
Method Article
Déchiffrer l’organisation de la chromatine 3D haute résolution via Capture Hi-C
Dans cet article
Résumé
Ce protocole décrit la méthode Capture Hi-C utilisée pour caractériser l’organisation 3D de régions génomiques ciblées de taille mégamétrique à haute résolution, y compris les limites des domaines topologiquement associés (TAD) et les interactions chromatines à longue distance entre les éléments de séquence d’ADN régulateurs et autres.
Résumé
L’organisation spatiale du génome contribue à sa fonction et à sa régulation dans de nombreux contextes, y compris la transcription, la réplication, la recombinaison et la réparation. Comprendre la causalité exacte entre la topologie et la fonction du génome est donc crucial et fait de plus en plus l’objet de recherches intensives. Les technologies de capture de conformation chromosomique (3C) permettent de déduire la structure 3D de la chromatine en mesurant la fréquence des interactions entre n’importe quelle région du génome. Nous décrivons ici un protocole rapide et simple pour effectuer Capture Hi-C, une méthode d’enrichissement de cible basée sur 3C qui caractérise l’organisation 3D spécifique à l’allèle de cibles génomiques de taille mégamétrique à haute résolution. Dans Capture Hi-C, les régions cibles sont capturées par un ensemble de sondes biotinylées avant le séquençage à haut débit en aval. Ainsi, une résolution et une spécificité allèle plus élevées sont obtenues tout en améliorant le temps et l’abordabilité de la technologie. Pour démontrer ses points forts, le protocole Capture Hi-C a été appliqué au centre d’inactivation X ( Xic) de la souris, le locus régulateur principal de l’inactivation du chromosome X (XCI).
Introduction
Le génome linéaire contient toutes les informations nécessaires pour qu’un organisme subisse un développement embryonnaire et survive tout au long de l’âge adulte. Cependant, demander à des cellules génétiquement identiques de remplir différentes fonctions est fondamental pour contrôler avec précision quelles informations sont utilisées dans des contextes spécifiques, y compris différents tissus et / ou stades de développement. On pense que l’organisation tridimensionnelle du génome participe à cette régulation spatio-temporelle précise de l’activité des gènes en facilitant ou en empêchant l’interaction physique entre des éléments régulateurs qui peuvent être séparés par plusieurs centaines de kilobases dans le génome linéaire (pour les revues 1,2,3). Au cours des 20 dernières années, notre compréhension de l’interaction entre le repliement du génome et l’activité s’est rapidement améliorée, en grande partie grâce au développement de technologies de capture de conformation chromosomique (3C) (pour la revue 4,5,6,7). Ces méthodes mesurent la fréquence des interactions entre toutes les régions du génome et reposent sur la ligature de séquences d’ADN qui sont à proximité 3D dans le noyau. Les protocoles 3C les plus courants commencent par la fixation des populations cellulaires avec un agent de réticulation tel que le formaldéhyde. La chromatine réticulée est ensuite digérée avec une enzyme de restriction, bien que la digestion de la MNase ait également été utilisée 8,9. Après la digestion, les extrémités de l’ADN libre à proximité spatiale sont re-ligaturées et la réticulation est inversée. Cette étape donne naissance à la « bibliothèque » 3C ou « modèle », un pool mixte de fragments hybrides dans lequel les séquences qui étaient à proximité 3D du noyau ont plus de chances d’être ligaturées dans le même fragment d’ADN. La quantification en aval de ces fragments hybrides permet de déduire la conformation 3D de régions génomiques situées à des milliers de paires de bases dans le génome linéaire, mais qui pourraient interagir dans l’espace 3D.
De nombreuses approches différentes ont été développées pour caractériser la bibliothèque 3C, différant à la fois en termes de sous-ensembles de fragments de ligature analysés et de technologie utilisée pour leur quantification en aval. Le protocole 3C original reposait sur la sélection de deux régions d’intérêt et la quantification de leur fréquence d’interaction « un contre un » par PCR10,11. L’approche 4C (capture circulaire de conformation chromosomique) mesure les interactions entre un seul locus d’intérêt (c.-à-d. le « point de vue ») et le reste du génome (« un contre tous »)12,13,14. En 4C, la bibliothèque 3C subit un deuxième cycle de digestion et de re-ligature pour générer de petites molécules d’ADN circulaires qui sont amplifiées par des amorces spécifiques au point de vue15. 5C (chromosome conformation capture carbon copy) permet la caractérisation des interactions 3D dans de plus grandes régions d’intérêt, fournissant des informations sur le repliement de la chromatine d’ordre supérieur dans cette région (« plusieurs contre plusieurs »)16. En 5C, la bibliothèque 3C est hybridée à un pool d’oligonucléotides chevauchant des sites de restriction qui peuvent ensuite être amplifiés par PCR multiplex avec amorces universelles15. Dans 4C et 5C, les fragments d’ADN informatifs ont d’abord été quantifiés par des puces à ADN et plus tard par séquençage de nouvelle génération (NGS)17,18,19. Ces stratégies caractérisent les régions d’intérêt ciblées, mais ne peuvent pas être appliquées pour cartographier les interactions à l’échelle du génome. Ce dernier objectif est atteint avec Hi-C, une stratégie à haut débit basée sur 3C dans laquelle le séquençage massivement parallèle du modèle 3C permet la caractérisation impartiale du repliement de la chromatine au niveau du génome (« tous contre tous »)20. Le protocole Hi-C comprend l’incorporation d’un résidu biotinylé aux extrémités des fragments digérés, qui est suivi d’une réduction des fragments de ligature avec des billes de streptavidine pour augmenter la récupération des fragments ligaturés20.
Hi-C a révélé que les génomes des mammifères sont structurellement organisés à plusieurs échelles dans le noyau 3D. À l’échelle de la mégabase, le génome est divisé en régions de chromatine active et inactive, les compartiments A et B, respectivement20,21. L’existence d’autres sous-compartiments représentés par différents états de chromatine et d’activité a également été démontrée par la suite22. À une résolution plus élevée, le génome est ensuite partitionné en domaines d’auto-interaction sous-mégabase appelés domaines d’association topologique (TAD), révélés pour la première fois par l’analyse Hi-C et 5C des génomes humain et murin23,24. Contrairement aux compartiments qui varient d’une manière spécifique aux tissus, les TAD ont tendance à être constants (bien qu’il existe de nombreuses exceptions). Il est important de noter que les limites TAD sont conservées pour toutes les espèces25. Dans les cellules de mammifères, les TAD englobent souvent des gènes partageant le même paysage régulateur et il a été démontré qu’ils représentent un cadre structurel qui facilite la corégulation des gènes tout en limitant les interactions avec les domaines régulateurs voisins (pour la revue 3,26,27,28). De plus, au sein des TAD, les interactions dues aux sites CTCF à la base des boucles extrudées de cohésine peuvent augmenter la probabilité d’interactions promoteur-amplificateur ou amplificateur-amplificateur (pour la revue29).
En Hi-C, les compartiments et les TAD peuvent être détectés à une résolution de 1 Mb à 40 kb, mais une résolution plus élevée peut être obtenue pour caractériser des contacts à plus petite échelle, tels que les interactions en boucle entre éléments distaux à l’échelle de 5 à 10 kb. Cependant, l’augmentation de la résolution pour pouvoir détecter efficacement de telles boucles par HiC nécessite une augmentation significative de la profondeur de séquençage et, par conséquent, des coûts de séquençage. Ceci est exacerbé si l’analyse doit être spécifique à l’allèle. En effet, une augmentation X de la résolution nécessite une augmentation X2 de la profondeur de séquençage, ce qui signifie que les approches à haute résolution et spécifiques à l’allèle à l’échelle du génome peuvent être prohibitives30.
Pour améliorer la rentabilité et l’abordabilité tout en maintenant une haute résolution, les régions cibles d’intérêt peuvent être physiquement extraites des bibliothèques 3C ou Hi-C à l’échelle du génome après leur hybridation avec des sondes oligonucléotidiques complémentaires marquées à la biotine avant le séquençage en aval. Ces stratégies d’enrichissement de la cible sont appelées méthodes de capture-C et permettent l’interrogation des interactions de centaines de loci cibles dispersés dans le génome (c.-à-d. Promoter Capture (PC) Hi-C; Capture C de nouvelle génération (NG); Capture à faible entrée (LI) en C ; Capture nucléaire titrée (NuTi) en C; Tri-C)31,32,33,34,35,36,37,38,39,40, ou dans des régions couvrant jusqu’à plusieurs mégabases (c.-à-d. Capture HiC; HYbrid Capture Hi-C (Hi-C2); Tuilé-C)41,42,43. Deux aspects peuvent varier dans les méthodes basées sur la capture : (1) la nature et la conception des oligonucléotides biotinylés (c.-à-d. ARN ou ADN, oligos uniques capturant des cibles génomiques dispersées ou oligos multiples marquant une région d’intérêt); et (2) le modèle utilisé pour abattre les cibles qui peuvent être la bibliothèque 3C ou Hi-C, cette dernière étant constituée de fragments de restriction biotinylés tirés de la bibliothèque 3C.
Ici, un protocole Capture Hi-C basé sur l’enrichissement des contacts cibles à partir de la bibliothèque 3C est décrit. Le protocole repose sur la conception d’un réseau de pavage sur mesure de sondes d’ARN biotinylées et peut être réalisé en 1 semaine de la préparation de la bibliothèque 3C au séquençage NGS. Le protocole est rapide, simple et permet de caractériser l’organisation 3D d’ordre supérieur des régions d’intérêt de la taille d’une mégabase à une résolution de 5 Ko tout en améliorant l’efficacité temporelle et l’abordabilité par rapport aux autres méthodes 3C. Le protocole Capture Hi-C a été appliqué au locus régulateur maître de l’inactivation du chromosome X (XCI), le centre d’inactivation X (Xic), qui héberge l’ARN non codant Xist. Le Xic a déjà fait l’objet d’analyses structurelles et fonctionnelles approfondies (pour examen44,45). Chez les mammifères, XCI compense le dosage des gènes liés à l’X entre les femelles (XX) et les mâles (XY) et implique le silence transcriptionnel de la quasi-totalité de l’un des deux chromosomes X dans les cellules femelles. Le Xic a représenté un puissant locus de référence pour les études sur la topologie du génome 3D et l’interaction avec la régulation des gènes44. L’analyse 5C de la Xic dans les cellules souches embryonnaires de souris (CSEm) a conduit à la découverte et à la dénomination des TAD, fournissant les premiers aperçus de la pertinence fonctionnelle de la partition topologique et de la corégulation génique24. L’organisation topologique du Xic s’est par la suite révélée être impliquée de manière critique dans le moment approprié du développement de la régulation positive de Xist et de XCI 46, et des éléments cis-régulateurs insoupçonnés qui peuvent influencer l’activité des gènes dans et entre les TAD ont également été récemment découverts dans le Xic47,48,49. L’application de Capture Hi-C à 3 Mo du chromosome X de la souris couvrant le Xic démontre la puissance de cette approche pour disséquer le repliement de la chromatine à grande échelle à haute résolution. Un protocole détaillé et facile à suivre est fourni, allant de la conception de la gamme de sondes biotinylées sur chaque site de restriction DpnII dans la région d’intérêt à la génération de la bibliothèque 3C à l’échelle du génome, à l’hybridation et à la capture des contacts cibles et à l’analyse des données en aval. Un aperçu des contrôles de qualité appropriés et des résultats attendus est également inclus, et les forces et les limites de l’approche sont discutées à la lumière de méthodes similaires existantes.
Protocole
Les cellules souches embryonnaires de souris (CSEm) utilisées dans cette étude proviennent d’un croisement d’une femelle TX/TX R26 rtTA/rtTA 50 avec un mâle Mus musculus castaneus selon les directives de soins aux animaux de l’Institut Curie (Paris)51.
1. Conception de la sonde
- Concevoir un réseau de sondes biotinylées (oligonucléotides à ARN 120 mères) couvrant la région cible d’intérêt.
- Mosaïquer la région d’intérêt avec des oligonucléotides qui se chevauchent de sorte qu’en moyenne chaque séquence de la cible soit couverte par deux sondes uniques (couverture 2x) (Figure 1).
- Exclure les séquences répétitives de la couverture de la sonde pour éviter l’enrichissement d’interactions non spécifiques.
REMARQUE: Pour maximiser l’enrichissement des fragments de ligature informative, des régions couvrant 300 bp en amont et en aval de chaque site de restriction DpnII à travers la cible ont été définies (ChrX: 102 475 000-105 475 000), et 28 913 sondes biotinylées ont été conçues selon la technologie d’enrichissement de cible ADN SureSelect via la plate-forme Sure Design52. Selon cette stratégie, jusqu’à un maximum de 40 bases de séquences répétitives sont autorisées dans chaque oligonucléotide pour minimiser l’enrichissement des interactions non spécifiques. Le réseau de sondes a été synthétisé par Agilent. Ici, DpnII est utilisé comme enzyme de restriction pour deux raisons: (1) il s’agit d’un quatre cutter couramment utilisé dans plusieurs méthodes basées sur 3C53; et (2) il maximise les chances de capturer des polymorphismes mononucléotidiques (SNP) informatifs à proximité des sites de coupe par rapport à d’autres enzymes de restriction qui ont été testées in silico dans des lignées hybrides F1 utilisées dans cette étude (C57BL/6J x CASTEi/J).
2. Procédure expérimentale
- Préparation cellulaire
- Ensemencer le nombre approprié de cellules sur une ou plusieurs plaques de culture cellulaire pour obtenir un nombre total de cellules de ≥ 5 x 107 cellules le jour de la fixation.
REMARQUE : Des cellules souches embryonnaires de souris (CSEm) ont été utilisées dans cette étude. Les CSEm sont plaquées sur des plaques de culture cellulaire gélatinisées (0,1 % de gélatine dans 1 x PBS - o/n à 37 °C, incubateur de CO2 à 5 %) dans un milieu mESCs contenant 2i + LIF et du sérum de veau fœtal testé par lots (DMEM, FBS à 15 %, 0,1 mM β-mercaptoéthanol, 1 000 U/mL−1 facteur inhibiteur de la leucémie (LIF), CHIR99021 (3 μM) et PD0325901 (1 μM)). Pour ce type de cellule, une plaque de 10 cm confluente à 80 % contient environ 2 x 107 cellules. - Préparez une plaque de culture cellulaire supplémentaire pour le comptage cellulaire.
REMARQUE: Une plaque de culture cellulaire plus petite peut être utilisée pour réduire l’utilisation du média. Dans ce cas, le nombre de cellules à ensemencer sur la plus petite plaque doit être ajusté en conséquence (p. ex., 3 fois moins de cellules sur une plaque de 10 cm par rapport à une plaque de 15 cm).
- Ensemencer le nombre approprié de cellules sur une ou plusieurs plaques de culture cellulaire pour obtenir un nombre total de cellules de ≥ 5 x 107 cellules le jour de la fixation.
- Fixation du formaldéhyde
- Estimer le nombre total de cellules à réticuler.
- Avant de commencer la réaction de réticulation, trypsiniser et compter les cellules de la plaque de contrôle préparée spécifiquement pour le comptage des cellules à l’aide d’un compteur de cellules automatisé conformément aux instructions du fabricant.
- Inclure une coloration de viabilité (p. ex., bleu de trypan) pour déterminer le pourcentage de cellules viables54. À partir de ce nombre de cellules, estimer le nombre total de cellules dans la ou les plaques préparées pour la réticulation.
- Retirer le milieu de culture des plaques préparées pour la réticulation et le remplacer par la quantité appropriée de solution de fixation (formaldéhyde à 2 % dans le milieu de culture cellulaire). Utiliser 10 mL sur une assiette de 10 cm (p. ex. ~20 mL pour une assiette de 15 cm).
REMARQUE: Ajouter un volume exact de solution de fixation. Si la fixation des cellules adhérentes n’est pas possible, cette étape peut être adaptée aux cellules trypsinisées et réalisée dans 30 mL de solution de fixation dans des tubes centrifugés coniques de 50 mL. Le formaldéhyde ne doit pas avoir plus de 1 an. Il est préférable d’utiliser des flacons à usage unique. La solution de fixation doit être portée à température ambiante (RT) avant utilisation.
ATTENTION : Le formaldéhyde est dangereux et doit être manipulé conformément aux réglementations appropriées en matière de santé et de sécurité. - Fixer pendant 10 min à TA sous un mélange doux sur un shaker.
- Éteindre la réaction de fixation par addition de 2,5 M glycine-1x PBS à une concentration finale de 0,125 M. Ajouter 530 μL de glycine-1x PBS 2,5 M à 10 mL sur une plaque de 10 cm (p. ex. 1060 μL à 20 mL sur une plaque de 15 cm).
REMARQUE: Si les cellules ont été fixées en solution, éteindre la réaction de fixation avec 1590 μL de glycine-1x PBS 2,5 M. - Incuber pendant 5 min à TA, en mélangeant délicatement sur un shaker.
- Transférer les plaques dans de la glace et incuber pendant 15 minutes supplémentaires sur de la glace tout en mélangeant doucement sur un shaker.
REMARQUE: À partir de maintenant, les cellules doivent être maintenues sur la glace et les tampons doivent être pré-réfrigérés pour éviter d’autres réticulations. Passez dans une chambre froide si de nombreuses plaques doivent être traitées. - Retirez la solution de fixation des cellules en la versant dans un bécher pour assurer une manipulation rapide.
REMARQUE : Assurez-vous d’éliminer les déchets liquides contenant du formaldéhyde conformément aux règlements de santé et de sécurité appropriés. - Rincer rapidement la plaque de 10 cm deux fois avec 5 mL de glycine-1x PBS froid 0,125 M (8 mL pour une assiette de 15 cm) pour éliminer les débris et les cellules mortes. Retirez le liquide de la plaque en le versant dans un bécher pour assurer une manipulation rapide.
- Ajouter 5 mL de glycine-1x PBS froid 0,125 M à la plaque de 10 cm (10 mL pour une plaque de 15 cm) et gratter rapidement les cellules de la plaque à l’aide d’un racleur de cellules en plastique.
- Transférer la suspension cellulaire dans un tube centrifuge conique prérefroidi de 50 ml à l’aide d’une pipette sérologique.
- Rincer la plaque deux fois avec 5 mL de glycine-1x PBS froid 0,125 M et ajouter la suspension cellulaire dans le tube centrifuge conique.
- Faire tourner vers le bas à 480 x g pendant 10 min à 4 °C.
NOTE: Si les cellules ont été fixées en solution, transférer la cellule dans un tube centrifuge conique pré-refroidi et faire tourner vers le bas à 480 x g pendant 10 minutes à 4 °C. Retirer la solution de fixation en la versant dans un bécher et laver trois fois dans 10 mL de glycine-1x PBS froid 0,125 M. Assurez-vous de remettre les cellules en suspension à chaque étape de lavage. - Retirez le surnageant en l’aspirant à l’aide d’un système d’aspiration de paillasse. Remettez les cellules en suspension dans 500 μL de 1x PBS pour 1 x 107 cellules en pipetant soigneusement de haut en bas avec une pipette P1000. Pour remettre en suspension des cellules dans le volume exact, se référer à l’estimation du nombre total de cellules obtenue au point 2.2.1.
- Aliquote 500 μL de la suspension cellulaire dans le nombre calculé de tubes microcentrifugeuses de 1,5 mL (1 x 107 cellules/tube).
- Faire tourner vers le bas à 480 x g pendant 10 min à 4 °C.
- Retirez le surnageant à l’aide d’un système d’aspiration de paillasse et congelez les pastilles cellulaires dans de l’azote liquide. Conserver les granulés de cellules sèches à -80 °C.
REMARQUE: Les échantillons peuvent être conservés pendant au moins 1 an.
- Estimer le nombre total de cellules à réticuler.
- Lyse cellulaire
- Décongeler le(s) granulé(s) congelé(s) sur la glace.
- Préparer 1,5 mL de tampon de lyse dans H 2 0 par échantillon : Ajouter 10 mM de Tris-HCl, pH 8,0, 10 mM de NaCl et0,2% de NP40.
- Ajouter 600 μL du tampon de lyse froide et bien remettre en suspension sur de la glace.
- Incuber sur de la glace pendant 15 min pour laisser les cellules gonfler.
- Faire tourner à 2655 x g pendant 5 min à 4 °C et retirer le surnageant à l’aide d’un système d’aspiration de paillasse.
- Pour enlever les débris, remettre la pastille en suspension dans 1 mL du tampon de lyse à froid, faire tourner vers le bas à 2655 x g pendant 5 min à 4 °C et retirer le surnageant.
- Tourner à nouveau brièvement à 2655 x g et 4 °C et enlever autant de surnageant restant que possible à l’aide d’un système d’aspiration de paillasse muni d’un embout P200.
- Resuspendre dans 100 μL de FDS à 0,5 % (vol/vol).
- Incuber dans un thermomélangeur à 62 °C, en tourbillonnant à 1400 tr/min pendant 10 min.
- Ajouter 290 μL deH2O+ 50 μL de TritonX-100 à 10% et bien mélanger, en évitant les bulles d’air.
- Incuber dans un thermomélangeur à 37 °C, en tourbillonnant à 1400 tr/min pendant 15 min.
- Ajouter 50 μL de tampon Dpnll 10x et retourner le tube pour mélanger.
- Prélevez 50 μL d’ADN non digéré pour le contrôle de la qualité dans un tube séparé. N’oubliez pas de prélever l’échantillon témoin non digéré.
- Digestion DpnII
- Ajouter 10 μL de Dpnll à haute concentration (500 U au total) et mélanger en retournant.
- Incuber les échantillons et le témoin non digéré dans un thermomélangeur à 37 °C, en tourbillonnant à 1400 tr/min pendant >4 h.
- Ajouter 10 μL de Dpnll à haute concentration (500 U au total) à la fin de la journée.
- Incuber les échantillons et le témoin non digéré à 37 °C, en tourbillonnant à 1400 tr/min pendant la nuit.
- Ajouter 10 μL de Dpnll à haute concentration (500 U au total) au début de la journée suivante aux échantillons.
- Incuber les échantillons et le témoin non digéré dans un thermomélangeur à 37 °C, en tourbillonnant à 1400 tr/min pendant 4 h.
- Ligature et inversion de la réticulation
- Incuber les tubes à 65 °C pendant 20 min à 1400 tr/min.
REMARQUE : N’ajoutez pas de FDS à ce stade. L’idée est de préserver l’intégrité nucléaire, de sorte que la ligature est effectuée à l’intérieur des noyaux, en contournant la nécessité d’une dilution extrême. - Refroidir les échantillons sur de la glace pendant un maximum de 5 à 10 minutes. Pour éviter les précipitations de la SDS, ne laissez pas les échantillons sur la glace plus longtemps que cela.
- Prélevez 50 μL de l’ADN digéré non ligaturé pour le contrôle de la qualité dans un tube séparé. Conserver les témoins non digérés et non ligaturés à -20 °C.
REMARQUE: N’oubliez pas de prélever l’échantillon de contrôle non ligaturé. - Ajouter 800 μL de cocktail de ligature : 122 μL de tampon ligase 10x, 8 μL de T4 ligase (30 U/μL) et 670 μL deH200.
- Incuber à 16 °C, en tourbillonnant à 1000 tr/min pendant la nuit.
- Ajouter 7,5 μL de protéinase K (20 mg/mL) aux échantillons et 2 μL aux témoins.
- Incuber à 65 °C pendant 4 h à 1000 tr/min.
- Incuber les tubes à 65 °C pendant 20 min à 1400 tr/min.
- Purification de l’ADN
- Transférer les échantillons sur de la glace dans des tubes à centrifuger coniques prérefroidis de 15 mL et ajouter 2 mL d’eau, 10,5 mL d’EtOH glacé et 583 μL de NaAC 3 M.
NOTE: L’eau supplémentaire vise à empêcher le transfert de DTT dans le granulé. - Ajouter 200 μL d’EtOH glacé, 10,8 μL de NaAC et 1 μL de coprécipitant aux contrôles de qualité non digérés et non ligaturés.
- Incuber à -80 °C pendant au moins 4 h jusqu’à la nuit.
- Faire tourner les tubes de 15 mL à 2200 x g à 4 °C pendant 45 min.
- Faire tourner les tubes de contrôle de 1,5 mL à 20 500 x g à 4 °C pendant 30 min.
- Laver une fois avec 3 mL (échantillons) et 1 mL (témoins) d’EtOH glacé à 70 %.
- Faire tourner à 2200 x g (échantillons) ou 20 500 x g (témoins) à 4 °C pendant 10 min.
- Retirer soigneusement l’EtOH et sécher à l’air libre à TA pendant 10-15 min; Ne pas trop sécher.
- Resuspendre les échantillons et les témoins dans 100 μL et 20 μL de H20, respectivement.
- Ajouter 1 μL d’ARNseA et incuber à 37 °C en tourbillonnant à 1400 tr/min pendant 30 min.
- Transférer les échantillons sur de la glace dans des tubes à centrifuger coniques prérefroidis de 15 mL et ajouter 2 mL d’eau, 10,5 mL d’EtOH glacé et 583 μL de NaAC 3 M.
- Contrôle de la qualité de la préparation du modèle 3C
- Quantifier chaque échantillon et contrôle à l’aide d’un kit de fluoromètre pour des mesures de concentration d’ADN à haute sensibilité.
- Charger 100-200 ng de chaque échantillon et de chaque témoin sur un gel d’agarose à 1 % / 1x TBE.
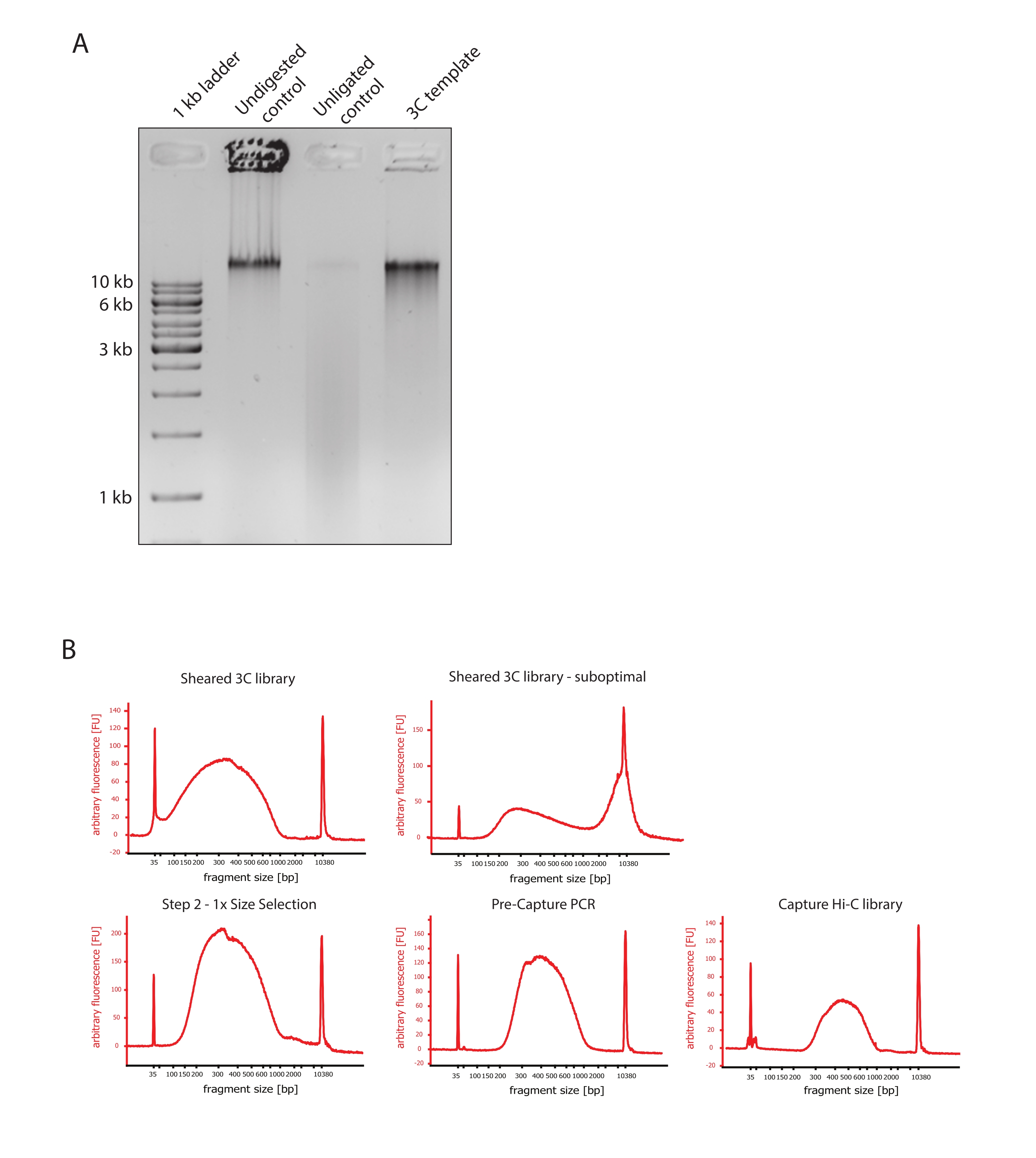
- Vérifiez que l’image du gel montre le résultat attendu en comparant les différences dans la taille des fragments d’ADN des témoins et du modèle 3C, comme illustré à la figure 2A.
- Conserver les échantillons et les témoins à -20 °C.
- Hybridation, capture et traitement d’échantillons pour le séquençage multiplexé
- Pour hybrider le réseau de sondes d’ARN biotinylées au modèle 3C, capturer les fragments de ligature ciblés et préparer les échantillons pour le séquençage multiplexé selon le système d’enrichissement cible utilisé dans cette étude pour le séquençage multiplexé à extrémités appariées (voir Tableau des matériaux). Suivez le protocole selon les instructions du fabricant tout en introduisant les modifications mineures suivantes:
- Section 2 du protocole du fabricant : Préparation de l’échantillon
- Suivez les instructions pour l’enrichissement de la cible à partir de 3 μg d’entrée d’ADNg.
- Cisailler l’ADN dans un sonicateur en utilisant les spécifications suivantes: 10% de rapport cyclique, 4 intensité, 200 cyc/burst et 130 s. Commencez avec 4 μg de matrice 3C remise en suspension dans 130 μL d’eau pour chaque réaction de capture afin d’assurer suffisamment de matériel pour poursuivre la préparation de l’échantillon avec 3 μg d’ADN cisaillé.
- Évaluer la qualité de l’ADN cisaillé. Exécutez 1 μL de l’ADN cisaillé sur un bioanalyseur d’ADN selon le protocole haute sensibilité. Attendez-vous à une distribution de la taille des fragments comprise entre 150 et 700 pb (Figure 2).
- Purifier l’échantillon à l’aide de billes d’immobilisation réversible en phase solide (SPRI). Ajouter 124 μL de billes SPRI à 124 μL de l’échantillon d’ADN pour effectuer une sélection de taille du côté gauche 1:1 selon les instructions du fabricant et éluer dans 25 μL d’eau sans nucléase. Cette étape de purification permettra d’enlever les fragments plus courts pour enrichir les fragments d’environ 300 pb (Figure 2).
REMARQUE : La quantité d’échantillons et de billes SPRI utilisée à cette étape tient compte de la perte de volume qui s’est produite lors du transfert des échantillons dans de nouveaux tubes et de l’exécution des contrôles de qualité au bioanalyseur. Toutes les étapes ultérieures de sélection de la taille sont effectuées selon les ratios recommandés par le protocole du fabricant. L’élution de l’ADN des billes SPRI est effectuée à RT tout au long du protocole. - Évaluer la qualité de l’ADN cisaillé de taille sélectionnée. Exécutez 1 μL de l’ADN cisaillé sur le bioanalyseur d’ADN selon le protocole haute sensibilité (HS). Attendez-vous à une distribution des tailles de fragments avec l’enrichissement le plus élevé à 300 pb (Figure 2). Allez de l’avant avec la quantification de l’ADN cisaillé si le cisaillement a réussi.
- Quantifier l’ADN cisaillé avec un kit de fluoromètre pour les mesures de concentration d’ADN HS.
REMARQUE: Si le cisaillement de l’ADN donne un rendement en ADN de <3 μg, effectuez un deuxième cycle de cisaillement de l’ADN avec 4 μg d’ADN supplémentaires et combinez les échantillons d’ADN cisaillés après la première étape de purification des billes SPRI pour obtenir un total de 3 μg d’ADN cisaillé. - Ajouter de l’eau exempte de nucléases à l’échantillon d’ADN nettoyé de taille sélectionnée (3 μg au total) jusqu’à un volume final de 48 μL et procéder à la réaction de réparation finale selon le protocole du fabricant.
- Après la ligature des adaptateurs appariés, amplifiez la bibliothèque en effectuant cinq cycles de PCR pré-capture selon les instructions du fabricant (les conditions de PCR et les amorces sont fournies dans le kit).
- Section 4 du protocole du fabricant : Hybridation et capture
- Pour hybrider les échantillons d’ADN préparés aux sondes d’ARN spécifiques à la cible, diluer 750 ng d’échantillons d’ADN dans un volume final de 3,4 μL, ce qui donne une concentration initiale de 221 ng / μL. Pour les échantillons d’ADN dilués dans de plus grands volumes, utiliser un concentrateur à vide rapide pour réduire au volume final. Une concentration de vide rapide (250 x g; ≤45 °C) pendant 15-20 min est normalement suffisante pour les échantillons remis en suspension dans 10 μL. Assurez-vous d’avoir le même volume d’entrée pour chaque échantillon avant de démarrer le concentrateur de vide rapide.
- Incuber le mélange d’hybridation pendant 16-18 h à 65 °C avec un couvercle chauffé à 105 °C selon les instructions du fabricant.
- Section 5 du protocole du fabricant : Indexation et traitement des échantillons pour le séquençage multiplexé
- Pour amplifier les bibliothèques capturées avec des amorces d’indexation, effectuez 12 cycles de PCR post-capture selon les instructions du fabricant (les conditions de PCR et les amorces sont fournies dans le kit).
- Section 2 du protocole du fabricant : Préparation de l’échantillon
- Pour hybrider le réseau de sondes d’ARN biotinylées au modèle 3C, capturer les fragments de ligature ciblés et préparer les échantillons pour le séquençage multiplexé selon le système d’enrichissement cible utilisé dans cette étude pour le séquençage multiplexé à extrémités appariées (voir Tableau des matériaux). Suivez le protocole selon les instructions du fabricant tout en introduisant les modifications mineures suivantes:
- Séquençage nouvelle génération
- Pour exécuter plusieurs bibliothèques Hi-C de capture sur la même cellule de flux, préparez un mélange équimolaire des bibliothèques de capture et séquencez 100 à 120 millions de lectures par bibliothèque.
- Si l’analyse spécifique à l’allèle est nécessaire, séquencer 150 pb à l’extrémité appariée pour assurer une couverture SNP suffisante.
3. Analyse des données
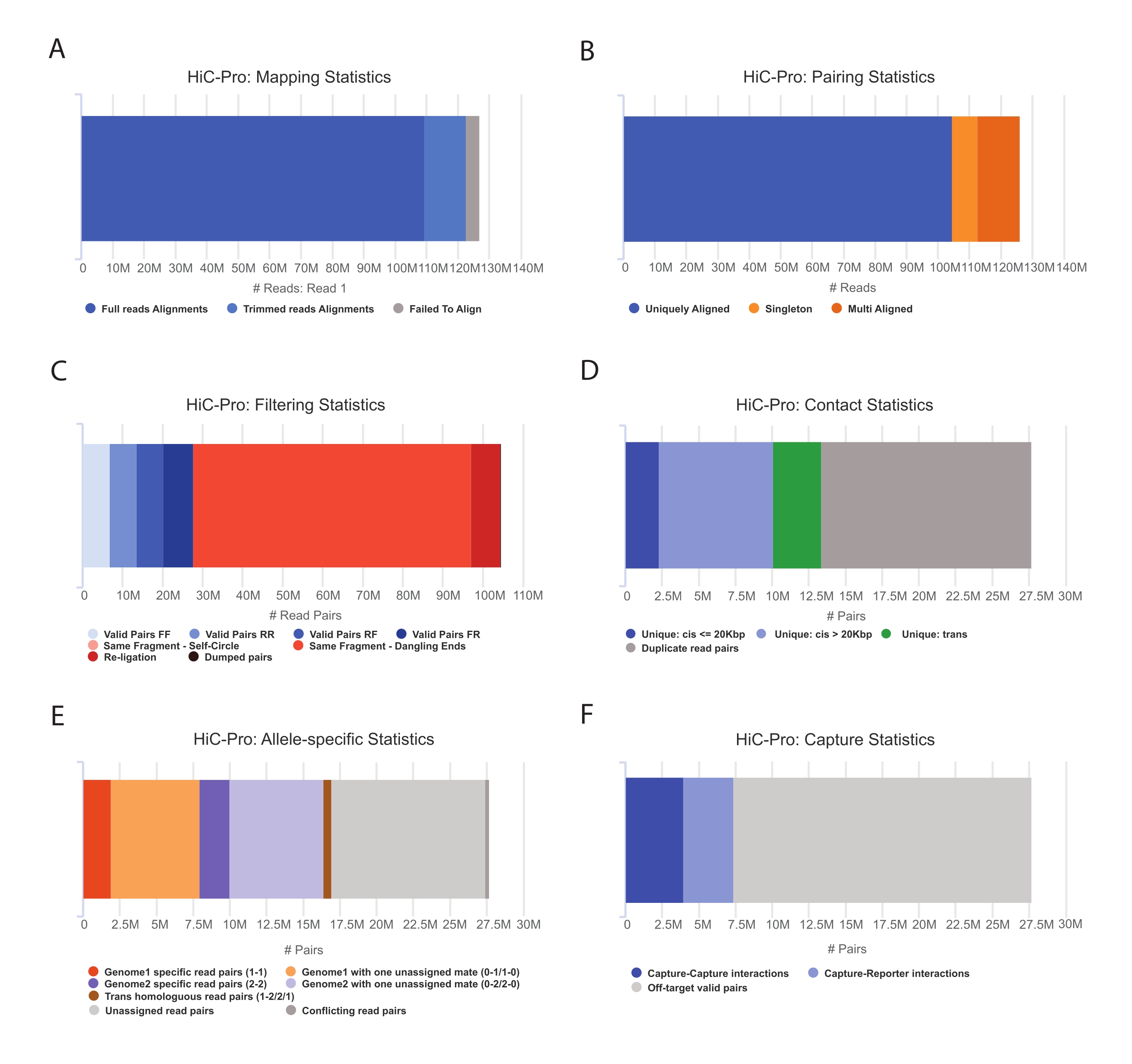
- Appliquez le pipeline HiC-Pro pour effectuer l’analyse des données Capture Hi-C55. HiC-Pro fournit des contrôles de qualité à chaque étape du traitement, notamment (Figure 3) :
(i) Le taux d’alignement sur le génome de référence spécifiant la fraction de lectures couvrant un site de ligature, ainsi que le nombre de paires et de singletons.
(ii) La fraction de produits de ligature valides et de paires de lecture non informatives (extrémité pendante, auto-ligature, etc.).
(iii) La fraction des contacts à courte / longue portée et intra / inter-chromosomiques.
(iv) Fraction des contacts sur cible pour Capture Hi-C.
(v) Fraction des lectures spécifiques aux allèles si spécifiée.
REMARQUE: HiC-Pro prend en charge une large gamme de protocoles, y compris Hi-C in situ et Capture Hi-C. Dans ce dernier cas, il suffit à l’utilisateur de spécifier la région ciblée (format BED) dans le fichier de configuration. Une fois les données traitées, les sorties HiC-Pro peuvent facilement être converties en un objet plus froid pour l’analyse en aval56. À cette étape, les cartes de contact à différentes résolutions sont normalisées à l’aide de la méthode ICE décrite précédemment par Imakaev et ses collègues57. Plusieurs analyses peuvent ensuite être effectuées pour appeler des compartiments chromosomiques, des TAD ou des boucles de chromatine (pour la revue58). Le flux de travail du protocole est illustré à la figure 4. Ici, la suite « cooltools » est appliquée pour calculer le score d’isolation et les limites des TAD, comme illustré à la figure 5 et à la figure 659.
Résultats
Le protocole Capture Hi-C décrit est basé sur la préparation du modèle 3C à l’échelle du génome à l’aide d’un cutter à quatre bases (DpnII). L’enrichissement ultérieur des fragments de ligature dans la région génomique d’intérêt est obtenu par hybridation d’un réseau de sondes à ARN en pavage et leur capture à base de streptavidine selon le système d’enrichissement de la cible utilisé dans cette étude (Figure 1). Les sondes d’ARN biotinylées ont été sélectionnées car elles montrent une affinité de liaison plus étroite avec leurs cibles par rapport aux sondes ADN52,60. Les bibliothèques capturées sont ensuite indexées et regroupées pour le séquençage multiplexé à haut débit. Les données Hi-C de capture peuvent être visualisées sous forme de cartes d’interaction Hi-C haute résolution, mais aussi sous forme de cartes de contact à point de vue unique de type 4C pour visualiser spécifiquement les interactions de séquences plus petites telles que des promoteurs ou des amplificateurs dans toute la région capturée. Le flux de travail du protocole est illustré à la figure 4. Les contrôles de qualité pré-séquençage sont illustrés à la figure 2 et comprennent l’évaluation de la digestion et de la religature appropriées du gabarit 3C ainsi que son cisaillement et sa purification efficaces à travers les différentes étapes du protocole. L’ADN du modèle 3C cisaillé devrait atteindre entre 150 et 700 pb, et aucun enrichissement de fragments de >2 kb ne devrait être détecté. Au cours des étapes suivantes, plusieurs étapes de nettoyage de l’ADN et de sélection de la taille à base de billes sont effectuées, d’abord après le cisaillement, puis après les PCR pré-capture et post-capture. Les bibliothèques nettoyées présentent un profil d’enrichissement de fragments distinct tel que visualisé sur un bioanalyseur d’ADN haute sensibilité (Figure 2). La taille moyenne des fragments augmente au cours de la préparation de la bibliothèque en raison de la ligature des adaptateurs, du séquençage et des amorces d’indexation. Les contrôles de qualité post-séquençage sont obtenus via Hi-C Pro et illustrés à la figure 3. De nombreuses applications logicielles bioinformatiques différentes ont été proposées pour le traitement et l’analyse de données de type 3C. Parmi elles, le pipeline HiC-Pro est l’une des solutions les plus populaires, permettant le traitement des données brutes de séquençage vers les cartes de contact finales à différentes résolutions55. HiC-Pro utilise une stratégie de cartographie en deux étapes pour aligner les lectures de séquençage sur le génome de référence. Les produits 3C sont ensuite reconstruits et filtrés pour supprimer les paires de contacts non informatives et générer les cartes de contacts. En outre, il est capable d’utiliser une liste de polymorphismes connus pour effectuer une analyse spécifique aux allèles et pour séparer les contacts provenant des deux allèles parentaux dans des cartes de contact distinctes. Plus récemment, HiC-Pro a été inclus et étendu dans le cadre nf-core (nf-core-hic), fournissant un pipeline hautement évolutif et reproductible piloté par la communauté61,62.
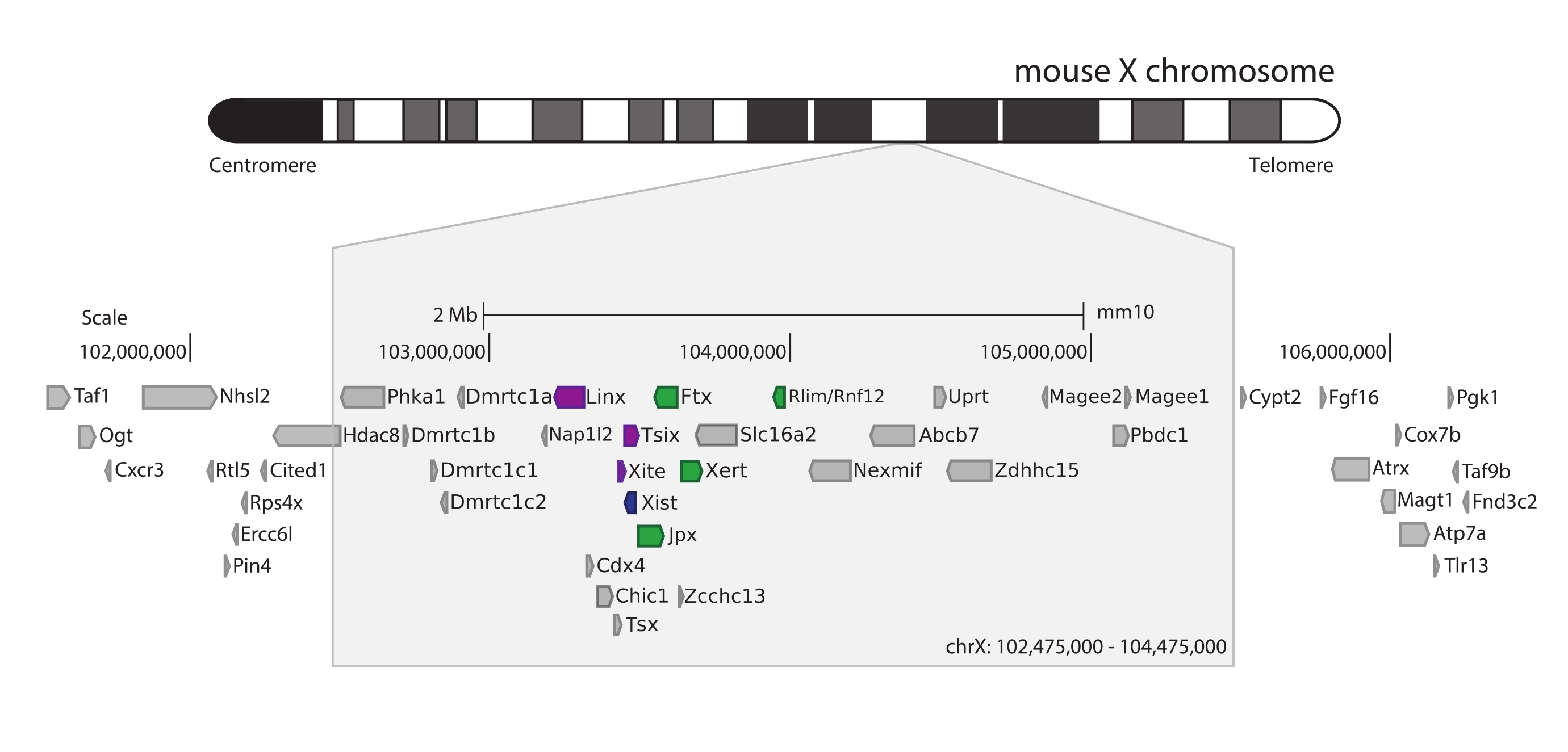
Pour capturer la souris Xic, un réseau de 28 913 sondes d’ARN tuilant 3 Mb du chromosome X a été conçu. Cette région comprend l’acteur clé de XCI, le gène long non codant Xist, et son paysage réglementaire connu de ~800 kb (Figure 5). Cette région ~800 kb est divisée en deux TAD: l’un incluant le promoteur Xist et ses régulateurs positifs connus (c’est-à-dire les transcrits non codants Ftx, Jpx et Xert et le gène codant pour la protéine Rnf12), et le TAD voisin englobant les cis-régulateurs négatifs de Xist (c’est-à-dire son transcrit antisens Tsix, l’élément amplificateur Xite et le transcrit non codant Linx) (pour examen44, 45).
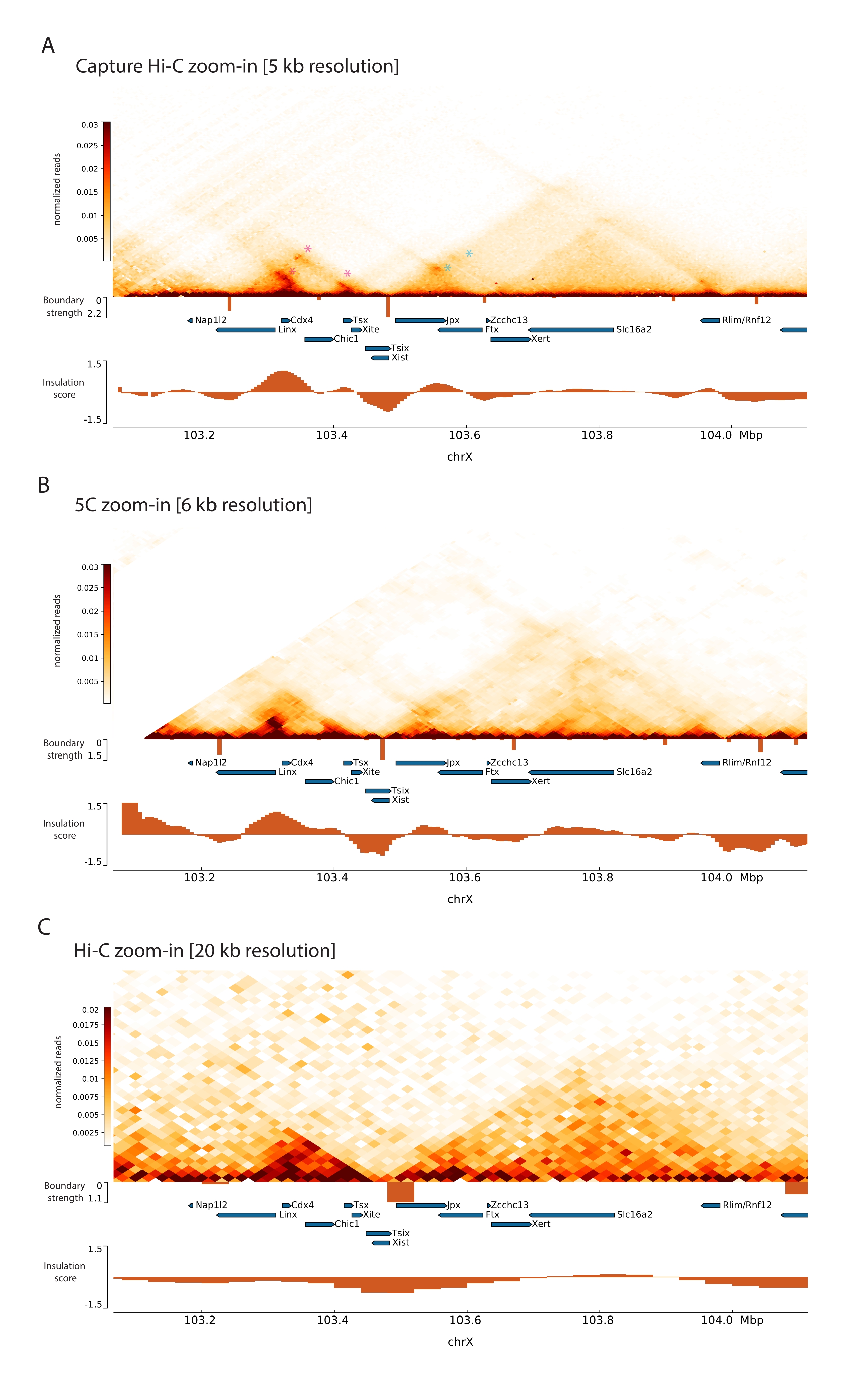
En appliquant le protocole Capture Hi-C décrit au Xic, l’organisation topologique de ce locus a été obtenue avec une résolution sans précédent (Figure 6 et Figure 7). Cela est particulièrement évident lorsque l’on compare le profil Capture Hi-C au 5C47 précédemment publié (Figure 6 et Figure 7; Tableau supplémentaire 1) et Hi-C61 (Figure 6 et Figure 7; Tableau supplémentaire 1) Profils. Par exemple, les structures sous-TAD sont plus évidentes — le TAD contenant le promoteur Xist (Xist-TAD) est clairement subdivisé en deux domaines plus petits (Figure 6A, pointe de flèche bleue). Auparavant, cela ne pouvait être visuellement « deviné » qu’à partir du profil 5C (Figure 6B), bien que la détection d’une limite dans cette région à l’aide de l’algorithme de score d’isolation. De même, la résolution du profil Capture Hi-C permet l’identification de deux domaines plus petits dans le TAD voisin (Figure 6A, B), qui contient le promoteur du locus Tsix ( Tsix-TAD ) ; cela n’avait pas été réalisé auparavant avec 5C (figure 6B). Il convient de noter que les limites topologiques déterminées par le score d’isolation des données de capture Hi-C et 5C sont généralement détectées à des endroits légèrement différents et avec des forces relatives différentes.
De plus, d’autres structures sous-TAD telles que les boucles de contact sont clairement visibles à partir des données de Capture Hi-C, telles que la boucle entre Xist et Ftx (Figure 7A), précédemment identifiée avec Capture-C63, et la boucle entre Xist et Xert (Figure 7B), récemment identifiée à l’aide d’un protocole similaire pour Capture Hi-C48. D’autres contacts peuvent également être cartographiés plus précisément en raison de la résolution accrue des profils Capture Hi-C, tels que ceux formant les points chauds de contact connus dans le Tsix-TAD entre les loci Linx, Chic1 et Xite (Figure 7A).
Par rapport aux données Hi-C présentées à la figure 7, Capture Hi-C a permis de multiplier par quatre la résolution, mais il n’a nécessité qu’un quart de la profondeur de séquençage (c.-à-d. 126 M de lectures contre 571 M) (tableau supplémentaire 1). Cette augmentation de la résolution permet de détecter les subTADs et les interactions en boucle qui n’ont pas pu être détectées par Hi-C à la profondeur de séquençage illustrée à la figure 6 et à la figure 7. Le protocole décrit pour Capture Hi-C permet donc une caractérisation beaucoup plus détaillée et à haute résolution d’une grande région génomique d’intérêt, par rapport aux approches précédentes.

Figure 1 : Conception de la sonde. Représentation schématique de la stratégie utilisée pour la conception de la sonde. Des régions de 300 pb en amont et en aval de chaque site de restriction DpnII dans la région cible de 3 Mb ont été sélectionnées et carrelées avec des sondes d’ARN biotinylées qui se chevauchent. L’une de ces régions sélectionnées est affichée, chrX : 102 474 805-102 475 500. Pas plus de 40 bases de séquences répétitives sont autorisées dans chaque sonde. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Capture des contrôles de qualité de pré-séquençage Hi-C. (A) Exemple représentatif de contrôles de qualité du modèle 3C. 200 ng d’ADN ont été chargés sur un gel d’agarose à 1%. Voie 1 : échelle de 1 ko. Voie 2 : La chromatine non digérée, réticulée et intacte fonctionne comme une bande pointue à >10 kb. Voie 3 : La chromatine réticulée digérée par DpnII se présente comme un frottis d’une taille comprise entre 1 kb et 3 kb. Lane 4: Bibliothèque ou modèle 3C final; les extrémités libres des fragments d’ADN réticulé digérés sont re-ligaturées. Le frottis d’ADN de taille moléculaire inférieure est presque indétectable et le produit de ligature est détecté comme une bande de >10 kb. (B) Exemples représentatifs de profils ADN de bioanalyseurs à haute sensibilité. En haut à gauche : bibliothèque 3C cisaillée avec succès montrant une distribution de la taille des fragments comprise entre 150 bp et 700 bp. En haut à droite : bibliothèque 3C cisaillée insatisfaisante. L’ADN non cisaillé est détecté comme un enrichissement large de fragments >2 kb. (C) En bas à gauche : échantillon d’ADN cisaillé suivant une sélection de taille latérale gauche 1:1 à l’aide de billes SPRI. Des fragments de ~300 pb sont enrichis. En bas au milieu : profil PCR pré-capture après ligature des adaptateurs d’extrémité appariés selon le protocole du fabricant. En bas à droite : bibliothèque Capture Hi-C finale comprenant les adaptateurs, le séquençage et les amorces d’indexation pour le séquençage multiplexé. Abréviations : bp = paires de bases, FU = unité de fluorescence arbitraire. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Capture des contrôles qualité post-séquençage Hi-C avec HiC-Pro. (A) Exemple de taux de cartographie sur le génome de référence pour le premier compagnon des paires de séquençage. La fraction bleu clair représente les lectures alignées par HiC-Pro et couvrant une jonction de ligature. Cette métrique peut ainsi être utilisée pour valider l’étape expérimentale de ligature. (B) Une fois que les partenaires de séquençage sont alignés sur le génome, seules les paires de lecture alignées de manière unique sont conservées pour analyse. (C) Les paires non valides (en rouge) telles que l’extrémité pendante, l’auto-cercle ou la religature sont écartées de l’analyse. La fraction de paires valides est un bon indicateur de l’efficacité de la ligature et de la pull-down. (D) Les paires valides peuvent être divisées en contacts intra/interchromosomiques et à court/longue portée. Les paires de lecture dupliquées susceptibles de représenter des artefacts de PCR sont éliminées de l’analyse. (E) Pour l’analyse spécifique aux allèles, HiC-Pro indique le nombre de lectures alléliques prises en charge par un ou deux partenaires pour chaque génome parental (c.-à-d. C57BL/6J x CASTEi/J). La même fraction de lectures attribuées à l’allèle maternel et paternel est attendue. (F) Enfin, seules les paires valides chevauchant la région de capture sont sélectionnées pour construire les cartes de contact. Les paires capture-capture représentent les contacts au sein de la région ciblée, tandis que les paires capture-rapporteur impliquent une interaction entre la région ciblée et une région hors cible. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

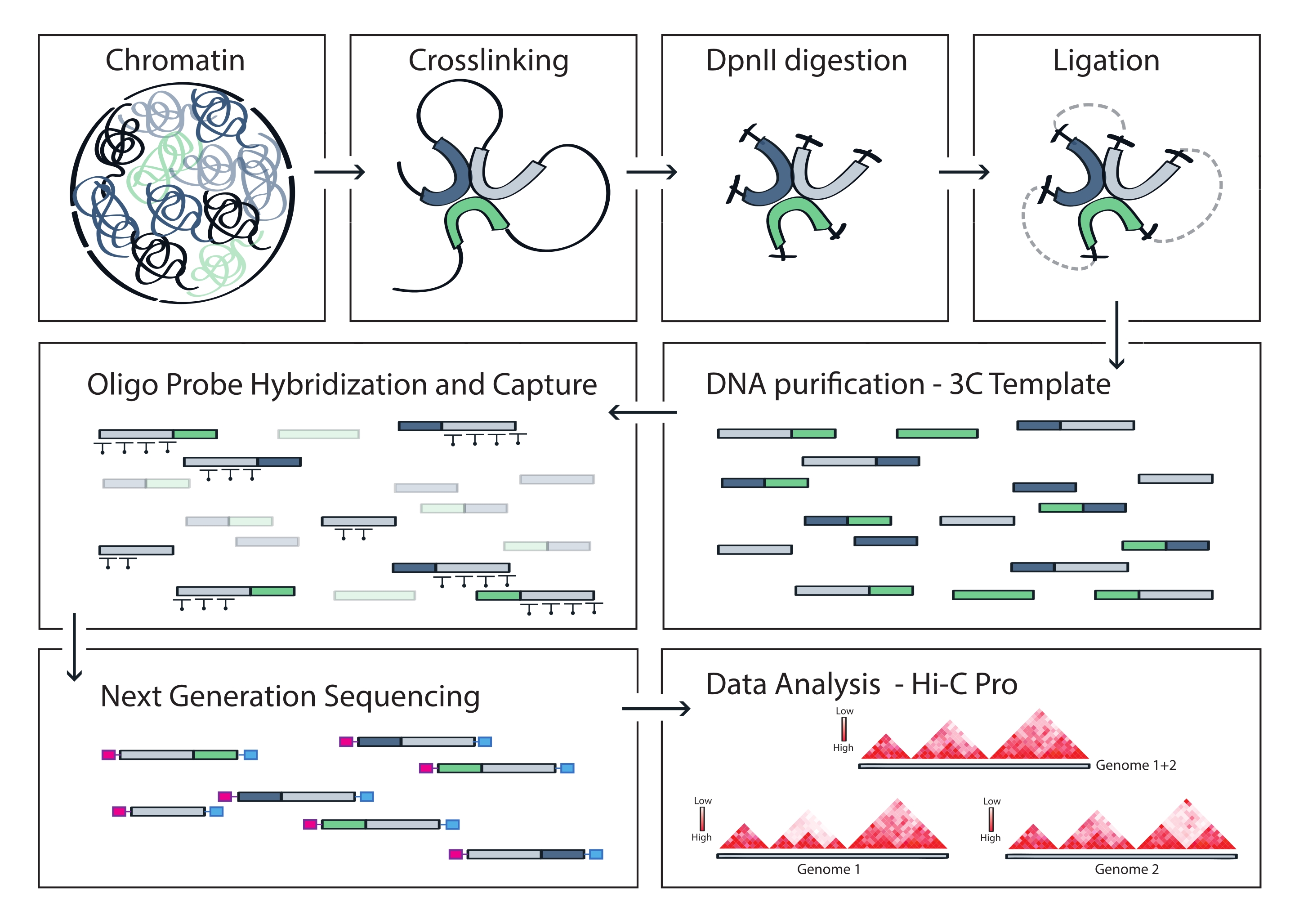
Figure 4 : Workflow du protocole Capture Hi-C. Représentation schématique des différentes étapes du protocole. Pour générer la matrice 3C à l’échelle du génome, la chromatine est d’abord réticulée avec le formaldéhyde, puis digérée avec l’enzyme de restriction DpnII. Les extrémités de l’ADN libre sont ensuite re-ligaturées, la réticulation est inversée et l’ADN est purifié. Pour enrichir les fragments englobant la région cible, un réseau de sondes d’ARN biotinylées est hybridé au modèle 3C et capturé par tirage médié par la streptavidine. Les bibliothèques de capture sont traitées pour le séquençage multiplexé, et des fragments de ligature valides sont quantifiés pour déduire la fréquence des contacts de chromatine à travers la cible, qui sont visualisés sous forme de cartes d’interaction haute résolution. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 5: Vue d’ensemble de la région englobant le Xic sur le chromosome X de la souris. Représentation schématique du chromosome X de la souris et zoom avant de la région capturée de 3 Mo (ChrX : 102 475 000-105 475 000). La région ciblée comprend ~800 kb d’ADN correspondant au Xic, le locus régulateur maître de XCI. Le Xic comprend les gènes longs non codants, Xist, un acteur clé de XCI, et son paysage réglementaire. Les régulateurs positifs de Xist sont représentés en vert et les régulateurs négatifs en violet. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 6 : Capturez les cartes d’interaction Hi-C, 5C et Hi-C dans la région capturée de 3 Mo. (A) Capturer la carte d’interaction Hi-C de la cible de 3 Mo englobant la souris Xic à une résolution de 10 kb (cette étude). (B) Carte d’interaction 5C de la même région cible que dans A à une résolution de 6 kb (données retraitées à partir de47). Les régions répétitives non incluses dans les analyses sont masquées en blanc. Les données 5C nécessitent leur propre traitement bioinformatique (voir47). Après le nettoyage et l’alignement, les cartes 5C à la résolution de l’amorce sont regroupées à l’aide d’une médiane courante (fenêtre = 30 kb, étape = 5) pour atteindre une résolution finale de 6 ko. (C) Carte d’interaction Hi-C de la même région génomique que dans A et B à une résolution de 40 kb (données retraitées à partir de64). Toutes les cartes d’interaction ont été générées à partir d’ESC de souris. Le score d’isolation a été calculé à l’aide de cooltools et est représenté sous forme d’histogrammes avec des minimaux d’isolation aux limites TAD. Les limites TAD sont indiquées sous forme de lignes verticales sous la carte. La hauteur de chaque ligne indique la force des limites. Les gènes sont représentés par des flèches pointant dans le sens de la transcription. Les limites des sous-TAD qui sont détectées exclusivement ou plus précisément dans les cartes Capture Hi-C sont indiquées par des pointes de flèches magenta et bleues pour les sous-TAD dans les TAD Tsix et Xist, respectivement. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 7 : Capturez les cartes d’interaction Hi-C, 5C et Hi-C sur 1 Mo dans la région capturée. (A) Capturer la carte d’interaction Hi-C de la région génomique de 1 Mb englobant la souris Xic à une résolution de 5 kb (cette étude). (B) Carte d’interaction 5C de la même région génomique que dans A. à une résolution de 6 Ko (données retraitées à partir de47). Les régions répétitives non incluses dans les analyses sont masquées en blanc. Il convient de noter que les données 5C nécessitent leur propre traitement bioinformatique (voir47). Après le nettoyage et l’alignement, les cartes 5C à la résolution de l’amorce sont regroupées à l’aide d’une médiane courante (fenêtre = 30 kb, étape = 5) pour atteindre une résolution finale de 6 ko. (C) Carte d’interaction Hi-C de la même région génomique que dans A et B de Hi-C à une résolution de 20 kb (données retraitées à partir de64). Toutes les cartes d’interaction ont été générées à partir de CSEm. Le score d’isolation a été calculé à l’aide de cooltools et est représenté sous forme d’histogrammes avec des minimaux d’isolation aux limites TAD. Les limites TAD sont indiquées sous forme de lignes verticales sous la carte. La hauteur de chaque ligne indique la force des limites. Les gènes sont représentés par des flèches pointant vers la direction de la transcription. Les boucles de contact détectées exclusivement ou plus précisément dans Capture Hi-C sont indiquées par des astérisques magenta et bleus pour les boucles dans les TAD Tsix et Xist, respectivement. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Tableau supplémentaire 1 : Statistiques post-séquençage pour les ensembles de données utilisés dans ce manuscrit : Capture Hi-C (cette étude), Hi-C64 et 5C47. Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Nous décrivons ici un protocole Capture Hi-C relativement rapide et facile pour caractériser l’organisation d’ordre supérieur de régions génomiques de la taille d’une mégabase à une résolution de 5 à 10 ko. Capture Hi-C appartient à la famille des technologies Capture-C conçues pour enrichir les interactions chromatines ciblées à partir de modèles 3C ou Hi-C à l’échelle du génome. À ce jour, la grande majorité des applications de Capture-C ont été exploitées pour cartographier les contacts chromatiniques d’éléments régulateurs relativement petits dispersés dans l’ensemble du génome. Dans le premier protocole Capture-C, plusieurs sondes biotinylées à ARN superposées ont été utilisées pour capturer >400 promoteurs présélectionnés dans des bibliothèques 3C préparées à partir de cellules érythroïdes31. La même stratégie a ensuite été améliorée dans Next Generation (NG) et Nuclear Titrated (NuTi) Capture-C pour obtenir des profils d’interaction haute résolution de >8 000 promoteurs en utilisant des appâts d’ADN uniques de 120 pb couvrant des sites de restriction uniques et deux cycles séquentiels de capture pour maximiser l’enrichissement des fragments de ligature informative32,40. Ces stratégies ont conduit à la dissection fonctionnelle d’éléments agissant sur cis dans de nombreux contextes différents, y compris le développement embryonnaire de souris, la différenciation cellulaire, l’inactivation du chromosome X et la mauvaise régulation des gènes dans des conditions pathologiques 46,63,65,66,67,68,69,70,71.
Dans Promoter Capture Hi-C (PCHi-C), > 22 000 promoteurs annotés contenant des fragments de restriction ont été extraits des bibliothèques Hi-C par hybridation de sondes biotinylées à ARN unique 120-mer à l’une ou l’autre ou aux deux extrémités du fragment de restriction34,72. Cette méthode a permis de disséquer l’interactome de milliers de promoteurs dans un nombre croissant de types cellulaires, y compris les cellules souches embryonnaires de souris, les cellules hépatiques fœtales et les adipocytes 34,35,72,73, mais aussi les lignées lymphoblastoïdes humaines, les progéniteurs hématopoïétiques, les kératinocytes épidermiques et les cellules pluripotentes 37,74,75,76,77 .
En comparaison avec ces technologies d’enrichissement cible, Capture Hi-C cible des régions génomiques contiguës jusqu’à l’échelle de la mégabase, couvrant ainsi un ou plusieurs TAD et englobant les paysages régulateurs des gènes. Toute la région d’intérêt doit être carrelée avec un ensemble de sondes biotinylées englobant chaque site de restriction DpnII dans la cible. L’hybridation de la matrice biotinylée au modèle 3C, sa capture ultérieure à base de streptavidine et le traitement pour le séquençage multiplexé sont effectués à l’aide d’un système d’enrichissement de cible pour le séquençage multiplexé Illumina Paired-End. L’ensemble du protocole est rapide, car il peut être effectué en 1 semaine, de la préparation de la bibliothèque 3C au séquençage NGS, et il ne nécessite que des adaptations mineures et / ou un dépannage personnalisé.
Le protocole offre également des avantages par rapport à d’autres méthodes basées sur 3C. Pour obtenir des cartes d’interaction à une résolution de 5 à 10 Ko, nous avons séquencé 100 à 120 M de lectures d’extrémités appariées. À titre de comparaison, nous avons utilisé ici un ensemble de données Hi-C de 571 M lectures pour atteindre une résolution de 20 kb64 (GSM2053973), et au moins 1 milliard de lectures seraient nécessaires pour atteindre une résolution de 5 kb avec Hi-C22 à l’échelle du chromosome.
La capture Hi-C telle qu’utilisée dans la présente étude atteint une résolution beaucoup plus élevée que la 5C précédemment publiée sur la base d’une enzyme de restriction de coupure47 à 6 pb (tableau supplémentaire 1). Il est important de noter que la stratégie conçue pour enrichir et amplifier les interactions ciblées dans 5C ne permet pas une analyse spécifique des allèles des interactions chromatines. Au contraire, les données Capture Hi-C peuvent être cartographiées de manière spécifique à l’allèle, permettant la dissection des paysages structurels 3D de paires de chromosomes homologues, par exemple dans des cellules humaines ou dans des lignées cellulaires hybrides F1 dérivées en croisant génétiquement différentes souches de souris78. Pour générer des cartes d’interaction Capture Hi-C spécifiques aux allèles à une résolution de 5 Ko, nous avons séquencé des lectures d’extrémité appariées de 150 pb pour augmenter la couverture SNP. Des approches similaires spécifiques aux allèles peuvent être appliquées aux lignées cellulaires humaines, pour lesquelles l’annotation des SNP est disponible22.
Il est important de noter que, bien que Capture Hi-C assure généralement une haute résolution tout en améliorant l’abordabilité des coûts de séquençage, la production d’oligonucléotides biotinylés sur mesure a un impact sur le coût global de cette méthode. Par conséquent, le choix de la méthode 3C la plus appropriée différera pour différentes applications et dépendra de la question biologique abordée et de la résolution requise, ainsi que de la taille de la région d’intérêt. D’autres protocoles Capture Hi-C développés partagent des fonctionnalités clés avec le protocole décrit ici. Par exemple, une stratégie Capture Hi-C a été appliquée pour caractériser des régions génomiques de ~50 kb à 1 Mb couvrant des variantes non codantes associées au risque de cancer du sein et colorectal; dans ce protocole, les régions cibles ont été retirées des bibliothèques Hi-C en hybridant des appâts à ARN 120-mère en marquant les régions cibles à une couverture3x 33,38,79. De même, HYbrid Capture Hi-C (Hi-C 2) a été utilisé pour cibler les interactions dans les régions d’intérêt jusqu’à2 Mb80. Dans les deux protocoles, l’utilisation d’un modèle Hi-C enrichi pour les fragments de ligature tirés vers le bas de biotine a augmenté le pourcentage de lectures informatives totales par rapport à notre protocole. Par exemple, dans l’ensemble de données Hi-C que nous avons utilisé ici pour la comparaison64 (GSM2053973), le pourcentage de paires valides après la suppression des doublons est 4,8 fois plus élevé que les paires valides obtenues dans Capture Hi-C comme décrit dans la figure 3 et le tableau supplémentaire 1. Cependant, l’extraction consécutive de fragments ligaturés biotinylés et de sondes hybrides rend le protocole beaucoup plus complexe et prend beaucoup plus de temps tout en réduisant éventuellement la complexité de la région capturée.
Une autre méthode disponible pour enrichir les modèles 3C avec des sondes de pavage est Tiled-C, qui a été appliquée pour étudier l’architecture de la chromatine à haute résolution spatiale et temporelle lors de la différenciation érythroïdede souris 43. En Tiled-C, un panel de sondes biotinylées de 70 pb est utilisé pour enrichir les contacts au sein de régions à grande échelle lors de deux cycles consécutifs de capture afin de générer des cartes à très haute résolution des interactions ciblées43,81. Le double enrichissement de capture rend également le protocole plus long et plus complexe par rapport à Capture Hi-C. Cependant, contrairement aux stratégies de capture C ciblant des sites de restriction uniques, dans Tiled-C, le deuxième cycle de capture ne semble pas augmenter de manière significative l’efficacité de la capture et peut donc probablement être omis43. Enfin, une approche de pavage similaire basée sur la même stratégie d’enrichissement cible utilisée dans cette étude a été appliquée à la dissection de paysages régulateurs englobant des variantes structurelles décrites chez des patients atteints de malformations congénitales et remaniées chez des souris transgéniques41,42. Dans ce cas, le réseau de sondes en mosaïque a été conçu sur l’ensemble de la cible plutôt qu’à proximité des sites de restriction DpnII41. Néanmoins, ce travail a été déterminant en soulignant la sensibilité et la puissance de cette stratégie pour parvenir à une caractérisation à haute résolution de grandes régions génomiques dans différents contextes41,42,48.
En conclusion, le protocole décrit ici représente une stratégie simple, robuste et puissante pour la caractérisation 3D haute résolution de toutes les régions génomiques d’intérêt. L’application de cette approche à différents systèmes modèles, types cellulaires, paysages de chromatine régulés par le développement et régulation génique dans des conditions saines et pathologiques est susceptible de faciliter notre compréhension de l’interaction et de la causalité entre la topologie du génome et la régulation des gènes, l’une des questions fondamentales ouvertes dans le domaine de l’épigénétique. En outre, l’application de Capture Hi-C pour cartographier les interactions à longue distance et le repliement de la chromatine d’ordre supérieur des variantes de risque identifiées par les études GWAS a le potentiel de révéler la pertinence fonctionnelle des loci génomiques non codants associés aux maladies humaines dans différents contextes, fournissant ainsi de nouvelles informations sur les processus potentiellement sous-jacents à la pathogenèse.
Déclarations de divulgation
Kai Hauschulz est Field Application Scientist chez Agilent Technologies - Diagnostic and Genomics Group. Tous les autres auteurs ne déclarent aucun intérêt concurrent.
Remerciements
Les travaux du laboratoire Heard ont été soutenus par une bourse Advanced Investigator du Conseil européen de la recherche (XPRESS - AdG671027). A.L. est soutenu par une bourse individuelle Marie Skłodowska-Curie Actions de l’Union européenne (IF-838408). A.H. est soutenu par le réseau innovant et interdisciplinaire ITN ChromDesign, dans le cadre de l’accord de subvention Marie Skłodowska-Curie 813327. Les auteurs remercient Daniel Ibrahim (MPI for Molecular Genetics, Berlin) pour ses conseils techniques utiles, la plateforme NGS de l’Institut Curie (Paris), ainsi que Vladimir Benes et le Genomics Core Facility de l’EMBL (Heidelberg) pour leur soutien et leur assistance.
matériels
| Name | Company | Catalog Number | Comments |
| 10x PBS pH 7.4 | Gibco | 10010-023 | |
| 37% (vol/vol) paraformaldehyde solution | Electron Microscopy Sciences | 15686 | single use glass-vials; do not reuse |
| 50 mL PP conical tube | Falcon | 352070 | |
| Agarose | Sigma | A9539-500g | |
| Bioanalyzer | Agilent | G2939BA | |
| Cell Scrapers - 25 cm Handle and 3.0 cm Blade | Falcon | 353089 | |
| CHIR99021 | Axon Medchem BV | Axon 1386 | |
| cOmplete Mini, Protease inhibitor cocktail (EDTA-free) | Merck | 11836170001 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | |
| Countess II FL | Invitrogen | ZGEXSCCOUNTESS2FL | Automated cell counter |
| Covaris S2 | Covaris | 500217 | Sonicator |
| DNA LoBind tube, 1.5 mL | Eppendorf | 30108051 | |
| DpnII (50000 units/mL) | New England Biolabs | R0543M | |
| Dulbecco's Modified Eagle Medium (DMEM) | Merck | D6429 | |
| Ethanol (100%) | Merck | 1.00983.2500 | |
| Fetal Bovine Serum (FBS) | Thermo Scientific | 10270106 | |
| gelatine from porcine skin | Sigma | G1890 | |
| GeneRuler 1 kb Plus DNA Ladder | Thermo Scientific | SM0313 | |
| GlycoBlue | Thermo Scientific | AM9516 | Coprecipitant |
| High-Sensitivity Bioanlayzer chips | Agilent | 5067-4626 | |
| Large Cooling Centrifuge 5920 R | Eppendorf | 5948000018 | |
| leukaemia inhibitory factor (LIF) | Merck | ESG1107 | |
| Liquiport | KNF | NF300 | Benchtop aspiration system |
| Low-binding filter tips | Biozym | VT0260U, VT0240, VT0220, VT0200U | |
| Molecular biology grade water | Merck | W3500-6x500ML | |
| Next Seq 500 | Illumina | SY-415-1001 | |
| Next Seq 500 High Output v2 Kit (300 cycles) | Illumina | FC-404-2004 | |
| Nonidet P40 Substitute (NP40) | Merck | 11332473001 | |
| PD0325901 | Axon Medchem BV | Axon 1408 | |
| Protease inhibitor cocktail (EDTA-free) | Merck | 11873580001 | |
| Proteinase K - recombinant, PCR-grade (20 mg/mL) | Thermo Scientific | EO0491 | |
| Qubit 2.0 | Thermo Scientific | Q32871 | |
| Qubit assay tubes | Thermo Scientific | Q32856 | |
| Qubit dsDNA High Sensitivity kit | Thermo Scientific | Q32851 | |
| RNase A (10 mg/mL) | Thermo Scientific | EN0531 | |
| Sodium acetate pH 5.2 (3M) | Merck | S7899 | |
| speed vacuum concentrator | Eppendorf | EP5305000100-1EA | |
| Agencourt AMPureXP | Beckman Coulter | A63881 | SPRI beads |
| SureSelect Target Enrichment Box 1 | Agilent | 5190-8645 | |
| SureSelect Target Enrichment Kit ILM Indexing Hyb Module Box 2 | Agilent | 5190-4455 | |
| SureSelect XT Library Prep Kit ILM | Agilent | 5500-0132 | |
| T4 ligase (30 units/µL) | Thermo Scientific | EL0013 | |
| table-top Centrifuge 5427 R | Eppendorf | 5409000012 | |
| Triton-X-100 (500 mL) | Merck | X100-500ML | |
| Trypan Blue | Invitrogen | T10282 | |
| Trypsine | Thermo Scientific | 25300054 | |
| UltraPure Glycine | Thermo Scientific | 15527013 | |
| β-mercaptoethanol | Thermo Scientific | 31350010 |
Références
- Ibrahim, D. M., Mundlos, S. The role of 3D chromatin domains in gene regulation: a multi-facetted view on genome organization. Current Opinion in Genetics & Development. 61, 1-8 (2020).
- Bolt, C. C., Duboule, D. The regulatory landscapes of developmental genes. Development. 147 (3), (2020).
- Glaser, J., Mundlos, S. 3D or not 3D: Shaping the genome during development. Cold Spring Harbor Perspectives in Biology. 14 (5), 040188 (2021).
- Denker, A., De Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Kempfer, R., Pombo, A. Methods for mapping 3D chromosome architecture. Nature Reviews Genetics. 21 (4), 207-226 (2020).
- McCord, R. P., Kaplan, N., Giorgetti, L. Chromosome conformation capture and beyond: Toward an integrative view of chromosome structure and function. Molecular Cell. 77 (4), 688-708 (2020).
- Jerkovic, I., Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nature ReviewsMolecular Cell Biology. 22 (8), 511-528 (2021).
- Hsieh, T. -. H. S., et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 162 (1), 108-119 (2015).
- Krietenstein, N., et al. Ultrastructural details of mammalian chromosome architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Naumova, N., Smith, E. M., Zhan, Y., Dekker, J. Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods. 58 (3), 192-203 (2012).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Zhao, Z., et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra-and interchromosomal interactions. Nature Genetics. 38 (11), 1341-1347 (2006).
- Würtele, H., Chartrand, P. Genome-wide scanning of HoxB1-associated loci in mouse ES cells using an open-ended Chromosome Conformation Capture methodology. Chromosome Research. 14 (5), 477-495 (2006).
- De Wit, E., De Laat, W. A decade of 3C technologies: insights into nuclear organization. Genes & Development. 26 (1), 11-24 (2012).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Splinter, E., et al. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes & Development. 25 (13), 1371-1383 (2011).
- Ferraiuolo, M. A., Sanyal, A., Naumova, N., Dekker, J., Dostie, J. From cells to chromatin: capturing snapshots of genome organization with 5C technology. Methods. 58 (3), 255-267 (2012).
- Kim, J. H., et al. 5C-ID: Increased resolution Chromosome-Conformation-Capture-Carbon-Copy with in situ 3C and double alternating primer design. Methods. 142, 39-46 (2018).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Zhang, Y., et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 148 (5), 908-921 (2012).
- Rao, S. S. P., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Dixon, J. R., et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 485 (7398), 376-380 (2012).
- Nora, E. P., et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 485 (7398), 381-385 (2012).
- Krefting, J., Andrade-Navarro, M. A., Ibn-Salem, J. Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biology. 16 (1), 87 (2018).
- Galupa, R., Heard, E. Topologically associating domains in chromosome architecture and gene regulatory landscapes during development, disease, and evolution. Cold Spring Harbor Symposia on Quantitative Biology. 82, 267-278 (2017).
- Tena, J. J., Santos-Pereira, J. M. Topologically associating domains and regulatory landscapes in development, evolution and disease. Frontiers in Cell and Developmental Biology. 9, 702787 (2021).
- Lupiáñez, D. G., Spielmann, M., Mundlos, S. Breaking TADs: How alterations of chromatin domains result in disease. Trends in Genetics. 32 (4), 225-237 (2016).
- Davidson, I. F., Peters, J. -. M. Genome folding through loop extrusion by SMC complexes. Nature Reviews Molecular Cell Biology. 22 (7), 445-464 (2021).
- Schmitt, A. D., Hu, M., Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nature Reviews Molecular Cell Biology. 17 (12), 743-755 (2016).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Davies, J. O. J., et al. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nature Methods. 13 (1), 74-80 (2016).
- Jäger, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178 (2015).
- Schoenfelder, S., et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Research. 25 (4), 582-597 (2015).
- Sahlén, P., et al. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology. 16, 156 (2015).
- Joshi, O., et al. Dynamic reorganization of extremely long-range promoter-promoter interactions between two states of pluripotency. Cell Stem Cell. 17 (6), 748-757 (2015).
- Mifsud, B., et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics. 47 (6), 598-606 (2015).
- Dryden, N. H., et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research. 24 (11), 1854-1868 (2014).
- Oudelaar, A. M., Davies, J. O. J., Downes, D. J., Higgs, D. R., Hughes, J. R. Robust detection of chromosomal interactions from small numbers of cells using low-input Capture-C. Nucleic Acids Research. 45 (22), 184 (2017).
- Oudelaar, A. M., et al. Single-allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nature Genetics. 50 (12), 1744-1751 (2018).
- Franke, M., et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 538 (7624), 265-269 (2016).
- Despang, A., et al. Functional dissection of the Sox9-Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nature Genetics. 51 (8), 1263-1271 (2019).
- Oudelaar, A. M., et al. Dynamics of the 4D genome during in vivo lineage specification and differentiation. Nature Communications. 11 (1), 1-12 (2020).
- Galupa, R., Heard, E. X-chromosome inactivation: A crossroads between chromosome architecture and gene regulation. Annual Review of Genetics. 52, 535-566 (2018).
- Loda, A., Collombet, S., Heard, E. Gene regulation in time and space during X-chromosome inactivation. Nature Reviews. Molecular Cell Biology. 23 (4), 231-249 (2022).
- van Bemmel, J. G., et al. The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nature Genetics. 51 (6), 1024-1034 (2019).
- Galupa, R., et al. A conserved noncoding locus regulates random monoallelic Xist expression across a topological boundary. Molecular Cell. 77 (2), 352-367 (2020).
- Gjaltema, R. A. F., et al. Distal and proximal cis-regulatory elements sense X chromosome dosage and developmental state at the Xist locus. Molecular Cell. 82 (1), 190-208 (2022).
- Galupa, R., et al. Inversion of a topological domain leads to restricted changes in its gene expression and affects inter-domain communication. Development. 149 (9), (2022).
- Savarese, F., Flahndorfer, K., Jaenisch, R., Busslinger, M., Wutz, A. Hematopoietic precursor cells transiently reestablish permissiveness for X inactivation. Molecular and Cellular Biology. 26 (19), 7167-7177 (2006).
- Schulz, E. G., et al. The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell. 14 (2), 203-216 (2014).
- Gnirke, A., et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology. 27 (2), 182-189 (2009).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Piccinini, F., Tesei, A., Arienti, C., Bevilacqua, A. Cell counting and viability assessment of 2D and 3D Cell cultures: Expected reliability of the trypan blue assay. Biological Procedures Online. 19 (1), 8 (2017).
- Servant, N., et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology. 16, 259 (2015).
- Abdennur, N., Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 36 (1), 311-316 (2020).
- Imakaev, M., et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nature Methods. 9 (10), 999-1003 (2012).
- Forcato, M., et al. Comparison of computational methods for Hi-C data analysis. Nature Methods. 14 (7), 679-685 (2017).
- Venev, S., et al. . open2c/cooltools: v0.4.1. , (2021).
- Wages, J. M. NUCLEIC ACIDS | Immunoassays. Encyclopedia of Analytical Science. , 408-417 (2005).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Servant, N., Peltzer, A. nf-core/hic: Initial release of nf-core/hic. Zenodo. , (2019).
- Furlan, G., et al. The Ftx noncoding locus controls X chromosome inactivation independently of its RNA products. Molecular Cell. 70 (3), 462-472 (2018).
- Giorgetti, L., et al. Structural organization of the inactive X chromosome in the mouse. Nature. 535 (7613), 575-579 (2016).
- Simon, C. S., et al. Functional characterisation of cis-regulatory elements governing dynamic Eomes expression in the early mouse embryo. Development. 144 (7), 1249-1260 (2017).
- Williams, R. M., et al. Reconstruction of the global neural crest gene regulatory network in vivo. Developmental Cell. 51 (2), 255-276 (2019).
- Godfrey, L., et al. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nature Communications. 10 (1), 2803 (2019).
- Hanssen, L. L. P., et al. Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nature Cell Biology. 19 (8), 952-961 (2017).
- Larke, M. S. C., et al. Enhancers predominantly regulate gene expression during differentiation via transcription initiation. Molecular Cell. 81 (5), 983-997 (2021).
- Oudelaar, A. M., et al. A revised model for promoter competition based on multi-way chromatin interactions at the α-globin locus. Nature Communications. 10 (1), 5412 (2019).
- Long, H. K., et al. Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell. 27 (5), 765-783 (2020).
- Schoenfelder, S., Javierre, B. -. M., Furlan-Magaril, M., Wingett, S. W., Fraser, P. Promoter Capture Hi-C: High-resolution, genome-wide profiling of promoter interactions. Journal of Visualized Experiments. (136), e57320 (2018).
- Siersbæk, R., et al. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Molecular Cell. 66 (3), 420-435 (2017).
- Rubin, A. J., et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nature Genetics. 49 (10), 1522-1528 (2017).
- Freire-Pritchett, P., et al. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. eLife. 6, 21926 (2017).
- Javierre, B. M., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 167 (5), 1369-1384 (2016).
- Miguel-Escalada, I., et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nature Genetics. 51 (7), 1137-1148 (2019).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Baxter, J. S., et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nature Communications. 9 (1), 1028 (2018).
- Sanborn, A. L., et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proceedings of the National Academy of Sciences. 112 (47), 6456-6465 (2015).
- Owens, D. D. G., et al. Dynamic Runx1 chromatin boundaries affect gene expression in hematopoietic development. Nature Communications. 13 (1), 773 (2022).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.