
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Análisis morfológico y composicional de trampas extracelulares de neutrófilos inducidas por estímulos microbianos y químicos
En este artículo
Resumen
Aquí se presenta un protocolo para la inducción y análisis de trampas extracelulares de neutrófilos (TNE) in vitro . La cuantificación del ADN, la catelicidina (LL37) y la actividad enzimática arrojó datos que muestran la variabilidad en la composición y morfología de los NET inducidos por estímulos microbianos y químicos en condiciones controladas similares.
Resumen
Los neutrófilos funcionan como la primera línea de defensa celular en una respuesta inmune innata mediante el empleo de diversos mecanismos, como la formación de trampas extracelulares de neutrófilos (TNE). Este estudio analiza los cambios morfológicos y de composición en los NET inducidos por estímulos microbianos y químicos utilizando metodologías estandarizadas in vitro para la inducción y caracterización de NET con células humanas. Los procedimientos descritos aquí permiten el análisis de la morfología NET (lítica o no lítica) y la composición (estructuras ADN-proteína y actividad enzimática), y el efecto de los factores solubles o el contacto celular sobre dichas características. Además, las técnicas descritas aquí podrían modificarse para evaluar el efecto de los factores solubles exógenos o el contacto celular en la composición de NET.
Las técnicas aplicadas incluyen la purificación de células polimorfonucleares de sangre periférica humana utilizando un gradiente de doble densidad (1.079-1.098 g/mL), garantizando una pureza y viabilidad óptimas (≥ 95%) como lo demuestra la tinción de Wright, la exclusión de azul de tripano y la citometría de flujo, incluido el análisis FSC versus SSC y la tinción 7AAD. La formación de NET se induce con estímulos microbianos (Pseudomonas aeruginosa, Staphylococcus aureus y Candida albicans) y químicos (acetato de forbol miristato, HOCl), y los NET se caracterizan por tinción de ADN-DAPI, inmunotinción para el péptido antimicrobiano catelicidina (LL37) y cuantificación de la actividad enzimática (elastasa de neutrófilos, catepsina G y mieloperoxidasa). Las imágenes se adquieren a través de microscopía de fluorescencia y se analizan con ImageJ.
Introducción
Los neutrófilos son los leucocitos más abundantes en el torrente sanguíneo, desempeñando un papel esencial durante la eliminación de agentes patógenos por varios mecanismos, incluyendo la liberación de grandes estructuras de cromatina compuestas de ADN y varias proteínas antibacterianas nucleares, citoplasmáticas y granulares 1,2. El antecedente directo que describe este papel antimicrobiano de los neutrófilos fue elaborado por Takei et al.3 en 1996. Estos autores relataron una nueva forma de muerte diferente de la apoptosis y necroptosis en neutrófilos, mostraron cambios morfológicos que exhibieron ruptura nuclear, seguido de derrame del nucleoplasma hacia el citoplasma, y un aumento en la permeabilidad de la membrana a partir de 3 h de incubación con acetato de forbol miristato (PMA)2,3. Sin embargo, no fue hasta 2004 que se utilizó el término "trampas extracelulares de neutrófilos (TNE)"4.
Se ha observado la formación de NET en diversas afecciones, como infecciones bacterianas, fúngicas5,virales 6 y parasitarias, para neutralizar, matar y prevenir la diseminación microbiana7. Otros estudios muestran que también puede ocurrir en condiciones no patógenas por estímulos estériles, como citoquinas, ácido úrico monosódico o cristales de colesterol, autoanticuerpos, complejos inmunes y plaquetas activadas7. El lipopolisacárido (LPS), la interleucina-8 (IL-8) y el PMA estuvieron entre los primeros estímulos in vitro descritos como inductores de NET, y la participación in vivo de NET en procesos patogénicos se demostró en dos modelos de inflamación aguda: disentería experimental y apendicitis humana espontánea4. El ADN es un componente NET esencial. Su estructura y composición apropiadas son necesarias para el secuestro y la muerte de microorganismos mediante la entrega de una alta concentración local de moléculas antimicrobianas hacia los microbios capturados, como lo demuestra un breve tratamiento con desoxirribonucleasa (DNasa) que desintegra los NET y sus propiedades microbicidas4. Además del ADN, los NET comprenden proteínas unidas como histonas, elastasa de neutrófilos (NE), catepsina G (CG), proteinasa 3, lactoferrina, gelatinasa, mieloperoxidasa (MPO) y péptidos antimicrobianos (AMP) como el péptido proinflamatorio catiónico catelicidina LL-37 entre otros 8,9. Tales agregados pueden formar hilos más grandes con diámetros de hasta 50 nm. Estos factores pueden alterar los factores de virulencia microbiana o la integridad de la membrana celular patógena; además, los AMPs pueden estabilizar el ADN derivado de NET contra la degradación por nucleasas bacterianas10.
Los mecanismos específicos que regulan la formación de NET aún no se han aclarado completamente. La vía mejor caracterizada que conduce a la liberación de NET es a través de la señalización ERK, que conduce a la activación de la NADPH oxidasa y a la producción de especies reactivas de oxígeno (ROS), así como al aumento del calcio intracelular que desencadena la activación de la vía MPO. Esto a su vez transforma el peróxido de hidrógeno en ácido hipocloroso, activando NE por oxidación11,12. NE es responsable de degradar los filamentos de actina del citoesqueleto para bloquear la fagocitosis y translocarlos al núcleo para su procesamiento por escisión proteolítica y desaminación por PAD4 que impulsan la desensibilización de las fibras de cromatina, que se asocian con proteínas granulares y citoplasmáticas, y luego se liberan extracelularmente7. Estas proteasas incluyen las liberadas por el complejo azurosómico de los gránulos azurófilos y otras proteasas como la catepsina G13.
Dependiendo de los cambios morfológicos en los neutrófilos, los TNE se clasifican en dos tipos: formación de NET suicida o lítica que conduce a la muerte celular4, y formación de NET vital o no lítica producida por células viables mediadas por una liberación vesicular de ADN nuclear o mitocondrial, con un remanente de un citoplasto anucleado con capacidad fagocítica14,15. Generalmente, los NET compuestos de ADN mitocondrial presentan una morfología de fibra alargada14, mientras que los estructurados de ADN nuclear tienen una apariencia de nube3. Sin embargo, no se sabe cómo el neutrófilo elige su origen de ADN. Contrariamente a estudios previos que describieron las vías canónicas de los NET como que requieren varias horas, la vía vital se activa rápidamente en solo 5-60 min15.
A pesar de estos avances, la composición NET varía dependiendo del estímulo; por ejemplo, diferentes cepas mucoides y no mucoides de P. aeruginosa inducen la formación de NET que contienen 33 proteínas comunes y hasta 50 proteínas variables7. Así, es necesario homogeneizar técnicas que permitan la generación de conclusiones objetivas en los grupos de investigación. Este artículo describe un protocolo con diversas técnicas que permiten comparar y evaluar la composición, estructura y morfología de los TNE inducidos con diferentes microorganismos: Staphylococcus aureus (bacteria grampositiva), Pseudomonas aeruginosa (bacteria gramnegativa) y Candida albicans (hongo), así como estímulos químicos (PMA, HOCl) en neutrófilos humanos de individuos sanos. Los resultados representativos demuestran la heterogeneidad de los TNE dependiendo de su estímulo inductor en condiciones in vitro comparables, caracterizadas por tinción de ADN-DAPI, inmunotinción para LL37 y cuantificación de la actividad enzimática (NE, CG y MPO).
Protocolo
Las muestras de sangre se obtuvieron como donaciones de participantes clínicamente sanos después del consentimiento informado. Todos los experimentos se realizaron con el permiso del Comité de Ética en Investigación Humana de la Facultad de Ciencias Bioquímicas de la Universidad Autónoma 'Benito Juárez' de Oaxaca.
NOTA: Los criterios de inclusión en el estudio fueron sexo y edad indistintos, y clínicamente sanos de acuerdo con las respuestas de los participantes a un cuestionario antes de tomar una muestra de sangre. Se realizó un análisis hematológico para determinar el recuento celular y descartar infecciones o anemia, así como la prueba de proteína C reactiva para descartar inflamación en el donante.
1. Extracción de sangre periférica y obtención del paquete de eritrocitos y leucocitos
- Recolectar 10 mL de sangre periférica por venopunción en tubos con 1.8 mg/mL de K2· EDTA como anticoagulante (ver Tabla de materiales) de individuos clínicamente sanos después de obtener el consentimiento informado. Luego, realice una biometría sanguínea estándar y una prueba de proteína C reactiva para descartar infección o inflamación, asegurando la calidad de la muestra.
- Centrifugar la muestra de sangre periférica a 82 x g durante 15 min para eliminar el plasma rico en plaquetas, seguido de una segunda centrifugación a 630 x g durante 5 min. Deseche el plasma restante para obtener el paquete de eritrocitos y leucocitos.
- Diluirlo en una proporción de 1:1 (v/v) con 1x solución salina tamponada con fosfato (DPBS) de Dulbecco.
2. Purificación polimorfonuclear de neutrófilos (PMN) utilizando un gradiente de doble densidad
NOTA: Realice la purificación de neutrófilos inmediatamente después de que se recolecte la sangre, ya que tienen una vida útil in vitro limitada de aproximadamente 8 h.
- Deposite lo siguiente en un tubo de vidrio estéril de 10 ml (consulte la Tabla de materiales) en orden: 1 ml de solución de densidad de 1.098 g/ml, 1 ml de solución de densidad de 1.079 g/ml (consulte la tabla de materiales) y luego 4 ml del paquete diluido de eritrocitos y leucocitos. Verter sobre las paredes sin romper la tensión superficial entre las capas para evitar que se mezclen.
- Centrifugar a 320 x g durante 20 min a 4 °C, evitando la aceleración/desaceleración para que las altas fuerzas de la centrífuga no perturben el gradiente.
- Aspirar la fase que corresponde a los granulocitos (Figura 1A) mediante pipeteo, y transferir a otro tubo de vidrio estéril de 10 mL. Lavar con 4 mL de 1x DPBS a 300 x g durante 10 min a 4 °C.
- Deseche el sobrenadante y trate las células con shock osmótico para eliminar los eritrocitos restantes. Añadir 4 ml de solución salina al 0,2% durante 2 min a 4 °C, y centrifugar a 300 x g durante 10 min a 4 °C. Deseche el sobrenadante. A continuación, añadir 4 ml de solución isotónica (solución salina al 0,65%) durante 5 min a 4 °C para restaurar la integridad de la membrana, y centrifugar a 300 x g durante 10 min a 4 °C.
NOTA: La solución salina al 0,2% es un medio hipotónico, con una concentración de soluto más baja en relación con la del medio intracelular RBC. El contacto con el medio hipotónico permite que el agua se difunda en el RBC, lo que lleva a su hinchazón y hemólisis. Esta eliminación de glóbulos rojos del sobrenadante fue confirmada por observación microscópica. - Retire el sobrenadante. Resuspender las células en 4 ml de 1x DPBS para eliminar los restos celulares, y luego centrifugar a 300 x g durante 10 min a 4 °C. Finalmente, resuspenda el pellet celular en 2 ml de tampón de solución salina balanceada de Hank (HBSS) frío.
3. Morfología y viabilidad de neutrófilos (Figura 1B)
- Prueba de exclusión de azul de tripano
- Diluir 5 μL de la suspensión celular en 20 μL de azul de tripano al 0,4% (relación 1:5). Contar las células en una cámara Neubauer y determinar la viabilidad celular mediante una prueba de exclusión. Considere viables las células que mantienen la integridad de su membrana sin permeabilizar el tinte.
- Monte 5 μL de la suspensión celular en un portaobjetos; seco y manchado con la tinción de Wright durante 15 s. Fijar inmediatamente la muestra con tampón fosfato pH 6,4 durante 30 s. Lavar con suficiente agua destilada y observar la morfología bajo un microscopio óptico (100x).
- 7Tinción AAD y análisis de citometría de flujo
- Añadir 1 x 105 células a los tubos de citometría de flujo y teñir con 1 μL de 7AAD en 100 μL de tampón FACS (1x DPBS, 0,1% de azida sódica y 10% de plasma autólogo descomplementado) durante 15 min a 4 °C en la oscuridad.
- Lavar con 500 μL de tampón FACS a 300 x g durante 10 min. Fijar las células con 500 μL de paraformaldehído al 2%, y almacenar a 4 °C hasta su análisis en el citómetro de flujo.
- Para un control de células muertas, fijar 1 x 105 células con 200 μL de paraformaldehído al 4% durante 30 min, y lavar con 500 μL de 1x PBS a 300 x g durante 10 min a 4 °C. Retire el sobrenadante y deséchelo. A continuación, añadir 200 μL de Triton X-100 al 0,1% durante 1 h a 4 °C. Lavar con 500 μL de 1x PBS y teñir con 7AAD como en el paso 3.2.1.
- Usando un citómetro de flujo (ver Tabla de materiales), realice análisis FSC versus SSC para analizar la pureza celular y SSC versus tinción 7AAD para analizar la viabilidad celular. Leer 3 x 104 eventos en 100 μL de volumen de captación a flujo medio (1.000 células/s) en los entornos polimorfonucleares (FSC, 400-490 y SSC, 300-320).
- Analice los datos capturados en el software del citómetro de flujo (ver Tabla de materiales) y determine el porcentaje de pureza y células positivas para 7AAD en la población polimorfonuclear, presentado a través de diagramas de puntos e histogramas.
4. Tinción CFSE de microorganismos
- Añadir 1 x 108 bacterias o 1 x 106 pseudohifas fúngicas en microtubos de 1,5 ml, y teñir con 200 μL de 5 μM de carboxifluoresceína succinimidil éster (CFSE) disuelto en 1x PBS. Mezclar durante unos segundos e incubar a 37 °C durante 10 minutos en la oscuridad.
- Detenga la reacción añadiendo 500 μL de plasma descomplementado y centrifugar a 620 x g durante 10 min para pseudohifas o a 1.800 x g durante 10 min para bacterias.
- Desechar los sobrenadantes y lavar los gránulos con 1 ml de 1x PBS con centrifugación como en el paso 4.2. Finalmente, resuspender los microorganismos en 250 μL de 1x PBS.
- Preparar alícuotas de 50 μL en microtubos de 1,5 mL con 2 x 107 bacterias (MOI: 100) o 2 x 105 pseudohifas (MOI:1) para la inducción NET.
5. Inducción NET
- Coloque los cubreobjetos de vidrio estéril de 10 mm x 10 mm en una placa de 24 pocillos y cubra con 10 μL de poli-L-lisina al 0,001% durante 1 h a temperatura ambiente. Lavar dos veces con 100 μL de 1x PBS, secar al aire e irradiar con luz UV durante 15 min.
- Reemplace la solución HBSS de la suspensión de neutrófilos en el paso 2.5 con RPMI 1640 medio suplementado con plasma autólogo al 10%. A la placa de 24 pocillos (paso 5.1), añadir 350 μL de esta suspensión celular, para obtener una concentración final de 2 x 105 neutrófilos/pocillo.
- Permitir que las células se adhieran al fondo de los pocillos incubando durante 20 min a 37 °C con 5% deCO2.
- Añadir los estímulos para inducir la formación de NET en 50 μL: estímulos microbianos-grampositivos de la bacteria S. aureus (ATCC 25923), la bacteria gramnegativa P. aeruginosa (ATCC 10145) en MOI 100, y pseudohifas de C. albicans (ATCC 10231) en MOI:1; estímulos bioquímicos-PMA (200 nM) y HOCl (4,5 mM), y control con estímulo ausente (50 μL de HBSS).
- Obtener un volumen final de 400 μL por pocillo. Mezclar en una agitadora de placas a 140 rpm durante 30 s, e incubar durante 4 h a 37 °C y 5% deCO2.
6. Visualización de NET por microscopía de fluorescencia
- Inmunotinción de ADN y LL37
- Después de la inducción NET, retire los sobrenadantes de los pocillos pipeteando cuidadosamente y fije las células con 300 μL de paraformaldehído al 4% durante 30 min.
- Lavar las células con 200 μL de 1x PBS sin centrifugar, y añadir 200 μL de tampón de bloqueo (10% de plasma descomplementado en 1x PBS) durante 30 min.
- Para la tinción LL-37, permeabilizar las células con 200 μL de Triton X-100 al 0,2% en 1x PBS durante 10 min para permitir que el anticuerpo entre en las células. Lave 2x cuidadosamente con 1x PBS para eliminar el exceso de detergente.
- Monte los cubreobjetos en portaobjetos de vidrio (cuatro cubreobjetos en cada portaobjetos). El ADN tiñe las células con 2 μL de DAPI (ver Tabla de materiales), sella los cubreobjetos y almacena a -20 °C hasta su análisis por microscopía de fluorescencia confocal.
- Adquisición y análisis de imágenes fluorescentes
- Tome imágenes NET para cuantificar sus componentes y use los filtros correspondientes en el microscopio de fluorescencia confocal (consulte la Tabla de materiales) para adquirir las imágenes con el software de la computadora.
NOTA: Considere que el ADN está teñido con DAPI (color azul), mostrando excitación a 360 nm y emisión a 460 nm. Los microorganismos se tiñen con CFSE (color verde), que tiene una excitación de 492 nm y una emisión de 521 nm. El péptido LL37 está marcado con anticuerpo anti-LL37 Alexa Fluor 594 (color rojo), que tiene una excitación de 594 nm y una emisión de 614 nm. - Calibre el microscopio. Coloque la diapositiva y enfoque utilizando el contraste de interferencia diferencial (DIC) con la luz normal encendida. Elija Live (En directo ) para proyectar la imagen en el monitor.
- Apague la luz y seleccione el canal de fluorocromo correspondiente. Por ejemplo, seleccione el filtro 365 nm/azul para DAPI, 43 HE DsRed para Alexa 594 o 38 HE GFP para CFSE.
- Ajuste la configuración con el anticuerpo de control de isotipo para LL37 y células no teñidas para DAPI y CFSE. Establezca el mismo tiempo de exposición, voltaje, contraste y configuración de lente para capturar todas las imágenes en las mismas condiciones.
NOTA: En este estudio, el tiempo de exposición, el voltaje y el contraste se establecieron en 1.0 ms, 4.0 V y 0.0, respectivamente, con un objetivo de 40x. Estos valores se pueden ajustar para facilitar la mejor captura de imágenes para las muestras. - Seleccione Ajustar para capturar la imagen. Guardar cinco imágenes (cuatro extremos y el centro) por pocillo, y de la colocalización (fusión) de DNA/LL37/CFSE.
- Defina las tres clases de píxeles como fondo con las imágenes independientes de cada color y analice el valor de la señal de gris medio por área con el software Image J.
- Tome imágenes NET para cuantificar sus componentes y use los filtros correspondientes en el microscopio de fluorescencia confocal (consulte la Tabla de materiales) para adquirir las imágenes con el software de la computadora.
7. Cuantificación de la actividad enzimática
- En una placa de 96 pocillos, añadir 90 μL de suspensión celular en HBSS que contenga 1 x 10 5 neutrófilos para la inducción NET e incubar durante 20 min a 37 °C y5 % deCO2.
- Añadir inmediatamente 10 μL de los estímulos correspondientes (concentración como en el paso 5.4) e incubar durante 4 h a 37 °C con 5% deCO2.
- Deseche los sobrenadantes y lave las células con 100 μL de HBSS. Tratar con 1 U/mL de DNasa durante 10 min a 37 °C para favorecer la liberación de estructuras ADN-proteína, y centrifugar a 1.800 x g durante 10 min.
- Recuperar los sobrenadantes y evaluar la actividad enzimática en el sobrenadante mediante reacciones colorimétricas descritas previamente por White et al.17.
- Determinar la actividad enzimática máxima de NE, CG y MPO en neutrófilos bajo las mismas condiciones experimentales sin agregar ningún estímulo para la inducción de NET. A continuación, congelar la muestra celular a -70 °C y descongelar a 37 °C en un baño maría, generando un choque de temperatura para favorecer la liberación de proteínas intracelulares por lisis celular. Centrifugar a 1.800 x g durante 10 min y recuperar los sobrenadantes.
- Añadir 50 μL del sobrenadante a cada pocillo en placas de 96 pocillos y, a continuación, añadir 50 μL de cada sustrato como se indica en el paso 7.7.
- Añadir 0,5 M de N-metoxisuccinil-Ala-Ala-Pro-Val-p-nitro anilina como sustrato para NE, y 1 mM de N-succinil-Ala-Ala-Pro-Phe-p-nitroanilida para CG. Incubar durante 3 h a temperatura ambiente. Para MPO, añadir 1,6 mM de 3,3', 5,5'-tetrametilbencidina (TMB) e incubar durante 30 min a temperatura ambiente.
- Después de la incubación, añadir 50 μL de la solución de parada (0,5 M H2SO4) para MPO y medir la absorbancia a 405 nm para NE y CG y 450 nm para MPO, utilizando un espectrofotómetro.
- Comparar los valores obtenidos con las curvas de calibración correspondientes y mostrar los resultados de cada condición en relación con la actividad enzimática máxima (100%).
8. Análisis estadístico
- Analizar los datos de medición por triplicado para cada experimento independiente (n = 10) y realizar un ANOVA para el análisis estadístico comparando grupos con un nivel de confianza del 95%.

Figura 1: Purificación de PMN y protocolo de inducción NET. (A) El plasma se extrajo de la sangre periférica para obtener el paquete de eritrocitos y leucocitos y se diluyó 1:1 (v/v) con 1x DPBS. A continuación, se añadieron 4 mL de dilución a lo largo de la pared hasta el tubo de gradiente de doble densidad, y se centrifugaron a 320 x g durante 20 min a 4 °C, obteniendo la separación de diferentes capas celulares y recuperando la correspondiente a PMN. (B) Se contaron las células purificadas y se analizó su morfología mediante tinción de Wright. La viabilidad se determinó mediante exclusión de azul de tripano y tinción de 7AAD mediante citometría de flujo. Una vez que se verificó la pureza y viabilidad óptimas de los neutrófilos, se indujo la formación de NET mediante la adición de microbios (S. aureus, P. aeruginosa y C. albicans) o productos químicos (PMA, HOCl) en placas de 24 pocillos para su análisis por microscopía de fluorescencia con DAPI-DNA, anti-LL37 Alexa Fluor 594 y tinción de microorganismos-CFSE. Para la cuantificación enzimática, los TNE se indujeron en placas de 96 pocillos durante 3 h y se trataron con DNasa, seguido de la adición de sustratos para cada enzima: NE, CG y MPO; Los cambios de color se cuantificaron mediante espectrofotometría. DPBS = solución salina tamponada con fosfato de Dulbecco; PBMC = células mononucleares de sangre periférica; PMN = neutrófilos polimorfonucleares; NE = elastasa de neutrófilos; CG = catepsina G; MPO = mieloperoxidasa; PMA = acetato de miristato de forbol; HOCl = ácido hipocloroso. Haga clic aquí para ver una versión más grande de esta figura.
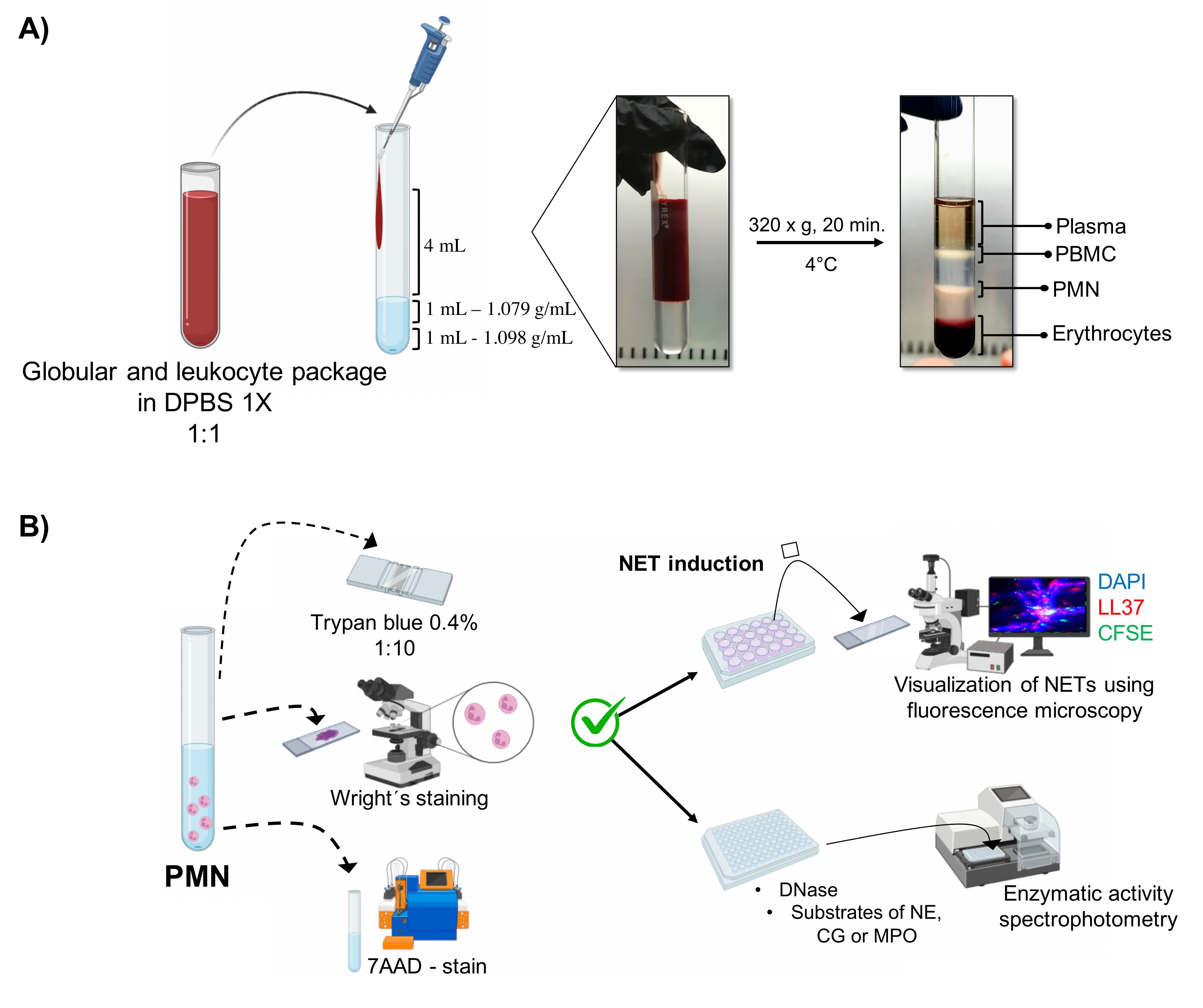{kind=link}
Resultados
Pureza y viabilidad de los neutrófilos
Las fases celulares dinámicas se visualizan en el tubo a partir de la purificación de gradiente de doble densidad. Dentro de estas capas, la capa correspondiente a los granulocitos está por encima de la capa de densidad de 1,079 g/ml, que se distingue de las fases de los mononucleocitos de sangre periférica (PBMC) y eritrocitos (Figura 1A). La morfología de las células purificadas se verificó con la tinción de Wright mediant...
Discusión
Se debe obtener una población altamente pura de neutrófilos viables para inducir la liberación de TNE, ya que estas células tienen una vida útil ex vivo limitada de 8 h en promedio, un período dentro del cual se deben realizar todos los experimentos. Para ello, la metodología ideal es el gradiente de doble densidad para optimizar el tiempo de purificación mediante el aislamiento de células no activadas más sensibles a la estimulación exógena, en contraste con el gradiente de Ficoll-Histopaque o las t...
Divulgaciones
Los autores declaran que no tienen conflictos de intereses.
Agradecimientos
Este trabajo fue apoyado por una beca de ciencias básicas (#285480) del CONACyT y por el Departamento de Investigación en Inmunología Clínica de la Facultad de Ciencias Bioquímicas de la Universidad Autónoma 'Benito Juárez' de Oaxaca. A.A.A, S.A.S.L, y W.J.R.R. tienen becas doctorales de CONACyT números #799779, #660793 y #827788, respectivamente.
Materiales
| Name | Company | Catalog Number | Comments |
| 24 Well plate for cell culture | Corning | 3526 | |
| 7-aminoactinomycin D (7-AAD) | BD Pharmingen | 51-668981E | |
| 96 Well plate for cell culture | Costar | 3596 | Flat bottom |
| Agitator | CRM Globe | CRM-OS1 | |
| Antibody LL37 | Santa Cruz Biotechnology | sc-166770 | |
| Blood collection tubes | BD VACUTAINER | 368171 | K2 EDTA 7.2 mg |
| Carboxyfluorescein succinimidyl ester (CFSE) | Sigma-Aldrich | 21878 | |
| Centrifuge | Hettich | 1406-01 | |
| Coverslip | Madesa | M03-CUB-22X22 | 22 mm x 22 mm |
| Dulbecco´s phosphate-buffered saline (DPBS) | Caisson | 1201022 | |
| Falcon tubes 50 mL | CORNING | 430829 | |
| Flow Cytometry Tubes | Miltenyi Biotec | 5 mL - Without caps | |
| FlowJo Software | BD Biosciences | Analyze flow cytometry data | |
| Fluorescence microscope | DM 2000 | LEICA | |
| Fluoroshield with DAPI | Sigma-Aldrich | F6057 | |
| Incubator | NUAIRE | UN-4750 | |
| MACSQuant Analyzer | Miltenyi Biotec | Flow cytometer | |
| Microplate reader photometer | Clarkson Laboratory - CL | ||
| Microtubes 1.5 mL | Zhejiang Runlab Tech | 35200N | wire snap |
| Minitab Software | Minitab | Statistical analysis | |
| Needles | BD VACUTAINER | 301746 | Diameter 1.34 mm |
| Optical microscope | VELAB | VE-B50 | |
| Percoll | GE Healthcare | 17-0891-01 | Solution for density gradient |
| Phosphate Buffered Saline (10x) | Caisson | PBL07-500ML | |
| Pyrex culture tubes | CORNING | CLS982025 | N°9820 |
| RPMI 1640 1x | Corning | 10-104-CV | contains Glutagro |
| Slides | Madesa | PDI257550 | 22 mm x 75 mm |
| Trypan Blue solution 0.4% | SIGMA | T8154-100ML |
Referencias
- De Buhr, N., Maren, K. B. How neutrophil extracellular traps become visible. Journal of Immunology Research. 2016, 4604713 (2016).
- Karlsson, A., Nixon, J. B., McPhail, L. C. Phorbol myristate acetate induces neutrophil NADPH-oxidase activity by two separate signal transduction pathways: dependent or independent of phosphatidylinositol 3-kinase. Journal of Leukocyte Biology. 67 (3), 396-404 (2000).
- Takei, H., Araki, A., Watanabe, H., Ichinose, A., Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. Journal of Leukocyte Biology. 59 (2), 229-240 (1996).
- Brinkmann, V., et al. Neutrophil extracellular traps kill bacteria. Science. 303 (5663), 1532-1535 (2004).
- Kenny, E. F., et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife. 6, 24437 (2017).
- Schultz, B. M., Acevedo, O. A., Kalergis, A. M., Bueno, S. M. Role of extracellular trap release during bacterial and viral infection. Frontiers in Microbiology. 13, 798853 (2022).
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nature Reviews Immunology. 18 (2), 134-147 (2018).
- Delgado-Rizo, V., et al. Neutrophil extracellular traps and its implications in inflammation: An overview. Frontiers in Immunology. 8, 81 (2017).
- Petretto, A., et al. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS One. 14 (7), 0218946 (2019).
- Neumann, A., et al. Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular traps against degradation by bacterial nucleases. Journal of Innate Immunity. 6 (6), 860-868 (2014).
- Hakkim, A., et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nature Chemical Biology. 7 (2), 75-77 (2011).
- Sabbatini, M., Magnelli, V., Renò, F. NETosis in wound healing: When enough Is enough. Cells. 10 (3), 494 (2021).
- Metzler, K. D., Goosmann, C., Lubojemska, A., Zychlinsky, A., Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Reports. 8 (3), 883-896 (2014).
- Clark, S. R., et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature Medicine. 13 (4), 463-469 (2007).
- Pilsczek, F. H., et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. Journal of Immunology. 185 (12), 7413-7425 (2010).
- Sosa, S. A., et al. Structural differences of neutrophil extracellular traps induced by biochemical and microbiologic stimuli under healthy and autoimmune milieus. Immunologic Research. 69 (3), 264-274 (2021).
- White, P. C., et al. Characterization, quantification, and visualization of neutrophil extracellular traps. Methods in Molecular Biology. 1537, 481-497 (2017).
- Boeltz, S., et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death and Differentiation. 26 (3), 395-408 (2019).
- Yousefi, S., Mihalache, C., Kozlowski, E., Schmid, I., Simon, H. U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death and Differentiation. 16 (11), 1438-1444 (2009).
- Brinkmann, V., Zychlinsky, A. Neutrophil extracellular traps: is immunity the second function of chromatin. The Journal of Cell Biology. 198 (5), 773-783 (2012).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados