
Method Article
Расшифровка организации 3D-хроматина высокого разрешения с помощью Capture Hi-C
В этой статье
Резюме
В этом протоколе описывается метод Capture Hi-C, используемый для характеристики 3D-организации мегаразмерных целевых областей генома с высоким разрешением, включая границы топологически ассоциирующих доменов (TAD) и дальнодействующих взаимодействий хроматина между регуляторными и другими элементами последовательности ДНК.
Аннотация
Пространственная организация генома способствует его функции и регуляции во многих контекстах, включая транскрипцию, репликацию, рекомбинацию и репарацию. Поэтому понимание точной причинно-следственной связи между топологией и функцией генома имеет решающее значение и становится все более предметом интенсивных исследований. Технологии захвата конформации хромосом (3C) позволяют вывести трехмерную структуру хроматина путем измерения частоты взаимодействий между любыми областями генома. Здесь мы описываем быстрый и простой протокол для выполнения Capture Hi-C, метода обогащения мишеней на основе 3C, который характеризует аллель-специфическую 3D-организацию геномных мишеней мегабазового размера с высоким разрешением. В Capture Hi-C целевые области захватываются массивом биотинилированных зондов перед последующим высокопроизводительным секвенированием. Таким образом, достигается более высокое разрешение и аллель-специфичность при одновременном повышении эффективности времени и доступности технологии. Чтобы продемонстрировать свои сильные стороны, протокол Capture Hi-C был применен к мышиному центру Х-инактивации ( Xic), главному регуляторному локусу инактивации Х-хромосомы (XCI).
Введение
Линейный геном содержит всю информацию, необходимую организму для эмбрионального развития и выживания на протяжении всей взрослой жизни. Тем не менее, инструктирование генетически идентичных клеток выполнять различные функции имеет основополагающее значение для точного контроля того, какая информация используется в определенных контекстах, включая различные ткани и / или стадии развития. Считается, что трехмерная организация генома участвует в этой точной пространственно-временной регуляции активности генов, облегчая или предотвращая физическое взаимодействие между регуляторными элементами, которые могут быть разделены несколькими сотнями килобаз в линейном геноме (для обзоров 1,2,3). За последние 20 лет наше понимание взаимодействия между сворачиванием генома и активностью быстро возросло, в основном благодаря развитию технологий захвата конформации хромосом (3C) (для обзора 4,5,6,7). Эти методы измеряют частоту взаимодействий между любыми областями генома и полагаются на лигирование последовательностей ДНК, которые находятся в непосредственной 3D-близости внутри ядра. Наиболее распространенные протоколы 3C начинаются с фиксации клеточных популяций с помощью сшивающего агента, такого как формальдегид. Затем сшитый хроматин расщепляют с помощью фермента рестрикции, хотя также используется расщепление MNазы 8,9. После переваривания свободные концы ДНК в тесной пространственной близости повторно лигируются, и сшивание происходит в обратном порядке. Этот шаг приводит к появлению «библиотеки» или «шаблона» 3C, смешанного пула гибридных фрагментов, в котором последовательности, которые находились в 3D-близости к ядру, имеют более высокие шансы быть лигированными в одном и том же фрагменте ДНК. Последующая количественная оценка этих гибридных фрагментов позволяет вывести 3D-конформацию геномных областей, которые расположены на расстоянии тысяч пар оснований друг от друга в линейном геноме, но могут взаимодействовать в 3D-пространстве.
Для характеристики библиотеки 3C было разработано множество различных подходов, различающихся как с точки зрения того, какие подмножества фрагментов лигирования анализируются, так и с точки зрения того, какая технология используется для их последующей количественной оценки. Первоначальный протокол 3C основывался на выборе двух областей интереса и количественной оценке частоты их взаимодействия «один против одного» с помощью ПЦР10,11. Подход 4C (захват кольцевой конформации хромосомы) измеряет взаимодействие между одним интересующим локусом (т. е. «точкой зрения») и остальной частью генома («один против всех»)12,13,14. В 4C библиотека 3C подвергается второму раунду переваривания и перелигирования для получения небольших кольцевых молекул ДНК, которые амплифицированы ПЦР с помощью праймеров, специфичных для точки зрения15. 5C (точная копия захвата конформации хромосомы) позволяет характеризовать 3D-взаимодействия в более крупных областях, представляющих интерес, обеспечивая понимание сворачивания хроматина более высокого порядка в этой области («многие против многих»)16. В 5С библиотека 3С гибридизуется с пулом олигонуклеотидов, перекрывающих сайты рестрикции, которые впоследствии могут быть амплифицированы с помощью мультиплексной ПЦР с универсальными праймерами15. Как в 4C, так и в 5C информативные фрагменты ДНК были первоначально количественно определены с помощью микрочипов, а затем с помощью секвенирования следующего поколения (NGS)17,18,19. Эти стратегии характеризуют целевые области, представляющие интерес, но не могут быть применены для картирования полногеномных взаимодействий. Эта последняя цель достигается с помощью Hi-C, высокопроизводительной стратегии на основе 3C, в которой массовое параллельное секвенирование шаблона 3C позволяет объективно охарактеризовать сворачивание хроматина на уровне всего генома («все против всех»)20. Протокол Hi-C включает включение биотинилированного остатка на концах переваренных фрагментов, после чего следует вытягивание фрагментов лигирования шариками стрептавидина для увеличения извлечения лигированных фрагментов20.
Hi-C показал, что геномы млекопитающих структурно организованы в нескольких масштабах в 3D-ядре. В мегабазовом масштабе геном делится на области активного и неактивного хроматина, компартменты А и В, соответственно20,21. Существование дополнительных субкомпартментов, представленных различными состояниями хроматина и активности, также было впоследствии показано22. При более высоком разрешении геном далее разделяется на субмегабазовые самовзаимодействующие домены, называемые топологически ассоциирующими доменами (TAD), впервые выявленные при анализе Hi-C и 5C геномов человека и мыши23,24. В отличие от компартментов, которые варьируются тканеспецифическим образом, TAD, как правило, постоянны (хотя есть много исключений). Важно отметить, что границы TAD сохраняются для всех видов25. В клетках млекопитающих TAD часто охватывают гены, разделяющие один и тот же регуляторный ландшафт, и, как было показано, представляют собой структурную основу, которая облегчает корегуляцию генов, ограничивая взаимодействие с соседними регуляторными доменами (для обзора 3,26,27,28). Кроме того, в TAD взаимодействия, обусловленные сайтами CTCF в основании когезин-экструдированных петель, могут увеличивать вероятность взаимодействий промотор-энхансер или энхансер-энхансер (для обзора29).
В Hi-C компартменты и TAD могут быть обнаружены с разрешением от 1 Мб до 40 кб, но более высокое разрешение может быть достигнуто для характеристики контактов меньшего масштаба, таких как петлевые взаимодействия между дистальными элементами в масштабе 5-10 кб. Однако увеличение разрешения для эффективного обнаружения таких петель с помощью HiC требует значительного увеличения глубины секвенирования и, следовательно, затрат на секвенирование. Это усугубляется, если анализ должен быть аллель-специфическим. Действительно, X-кратное увеличение разрешения требует увеличения глубины секвенирования наX2 , а это означает, что подходы с высоким разрешением и аллель-специфическими для всего генома могут быть непомерно дорогими30.
Для повышения экономической эффективности и доступности при сохранении высокого разрешения целевые области, представляющие интерес, могут быть физически извлечены из полногеномных библиотек 3C или Hi-C после их гибридизации с комплементарными олигонуклеотидными зондами, меченными биотином, перед последующим секвенированием. Эти стратегии обогащения мишеней называются методами Capture-C и позволяют исследовать взаимодействия сотен целевых локусов, разбросанных по всему геному (т. е. Promoter Capture (PC) Hi-C; Захват нового поколения (NG) Capture-C; Захват с низким входом (LI); Ядерно-титрованный (NuTi) Capture-C; Tri-C)31,32,33,34,35,36,37,38,39,40 или между регионами, охватывающими до нескольких мегабаз (т. е. Capture HiC; HYbrid Capture Hi-C (Hi-C2); Плитка-С)41,42,43. Два аспекта могут различаться в методах, основанных на захвате: (1) природа и дизайн биотинилированных олигонуклеотидов (т.е. РНК или ДНК, одиночные олиго, захватывающие дисперсные геномные мишени, или несколько олиго, образующих интересующую область); и (2) шаблон, который используется для извлечения мишеней, которые могут быть библиотекой 3C или Hi-C, последняя состоит из биотинилированных фрагментов рестрикции, извлеченных из библиотеки 3C.
Здесь описан протокол Capture Hi-C, основанный на обогащении целевых контактов из библиотеки 3C. Протокол основан на разработке специально разработанного набора биотинилированных РНК-зондов и может быть выполнен за 1 неделю от подготовки библиотеки 3C до секвенирования NGS. Протокол быстрый, простой и позволяет охарактеризовать 3D-организацию более высокого порядка интересующих областей мегабазы с разрешением 5 кб, одновременно повышая эффективность времени и доступность по сравнению с другими методами 3C. Протокол Capture Hi-C был применен к главному регуляторному локусу инактивации Х-хромосомы (XCI), центру Х-инактивации (Xic), в котором находится некодирующая РНК Xist. Xic ранее был предметом обширного структурного и функционального анализа (для обзора44,45). У млекопитающих XCI компенсирует дозировку Х-сцепленных генов между самками (XX) и самцами (XY) и включает в себя подавление транскрипции почти всей одной из двух Х-хромосом в женских клетках. Xic представляет собой мощный, золотой стандарт локуса для исследований в области топологии 3D-генома и взаимодействия с регуляциейгенов 44. Анализ 5C Xic в эмбриональных стволовых клетках мыши (mESCs) привел к открытию и наименованию TAD, что позволило впервые понять функциональную значимость топологического разделения и корегуляции генов24. Впоследствии было показано, что топологическая организация Xic критически влияет на соответствующие сроки развития апрегуляции Xist и XCI 46, и неожиданные цис-регуляторные элементы, которые могут влиять на активность генов внутри и между TAD, также были недавно обнаружены в Xic47,48,49. Применение Capture Hi-C к 3 Мб Х-хромосомы мыши, охватывающей Xic, демонстрирует мощь этого подхода при анализе крупномасштабного сворачивания хроматина с высоким разрешением. Предоставляется подробный и простой для понимания протокол, начиная с разработки массива биотинилированных зондов на каждом сайте рестрикции DpnII в интересующей области до создания полногеномной библиотеки 3C, гибридизации и захвата целевых контактов и последующего анализа данных. Также включен обзор соответствующих мер контроля качества и ожидаемых результатов, а также обсуждаются как сильные, так и слабые стороны подхода в свете аналогичных существующих методов.
протокол
Эмбриональные стволовые клетки мыши (mESCs), использованные в этом исследовании, были получены в результате скрещивания самки TX/TX R26rtTA/rtTA 50 с самцом Mus musculus castaneus в соответствии с рекомендациями по уходу за животными Института Кюри (Париж)51.
1. Конструкция зонда
- Разработайте массив биотинилированных зондов (120-мерных олигонуклеотидов РНК), охватывающих интересующую целевую область.
- Разбейте интересующую область перекрывающимися олигонуклеотидами так, чтобы в среднем каждая последовательность в мишени была покрыта двумя уникальными зондами (2-кратное покрытие) (рис. 1).
- Исключите повторяющиеся последовательности из покрытия зонда, чтобы избежать обогащения неспецифическими взаимодействиями.
ПРИМЕЧАНИЕ: Для максимального обогащения информативных фрагментов лигирования были определены области, охватывающие 300 п.н. вверх и вниз по течению от каждого сайта рестрикции DpnII по мишени (ChrX: 102 475 000-105 475 000), и 28 913 биотинилированных зондов были разработаны в соответствии с технологией обогащения мишени ДНК SureSelect через платформуSure Design 52. Согласно этой стратегии, в каждом олигонуклеотиде допускается не более 40 оснований повторяющихся последовательностей, чтобы свести к минимуму обогащение неспецифических взаимодействий. Массив зондов был синтезирован компанией Agilent. Здесь DpnII используется в качестве рестрикционного фермента по двум причинам: (1) это четырехрезак, обычно используемый в нескольких методахна основе 3C 53; и (2) он максимизирует шансы на захват информативных однонуклеотидных полиморфизмов (SNP) в непосредственной близости от мест разреза по сравнению с другими ферментами рестрикции, которые были протестированы in silico в гибридных линиях F1, используемых в этом исследовании (C57BL / 6J x CASTEi / J).
2. Экспериментальная процедура
- Подготовка клеток
- Засейте соответствующее количество клеток на одной или нескольких пластинах для культивирования клеток, чтобы достичь общего количества клеток ≥ 5 x 107 клеток в день фиксации.
ПРИМЕЧАНИЕ: В этом исследовании использовались мышиные эмбриональные стволовые клетки (мЭСК). мЭСК наносят покрытие на желатинизированные (0,1% желатина в 1x PBS - o/n при 37 °C, 5% CO2-инкубаторе) планшеты для культивирования клеток в среде мЭСК, содержащей 2i + LIF и протестированную сыворотку плода (DMEM, 15% FBS, 0,1 мМ β-меркаптоэтанол, 1,000 ЕД / мл-1 фактор ингибирования лейкемии (LIF), CHIR99021 (3 мкМ) и PD0325901 (1 мкМ)). Для этого типа ячеек одна 80% сливающаяся 10-сантиметровая пластина содержит примерно 2 x 107 ячеек. - Подготовьте одну дополнительную тарелку для клеточной культуры для подсчета клеток.
ПРИМЕЧАНИЕ: Меньшая пластина для культивирования клеток может быть использована для уменьшения использования среды. В этом случае количество ячеек для посева на меньшую тарелку необходимо соответствующим образом отрегулировать (например, в 3 раза меньше ячеек на 10-сантиметровой тарелке по сравнению с 15-сантиметровой тарелкой).
- Засейте соответствующее количество клеток на одной или нескольких пластинах для культивирования клеток, чтобы достичь общего количества клеток ≥ 5 x 107 клеток в день фиксации.
- Фиксация формальдегида
- Оцените общее количество ячеек, которые необходимо сшить.
- Перед началом реакции сшивания трипсинизировать и подсчитывать клетки с контрольной пластины, подготовленной специально для подсчета клеток, с помощью автоматизированного счетчика клеток в соответствии с инструкциями производителя.
- Включают окрашивание жизнеспособности (например, Trypan Blue) для определения процентного содержания жизнеспособных клеток54. Исходя из этого количества ячеек, оцените общее количество ячеек в планшете (планшетах), подготовленных для сшивания.
- Культуральную среду удаляют из пластин, подготовленных для сшивания, и заменяют ее соответствующим количеством фиксирующего раствора (2% формальдегида в среде для культивирования клеток). Используйте 10 мл на 10-сантиметровой тарелке (например, ~20 мл на 15-сантиметровую тарелку).
ПРИМЕЧАНИЕ: Добавьте точный объем фиксирующего раствора. Если фиксация адгезивных клеток невозможна, этот этап может быть адаптирован к трипсинизированным клеткам и выполнен в 30 мл фиксирующего раствора в конических центрифужных пробирках объемом 50 мл. Формальдегид не должен быть старше 1 года. Предпочтительно использовать одноразовые флаконы. Фиксирующий раствор перед использованием необходимо довести до комнатной температуры (ОТ).
ВНИМАНИЕ: Формальдегид опасен и должен обрабатываться в соответствии с соответствующими правилами охраны труда и техники безопасности. - Зафиксируйте на 10 минут при RT при аккуратном перемешивании на шейкере.
- Погасите реакцию фиксации добавлением 2,5 М глицина-1x PBS до конечной концентрации 0,125 М. Добавьте 530 мкл 2,5 М глицина-1x PBS к 10 мл на 10-сантиметровой пластине (например, от 1060 мкл до 20 мл на 15-сантиметровой пластине).
ПРИМЕЧАНИЕ: Если клетки были зафиксированы в растворе, погасите реакцию фиксации 1590 мкл 2,5 М глицина-1x PBS. - Выдерживать 5 минут при ЛТ, аккуратно перемешивая на шейкере.
- Переложите тарелки на лед и выдерживайте еще 15 минут на льду, аккуратно перемешивая на шейкере.
ПРИМЕЧАНИЕ: Отныне ячейки должны храниться на льду, а буферы должны быть предварительно охлаждены, чтобы избежать дальнейшего сшивания. Переместите в холодное помещение, если нужно обработать много тарелок. - Удалите фиксирующий раствор из ячеек, налив его в стакан, чтобы обеспечить быструю обработку.
ПРИМЕЧАНИЕ: Обязательно утилизируйте жидкие отходы, содержащие формальдегид, в соответствии с соответствующими правилами охраны труда и техники безопасности. - Быстро промойте 10-сантиметровую пластину два раза 5 мл холодного 0,125 М глицина-1x PBS (8 мл для 15-сантиметровой тарелки), чтобы смыть мусор и мертвые клетки. Удалите жидкость с тарелки, налив ее в стакан, чтобы обеспечить быструю обработку.
- Добавьте 5 мл холодного 0,125 М глицина-1x PBS в 10-сантиметровую пластину (10 мл для 15-сантиметровой пластины) и быстро соскребите клетки с пластины с помощью пластикового скребка для клеток.
- Переведите клеточную суспензию в предварительно охлажденную коническую центрифужную пробирку объемом 50 мл с помощью серологической пипетки.
- Дважды промойте пластину 5 мл холодного 0,125 М глицина-1x PBS и добавьте клеточную суспензию в коническую центрифужную пробирку.
- Отжим при 480 x g в течение 10 мин при 4 °C.
ПРИМЕЧАНИЕ: Если ячейки были закреплены в растворе, перенесите ячейку в предварительно охлажденную коническую центрифужную пробирку и открутите при 480 x g в течение 10 мин при 4 °C. Удалите фиксирующий раствор, налив его в стакан, и промойте три раза в 10 мл холодного 0,125 М глицина-1x PBS. Обязательно ресуспендируйте ячейки на каждом этапе стирки. - Удалите надосадочную жидкость путем аспирации с помощью настольной системы аспирации. Ресуспендируют клетки в 500 мкл 1x PBS на 1 x 107 клеток, осторожно пипетируя вверх и вниз пипеткой P1000. Чтобы ресуспендировать клетки в точном объеме, обратитесь к оценке общего количества клеток, полученной в 2.2.1.
- Аликвоту 500 мкл клеточной суспензии в расчетное количество микроцентрифужных пробирок по 1,5 мл (1 х 107 клеток /пробирка).
- Отжим при 480 x g в течение 10 мин при 4 °C.
- Удалите надосадочную жидкость с помощью настольной системы аспирации и заморозьте гранулы ячейки в жидком азоте. Храните гранулы с сухими ячейками при температуре -80 °C.
ПРИМЕЧАНИЕ: Образцы могут храниться не менее 1 года.
- Оцените общее количество ячеек, которые необходимо сшить.
- Лизис клеток
- Разморозьте замороженные гранулы на льду.
- Приготовьте 1,5 мл буфера для лизиса в H 2 0 на образец: добавьте 10 мМ Tris-HCl, pH 8,0, 10 мМ NaCl и0,2% NP40.
- Добавьте 600 мкл буфера холодного лизиса и хорошо ресуспендируйте на льду.
- Инкубируйте на льду в течение 15 минут, чтобы клетки набухли.
- Отжим при 2655 x g в течение 5 минут при 4 °C и удалите надосадочную жидкость с помощью настольной системы аспирации.
- Чтобы удалить мусор, ресуспендируют гранулу в 1 мл буфера холодного лизиса, отжимают при 2655 x g в течение 5 мин при 4 ° C и удаляют надосадочную жидкость.
- Вскоре снова отжмите при 2655 x g и 4 °C и удалите как можно больше оставшейся надосадочной жидкости с помощью настольной аспирационной системы, оснащенной наконечником P200.
- Ресуспендировать в 100 мкл 0,5% (об/об.) SDS.
- Инкубировать в термомиксере при 62 °C, взбалтывать при 1400 об/мин в течение 10 мин.
- Добавьте 290 мкл H2O + 50 мкл 10% TritonX-100 и хорошо перемешайте, избегая пузырьков воздуха.
- Инкубировать в термомиксере при 37 °C, взбалтывать при 1400 об/мин в течение 15 мин.
- Добавьте 50 мкл 10-кратного буфера Dpnll и переверните пробирку для перемешивания.
- Возьмите 50 мкл непереваренной ДНК для контроля качества в отдельную пробирку. Не забудьте взять непереваренную контрольную пробу.
- Пищеварение DpnII
- Добавьте 10 мкл высокой концентрации Dpnll (всего 500 ЕД) и перемешайте инвертированием.
- Инкубируют образцы и непереваренный контроль в термомиксере при 37 °C, вращая при 1400 об/мин в течение >4 ч.
- Добавьте 10 мкл высокой концентрации Dpnll (всего 500 ЕД) в конце дня.
- Инкубируют образцы и непереваренный контроль при 37 °C, перемешивая при 1400 об/мин в течение ночи.
- Добавьте 10 мкл высокой концентрации Dpnll (всего 500 ЕД) в начале следующего дня в образцы.
- Инкубируйте образцы и непереваренный контроль в термомиксере при 37 °C, перемешивая при 1400 об/мин в течение 4 часов.
- Лигирование и реверсирование сшивки
- Инкубировать пробирки при 65 °C в течение 20 мин при 1400 об/мин.
ПРИМЕЧАНИЕ: Не добавляйте паспорт безопасности на этом этапе. Идея состоит в том, чтобы сохранить целостность ядра, поэтому лигирование проводится внутри ядер, обходя необходимость экстремального разбавления. - Охладите образцы на льду максимум 5-10 минут. Чтобы избежать осаждения SDS, не оставляйте образцы на льду дольше этого срока.
- Возьмите 50 мкл нелигированной переваренной ДНК для контроля качества в отдельную пробирку. Храните непереваренные и нелигированные регуляторы при температуре -20 °C.
ПРИМЕЧАНИЕ: Не забудьте взять нелигированный контрольный образец. - Добавьте 800 мкл лигационного коктейля: 122 мкл 10-кратного лигазного буфера, 8 мкл Т4-лигазы (30 ЕД/мкл) и 670 мкл H20.
- Инкубировать при 16 °C, закручивая при 1000 об/мин в течение ночи.
- Добавьте 7,5 мкл протеиназы К (20 мг / мл) в образцы и 2 мкл в контрольную группу.
- Инкубировать при 65 °C в течение 4 ч при 1000 об/мин.
- Инкубировать пробирки при 65 °C в течение 20 мин при 1400 об/мин.
- Очистка ДНК
- Перенесите образцы на льду в предварительно охлажденные конические центрифужные пробирки объемом 15 мл и добавьте 2 мл воды, 10,5 мл ледяного EtOH и 583 мкл 3 М NaAC.
ПРИМЕЧАНИЕ: Дополнительная вода предназначена для предотвращения переноса DTT в гранулы. - Добавьте 200 мкл ледяного EtOH, 10,8 мкл NaAC и 1 мкл соосаждителя к непереваренному и нелигированному контролю качества.
- Инкубировать при температуре -80 °C не менее 4 ч до ночи.
- Отжимайте пробирки объемом 15 мл при 2200 x g при 4 °C в течение 45 мин.
- Вращайте контрольные трубки объемом 1,5 мл при 20 500 x g при 4 ° C в течение 30 мин.
- Промойте один раз 3 мл (образцы) и 1 мл (контрольная группа) ледяного 70% EtOH.
- Отжим при 2200 x g (образцы) или 20 500 x g (контроль) при 4 °C в течение 10 мин.
- Осторожно удалите EtOH и высушите на воздухе при RT в течение 10-15 минут; Не пересушивайте.
- Ресуспендировать образцы и контрольную группу в 100 мкл и 20 мкл H20 соответственно.
- Добавьте 1 мкл РНКaseA и инкубируйте при 37 °C, вращая при 1400 об/мин в течение 30 мин.
- Перенесите образцы на льду в предварительно охлажденные конические центрифужные пробирки объемом 15 мл и добавьте 2 мл воды, 10,5 мл ледяного EtOH и 583 мкл 3 М NaAC.
- Контроль качества подготовки шаблонов 3С
- Количественно оцените каждый образец и контролируйте его с помощью комплекта флуорометра для высокочувствительных измерений концентрации ДНК.
- Загрузите 100-200 нг каждого образца и каждого контроля на 1% агарозный / 1x КЭ-гель.
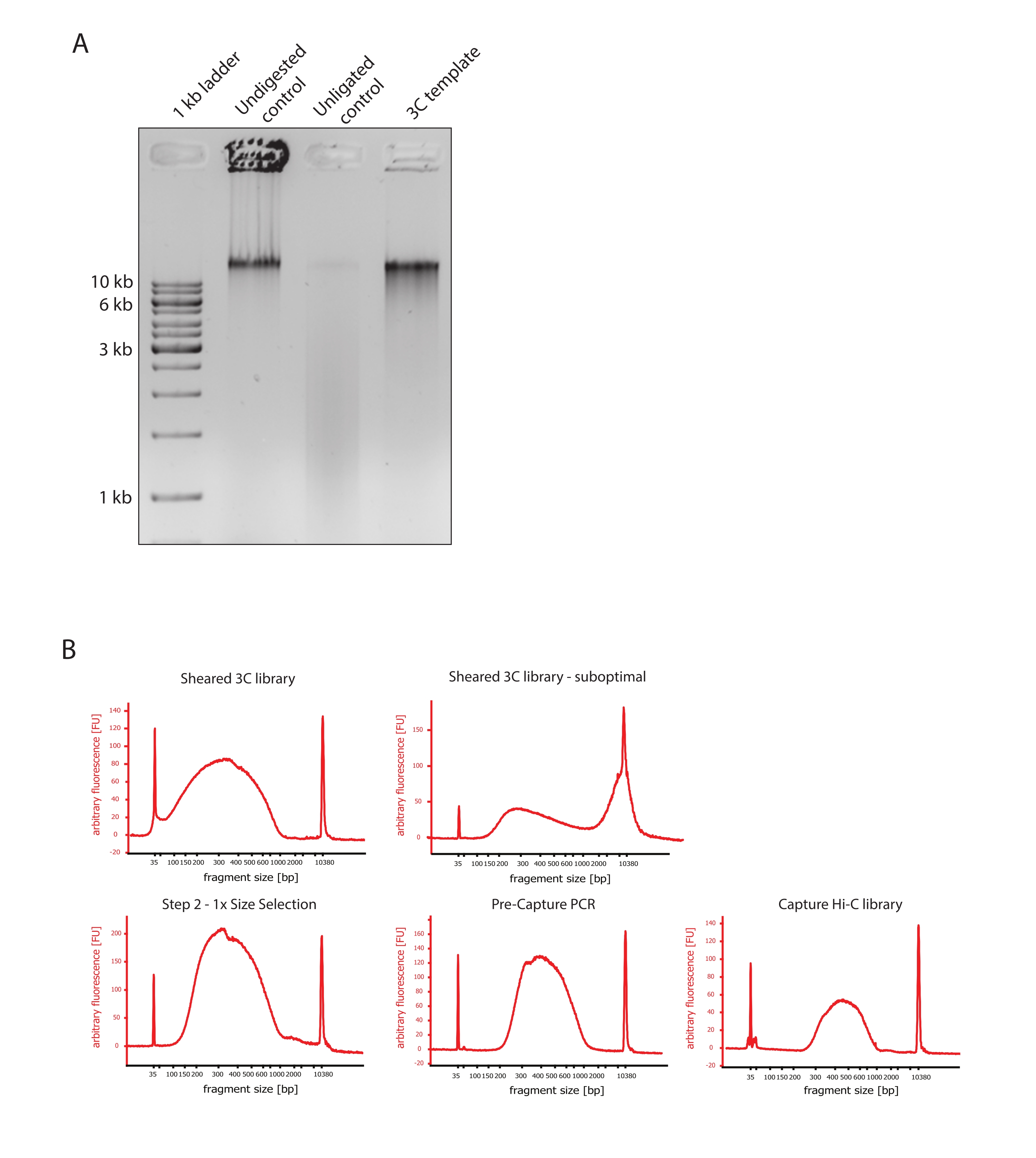
- Убедитесь, что изображение геля показывает ожидаемый результат, сравнив различия в размерах фрагментов ДНК контрольной группы и шаблона 3C, как показано на рисунке 2A.
- Храните образцы и контрольные элементы при температуре -20 °C.
- Гибридизация, захват и обработка образцов для мультиплексного секвенирования
- Чтобы гибридизировать массив биотинилированных РНК-зондов с шаблоном 3C, захватите целевые фрагменты лигирования и подготовьте образцы для мультиплексного секвенирования в соответствии с целевой системой обогащения, используемой в этом исследовании для мультиплексного секвенирования с парным концом (см. Таблица материалов). Следуйте протоколу в соответствии с инструкциями производителя, внося следующие незначительные изменения:
- Раздел 2 протокола завода-изготовителя: Подготовка образцов
- Следуйте инструкциям по обогащению мишени, начиная с ввода 3 мкг гДНК.
- Срежьте ДНК в ультразвуковом аппарате, используя следующие характеристики: рабочий цикл 10%, интенсивность 4, 200 cyc / burst и 130 с. Начните с 4 мкг матрицы 3C, ресуспендированной в 130 мкл воды для каждой реакции захвата, чтобы обеспечить достаточное количество материала для продолжения пробоподготовки с 3 мкг срезанной ДНК.
- Оцените качество срезанной ДНК. Запустите 1 мкл срезанной ДНК на биоанализаторе ДНК в соответствии с высокочувствительным протоколом. Ожидайте распределение размера фрагмента в пределах 150-700.н. (рис. 2).
- Очистите образец с помощью твердофазной обратимой иммобилизации (SPRI). Добавьте 124 мкл шариков SPRI к 124 мкл образца ДНК, чтобы выполнить выбор размера левой стороны 1:1 в соответствии с инструкциями производителя и разбавить 25 мкл воды, не содержащей нуклеаз. На этом этапе очистки будут удалены более короткие фрагменты, чтобы обогатить фрагменты примерно до 300.н. (рис. 2).
ПРИМЕЧАНИЕ: Количество образцов и шариков SPRI, используемых на этом этапе, учитывает потерю объема, которая произошла при переносе образцов в новые пробирки и выполнении контроля качества в биоанализаторе. Все последующие этапы подбора размеров выполняются в соответствии с соотношениями, рекомендованными протоколом производителя. Элюирование ДНК из шариков SPRI выполняется при ЛТ на протяжении всего протокола. - Оцените качество выбранной по размеру срезанной ДНК. Запустите 1 мкл срезанной ДНК на биоанализаторе ДНК в соответствии с протоколом высокой чувствительности (HS). Ожидается распределение размеров фрагментов с наибольшим обогащением при 300.н. (рис. 2). Продолжайте количественную оценку срезанной ДНК, если стрижка прошла успешно.
- Количественно определите срезанную ДНК с помощью набора флуорометра для измерения концентрации ДНК HS.
ПРИМЕЧАНИЕ: Если сдвиг ДНК приводит к выходу ДНК <3 мкг, проведите второй раунд сдвига ДНК с еще 4 мкг ДНК и объедините срезанные образцы ДНК после первого этапа очистки шариков SPRI, чтобы получить в общей сложности 3 мкг срезанной ДНК. - Добавьте воду, не содержащую нуклеазы, в очищенный образец ДНК выбранного размера (всего 3 мкг) до конечного объема 48 мкл и приступайте к конечной реакции репарации в соответствии с протоколом производителя.
- После лигирования адаптеров с парными концами амплифицируйте библиотеку, выполнив пять циклов ПЦР перед захватом в соответствии с инструкциями производителя (условия для ПЦР и праймеров приведены в наборе).
- Раздел 4 протокола производителя: Гибридизация и захват
- Чтобы гибридизировать подготовленные образцы ДНК с целевыми РНК-зондами, разбавьте 750 нг образцов ДНК в конечном объеме 3,4 мкл, в результате чего начальная концентрация составит 221 нг/мкл. Для образцов ДНК, разбавленных в больших объемах, используйте концентратор скоростного вакуума, чтобы уменьшить их до конечного объема. Концентрация в вакууме (250 x g; ≤45 °C) в течение 15-20 мин обычно достаточна для ресуспендированных образцов в 10 мкл. Перед запуском концентратора скоростного вакуума убедитесь, что для каждого образца имеется одинаковый входной объем.
- Инкубировать гибридизационную смесь в течение 16-18 ч при 65 °C с нагретой крышкой при 105 °C в соответствии с инструкциями производителя.
- Раздел 5 протокола производителя: Индексация и обработка образцов для мультиплексного секвенирования
- Для амплификации захваченных библиотек индексирующими праймерами выполните 12 циклов постзахватной ПЦР согласно инструкции производителя (условия для ПЦР и праймеров предусмотрены в наборе).
- Раздел 2 протокола завода-изготовителя: Подготовка образцов
- Чтобы гибридизировать массив биотинилированных РНК-зондов с шаблоном 3C, захватите целевые фрагменты лигирования и подготовьте образцы для мультиплексного секвенирования в соответствии с целевой системой обогащения, используемой в этом исследовании для мультиплексного секвенирования с парным концом (см. Таблица материалов). Следуйте протоколу в соответствии с инструкциями производителя, внося следующие незначительные изменения:
- Секвенирование нового поколения
- Чтобы запустить несколько библиотек захвата Hi-C на одной и той же ячейке потока, подготовьте эквимолярную смесь библиотек захвата и последовательность 100-120 млн чтений на библиотеку.
- Если необходим аллель-специфический анализ, секвенируйте парный конец 150.н., чтобы обеспечить достаточное покрытие SNP.
3. Анализ данных
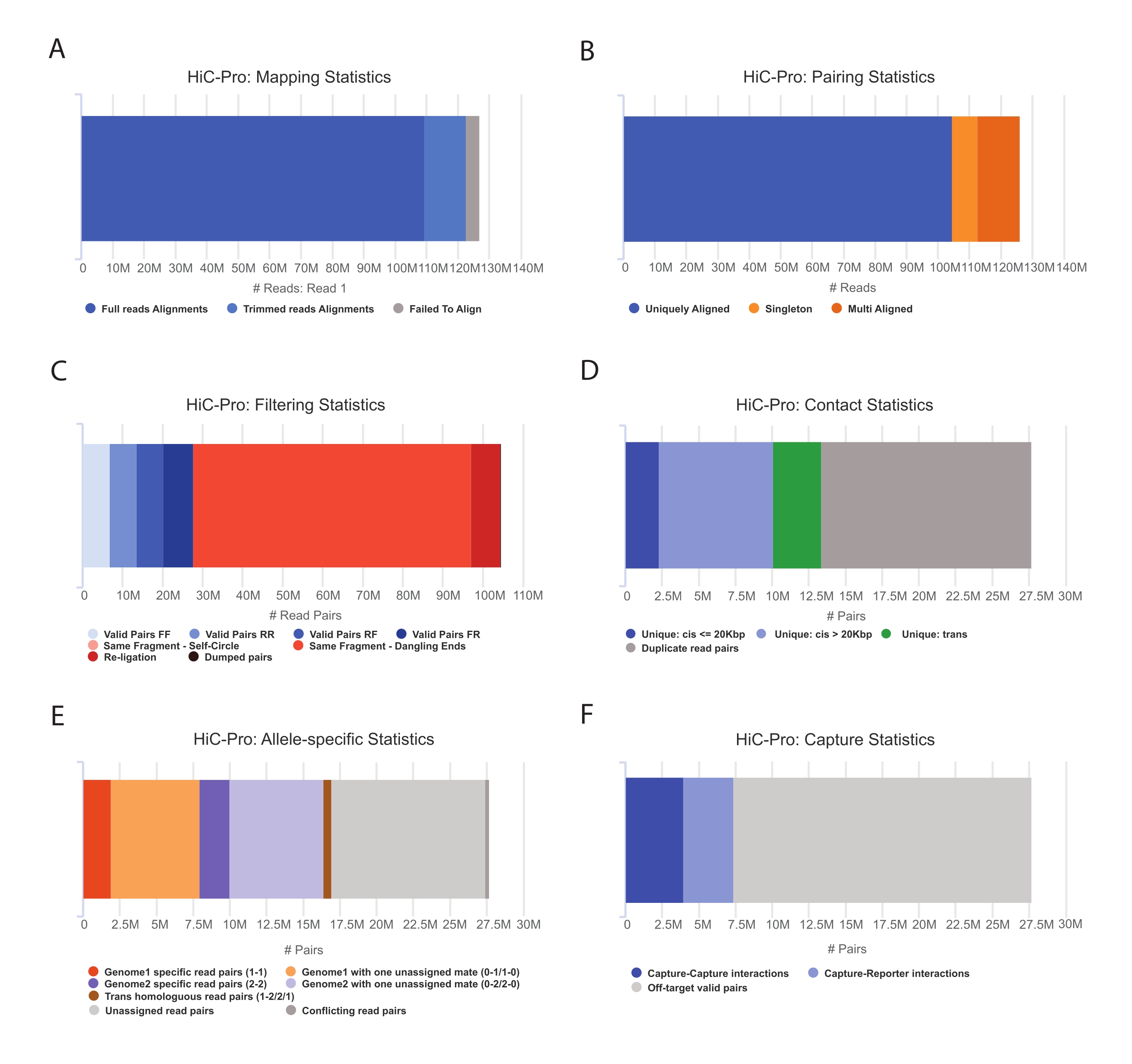
- Примените конвейер HiC-Pro для выполнения анализа данных Capture Hi-C55. HiC-Pro обеспечивает контроль качества на каждом этапе обработки, включая (рис. 3):
(i) Скорость выравнивания на эталонном геноме, определяющая долю считываний, охватывающих сайт лигирования, а также количество пар и синглтонов.
(ii) Доля допустимых продуктов лигирования и неинформативных пар считывания (свисающий конец, самолигирование и т. д.).
(iii) Фракция ближних/дальних и внутри/межхромосомных контактов.
(iv) Доля целевых контактов для захвата Hi-C.
(v) Доля аллель-специфических считывается, если указано.
ПРИМЕЧАНИЕ: HiC-Pro поддерживает широкий спектр протоколов, включая in situ Hi-C и Capture Hi-C. В последнем случае пользователю достаточно указать целевой регион (формат BED) в конфигурационном файле. После обработки данных выходы HiC-Pro могут быть легко преобразованы в более холодный объект для последующего анализа56. На этом этапе карты контактов при различных разрешениях нормализуются с помощью метода ICE, ранее описанного Имакаевым и его коллегами57. Затем можно провести несколько анализов, чтобы вызвать хромосомные компартменты, TAD или хроматиновые петли (для обзора58). Рабочий процесс протокола показан на рисунке 4. Здесь набор «cooltools» применяется для расчета оценки изоляции и границ TAD, как показано на рисунке 5 и рисунке 659.
Результаты
Описанный протокол Capture Hi-C основан на подготовке полногеномной 3C-матрицы с помощью четырехбазового резака (DpnII). Последующее обогащение фрагментов лигирования по интересующей области генома получается путем гибридизации массива тайлинговых РНК-зондов и их захвата на основе стрептавидина в соответствии с целевой системой обогащения, используемой в данном исследовании (рис. 1). Были выбраны биотинилированные РНК-зонды, поскольку они демонстрируют более плотное сродство к своим мишеням по сравнению с ДНК-зондами52,60. Затем захваченные библиотеки индексируются и объединяются в пулы для мультиплексирования с высокой пропускной способностью. Данные захвата Hi-C могут быть визуализированы в виде карт взаимодействия Hi-C с высоким разрешением, а также в виде карт контактов с одной точкой обзора в стиле 4C, чтобы специально визуализировать взаимодействия меньших последовательностей, таких как промоторы или усилители, во всей области захвата. Рабочий процесс протокола показан на рисунке 4. Контроль качества предварительного секвенирования показан на рисунке 2 и включает оценку правильного разложения и повторного лигирования шаблона 3C, а также его эффективную резку и очистку на различных этапах протокола. Ожидается, что срезанная матричная ДНК 3C будет работать в диапазоне от 150 до 700.н., и обогащение фрагментов >2 кб не должно быть обнаружено. Во время следующих этапов выполняется несколько этапов очистки ДНК на основе шариков и выбора размера, сначала после сдвига, затем после ПЦР до и после захвата. Очищенные библиотеки показывают отчетливый профиль обогащения фрагментов, визуализированный на высокочувствительном биоанализаторе ДНК (рис. 2). Средний размер фрагмента увеличивается в процессе подготовки библиотеки за счет лигирования адаптеров, секвенирования и индексации праймеров. Контроль качества после секвенирования осуществляется с помощью Hi-C Pro и показан на рисунке 3. Для обработки и анализа данных, подобных 3C, было предложено множество различных программных приложений для биоинформатики. Среди них конвейер HiC-Pro является одним из самых популярных решений, позволяющих обрабатывать необработанные данные секвенирования до конечных карт контактов при различных разрешениях55. HiC-Pro использует двухэтапную стратегию картирования для выравнивания чтений секвенирования в эталонном геноме. Затем продукты 3C реконструируются и отфильтровываются, чтобы удалить неинформативные пары контактов и создать карты контактов. Кроме того, он может использовать список известных полиморфизмов для проведения аллель-специфического анализа и разделения контактов, исходящих от двух родительских аллелей, в различных контактных картах. Совсем недавно HiC-Pro был включен и расширен в структуру nf-core (nf-core-hic), обеспечивая высокомасштабируемый и воспроизводимый конвейер, управляемый сообществом61,62.
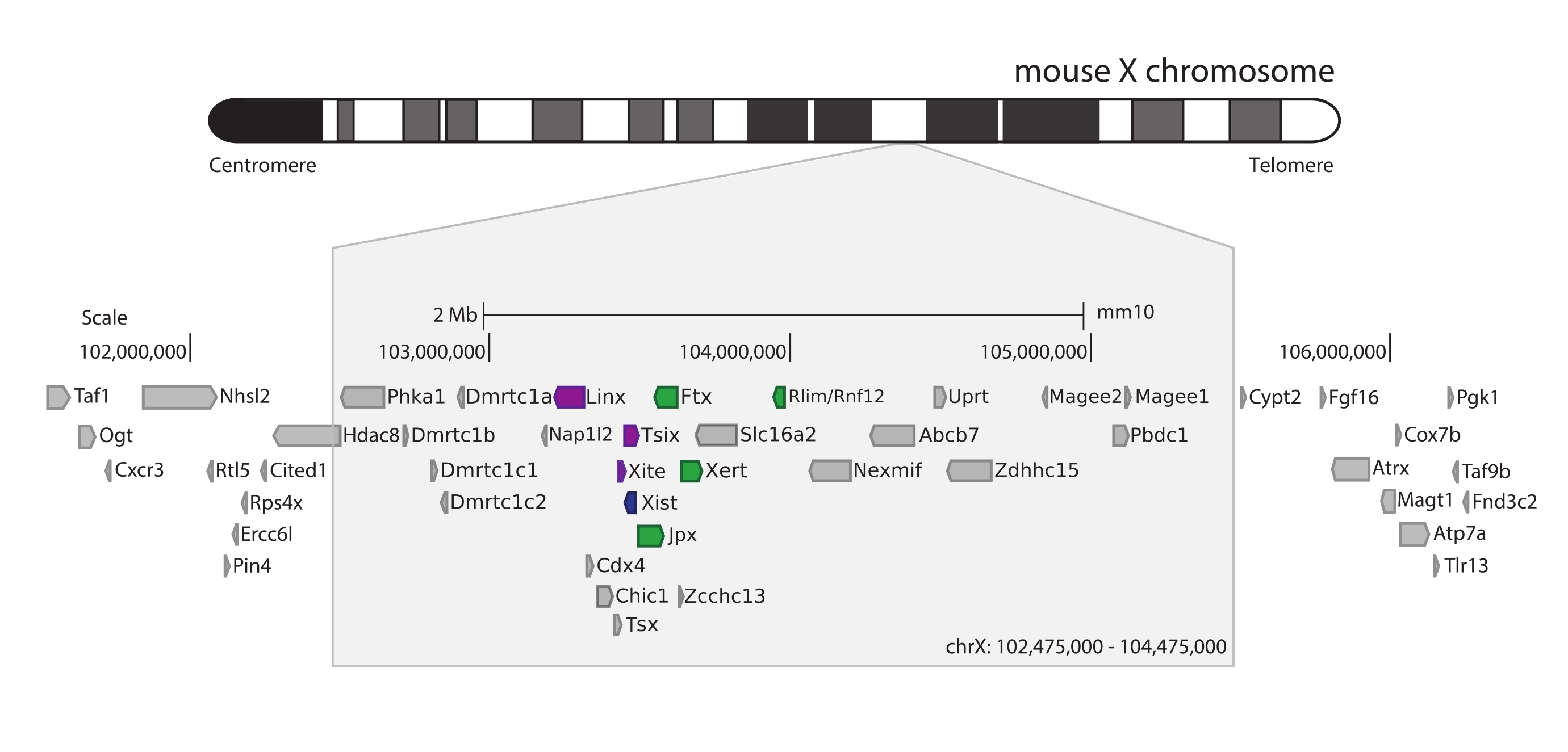
Для захвата мыши Xic был разработан массив из 28 913 РНК-зондов, разбивающих 3 Мб Х-хромосомы. Эта область включает в себя ключевого игрока в XCI, длинный некодирующий ген Xist, и его известный регуляторный ландшафт ~ 800 кб (рис. 5). Эта область размером ~800 кб разделена на два TAD: один, включающий промотор Xist и его известные положительные регуляторы (т.е. некодирующие транскрипты Ftx, Jpx и Xert и ген, кодирующий белок Rnf12), и соседний TAD, охватывающий отрицательные цис-регуляторы Xist (т.е. его антисмысловой транскрипт Tsix, элемент энхансера Xite и некодирующий транскрипт Linx) (для обзора44,45).
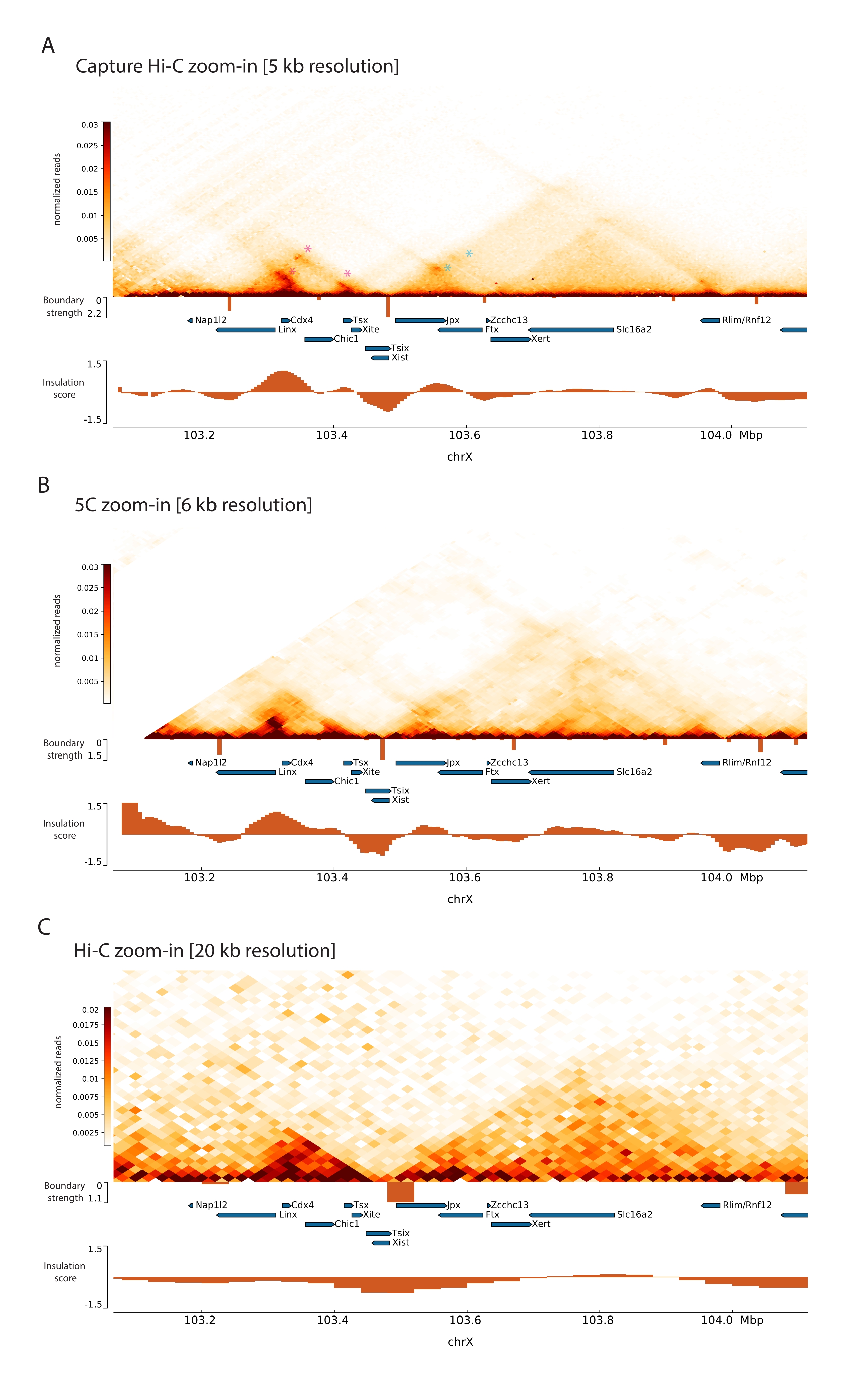
Применив описанный протокол Capture Hi-C к Xic, топологическая организация этого локуса была получена с беспрецедентным разрешением (рис. 6 и рис. 7). Это особенно ясно при сравнении профиля Capture Hi-C с ранее опубликованным профилем 5C47 (рис. 6 и рис. 7; Дополнительная таблица 1) и Hi-C61 (рис. 6 и рис. 7; Дополнительная таблица 1) Профили. Например, структуры суб-TAD более очевидны — TAD, содержащий промотор Xist ( Xist-TAD ), четко подразделяется на два меньших домена (рис. 6A, синяя стрелка). Ранее об этом можно было только визуально «догадаться» по профилю 5С (рис. 6В), правда, обнаружение границы в этой области с помощью алгоритма оценки изоляции. Аналогичным образом, разрешение профиля Capture Hi-C позволяет идентифицировать два меньших домена в соседнем TAD (рис. 6A, B), который содержит промотор локуса Tsix ( Tsix-TAD ); ранее это не достигалось с помощью 5C (рис. 6B). Следует отметить, что топологические границы, определяемые оценкой изоляции по данным Capture Hi-C и 5C, обычно обнаруживаются в несколько разных местах и с разной относительной прочностью.
Более того, другие структуры суб-TAD, такие как контактные петли, хорошо видны из данных Capture Hi-C, такие как петля между Xist и Ftx (рис. 7A), ранее идентифицированная с Capture-C63, и петля между Xist и Xert (рис. 7B), недавно идентифицированная с использованием аналогичного протокола для Capture Hi-C48. Другие контакты также могут быть отображены более точно из-за увеличенного разрешения профилей Capture Hi-C, таких как те, которые образуют известные горячие точки контакта в Tsix-TAD между локусами Linx, Chic1 и Xite (рис. 7A).
По сравнению с данными Hi-C, показанными на рисунке 7, Capture Hi-C позволил увеличить разрешение в четыре раза, но потребовал только одну четвертую глубины секвенирования (т. е. 126 М считываний против 571 М) (Дополнительная таблица 1). Это увеличение разрешения позволяет обнаруживать субТАД и циклические взаимодействия, которые не могут быть обнаружены Hi-C на глубине секвенирования, показанной на рисунках 6 и 7. Таким образом, описанный протокол для Capture Hi-C позволяет получить гораздо более подробную характеристику с высоким разрешением большой интересующей области генома по сравнению с предыдущими подходами.

Рисунок 1: Конструкция зонда. Схематическое изображение стратегии, используемой для проектирования зонда. Области 300.н. вверх и вниз по течению от каждого сайта рестрикции DpnII в целевой области 3 Mb были выбраны и покрыты перекрывающимися биотинилированными РНК-зондами. Показан один из этих выбранных регионов, chrX: 102 474 805-102 475 500. В каждом зонде допускается не более 40 оснований повторяющихся последовательностей. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 2: Контроль качества предварительного секвенирования Hi-C . (A) Репрезентативный пример контроля качества шаблонов 3C. 200 нг ДНК были загружены на 1% агарозный гель. Переулок 1: лестница 1 кб. Полоса 2: Непереваренный, сшитый и интактный хроматин работает в виде резкой полосы со скоростью >10 кб. Полоса 3: Сшитый хроматин, переваренный DpnII, представляет собой мазок размером от 1 до 3 кб. Полоса 4: Окончательная библиотека или шаблон 3C; свободные концы переваренных сшитых фрагментов ДНК перелигируются. Мазок ДНК с меньшим молекулярным размером практически не обнаруживается, а продукт лигирования обнаруживается в виде полосы размером >10 кб. (B) Репрезентативные примеры высокочувствительных профилей ДНК биоанализатора. Слева вверху: успешно срезанная библиотека 3C, показывающая распределение размера фрагмента от 150 до 700.н. Вверху справа: неудовлетворительно срезанная библиотека 3C. Несрезанная ДНК обнаруживается как широкое обогащение фрагментов >2 кб. (C) Внизу слева: срезанный образец ДНК после отбора размера левой стороны 1:1 с использованием шариков SPRI. Фрагменты ~300.н. обогащены. Нижняя середина: ПЦР-профиль предварительного захвата после лигирования адаптеров с парными концами в соответствии с протоколом производителя. Внизу справа: окончательная библиотека Capture Hi-C, включающая адаптеры, праймеры для секвенирования и индексирования для мультиплексного секвенирования. Сокращения: bp = пары оснований, FU = произвольная единица флуоресценции. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 3: Захват контроля качества Hi-C после секвенирования с помощью HiC-Pro . (A) Пример скорости картирования эталонного генома для первого помощника пар секвенирования. Светло-голубая дробь представляет собой показания, выровненные HiC-Pro и охватывающие соединение лигирования. Таким образом, эта метрика может быть использована для проверки экспериментального этапа лигирования. (B) После того, как партнеры секвенирования выровнены в геноме, для анализа сохраняются только однозначно выровненные пары считывания. (C) Недопустимые пары (выделены красным цветом), такие как свисающий конец, автокруг или перевязка, исключаются из анализа. Доля допустимых пар является хорошим показателем эффективности лигирования и вытягивания. (D) Действительные пары могут быть дополнительно разделены на внутрихромосомные и ближние/дальние контакты. Дублированные пары чтения, которые, вероятно, представляют артефакты ПЦР, отбрасываются из анализа. (E) Для аллель-специфического анализа HiC-Pro сообщает о количестве аллельных считываний, поддерживаемых одним или двумя партнерами для каждого родительского генома (т.е. C57BL/6J x CASTEi/J). Ожидается одинаковая доля считываний, отнесенных к материнскому и отцовскому аллелю. (F) Наконец, для построения карт контактов выбираются только допустимые пары, перекрывающие область захвата. Пары захват-захват представляют контакты в целевом регионе, в то время как пары захват-репортер предполагают взаимодействие между целевым регионом и нецелевым регионом. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

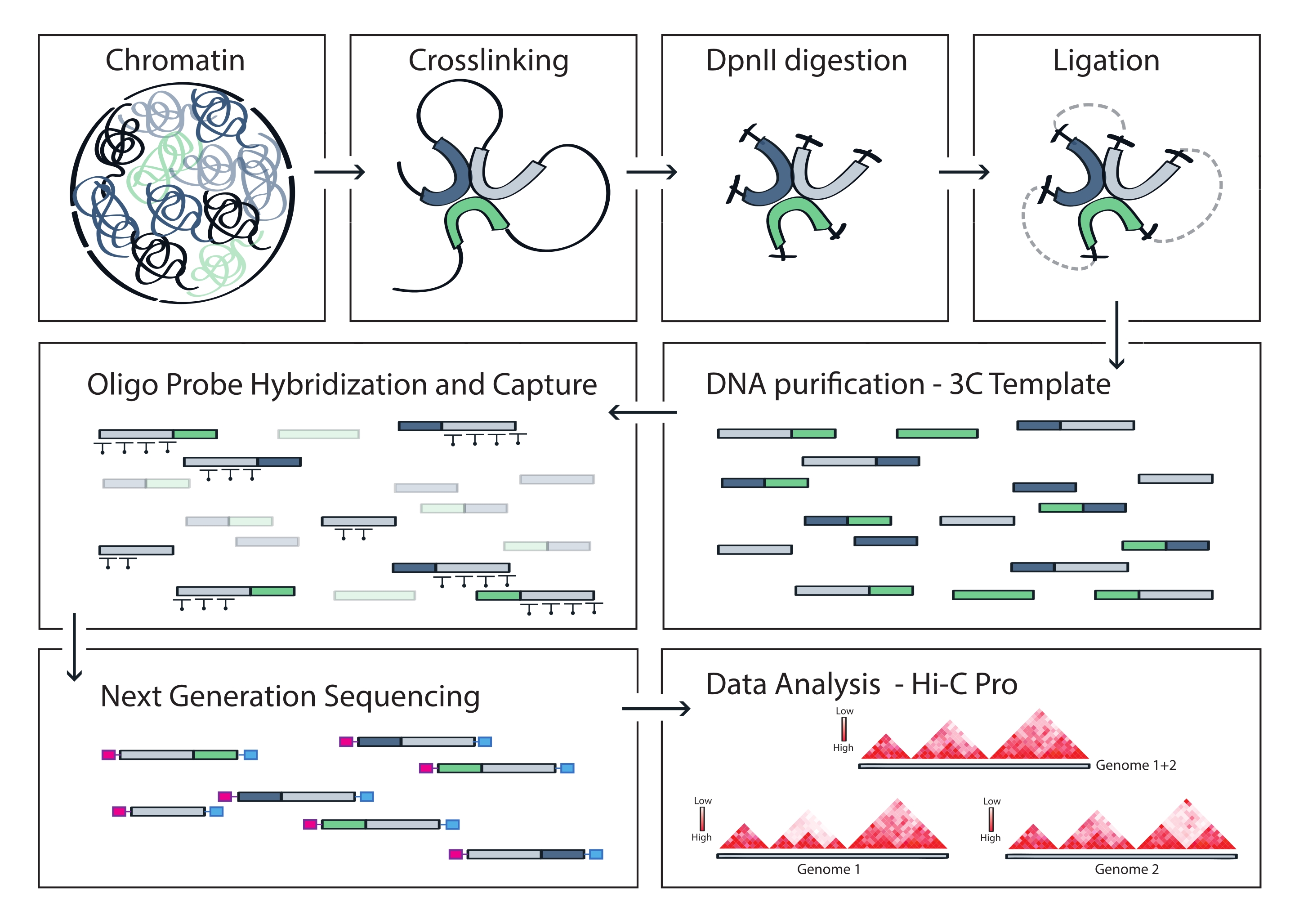
Рисунок 4: Рабочий процесс протокола Capture Hi-C. Схематическое изображение различных шагов протокола. Для создания полногеномного шаблона 3C хроматин сначала сшивают с формальдегидом, а затем расщепляют ферментом рестрикции DpnII. Затем свободные концы ДНК повторно лигируются, сшивание обращается вспять, и ДНК очищается. Чтобы обогатить фрагменты, охватывающие целевую область, массив биотинилированных РНК-зондов гибридизуют с матрицей 3C и захватывают с помощью стрептавидин-опосредованного вытягивания. Библиотеки захвата обрабатываются для мультиплексного секвенирования, а действительные фрагменты лигирования количественно оцениваются, чтобы определить частоту контактов хроматина по мишени, которые визуализируются в виде карт взаимодействия с высоким разрешением. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 5: Обзор области, охватывающей Xic на Х-хромосоме мыши. Схематическое изображение Х-хромосомы мыши и увеличение захваченной области размером 3 Мб (ChrX: 102 475 000-105 475 000). Целевая область включает ~ 800 кб ДНК, соответствующей Xic, главному регуляторному локусу XCI. Xic включает в себя длинные некодирующие гены, Xist, ключевого игрока XCI, и его регуляторный ландшафт. Положительные регуляторы Xist показаны зеленым цветом, а отрицательные - фиолетовым. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 6: Захват карт взаимодействия Hi-C, 5C и Hi-C в захваченной области размером 3 Мб. (A) Захват карты взаимодействия Hi-C целевого объекта размером 3 Мб, охватывающего Xic мыши с разрешением 10 кб (это исследование). (B) Карта взаимодействия 5C того же целевого региона, что и в A, с разрешением 6 кб (данные, обработанные из47). Повторяющиеся области, не включенные в анализы, замаскированы белым цветом. Данные 5C требуют собственной биоинформационной обработки (см.47). После очистки и выравнивания карты 5C с разрешением праймера объединяются с использованием бегущей медианы (окно = 30 кб, шаг = 5) для достижения окончательного разрешения 6 кб. (C) Карта взаимодействия Hi-C той же области генома, что и в A и B, с разрешением 40 кб (данные, обработанные из64). Все карты взаимодействия были сгенерированы с помощью ЭСК мыши. Оценка изоляции была рассчитана с помощью cooltools и представлена в виде гистограмм с минимумами изоляции на границах TAD. Границы TAD отображаются в виде вертикальных линий под картой. Высота каждой линии указывает на граничную прочность. Гены показаны в виде стрелок, указывающих в направлении транскрипции. Границы суб-TAD, которые определяются исключительно или более точно на картах Capture Hi-C, обозначаются пурпурными и синими стрелками для суб-TAD в Tsix и Xist TAD соответственно. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 7: Захват карт взаимодействия Hi-C, 5C и Hi-C на 1 Мб в захваченной области. (A) Захват карты взаимодействия Hi-C геномной области размером 1 Мб, охватывающей Xic мыши, с разрешением 5 кб (это исследование). (B) Карта взаимодействия 5C той же области генома, что и в A. при разрешении 6 КБ (данные обработаны из47). Повторяющиеся области, не включенные в анализы, замаскированы белым цветом. Следует отметить, что данные 5C требуют собственной биоинформационной обработки (см.47). После очистки и выравнивания карты 5C с разрешением праймера объединяются с использованием бегущей медианы (окно = 30 кб, шаг = 5) для достижения окончательного разрешения 6 кб. (C) Карта взаимодействия Hi-C той же области генома, что и в A и B Hi-C, с разрешением 20 кб (данные, обработанные из64). Все карты взаимодействия были сгенерированы с помощью mESCs. Оценка изоляции была рассчитана с помощью cooltools и представлена в виде гистограмм с минимумами изоляции на границах TAD. Границы TAD отображаются в виде вертикальных линий под картой. Высота каждой линии указывает на граничную прочность. Гены показаны в виде стрелок, указывающих на направление транскрипции. Контактные петли, которые обнаруживаются исключительно или более точно в Capture Hi-C, обозначаются пурпурными и синими звездочками для петель в Tsix и Xist TAD соответственно. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Дополнительная таблица 1: Статистика после секвенирования для наборов данных, используемых в этой рукописи: Capture Hi-C (это исследование), Hi-C64 и 5C47. Пожалуйста, нажмите здесь, чтобы загрузить этот файл.
Обсуждение
Здесь мы описываем относительно быстрый и простой протокол Capture Hi-C для характеристики организации геномных областей мегабазового размера более высокого порядка при разрешении 5-10 кб. Capture Hi-C принадлежит к семейству технологий Capture-C, которые предназначены для обогащения целевых взаимодействий хроматина из полногеномных шаблонов 3C или Hi-C. На сегодняшний день подавляющее большинство приложений Capture-C было использовано для картирования хроматиновых контактов относительно небольших регуляторных элементов, разбросанных по всему геному. В первом протоколе Capture-C несколько перекрывающихся биотинилированных зондов РНК использовались для захвата >400 предварительно отобранных промоторов в библиотеках 3C, полученных из эритроидных клеток31. Та же стратегия была впоследствии усовершенствована в Next Generation (NG) и Nuclear Titrated (NuTi) Capture-C для достижения профилей взаимодействия с высоким разрешением >8,000 промоторов за счет использования одиночных приманок ДНК 120.н., охватывающих одиночные сайты рестрикции, и двух последовательных раундов захвата для максимального обогащения информативных фрагментов лигирования32,40. Эти стратегии привели к функциональному рассечению цис-действующих элементов во многих различных контекстах, включая эмбриональное развитие мыши, дифференцировку клеток, инактивацию Х-хромосомы и неправильную регуляцию генов при патологических состояниях 46,63,65,66,67,68,69,70,71.
В Promoter Capture Hi-C (PCHi-C) >22 000 аннотированных промоторов, содержащих фрагменты рестрикции, были извлечены из библиотек Hi-C путем гибридизации одиночных 120-мерных биотинилированных зондов РНК на одном или обоих концах фрагмента рестрикции34,72. Этот метод позволил рассечь интерактом тысяч промоторов в быстро растущем числе типов клеток, включая эмбриональные стволовые клетки мыши, клетки печени плода и адипоциты 34,35,72,73, а также лимфобластоидные линии человека, гемопоэтические предшественники, эпидермальные кератиноциты и плюрипотентные клетки 37,74,75,76,77.
По сравнению с этими технологиями целевого обогащения, Capture Hi-C нацелен на смежные области генома вплоть до мегабазового масштаба, тем самым охватывая один или несколько TAD и охватывая регуляторные ландшафты генов. Вся интересующая область должна быть покрыта массивом биотинилированных зондов, охватывающих каждый сайт рестрикции DpnII в мишени. Гибридизация биотинилированного массива с шаблоном 3C, его последующий захват на основе стрептавидина и обработка для мультиплексного секвенирования выполняются с использованием системы обогащения мишени для мультиплексного секвенирования Illumina Paired-End. Весь протокол является быстрым, так как он может быть выполнен за 1 неделю от подготовки библиотеки 3C до секвенирования NGS, и требует лишь незначительных адаптаций и/или устранения неполадок с учетом специфики.
Протокол также дает преимущества по сравнению с другими методами на основе 3C. Чтобы получить карты взаимодействия с разрешением 5-10 кб, мы секвенировали 100-120 М парных считываний. В качестве сравнения мы использовали здесь набор данных Hi-C из 571 млн чтений для достижения разрешения20 КБ 64 (GSM2053973), и потребовалось бы не менее 1 миллиарда чтений для достижения разрешения 5 КБ с Hi-C22 по всей хромосоме.
Захват Hi-C, используемый в настоящем исследовании, достигает гораздо более высокого разрешения, чем ранее опубликованный 5C на основе рестрикционного фермента47 6-bp (Дополнительная таблица 1). Важно отметить, что стратегия, разработанная для обогащения и усиления целевых взаимодействий в 5C, не позволяет проводить аллель-специфический анализ взаимодействий хроматина. Напротив, данные Capture Hi-C могут быть картированы в частности, в частности, что позволяет вскрывать 3D-структурные ландшафты пар гомологичных хромосом, например, в клетках человека или в гибридных клеточных линиях F1, полученных путем скрещивания генетически разных штаммовмышей 78. Для создания аллель-специфических карт взаимодействия Capture Hi-C с разрешением 5 кб мы секвенировали парные чтения 150.н., чтобы увеличить охват SNP. Аналогичные аллель-специфические подходы могут быть применены к клеточным линиям человека, для которых доступна аннотация SNP22.
Важно отметить, что, хотя Capture Hi-C обычно обеспечивает высокое разрешение при одновременном повышении доступности затрат на секвенирование, производство специально адаптированных биотинилированных олигонуклеотидов оказывает влияние на общую стоимость этого метода. Таким образом, выбор наиболее подходящего метода 3C будет отличаться для разных применений и будет зависеть от решаемого биологического вопроса и требуемого разрешения, а также от размера интересующей области. Другие разработанные протоколы Capture Hi-C имеют общие ключевые функции с протоколом, описанным здесь. Например, стратегия Capture Hi-C была применена для характеристики геномных областей размером от ~ 50 кб до 1 Мб, охватывающих некодирующие варианты, связанные с риском рака молочной железы и колоректального рака; в этом протоколе целевые области были извлечены из библиотек Hi-C путем гибридизации 120-мерных приманок РНК, покрывающих целевые области с 3-кратным покрытием33,38,79. Аналогичным образом, HYbrid Capture Hi-C (Hi-C 2) использовался для таргетинга взаимодействий в интересующих областях до2 Мб80. В обоих протоколах использование шаблона Hi-C, обогащенного фрагментами лигирования, вытянутыми биотином, увеличило процент общего информативного чтения по сравнению с нашим протоколом. Например, в наборе данных Hi-C, который мы использовали здесь для сравнения64 (GSM2053973), процент допустимых пар после удаления дубликатов в 4,8 раза выше, чем допустимые пары, полученные в Capture Hi-C, как описано на рисунке 3 и в дополнительной таблице 1. Тем не менее, последовательное вытягивание биотинилированных лигированных фрагментов и гибридизованных зондов делает протокол значительно более сложным и трудоемким, при этом, возможно, уменьшая сложность захваченной области.
Другим доступным методом обогащения 3C-шаблонов тайлинговыми зондами является Tiled-C, который был применен для изучения архитектуры хроматина с высоким пространственным и временным разрешением во время дифференцировки эритроидов мыши43. В Tiled-C панель из биотинилированных зондов 70.н. используется для обогащения контактов в крупномасштабных областях в двух последовательных раундах захвата для создания карт целевых взаимодействий с очень высоким разрешением43,81. Двойное обогащение захвата также делает протокол более длинным и сложным по сравнению с Capture Hi-C. Однако, в отличие от стратегий Capture-C, нацеленных на сайты с одним ограничением, в Tiled-C второй раунд захвата, по-видимому, существенно не повышает эффективность захвата, и поэтому, вероятно, может быть опущен43. Наконец, аналогичный подход к тайлингу, основанный на той же стратегии целевого обогащения, которая использовалась в этом исследовании, был применен к вскрытию регуляторных ландшафтов, охватывающих структурные варианты, описанные у пациентов с врожденными пороками развития и реконструированные у трансгенных мышей41,42. В этом случае тайловый массив зондов был спроектирован по всей цели, а не в непосредственной близости от участков41 ограничения DpnII. Тем не менее, эта работа была плодотворной в освещении чувствительности и мощи этой стратегии для достижения характеристики больших геномных областей с высоким разрешением в различных контекстах41,42,48.
В заключение, описанный здесь протокол представляет собой простую, надежную и мощную стратегию для 3D-характеристики с высоким разрешением любых интересующих областей генома. Применение этого подхода к различным модельным системам, типам клеток, регулируемым развитием хроматиновым ландшафтам и регуляции генов в здоровых и патологических состояниях, вероятно, облегчит наше понимание взаимодействия и причинно-следственной связи между топологией генома и регуляцией генов, что является одним из фундаментальных открытых вопросов в области эпигенетики. Кроме того, применение Capture Hi-C для картирования дальних взаимодействий и сворачивания хроматина более высокого порядка вариантов риска, выявленных в исследованиях GWAS, может выявить функциональную значимость некодирующих геномных локусов, связанных с заболеваниями человека в различных контекстах, тем самым обеспечивая новое понимание процессов, потенциально лежащих в основе патогенеза.
Раскрытие информации
Кай Хаушульц (Kai Hauschulz) — специалист по полевым применениям в Agilent Technologies - Diagnostic and Genomics Group. Все остальные авторы заявляют об отсутствии конкурирующих интересов.
Благодарности
Работа в лаборатории Херда была поддержана премией Европейского исследовательского совета Advanced Investigator (XPRESS - AdG671027). А.Л. поддерживается индивидуальной стипендией Европейского Союза Марии Склодовской-Кюри (IF-838408). A.H. поддерживается инновационной и междисциплинарной сетью ITN ChromDesign в рамках грантового соглашения Марии Склодовской-Кюри 813327. Авторы благодарны Даниэлю Ибрагиму (MPI for Molecular Genetics, Берлин) за полезные технические советы, платформе NGS в Институте Кюри (Париж), а также Владимиру Бенешу и Центру геномики в EMBL (Гейдельберг) за поддержку и помощь.
Материалы
| Name | Company | Catalog Number | Comments |
| 10x PBS pH 7.4 | Gibco | 10010-023 | |
| 37% (vol/vol) paraformaldehyde solution | Electron Microscopy Sciences | 15686 | single use glass-vials; do not reuse |
| 50 mL PP conical tube | Falcon | 352070 | |
| Agarose | Sigma | A9539-500g | |
| Bioanalyzer | Agilent | G2939BA | |
| Cell Scrapers - 25 cm Handle and 3.0 cm Blade | Falcon | 353089 | |
| CHIR99021 | Axon Medchem BV | Axon 1386 | |
| cOmplete Mini, Protease inhibitor cocktail (EDTA-free) | Merck | 11836170001 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | |
| Countess II FL | Invitrogen | ZGEXSCCOUNTESS2FL | Automated cell counter |
| Covaris S2 | Covaris | 500217 | Sonicator |
| DNA LoBind tube, 1.5 mL | Eppendorf | 30108051 | |
| DpnII (50000 units/mL) | New England Biolabs | R0543M | |
| Dulbecco's Modified Eagle Medium (DMEM) | Merck | D6429 | |
| Ethanol (100%) | Merck | 1.00983.2500 | |
| Fetal Bovine Serum (FBS) | Thermo Scientific | 10270106 | |
| gelatine from porcine skin | Sigma | G1890 | |
| GeneRuler 1 kb Plus DNA Ladder | Thermo Scientific | SM0313 | |
| GlycoBlue | Thermo Scientific | AM9516 | Coprecipitant |
| High-Sensitivity Bioanlayzer chips | Agilent | 5067-4626 | |
| Large Cooling Centrifuge 5920 R | Eppendorf | 5948000018 | |
| leukaemia inhibitory factor (LIF) | Merck | ESG1107 | |
| Liquiport | KNF | NF300 | Benchtop aspiration system |
| Low-binding filter tips | Biozym | VT0260U, VT0240, VT0220, VT0200U | |
| Molecular biology grade water | Merck | W3500-6x500ML | |
| Next Seq 500 | Illumina | SY-415-1001 | |
| Next Seq 500 High Output v2 Kit (300 cycles) | Illumina | FC-404-2004 | |
| Nonidet P40 Substitute (NP40) | Merck | 11332473001 | |
| PD0325901 | Axon Medchem BV | Axon 1408 | |
| Protease inhibitor cocktail (EDTA-free) | Merck | 11873580001 | |
| Proteinase K - recombinant, PCR-grade (20 mg/mL) | Thermo Scientific | EO0491 | |
| Qubit 2.0 | Thermo Scientific | Q32871 | |
| Qubit assay tubes | Thermo Scientific | Q32856 | |
| Qubit dsDNA High Sensitivity kit | Thermo Scientific | Q32851 | |
| RNase A (10 mg/mL) | Thermo Scientific | EN0531 | |
| Sodium acetate pH 5.2 (3M) | Merck | S7899 | |
| speed vacuum concentrator | Eppendorf | EP5305000100-1EA | |
| Agencourt AMPureXP | Beckman Coulter | A63881 | SPRI beads |
| SureSelect Target Enrichment Box 1 | Agilent | 5190-8645 | |
| SureSelect Target Enrichment Kit ILM Indexing Hyb Module Box 2 | Agilent | 5190-4455 | |
| SureSelect XT Library Prep Kit ILM | Agilent | 5500-0132 | |
| T4 ligase (30 units/µL) | Thermo Scientific | EL0013 | |
| table-top Centrifuge 5427 R | Eppendorf | 5409000012 | |
| Triton-X-100 (500 mL) | Merck | X100-500ML | |
| Trypan Blue | Invitrogen | T10282 | |
| Trypsine | Thermo Scientific | 25300054 | |
| UltraPure Glycine | Thermo Scientific | 15527013 | |
| β-mercaptoethanol | Thermo Scientific | 31350010 |
Ссылки
- Ibrahim, D. M., Mundlos, S. The role of 3D chromatin domains in gene regulation: a multi-facetted view on genome organization. Current Opinion in Genetics & Development. 61, 1-8 (2020).
- Bolt, C. C., Duboule, D. The regulatory landscapes of developmental genes. Development. 147 (3), (2020).
- Glaser, J., Mundlos, S. 3D or not 3D: Shaping the genome during development. Cold Spring Harbor Perspectives in Biology. 14 (5), 040188 (2021).
- Denker, A., De Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Kempfer, R., Pombo, A. Methods for mapping 3D chromosome architecture. Nature Reviews Genetics. 21 (4), 207-226 (2020).
- McCord, R. P., Kaplan, N., Giorgetti, L. Chromosome conformation capture and beyond: Toward an integrative view of chromosome structure and function. Molecular Cell. 77 (4), 688-708 (2020).
- Jerkovic, I., Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nature ReviewsMolecular Cell Biology. 22 (8), 511-528 (2021).
- Hsieh, T. -. H. S., et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 162 (1), 108-119 (2015).
- Krietenstein, N., et al. Ultrastructural details of mammalian chromosome architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Naumova, N., Smith, E. M., Zhan, Y., Dekker, J. Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods. 58 (3), 192-203 (2012).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Zhao, Z., et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra-and interchromosomal interactions. Nature Genetics. 38 (11), 1341-1347 (2006).
- Würtele, H., Chartrand, P. Genome-wide scanning of HoxB1-associated loci in mouse ES cells using an open-ended Chromosome Conformation Capture methodology. Chromosome Research. 14 (5), 477-495 (2006).
- De Wit, E., De Laat, W. A decade of 3C technologies: insights into nuclear organization. Genes & Development. 26 (1), 11-24 (2012).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Splinter, E., et al. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes & Development. 25 (13), 1371-1383 (2011).
- Ferraiuolo, M. A., Sanyal, A., Naumova, N., Dekker, J., Dostie, J. From cells to chromatin: capturing snapshots of genome organization with 5C technology. Methods. 58 (3), 255-267 (2012).
- Kim, J. H., et al. 5C-ID: Increased resolution Chromosome-Conformation-Capture-Carbon-Copy with in situ 3C and double alternating primer design. Methods. 142, 39-46 (2018).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Zhang, Y., et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 148 (5), 908-921 (2012).
- Rao, S. S. P., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Dixon, J. R., et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 485 (7398), 376-380 (2012).
- Nora, E. P., et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 485 (7398), 381-385 (2012).
- Krefting, J., Andrade-Navarro, M. A., Ibn-Salem, J. Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biology. 16 (1), 87 (2018).
- Galupa, R., Heard, E. Topologically associating domains in chromosome architecture and gene regulatory landscapes during development, disease, and evolution. Cold Spring Harbor Symposia on Quantitative Biology. 82, 267-278 (2017).
- Tena, J. J., Santos-Pereira, J. M. Topologically associating domains and regulatory landscapes in development, evolution and disease. Frontiers in Cell and Developmental Biology. 9, 702787 (2021).
- Lupiáñez, D. G., Spielmann, M., Mundlos, S. Breaking TADs: How alterations of chromatin domains result in disease. Trends in Genetics. 32 (4), 225-237 (2016).
- Davidson, I. F., Peters, J. -. M. Genome folding through loop extrusion by SMC complexes. Nature Reviews Molecular Cell Biology. 22 (7), 445-464 (2021).
- Schmitt, A. D., Hu, M., Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nature Reviews Molecular Cell Biology. 17 (12), 743-755 (2016).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Davies, J. O. J., et al. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nature Methods. 13 (1), 74-80 (2016).
- Jäger, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178 (2015).
- Schoenfelder, S., et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Research. 25 (4), 582-597 (2015).
- Sahlén, P., et al. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology. 16, 156 (2015).
- Joshi, O., et al. Dynamic reorganization of extremely long-range promoter-promoter interactions between two states of pluripotency. Cell Stem Cell. 17 (6), 748-757 (2015).
- Mifsud, B., et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics. 47 (6), 598-606 (2015).
- Dryden, N. H., et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research. 24 (11), 1854-1868 (2014).
- Oudelaar, A. M., Davies, J. O. J., Downes, D. J., Higgs, D. R., Hughes, J. R. Robust detection of chromosomal interactions from small numbers of cells using low-input Capture-C. Nucleic Acids Research. 45 (22), 184 (2017).
- Oudelaar, A. M., et al. Single-allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nature Genetics. 50 (12), 1744-1751 (2018).
- Franke, M., et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 538 (7624), 265-269 (2016).
- Despang, A., et al. Functional dissection of the Sox9-Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nature Genetics. 51 (8), 1263-1271 (2019).
- Oudelaar, A. M., et al. Dynamics of the 4D genome during in vivo lineage specification and differentiation. Nature Communications. 11 (1), 1-12 (2020).
- Galupa, R., Heard, E. X-chromosome inactivation: A crossroads between chromosome architecture and gene regulation. Annual Review of Genetics. 52, 535-566 (2018).
- Loda, A., Collombet, S., Heard, E. Gene regulation in time and space during X-chromosome inactivation. Nature Reviews. Molecular Cell Biology. 23 (4), 231-249 (2022).
- van Bemmel, J. G., et al. The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nature Genetics. 51 (6), 1024-1034 (2019).
- Galupa, R., et al. A conserved noncoding locus regulates random monoallelic Xist expression across a topological boundary. Molecular Cell. 77 (2), 352-367 (2020).
- Gjaltema, R. A. F., et al. Distal and proximal cis-regulatory elements sense X chromosome dosage and developmental state at the Xist locus. Molecular Cell. 82 (1), 190-208 (2022).
- Galupa, R., et al. Inversion of a topological domain leads to restricted changes in its gene expression and affects inter-domain communication. Development. 149 (9), (2022).
- Savarese, F., Flahndorfer, K., Jaenisch, R., Busslinger, M., Wutz, A. Hematopoietic precursor cells transiently reestablish permissiveness for X inactivation. Molecular and Cellular Biology. 26 (19), 7167-7177 (2006).
- Schulz, E. G., et al. The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell. 14 (2), 203-216 (2014).
- Gnirke, A., et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology. 27 (2), 182-189 (2009).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Piccinini, F., Tesei, A., Arienti, C., Bevilacqua, A. Cell counting and viability assessment of 2D and 3D Cell cultures: Expected reliability of the trypan blue assay. Biological Procedures Online. 19 (1), 8 (2017).
- Servant, N., et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology. 16, 259 (2015).
- Abdennur, N., Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 36 (1), 311-316 (2020).
- Imakaev, M., et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nature Methods. 9 (10), 999-1003 (2012).
- Forcato, M., et al. Comparison of computational methods for Hi-C data analysis. Nature Methods. 14 (7), 679-685 (2017).
- Venev, S., et al. . open2c/cooltools: v0.4.1. , (2021).
- Wages, J. M. NUCLEIC ACIDS | Immunoassays. Encyclopedia of Analytical Science. , 408-417 (2005).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Servant, N., Peltzer, A. nf-core/hic: Initial release of nf-core/hic. Zenodo. , (2019).
- Furlan, G., et al. The Ftx noncoding locus controls X chromosome inactivation independently of its RNA products. Molecular Cell. 70 (3), 462-472 (2018).
- Giorgetti, L., et al. Structural organization of the inactive X chromosome in the mouse. Nature. 535 (7613), 575-579 (2016).
- Simon, C. S., et al. Functional characterisation of cis-regulatory elements governing dynamic Eomes expression in the early mouse embryo. Development. 144 (7), 1249-1260 (2017).
- Williams, R. M., et al. Reconstruction of the global neural crest gene regulatory network in vivo. Developmental Cell. 51 (2), 255-276 (2019).
- Godfrey, L., et al. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nature Communications. 10 (1), 2803 (2019).
- Hanssen, L. L. P., et al. Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nature Cell Biology. 19 (8), 952-961 (2017).
- Larke, M. S. C., et al. Enhancers predominantly regulate gene expression during differentiation via transcription initiation. Molecular Cell. 81 (5), 983-997 (2021).
- Oudelaar, A. M., et al. A revised model for promoter competition based on multi-way chromatin interactions at the α-globin locus. Nature Communications. 10 (1), 5412 (2019).
- Long, H. K., et al. Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell. 27 (5), 765-783 (2020).
- Schoenfelder, S., Javierre, B. -. M., Furlan-Magaril, M., Wingett, S. W., Fraser, P. Promoter Capture Hi-C: High-resolution, genome-wide profiling of promoter interactions. Journal of Visualized Experiments. (136), e57320 (2018).
- Siersbæk, R., et al. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Molecular Cell. 66 (3), 420-435 (2017).
- Rubin, A. J., et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nature Genetics. 49 (10), 1522-1528 (2017).
- Freire-Pritchett, P., et al. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. eLife. 6, 21926 (2017).
- Javierre, B. M., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 167 (5), 1369-1384 (2016).
- Miguel-Escalada, I., et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nature Genetics. 51 (7), 1137-1148 (2019).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Baxter, J. S., et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nature Communications. 9 (1), 1028 (2018).
- Sanborn, A. L., et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proceedings of the National Academy of Sciences. 112 (47), 6456-6465 (2015).
- Owens, D. D. G., et al. Dynamic Runx1 chromatin boundaries affect gene expression in hematopoietic development. Nature Communications. 13 (1), 773 (2022).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены