della risoluzione, ma ha richiesto solo un quarto della profondità di sequenziamento (cioè 126 M letture contro 571 M) (Tabella supplementare 1). Questo aumento della risoluzione consente il rilevamento di subTAD e interazioni cicliche che non possono essere rilevate da Hi-C alla profondità di sequenziamento mostrata in Figura 6 e Figura 7. Il protocollo descritto per Capture Hi-C consente quindi una caratterizzazione molto più dettagliata e ad alta risoluzione di una grande regione genomica di interesse, rispetto agli approcci precedenti.

Figura 1: Progettazione della sonda. Rappresentazione schematica della strategia utilizzata per la progettazione della sonda. Le regioni di 300 bp a monte e a valle di ciascun sito di restrizione DpnII attraverso la regione target di 3 Mb sono state selezionate e affiancate con sonde di RNA biotinilato sovrapposte. Viene mostrata una di queste regioni selezionate, chrX: 102.474.805-102.475.500. Non sono consentite più di 40 basi di sequenze ripetitive in ogni sonda. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 2: Acquisizione dei controlli di qualità di pre-sequenziamento Hi-C. (A) Esempio rappresentativo di controlli di qualità del modello 3C. 200 ng di DNA sono stati caricati su un gel di agarosio all'1%. Corsia 1: scala da 1 kb. Corsia 2: La cromatina non digerita, reticolata e intatta corre come una banda affilata a >10 kb. Corsia 3: la cromatina reticolata digerita con DpnII funziona come uno striscio di dimensioni comprese tra 1 kb e 3 kb. Corsia 4: libreria o modello 3C finale; le estremità libere dei frammenti di DNA reticolato digeriti vengono ri-legate. Lo striscio di DNA di dimensioni molecolari inferiori è quasi impercettibile e il prodotto di legatura viene rilevato come una banda di >10 kb. (B) Esempi rappresentativi di profili DNA di bioanalizzatori ad alta sensibilità. In alto a sinistra: libreria 3C tagliata con successo che mostra una distribuzione della dimensione del frammento tra 150 bp e 700 bp. In alto a destra: libreria 3C tranciata insoddisfacente. Il DNA non tagliato viene rilevato come ampio arricchimento di frammenti >2 kb. (C) In basso a sinistra: campione di DNA tranciato a seguito di una selezione 1:1 delle dimensioni del lato sinistro utilizzando perline SPRI. Frammenti di ~ 300 bp sono arricchiti. In basso al centro: profilo PCR pre-acquisizione dopo la legatura degli adattatori accoppiati secondo il protocollo del produttore. In basso a destra: libreria finale Capture Hi-C che include adattatori, sequenziamento e primer di indicizzazione per il sequenziamento multiplexato. Abbreviazioni: bp = coppie di basi, FU = unità di fluorescenza arbitraria. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 3: Acquisizione di controlli di qualità post-sequenziamento Hi-C con HiC-Pro . (A) Esempio di velocità di mappatura sul genoma di riferimento per il primo ufficiale delle coppie di sequenziamento. La frazione azzurra rappresenta le letture allineate da HiC-Pro e che attraversano una giunzione di legatura. Questa metrica può quindi essere utilizzata per convalidare la fase sperimentale di legatura. (B) Una volta che i compagni di sequenziamento sono allineati sul genoma, solo le coppie di lettura allineate in modo univoco vengono conservate per l'analisi. (C) Le coppie non valide (in rosso) come l'estremità penzolante, l'autocerchio o la legatura vengono scartate dall'analisi. La frazione di coppie valide è un buon indicatore dell'efficienza di legatura e pull-down. (D) Le coppie valide possono essere ulteriormente suddivise in contatti intra/intercromosomici e contatti a corto/lungo raggio. Le coppie di lettura duplicate che potrebbero rappresentare artefatti PCR vengono eliminate dall'analisi. (E) Per l'analisi allele-specifica, HiC-Pro riporta il numero di letture alleliche supportate da uno o due compagni per ciascun genoma parentale (cioè C57BL / 6J x CASTEi / J). Sono attese la stessa frazione di letture assegnate all'allele materno e paterno. (F) Infine, vengono selezionate solo coppie valide che si sovrappongono alla regione di acquisizione per costruire le mappe di contatto. Le coppie acquisizione-acquisizione rappresentano i contatti all'interno della regione di destinazione, mentre le coppie acquisizione-reporter implicano l'interazione tra la regione di destinazione e una fuori bersaglio. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

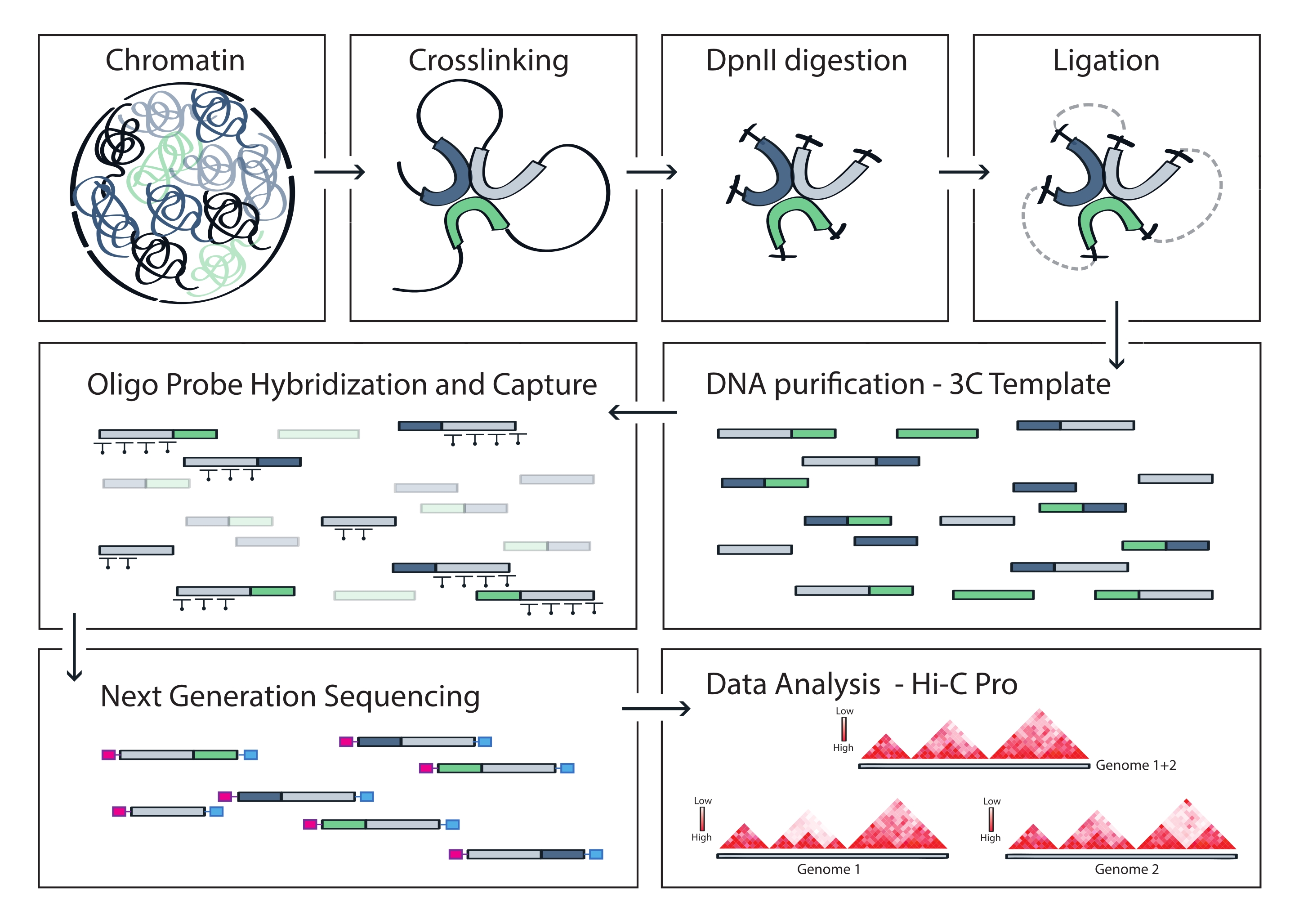
Figura 4: Flusso di lavoro del protocollo Capture Hi-C. Rappresentazione schematica di diversi passaggi del protocollo. Per generare il modello 3C dell'intero genoma, la cromatina viene prima reticolata con la formaldeide e poi digerita con l'enzima di restrizione DpnII. Le estremità libere del DNA vengono quindi ri-legate, la reticolazione viene invertita e il DNA viene purificato. Per arricchire i frammenti che comprendono la regione bersaglio, una serie di sonde di RNA biotinilato viene ibridata al modello 3C e catturata mediante pull-down mediato dalla streptavidina. Le librerie di acquisizione vengono elaborate per il sequenziamento multiplex e i frammenti di legatura validi vengono quantificati per dedurre la frequenza dei contatti della cromatina attraverso il bersaglio, che vengono visualizzati come mappe di interazione ad alta risoluzione. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 5: Panoramica della regione che comprende lo Xic sul cromosoma X del topo. Rappresentazione schematica del cromosoma X del topo e zoom della regione catturata di 3 Mb (ChrX: 102.475.000-105.475.000). La regione bersaglio include ~ 800 kb di DNA corrispondente allo Xic, il locus regolatore principale di XCI. Xic include i lunghi geni non codificanti, Xist, un attore chiave di XCI, e il suo panorama regolatorio. I regolatori positivi di Xist sono mostrati in verde e i regolatori negativi in viola. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 6: Acquisizione di mappe di interazione Hi-C, 5C e Hi-C nella regione catturata di 3 Mb. (A) Cattura la mappa di interazione Hi-C del target da 3 Mb che comprende il mouse Xic con una risoluzione di 10 kb (questo studio). (B) Mappa di interazione 5C della stessa regione target di A con risoluzione di 6 kb (dati rielaborati da47). Le regioni ripetitive non incluse nelle analisi sono mascherate in bianco. I dati 5C richiedono una propria elaborazione bioinformatica (cfr.47). Dopo la pulizia e l'allineamento, le mappe 5C alla risoluzione di primer vengono binned utilizzando una mediana corrente (finestra = 30 kb, passo = 5) per raggiungere una risoluzione finale di 6 kb. (C) Mappa di interazione Hi-C della stessa regione genomica di A e B con risoluzione di 40 kb (dati rielaborati da64). Tutte le mappe di interazione sono state generate da ESC del mouse. Il punteggio di isolamento è stato calcolato utilizzando cooltools ed è rappresentato come istogrammi con minimi di isolamento ai confini TAD. I confini TAD sono mostrati come linee verticali sotto la mappa. L'altezza di ogni linea indica la resistenza al limite. I geni sono mostrati come frecce che puntano nella direzione della trascrizione. I confini sub-TAD rilevati esclusivamente o più precisamente nelle mappe Capture Hi-C sono indicati da punte di freccia magenta e blu per i sub-TAD rispettivamente nei TAD Tsix e Xist. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 7: Cattura di mappe di interazione Hi-C, 5C e Hi-C su 1 Mb all'interno della regione acquisita. (A) Capture Hi-C interaction map della regione genomica di 1 Mb comprendente il topo Xic con una risoluzione di 5 kb (questo studio). (B) Mappa di interazione 5C della stessa regione genomica di cui ad A. con risoluzione di 6 KB (dati rielaborati da47). Le regioni ripetitive non incluse nelle analisi sono mascherate in bianco. Da notare che i dati 5C richiedono una propria elaborazione bioinformatica (cfr.47). Dopo la pulizia e l'allineamento, le mappe 5C alla risoluzione di primer vengono binned utilizzando una mediana corrente (finestra = 30 kb, passo = 5) per raggiungere una risoluzione finale di 6 kb. (C) Mappa di interazione Hi-C della stessa regione genomica di A e B di Hi-C con risoluzione di 20 kb (dati rielaborati da64). Tutte le mappe di interazione sono state generate da mESC. Il punteggio di isolamento è stato calcolato utilizzando cooltools ed è rappresentato come istogrammi con minimi di isolamento ai confini TAD. I confini TAD sono mostrati come linee verticali sotto la mappa. L'altezza di ogni linea indica la resistenza al limite. I geni sono mostrati come frecce che indicano la direzione della trascrizione. I loop di contatto rilevati esclusivamente o più precisamente in Capture Hi-C sono indicati da magenta e asterischi blu per i loop rispettivamente nei TAD Tsix e Xist. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
Tabella supplementare 1: Statistiche post-sequenziamento per i set di dati utilizzati in questo manoscritto: Capture Hi-C (questo studio), Hi-C64 e 5C47. Clicca qui per scaricare questo file.
Discussione
Qui descriviamo un protocollo Capture Hi-C relativamente semplice e veloce per caratterizzare l'organizzazione di ordine superiore di regioni genomiche di dimensioni megabase con una risoluzione di 5-10 kb. Capture Hi-C appartiene alla famiglia di tecnologie Capture-C progettate per arricchire le interazioni mirate della cromatina da modelli 3C o Hi-C a livello di genoma. Ad oggi, la grande maggioranza delle applicazioni di Capture-C sono state sfruttate per mappare i contatti della cromatina di elementi regolatori relativamente piccoli sparsi nell'intero genoma. Nel primo protocollo Capture-C, sono state utilizzate più sonde biotinilate di RNA sovrapposte per catturare >400 promotori preselezionati in librerie 3C preparate da cellule eritroidi31. La stessa strategia è stata successivamente migliorata in Next Generation (NG) e Nuclear Titrated (NuTi) Capture-C per ottenere profili di interazione ad alta risoluzione di >8.000 promotori utilizzando singole esche di DNA da 120 bp che coprono singoli siti di restrizione e due cicli sequenziali di cattura per massimizzare l'arricchimento dei frammenti di legatura informativi32,40. Queste strategie hanno portato alla dissezione funzionale degli elementi cis-agenti in molti contesti diversi, tra cui lo sviluppo embrionale del topo, la differenziazione cellulare, l'inattivazione del cromosoma X e la cattiva regolazione genica in condizioni patologiche 46,63,65,66,67,68,69,70,71.
In Promoter Capture Hi-C (PCHi-C), >22.000 promotori annotati contenenti frammenti di restrizione sono stati estratti dalle librerie Hi-C mediante ibridazione di singole sonde biotinilate RNA 120-mer a una o entrambe le estremità del frammento di restrizione34,72. Questo metodo ha permesso la dissezione dell'interattoma di migliaia di promotori in un numero rapidamente crescente di tipi cellulari, tra cui cellule staminali embrionali di topo, cellule epatiche fetali e adipociti 34,35,72,73, ma anche linee linfoblastoidi umane, progenitori ematopoietici, cheratinociti epidermici e cellule pluripotenti 37,74,75,76,77 .
Rispetto a queste tecnologie di arricchimento target, Capture Hi-C si rivolge a regioni genomiche contigue fino alla scala megabase, estendendo così uno o più TAD e comprendendo paesaggi regolatori di geni. L'intera regione di interesse deve essere piastrellata con una serie di sonde biotinilate che comprendano ogni sito di restrizione DpnII all'interno del target. L'ibridazione dell'array biotinilato al modello 3C, la sua successiva cattura basata sulla streptavidina e l'elaborazione per il sequenziamento multiplexato vengono eseguite utilizzando un sistema di arricchimento target per il sequenziamento multiplexato Illumina Paired-End. L'intero protocollo è veloce, in quanto può essere eseguito in 1 settimana dalla preparazione della libreria 3C fino al sequenziamento NGS e richiede solo piccoli adattamenti e / o risoluzione dei problemi specifici personalizzati.
Il protocollo offre anche vantaggi rispetto ad altri metodi basati su 3C. Per ottenere mappe di interazione con una risoluzione di 5-10 kb, abbiamo sequenziato letture accoppiate 100-120 M. A titolo di confronto, abbiamo usato qui un set di dati Hi-C di 571 M letture per raggiungere una risoluzione di 20 kb64 (GSM2053973), e almeno 1 miliardo di letture sarebbero necessarie per raggiungere una risoluzione di 5 kb con Hi-C22 cromosomico.
L'Hi-C di cattura utilizzato nel presente studio raggiunge una risoluzione molto più elevata rispetto al 5C precedentemente pubblicato sulla base di un enzima di restrizione della fresa 6-bp47 (Tabella supplementare 1). È importante sottolineare che la strategia progettata per arricchire e amplificare le interazioni mirate in 5C non consente un'analisi allele-specifica delle interazioni della cromatina. Al contrario, i dati di Capture Hi-C possono essere mappati allele-specific, consentendo la dissezione dei paesaggi strutturali 3D di coppie di cromosomi omologhi, ad esempio in cellule umane o in linee cellulari ibride F1 derivate incrociando ceppi di topo geneticamente diversi78. Per generare mappe di interazione Capture Hi-C specifiche per allele con una risoluzione di 5 kb, abbiamo sequenziato letture paired-end a 150 bp per aumentare la copertura SNP. Simili approcci allele-specifici possono essere applicati a linee cellulari umane, per le quali è disponibile l'annotazione degli SNP22.
È importante sottolineare che, sebbene Capture Hi-C garantisca generalmente un'alta risoluzione migliorando al contempo l'accessibilità economica dei costi di sequenziamento, la produzione di oligonucleotidi biotinilati su misura ha un impatto sul costo complessivo di questo metodo. Pertanto, la scelta del metodo 3C più adatto sarà diversa per le diverse applicazioni e dipenderà dalla questione biologica che viene affrontata e dalla risoluzione richiesta, nonché dalle dimensioni della regione di interesse. Altri protocolli Capture Hi-C sviluppati condividono le caratteristiche chiave con il protocollo descritto qui. Ad esempio, è stata applicata una strategia Capture Hi-C per caratterizzare regioni genomiche da ~ 50 kb a 1 Mb che coprono varianti non codificanti associate al rischio di cancro al seno e al colon-retto; in questo protocollo, le regioni bersaglio sono state estratte dalle librerie Hi-C ibridando esche di RNA 120-mer che piastrellano le regioni bersaglio con una copertura3x 33,38,79. Allo stesso modo, HYbrid Capture Hi-C (Hi-C 2) è stato utilizzato per indirizzare le interazioni all'interno delle regioni di interesse fino a2 Mb80. In entrambi i protocolli, l'uso di un modello Hi-C arricchito per frammenti di legatura tirati verso il basso della biotina ha aumentato la percentuale di letture informative totali rispetto al nostro protocollo. Ad esempio, nel set di dati Hi-C che abbiamo usato qui per il confronto64 (GSM2053973), la percentuale di coppie valide dopo la rimozione dei duplicati è 4,8 volte superiore alle coppie valide ottenute in Capture Hi-C come descritto nella Figura 3 e nella Tabella supplementare 1. Tuttavia, il pull-down consecutivo di frammenti legati biotinilati e sonde ibridate rende il protocollo significativamente più complesso e dispendioso in termini di tempo, riducendo al contempo la complessità della regione catturata.
Un altro metodo disponibile per arricchire i modelli 3C con sonde di piastrellatura è Tiled-C, che è stato applicato per studiare l'architettura della cromatina ad alta risoluzione spaziale e temporale durante la differenziazione eritroide del topo43. In Tiled-C, un pannello di sonde biotinilate da 70 bp viene utilizzato per arricchire i contatti all'interno di regioni su larga scala in due cicli consecutivi di cattura per generare mappe ad altissima risoluzione di interazioni mirate43,81. Il doppio arricchimento di cattura rende anche il protocollo più lungo e complesso rispetto a Capture Hi-C. Tuttavia, a differenza delle strategie Capture-C mirate a singoli siti di restrizione, in Tiled-C il secondo round di cattura non sembra aumentare significativamente l'efficienza di cattura, e quindi può probabilmente essere omesso43. Infine, un approccio simile basato sulla stessa strategia di arricchimento target utilizzata in questo studio è stato applicato alla dissezione di paesaggi regolatori che comprendono varianti strutturali descritte in pazienti con malformazioni congenite e reingegnerizzate in topi transgenici41,42. In questo caso, la serie di sonde di affiancamento è stata progettata su tutto il bersaglio piuttosto che in prossimità dei siti di restrizione DpnII41. Tuttavia, questo lavoro è stato fondamentale per evidenziare la sensibilità e il potere di questa strategia per ottenere una caratterizzazione ad alta risoluzione di grandi regioni genomiche in diversi contesti41,42,48.
In conclusione, il protocollo qui descritto rappresenta una strategia semplice, robusta e potente per la caratterizzazione 3D ad alta risoluzione di qualsiasi regione genomica di interesse. L'applicazione di questo approccio a diversi sistemi modello, tipi di cellule, paesaggi di cromatina regolati dallo sviluppo e regolazione genica in condizioni sane e patologiche è probabile che faciliti la nostra comprensione dell'interazione e della causalità tra topologia del genoma e regolazione genica, una delle questioni aperte fondamentali nel campo dell'epigenetica. Inoltre, l'applicazione di Capture Hi-C per mappare le interazioni a lungo raggio e il ripiegamento della cromatina di ordine superiore delle varianti di rischio identificate dagli studi GWAS ha il potenziale per rivelare la rilevanza funzionale dei loci genomici non codificanti associati a malattie umane in diversi contesti, fornendo così nuove informazioni sui processi potenzialmente alla base della patogenesi.
Divulgazioni
Kai Hauschulz è Field Application Scientist presso Agilent Technologies - Diagnostic and Genomics Group. Tutti gli altri autori non dichiarano interessi concorrenti.
Riconoscimenti
Il lavoro nel laboratorio Heard è stato sostenuto da un premio Advanced Investigator del Consiglio europeo della ricerca (XPRESS - AdG671027). A.L. è sostenuta da una borsa di studio individuale Marie Skłodowska-Curie Actions dell'Unione Europea (IF-838408). A.H. è supportato dalla rete innovativa e interdisciplinare ITN ChromDesign, nell'ambito dell'accordo Marie Skłodowska-Curie Grant 813327. Gli autori sono grati a Daniel Ibrahim (MPI for Molecular Genetics, Berlino) per gli utili consigli tecnici, alla piattaforma NGS dell'Institut Curie (Parigi), a Vladimir Benes e alla Genomics Core Facility dell'EMBL (Heidelberg), per il supporto e l'assistenza.
Materiali
| Name | Company | Catalog Number | Comments |
| 10x PBS pH 7.4 | Gibco | 10010-023 | |
| 37% (vol/vol) paraformaldehyde solution | Electron Microscopy Sciences | 15686 | single use glass-vials; do not reuse |
| 50 mL PP conical tube | Falcon | 352070 | |
| Agarose | Sigma | A9539-500g | |
| Bioanalyzer | Agilent | G2939BA | |
| Cell Scrapers - 25 cm Handle and 3.0 cm Blade | Falcon | 353089 | |
| CHIR99021 | Axon Medchem BV | Axon 1386 | |
| cOmplete Mini, Protease inhibitor cocktail (EDTA-free) | Merck | 11836170001 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | |
| Countess II FL | Invitrogen | ZGEXSCCOUNTESS2FL | Automated cell counter |
| Covaris S2 | Covaris | 500217 | Sonicator |
| DNA LoBind tube, 1.5 mL | Eppendorf | 30108051 | |
| DpnII (50000 units/mL) | New England Biolabs | R0543M | |
| Dulbecco's Modified Eagle Medium (DMEM) | Merck | D6429 | |
| Ethanol (100%) | Merck | 1.00983.2500 | |
| Fetal Bovine Serum (FBS) | Thermo Scientific | 10270106 | |
| gelatine from porcine skin | Sigma | G1890 | |
| GeneRuler 1 kb Plus DNA Ladder | Thermo Scientific | SM0313 | |
| GlycoBlue | Thermo Scientific | AM9516 | Coprecipitant |
| High-Sensitivity Bioanlayzer chips | Agilent | 5067-4626 | |
| Large Cooling Centrifuge 5920 R | Eppendorf | 5948000018 | |
| leukaemia inhibitory factor (LIF) | Merck | ESG1107 | |
| Liquiport | KNF | NF300 | Benchtop aspiration system |
| Low-binding filter tips | Biozym | VT0260U, VT0240, VT0220, VT0200U | |
| Molecular biology grade water | Merck | W3500-6x500ML | |
| Next Seq 500 | Illumina | SY-415-1001 | |
| Next Seq 500 High Output v2 Kit (300 cycles) | Illumina | FC-404-2004 | |
| Nonidet P40 Substitute (NP40) | Merck | 11332473001 | |
| PD0325901 | Axon Medchem BV | Axon 1408 | |
| Protease inhibitor cocktail (EDTA-free) | Merck | 11873580001 | |
| Proteinase K - recombinant, PCR-grade (20 mg/mL) | Thermo Scientific | EO0491 | |
| Qubit 2.0 | Thermo Scientific | Q32871 | |
| Qubit assay tubes | Thermo Scientific | Q32856 | |
| Qubit dsDNA High Sensitivity kit | Thermo Scientific | Q32851 | |
| RNase A (10 mg/mL) | Thermo Scientific | EN0531 | |
| Sodium acetate pH 5.2 (3M) | Merck | S7899 | |
| speed vacuum concentrator | Eppendorf | EP5305000100-1EA | |
| Agencourt AMPureXP | Beckman Coulter | A63881 | SPRI beads |
| SureSelect Target Enrichment Box 1 | Agilent | 5190-8645 | |
| SureSelect Target Enrichment Kit ILM Indexing Hyb Module Box 2 | Agilent | 5190-4455 | |
| SureSelect XT Library Prep Kit ILM | Agilent | 5500-0132 | |
| T4 ligase (30 units/µL) | Thermo Scientific | EL0013 | |
| table-top Centrifuge 5427 R | Eppendorf | 5409000012 | |
| Triton-X-100 (500 mL) | Merck | X100-500ML | |
| Trypan Blue | Invitrogen | T10282 | |
| Trypsine | Thermo Scientific | 25300054 | |
| UltraPure Glycine | Thermo Scientific | 15527013 | |
| β-mercaptoethanol | Thermo Scientific | 31350010 |
Riferimenti
- Ibrahim, D. M., Mundlos, S. The role of 3D chromatin domains in gene regulation: a multi-facetted view on genome organization. Current Opinion in Genetics & Development. 61, 1-8 (2020).
- Bolt, C. C., Duboule, D. The regulatory landscapes of developmental genes. Development. 147 (3), (2020).
- Glaser, J., Mundlos, S. 3D or not 3D: Shaping the genome during development. Cold Spring Harbor Perspectives in Biology. 14 (5), 040188(2021).
- Denker, A., De Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Kempfer, R., Pombo, A. Methods for mapping 3D chromosome architecture. Nature Reviews Genetics. 21 (4), 207-226 (2020).
- McCord, R. P., Kaplan, N., Giorgetti, L. Chromosome conformation capture and beyond: Toward an integrative view of chromosome structure and function. Molecular Cell. 77 (4), 688-708 (2020).
- Jerkovic, I., Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nature ReviewsMolecular Cell Biology. 22 (8), 511-528 (2021).
- Hsieh, T. -H. S., et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 162 (1), 108-119 (2015).
- Krietenstein, N., et al. Ultrastructural details of mammalian chromosome architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Naumova, N., Smith, E. M., Zhan, Y., Dekker, J. Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods. 58 (3), 192-203 (2012).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Zhao, Z., et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra-and interchromosomal interactions. Nature Genetics. 38 (11), 1341-1347 (2006).
- Würtele, H., Chartrand, P. Genome-wide scanning of HoxB1-associated loci in mouse ES cells using an open-ended Chromosome Conformation Capture methodology. Chromosome Research. 14 (5), 477-495 (2006).
- De Wit, E., De Laat, W. A decade of 3C technologies: insights into nuclear organization. Genes & Development. 26 (1), 11-24 (2012).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Splinter, E., et al. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes & Development. 25 (13), 1371-1383 (2011).
- Ferraiuolo, M. A., Sanyal, A., Naumova, N., Dekker, J., Dostie, J. From cells to chromatin: capturing snapshots of genome organization with 5C technology. Methods. 58 (3), 255-267 (2012).
- Kim, J. H., et al. 5C-ID: Increased resolution Chromosome-Conformation-Capture-Carbon-Copy with in situ 3C and double alternating primer design. Methods. 142, 39-46 (2018).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Zhang, Y., et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 148 (5), 908-921 (2012).
- Rao, S. S. P., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Dixon, J. R., et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 485 (7398), 376-380 (2012).
- Nora, E. P., et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 485 (7398), 381-385 (2012).
- Krefting, J., Andrade-Navarro, M. A., Ibn-Salem, J. Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biology. 16 (1), 87(2018).
- Galupa, R., Heard, E. Topologically associating domains in chromosome architecture and gene regulatory landscapes during development, disease, and evolution. Cold Spring Harbor Symposia on Quantitative Biology. 82, 267-278 (2017).
- Tena, J. J., Santos-Pereira, J. M. Topologically associating domains and regulatory landscapes in development, evolution and disease. Frontiers in Cell and Developmental Biology. 9, 702787(2021).
- Lupiáñez, D. G., Spielmann, M., Mundlos, S. Breaking TADs: How alterations of chromatin domains result in disease. Trends in Genetics. 32 (4), 225-237 (2016).
- Davidson, I. F., Peters, J. -M. Genome folding through loop extrusion by SMC complexes. Nature Reviews Molecular Cell Biology. 22 (7), 445-464 (2021).
- Schmitt, A. D., Hu, M., Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nature Reviews Molecular Cell Biology. 17 (12), 743-755 (2016).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Davies, J. O. J., et al. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nature Methods. 13 (1), 74-80 (2016).
- Jäger, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178(2015).
- Schoenfelder, S., et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Research. 25 (4), 582-597 (2015).
- Sahlén, P., et al. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology. 16, 156(2015).
- Joshi, O., et al. Dynamic reorganization of extremely long-range promoter-promoter interactions between two states of pluripotency. Cell Stem Cell. 17 (6), 748-757 (2015).
- Mifsud, B., et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics. 47 (6), 598-606 (2015).
- Dryden, N. H., et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research. 24 (11), 1854-1868 (2014).
- Oudelaar, A. M., Davies, J. O. J., Downes, D. J., Higgs, D. R., Hughes, J. R. Robust detection of chromosomal interactions from small numbers of cells using low-input Capture-C. Nucleic Acids Research. 45 (22), 184(2017).
- Oudelaar, A. M., et al. Single-allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nature Genetics. 50 (12), 1744-1751 (2018).
- Franke, M., et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 538 (7624), 265-269 (2016).
- Despang, A., et al. Functional dissection of the Sox9-Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nature Genetics. 51 (8), 1263-1271 (2019).
- Oudelaar, A. M., et al. Dynamics of the 4D genome during in vivo lineage specification and differentiation. Nature Communications. 11 (1), 1-12 (2020).
- Galupa, R., Heard, E. X-chromosome inactivation: A crossroads between chromosome architecture and gene regulation. Annual Review of Genetics. 52, 535-566 (2018).
- Loda, A., Collombet, S., Heard, E. Gene regulation in time and space during X-chromosome inactivation. Nature Reviews. Molecular Cell Biology. 23 (4), 231-249 (2022).
- van Bemmel, J. G., et al. The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nature Genetics. 51 (6), 1024-1034 (2019).
- Galupa, R., et al. A conserved noncoding locus regulates random monoallelic Xist expression across a topological boundary. Molecular Cell. 77 (2), 352-367 (2020).
- Gjaltema, R. A. F., et al. Distal and proximal cis-regulatory elements sense X chromosome dosage and developmental state at the Xist locus. Molecular Cell. 82 (1), 190-208 (2022).
- Galupa, R., et al. Inversion of a topological domain leads to restricted changes in its gene expression and affects inter-domain communication. Development. 149 (9), (2022).
- Savarese, F., Flahndorfer, K., Jaenisch, R., Busslinger, M., Wutz, A. Hematopoietic precursor cells transiently reestablish permissiveness for X inactivation. Molecular and Cellular Biology. 26 (19), 7167-7177 (2006).
- Schulz, E. G., et al. The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell. 14 (2), 203-216 (2014).
- Gnirke, A., et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology. 27 (2), 182-189 (2009).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Piccinini, F., Tesei, A., Arienti, C., Bevilacqua, A. Cell counting and viability assessment of 2D and 3D Cell cultures: Expected reliability of the trypan blue assay. Biological Procedures Online. 19 (1), 8(2017).
- Servant, N., et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology. 16, 259(2015).
- Abdennur, N., Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 36 (1), 311-316 (2020).
- Imakaev, M., et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nature Methods. 9 (10), 999-1003 (2012).
- Forcato, M., et al. Comparison of computational methods for Hi-C data analysis. Nature Methods. 14 (7), 679-685 (2017).
- Venev, S., et al. open2c/cooltools: v0.4.1. , (2021).
- Wages, J. M. NUCLEIC ACIDS | Immunoassays. Encyclopedia of Analytical Science. , Elsevier. 408-417 (2005).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Servant, N., Peltzer, A. nf-core/hic: Initial release of nf-core/hic. Zenodo. , (2019).
- Furlan, G., et al. The Ftx noncoding locus controls X chromosome inactivation independently of its RNA products. Molecular Cell. 70 (3), 462-472 (2018).
- Giorgetti, L., et al. Structural organization of the inactive X chromosome in the mouse. Nature. 535 (7613), 575-579 (2016).
- Simon, C. S., et al. Functional characterisation of cis-regulatory elements governing dynamic Eomes expression in the early mouse embryo. Development. 144 (7), 1249-1260 (2017).
- Williams, R. M., et al. Reconstruction of the global neural crest gene regulatory network in vivo. Developmental Cell. 51 (2), 255-276 (2019).
- Godfrey, L., et al. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nature Communications. 10 (1), 2803(2019).
- Hanssen, L. L. P., et al. Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nature Cell Biology. 19 (8), 952-961 (2017).
- Larke, M. S. C., et al. Enhancers predominantly regulate gene expression during differentiation via transcription initiation. Molecular Cell. 81 (5), 983-997 (2021).
- Oudelaar, A. M., et al. A revised model for promoter competition based on multi-way chromatin interactions at the α-globin locus. Nature Communications. 10 (1), 5412(2019).
- Long, H. K., et al. Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell. 27 (5), 765-783 (2020).
- Schoenfelder, S., Javierre, B. -M., Furlan-Magaril, M., Wingett, S. W., Fraser, P. Promoter Capture Hi-C: High-resolution, genome-wide profiling of promoter interactions. Journal of Visualized Experiments. (136), e57320(2018).
- Siersbæk, R., et al. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Molecular Cell. 66 (3), 420-435 (2017).
- Rubin, A. J., et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nature Genetics. 49 (10), 1522-1528 (2017).
- Freire-Pritchett, P., et al. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. eLife. 6, 21926(2017).
- Javierre, B. M., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 167 (5), 1369-1384 (2016).
- Miguel-Escalada, I., et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nature Genetics. 51 (7), 1137-1148 (2019).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Baxter, J. S., et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nature Communications. 9 (1), 1028(2018).
- Sanborn, A. L., et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proceedings of the National Academy of Sciences. 112 (47), 6456-6465 (2015).
- Owens, D. D. G., et al. Dynamic Runx1 chromatin boundaries affect gene expression in hematopoietic development. Nature Communications. 13 (1), 773(2022).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati