
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Analisi morfologica e composizionale di trappole extracellulari neutrofile indotte da stimoli microbici e chimici
In questo articolo
Riepilogo
Presentato qui è un protocollo per l'induzione e l'analisi di trappole extracellulari (NET) di neutrofili in vitro . La quantificazione del DNA, della catelicidina (LL37) e dell'attività enzimatica ha prodotto dati che mostrano la variabilità nella composizione e morfologia dei NET indotta da stimoli microbici e chimici in condizioni controllate simili.
Abstract
I neutrofili funzionano come la prima linea di difesa cellulare in una risposta immunitaria innata impiegando diversi meccanismi, come la formazione di trappole extracellulari per neutrofili (NET). Questo studio analizza i cambiamenti morfologici e composizionali nei NET indotti da stimoli microbici e chimici utilizzando metodologie standardizzate in vitro per l'induzione e la caratterizzazione del NET con cellule umane. Le procedure qui descritte consentono l'analisi della morfologia NET (litica o non litica) e della composizione (strutture DNA-proteina e attività enzimatica), e l'effetto di fattori solubili o di contatto cellulare su tali caratteristiche. Inoltre, le tecniche qui descritte potrebbero essere modificate per valutare l'effetto di fattori solubili esogeni o del contatto cellulare sulla composizione NET.
Le tecniche applicate includono la purificazione di cellule polimorfonucleate dal sangue periferico umano utilizzando un gradiente a doppia densità (1,079-1,098 g/ml), garantendo purezza e vitalità ottimali (≥ 95%) come dimostrato dalla colorazione di Wright, dall'esclusione del blu di tripano e dalla citometria a flusso, compresa l'analisi FSC versus SSC e la colorazione 7AAD. La formazione di NET è indotta da stimoli microbici (Pseudomonas aeruginosa, Staphylococcus aureus e Candida albicans) e chimici (phorbol myristate acetate, HOCl) e i NET sono caratterizzati da colorazione DNA-DAPI, immunocolorazione per il peptide antimicrobico catelicidina (LL37) e quantificazione dell'attività enzimatica (elastasi neutrofila, catepsina G e mieloperossidasi). Le immagini vengono acquisite al microscopio a fluorescenza e analizzate con ImageJ.
Introduzione
I neutrofili sono i leucociti più abbondanti nel flusso sanguigno, svolgendo un ruolo essenziale durante la clearance degli agenti patogeni attraverso diversi meccanismi, tra cui il rilascio di grandi strutture cromatiniche composte da DNA e diverse proteine antibatteriche nucleari, citoplasmatiche e granulari 1,2. L'antecedente diretto che descrive questo ruolo antimicrobico dei neutrofili è stato fatto da Takei et al.3 nel 1996. Questi autori hanno riportato una nuova forma di morte diversa dall'apoptosi e dalla necroptosi nei neutrofili, hanno mostrato cambiamenti morfologici che mostrano rottura nucleare, seguita da fuoriuscita dal nucleoplasma nel citoplasma e un aumento della permeabilità della membrana da 3 ore di incubazione con forbolo miristato acetato (PMA)2,3. Tuttavia, non è stato fino al 2004 che è stato utilizzato il termine "trappole extracellulari di neutrofili (NET)"4.
La formazione di NET è stata osservata in varie condizioni, come infezioni batteriche, fungine5, virali6 e parassitarie, per neutralizzare, uccidere e prevenire la diffusione microbica7. Altri studi dimostrano che può verificarsi anche in condizioni non patogene da stimoli sterili, come citochine, acido urico monosodico o cristalli di colesterolo, autoanticorpi, immunocomplessi e piastrine attivate7. Il lipopolisaccaride (LPS), l'interleuchina-8 (IL-8) e la PMA sono stati tra i primi stimoli in vitro descritti come induttori NET e il coinvolgimento in vivo del NET nei processi patogeni è stato dimostrato in due modelli di infiammazione acuta: dissenteria sperimentale e appendicite umana spontanea4. Il DNA è un componente essenziale di NET. La sua struttura e composizione appropriate sono necessarie per il sequestro e l'uccisione dei microrganismi fornendo un'alta concentrazione locale di molecole antimicrobiche verso i microbi catturati, come dimostrato da un breve trattamento con desossiribonucleasi (DNasi) che disintegra i NET e le loro proprietà microbicide4. Oltre al DNA, i NET comprendono proteine attaccate come istoni, elastasi neutrofila (NE), catepsina G (CG), proteinasi 3, lattoferrina, gelatinasi, mieloperossidasi (MPO) e peptidi antimicrobici (AMP) come il peptide pro-infiammatorio cationico catelicidina LL-37 tra gli altri 8,9. Tali aggregati possono formare fili più grandi con diametri fino a 50 nm. Questi fattori possono interrompere i fattori di virulenza microbica o l'integrità della membrana cellulare patogena; inoltre, gli AMP possono stabilizzare il DNA derivato dal NET contro la degradazione da parte delle nucleasi batteriche10.
I meccanismi specifici che regolano la formazione di NET non sono ancora stati completamente chiariti. La via meglio caratterizzata che porta al rilascio di NET è attraverso la segnalazione ERK, che porta all'attivazione della NADPH ossidasi e alla produzione di specie reattive dell'ossigeno (ROS), nonché all'aumento del calcio intracellulare che innesca l'attivazione della via MPO. Questo a sua volta trasforma il perossido di idrogeno in acido ipocloroso, attivando NE per ossidazione11,12. NE è responsabile della degradazione dei filamenti di actina del citoscheletro per bloccare la fagocitosi e della loro traslocazione al nucleo per l'elaborazione mediante scissione proteolitica e deaminazione da parte di PAD4 che guidano la desensibilizzazione delle fibre di cromatina, che si associano a proteine granulari e citoplasmatiche, e vengono quindi rilasciate extracellulare7. Queste proteasi includono quelle rilasciate dal complesso degli azurosomi dei granuli azurofili e altre proteasi come la catepsina G13.
A seconda dei cambiamenti morfologici nei neutrofili, i NET sono classificati in due tipi: formazione di NET suicidaria o litica che porta alla morte cellulare4 e formazione di NET vitale o non litica prodotta da cellule vitali mediate da un rilascio vescicolare di DNA nucleare o mitocondriale, con un residuo di un citoplasto anucleato con capacità fagocitaria14,15. Generalmente, i NET composti da DNA mitocondriale presentano una morfologia allungata della fibra14, mentre quelli strutturati di DNA nucleare hanno un aspetto simile a una nuvola3. Tuttavia, non è noto come il neutrofilo scelga la sua origine del DNA. Contrariamente a studi precedenti che descrivevano i percorsi canonici dei NET come se richiedessero diverse ore, il percorso vitale viene rapidamente attivato in soli 5-60 minutie 15.
Nonostante questi progressi, la composizione NET varia a seconda dello stimolo; ad esempio, diversi ceppi mucoidi e non mucoidi di P. aeruginosa inducono la formazione di NET contenenti 33 proteine comuni e fino a 50 proteine variabili7. Pertanto, è necessario omogeneizzare le tecniche che consentono la generazione di conclusioni oggettive nei gruppi di ricerca. Questo articolo descrive un protocollo con varie tecniche che consentono il confronto e la valutazione della composizione, della struttura e della morfologia dei NET indotti con diversi microrganismi: Staphylococcus aureus (batterio gram-positivo), Pseudomonas aeruginosa (batterio gram-negativo) e Candida albicans (fungo), nonché stimoli chimici (PMA, HOCl) in neutrofili umani da individui sani. I risultati rappresentativi dimostrano l'eterogeneità dei NET a seconda del loro stimolo inducente in condizioni comparabili in vitro, caratterizzate da colorazione DNA-DAPI, immunocolorazione per LL37 e quantificazione dell'attività enzimatica (NE, CG e MPO).
Protocollo
I campioni di sangue sono stati ottenuti come donazioni da partecipanti clinicamente sani dopo il consenso informato. Tutti gli esperimenti sono stati eseguiti con il permesso del Comitato Etico di Ricerca Umana della Facoltà di Scienze Biochimiche, Universidad Autónoma 'Benito Juárez' di Oaxaca.
NOTA: I criteri di inclusione nello studio erano sesso ed età indistinti e clinicamente sani in base alle risposte dei partecipanti a un questionario prima di prelevare un campione di sangue. È stata eseguita un'analisi ematologica per determinare la conta cellulare ed escludere infezioni o anemia, nonché il test della proteina C-reattiva per escludere l'infiammazione nel donatore.
1. Raccolta del sangue periferico e ottenimento del pacchetto di eritrociti e leucociti
- Raccogliere 10 ml di sangue periferico mediante venipuntura in provette con 1,8 mg/ml di K2· EDTA come anticoagulante (vedi Tabella dei materiali) da individui clinicamente sani dopo aver ottenuto il consenso informato. Quindi, eseguire la biometria del sangue standard e il test della proteina C-reattiva per escludere infezioni o infiammazioni, garantendo la qualità del campione.
- Centrifugare il campione di sangue periferico a 82 x g per 15 minuti per rimuovere il plasma ricco di piastrine, seguito da una seconda centrifugazione a 630 x g per 5 minuti. Eliminare il plasma rimanente per ottenere il pacchetto eritrocitario e leucocitario.
- Diluirlo in rapporto 1:1 (v/v) con 1x soluzione salina tamponata fosfato di Dulbecco (DPBS).
2. Purificazione di neutrofili polimorfonucleati (PMN) utilizzando un gradiente a doppia densità
NOTA: Eseguire la purificazione dei neutrofili immediatamente dopo la raccolta del sangue, perché hanno una durata limitata in vitro di circa 8 ore.
- Depositare quanto segue in un tubo di vetro sterile da 10 mL (vedere Tabella dei materiali) nell'ordine: 1 mL di soluzione di densità 1,098 g/mL, 1 mL di soluzione di densità 1,079 g/mL (vedere Tabella dei materiali) e quindi 4 mL del pacchetto diluito di eritrociti e leucociti. Versare sulle pareti senza rompere la tensione superficiale tra gli strati per evitare che si mescolino.
- Centrifugare a 320 x g per 20 minuti a 4 °C, evitando accelerazioni/decelerazioni in modo che le elevate forze della centrifuga non disturbino la pendenza.
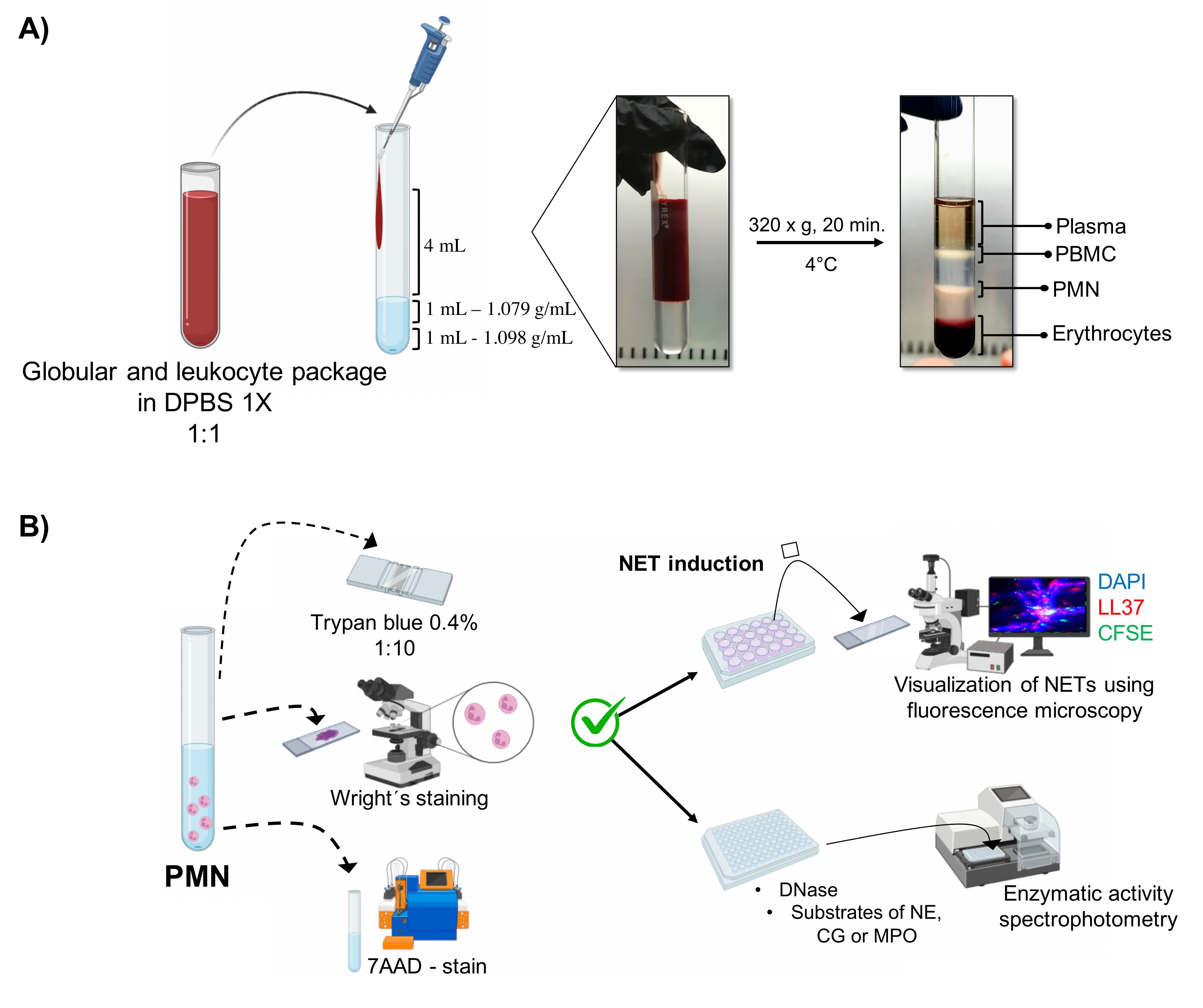
- Aspirare la fase corrispondente ai granulociti (Figura 1A) mediante pipettaggio e trasferire in un altro tubo di vetro sterile da 10 ml. Lavare con 4 mL di 1x DPBS a 300 x g per 10 minuti a 4 °C.
- Scartare il surnatante e trattare le cellule con shock osmotico per rimuovere gli eritrociti rimanenti. Aggiungere 4 mL di soluzione salina allo 0,2% per 2 min a 4 °C e centrifugare a 300 x g per 10 min a 4 °C. Scartare il surnatante. Quindi aggiungere 4 mL della soluzione isotonica (soluzione salina allo 0,65%) per 5 minuti a 4 °C per ripristinare l'integrità della membrana e centrifugare a 300 x g per 10 minuti a 4 °C.
NOTA: La soluzione salina allo 0,2% è un mezzo ipotonico, con una concentrazione di soluto inferiore rispetto a quella del mezzo intracellulare RBC. Il contatto con il mezzo ipotonico consente all'acqua di diffondersi nei globuli rossi, portando al loro gonfiore ed emolisi. Questa rimozione di RBC dal surnatante è stata confermata dall'osservazione microscopica. - Rimuovere il surnatante. Risospendere le cellule in 4 ml di 1x DPBS per rimuovere i detriti cellulari, quindi centrifugare a 300 x g per 10 minuti a 4 °C. Infine, risospendere il pellet cellulare in 2 ml di tampone freddo della soluzione salina bilanciata di Hank (HBSS).
3. Morfologia e vitalità dei neutrofili (Figura 1B)
- Test di esclusione del blu di Trypan
- Diluire 5 μL della sospensione cellulare in 20 μL di blu tripano allo 0,4% (rapporto 1:5). Contare le cellule in una camera di Neubauer e determinare la vitalità cellulare utilizzando un test di esclusione. Considera le cellule che mantengono l'integrità della loro membrana senza permeabilizzare il colorante come vitali.
- Montare 5 μL della sospensione cellulare su un vetrino; asciugare e macchiare con la macchia di Wright per 15 s. Fissare immediatamente il campione con tampone fosfato pH 6,4 per 30 s. Lavare con acqua distillata sufficiente e osservare la morfologia al microscopio ottico (100x).
- 7AAD-colorazione e analisi citometrica a flusso
- Aggiungere 1 x 105 cellule ai tubi per citometria a flusso e colorare con 1 μL di 7AAD in 100 μL di tampone FACS (1x DPBS, 0,1% di azoturo di sodio e 10% di plasma autologo decomplementato) per 15 minuti a 4 °C al buio.
- Lavare con 500 μL di tampone FACS a 300 x g per 10 min. Fissare le cellule con 500 μL di paraformaldeide al 2% e conservare a 4 °C fino alla loro analisi nel citometro a flusso.
- Per un controllo delle cellule morte, fissare 1 x 105 cellule con 200 μL di paraformaldeide al 4% per 30 minuti e lavare con 500 μL di 1x PBS a 300 x g per 10 minuti a 4 °C. Estrarre il surnatante e scartare. Quindi aggiungere 200 μL di Triton X-100 allo 0,1% per 1 ora a 4 °C. Lavare con 500 μL di 1x PBS e colorare con 7AAD come al punto 3.2.1.
- Utilizzando un citometro a flusso (vedere la tabella dei materiali), eseguire l'analisi FSC rispetto a SSC per analizzare la purezza cellulare e la colorazione SSC rispetto a 7AAD per analizzare la vitalità cellulare. Lettura di 3 x 104 eventi in 100 μL di volume di assorbimento a flusso medio (1.000 cellule/s) nelle impostazioni polimorfonucleate (FSC, 400-490 e SSC, 300-320).
- Analizzare i dati acquisiti nel software del citometro a flusso (vedi Tabella dei materiali) e determinare la percentuale di purezza e cellule positive per 7AAD nella popolazione polimorfonucleata, presentata attraverso dot plot e istogrammi.
4. Colorazione CFSE di microrganismi
- Aggiungere 1 x 108 batteri o 1 x 106 pseudoife fungine in microtubi da 1,5 mL e colorare con 200 μL di carbossimetilestere succinimidil (CFSE) 5 μM disciolto in 1x PBS. Mescolare per alcuni secondi e incubare a 37 °C per 10 minuti al buio.
- Interrompere la reazione aggiungendo 500 μL di plasma decomplementato e centrifugare a 620 x g per 10 minuti per le pseudoife o a 1.800 x g per 10 minuti per i batteri.
- Scartare i surnatanti e lavare il pellet con 1 mL di 1x PBS con centrifugazione come al punto 4.2. Infine, risospendere i microrganismi in 250 μL di 1x PBS.
- Preparare 50 μL di aliquote in microtubi da 1,5 mL con 2 x 107 batteri (MOI: 100) o 2 x 105 pseudoife (MOI:1) per l'induzione NET.
5. Induzione NET
- Posizionare i vetrini sterili da 10 x 10 mm in una piastra a 24 pozzetti e coprire con 10 μL di poli-L-lisina allo 0,001% per 1 ora a temperatura ambiente. Lavare due volte con 100 μL di 1x PBS, asciugare all'aria e irradiare con luce UV per 15 minuti.
- Sostituire la soluzione HBSS della sospensione di neutrofili nella fase 2.5 con RPMI 1640 mezzo integrato con plasma autologo al 10%. Alla piastra a 24 pozzetti (punto 5.1), aggiungere 350 μL di questa sospensione cellulare, per una concentrazione finale di 2 x 105 neutrofili/pozzetto.
- Lasciare che le cellule aderiscano al fondo dei pozzetti incubando per 20 minuti a 37 °C con il 5% di CO2.
- Aggiungere gli stimoli per indurre la formazione di NET in 50 μL: stimolo microbico batterio gram-positivo S. aureus (ATCC 25923), batterio gram-negativo P. aeruginosa (ATCC 10145) a MOI 100 e pseudoife di C. albicans (ATCC 10231) a MOI:1; stimoli biochimici-PMA (200 nM) e HOCl (4,5 mM), e controllo con stimolo assente (50 μL di HBSS).
- Ottenere un volume finale di 400 μL per pozzetto. Mescolare su uno scuotitore a 140 giri/min per 30 s e incubare per 4 ore a 37 °C e 5% di CO2.
6. Visualizzazione di NET mediante microscopia a fluorescenza
- DNA e immunocolorazione LL37
- Dopo l'induzione NET, rimuovere i surnatanti dai pozzetti pipettando accuratamente e fissare le cellule con 300 μL di paraformaldeide al 4% per 30 minuti.
- Lavare le celle con 200 μL di 1x PBS senza centrifugazione e aggiungere 200 μL di tampone bloccante (plasma decomplementato al 10% in 1x PBS) per 30 minuti.
- Per la colorazione LL-37, permeabilizzare le cellule con 200 μL di Triton X-100 allo 0,2% in 1x PBS per 10 minuti per consentire all'anticorpo di entrare nelle cellule. Lavare accuratamente 2 volte con 1x PBS per rimuovere il detersivo in eccesso.
- Montare i vetrini su vetrini (quattro vetrini su ogni vetrino). Colorare le cellule con 2 μL di DAPI (vedi Tabella dei materiali), sigillare i vetrini di copertura e conservarli a -20 °C fino alla loro analisi mediante microscopia a fluorescenza confocale.
- Acquisizione e analisi di immagini fluorescenti
- Prendere immagini NET per quantificare i loro componenti e utilizzare i filtri corrispondenti nel microscopio a fluorescenza confocale (vedi Tabella dei materiali) per acquisire le immagini con il software del computer.
NOTA: Si consideri che il DNA è colorato con DAPI (colore blu), mostrando eccitazione a 360 nm ed emissione a 460 nm. I microrganismi sono colorati con CFSE (colore verde), che ha un'eccitazione di 492 nm e un'emissione di 521 nm. Il peptide LL37 è etichettato con anticorpo anti-LL37 Alexa Fluor 594 (colore rosso), che ha un'eccitazione di 594 nm e un'emissione di 614 nm. - Calibrare il microscopio. Posiziona la diapositiva e la messa a fuoco utilizzando il contrasto di interferenza differenziale (DIC) con la luce normale attivata. Scegliere Live per proiettare l'immagine sul monitor.
- Spegnere la luce e selezionare il canale corrispondente fluorocromo. Ad esempio, selezionare il filtro 365 nm/blu per DAPI, 43 HE DsRed per Alexa 594 o 38 HE GFP per CFSE.
- Regolare le impostazioni con l'anticorpo di controllo isotipo per LL37 e le cellule non colorate per DAPI e CFSE. Imposta le stesse impostazioni del tempo di esposizione, della tensione, del contrasto e dell'obiettivo per acquisire tutte le immagini nelle stesse condizioni.
NOTA: In questo studio, il tempo di esposizione, la tensione e il contrasto sono stati impostati rispettivamente a 1,0 ms, 4,0 V e 0,0, con un obiettivo 40x. Questi valori possono essere regolati per facilitare la migliore acquisizione delle immagini per i campioni. - Selezionare Snap per acquisire l'immagine. Salvare cinque immagini (quattro estremi e il centro) per pozzetto, e della colocalizzazione (unione) di DNA/LL37/CFSE.
- Definisci le tre classi di pixel come sfondo con le immagini indipendenti di ciascun colore e analizza il valore del segnale grigio medio per area con il software Image J.
- Prendere immagini NET per quantificare i loro componenti e utilizzare i filtri corrispondenti nel microscopio a fluorescenza confocale (vedi Tabella dei materiali) per acquisire le immagini con il software del computer.
7. Quantificazione dell'attività enzimatica
- In una piastra a 96 pozzetti, aggiungere 90 μL di sospensione cellulare in HBSS contenente 1 x 10 5 neutrofili per l'induzione NET e incubare per 20 minuti a 37 °C e5 % di CO2.
- Aggiungere immediatamente 10 μL degli stimoli corrispondenti (concentrazione come al punto 5.4) e incubare per 4 ore a 37 °C con il 5% di CO2.
- Scartare i surnatanti e lavare le cellule con 100 μL di HBSS. Trattare con 1 U/mL di DNasi per 10 minuti a 37 °C per favorire il rilascio di strutture DNA-proteine e centrifugare a 1.800 x g per 10 minuti.
- Recuperare i surnatanti e valutare l'attività enzimatica nel surnatante utilizzando reazioni colorimetriche come precedentemente descritto da White et al.17.
- Determinare la massima attività enzimatica di NE, CG e MPO nei neutrofili nelle stesse condizioni sperimentali senza aggiungere alcuno stimolo per l'induzione di NET. Quindi, congelare il campione cellulare a -70 °C e scongelarlo a 37 °C a bagnomaria, generando uno shock termico per favorire il rilascio di proteine intracellulari mediante lisi cellulare. Centrifugare a 1.800 x g per 10 minuti e recuperare i surnatanti
- Aggiungere 50 μL di surnatante a ciascun pozzetto in piastre da 96 pozzetti, quindi aggiungere 50 μL di ciascun substrato come indicato al punto 7.7.
- Aggiungere 0,5 M di N-metossisuccinil-Ala-Ala-Pro-Val-p-nitro anilina come substrato per NE, e 1 mM di N-succinil-Ala-Ala-Pro-Phe-p-nitroanilide per CG. Incubare per 3 ore a temperatura ambiente. Per MPO, aggiungere 1,6 mM di 3,3', 5,5'-tetrametilbenzidina (TMB) e incubare per 30 minuti a temperatura ambiente.
- Dopo l'incubazione, aggiungere 50 μL della soluzione di arresto (0,5 M H2SO4) per MPO e misurare l'assorbanza a 405 nm per NE e CG e 450 nm per MPO, utilizzando uno spettrofotometro.
- Confrontare i valori ottenuti con le curve di calibrazione corrispondenti e mostrare i risultati di ciascuna condizione relativi alla massima attività enzimatica (100%).
8. Analisi statistica
- Analizzare i dati di misura in triplice copia per ogni esperimento indipendente (n = 10) ed eseguire un ANOVA per l'analisi statistica confrontando gruppi con un livello di confidenza del 95%.

Figura 1: Purificazione PMN e protocollo di induzione NET. (A) Il plasma è stato rimosso dal sangue periferico per ottenere il pacchetto eritrocitario e leucocitario e diluito 1:1 (v/v) con 1x DPBS. Quindi, 4 mL della diluizione sono stati aggiunti lungo la parete al tubo a gradiente a doppia densità e centrifugati a 320 x g per 20 minuti a 4 °C, ottenendo la separazione di diversi strati cellulari e recuperando quello corrispondente al PMN. (B) Le cellule purificate sono state contate e la loro morfologia è stata analizzata dalla colorazione di Wright. La vitalità è stata determinata mediante esclusione del blu di tripano e colorazione 7AAD mediante citometria a flusso. Una volta verificata la purezza e la vitalità ottimali dei neutrofili, la formazione di NET è stata indotta aggiungendo microbi (S. aureus, P. aeruginosa e C. albicans) o sostanze chimiche (PMA, HOCl) in piastre a 24 pozzetti per l'analisi mediante microscopia a fluorescenza con DAPI-DNA, anti-LL37 Alexa Fluor 594 e colorazione microorganismo-CFSE. Per la quantificazione enzimatica, i NET sono stati indotti in piastre a 96 pozzetti per 3 ore e trattati con DNasi, seguiti dall'aggiunta di substrati per ciascun enzima: NE, CG e MPO; I cambiamenti di colore sono stati quantificati mediante spettrofotometria. DPBS = soluzione salina tamponata fosfato di Dulbecco; PBMC = cellule mononucleate del sangue periferico; PMN = Neutrofili polimorfonucleati; NE = elastasi neutrofila; CG = catepsina G; MPO = Mieloperossidasi; PMA = Phorbol miristato acetato; HOCl = Acido ipocloroso. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
Risultati
Purezza e vitalità dei neutrofili
Le fasi cellulari dinamiche sono visualizzate nel tubo dalla purificazione a gradiente a doppia densità. All'interno di questi strati, lo strato corrispondente ai granulociti è al di sopra dello strato di densità di 1,079 g/ml, distinto dalle fasi dei mononucleociti del sangue periferico (PBMC) e degli eritrociti (Figura 1A). La morfologia delle cellule purificate è stata verificata con la colorazione di Wright osservando cellule con...
Discussione
Una popolazione altamente pura di neutrofili vitali deve essere ottenuta per indurre il rilascio di NET poiché queste cellule hanno una durata ex vivo limitata di 8 ore in media, un periodo entro il quale devono essere eseguiti tutti gli esperimenti. A tal fine, la metodologia ideale è il gradiente a doppia densità per ottimizzare il tempo di purificazione isolando cellule non attivate più sensibili alla stimolazione esogena, in contrasto con le tecniche di sedimentazione del gradiente di Ficoll-Histopaque o...
Divulgazioni
Gli autori dichiarano di non avere conflitti di interesse.
Riconoscimenti
Questo lavoro è stato supportato da una borsa di studio scientifica di base (# 285480) da CONACyT e dal Dipartimento di ricerca immunologica clinica della Facoltà di Scienze Biochimiche, Universidad Autónoma 'Benito Juárez' de Oaxaca. A.A.A, S.A.S.L e W.J.R.R. hanno borse di dottorato dei numeri CONACyT #799779, #660793 e #827788, rispettivamente.
Materiali
| Name | Company | Catalog Number | Comments |
| 24 Well plate for cell culture | Corning | 3526 | |
| 7-aminoactinomycin D (7-AAD) | BD Pharmingen | 51-668981E | |
| 96 Well plate for cell culture | Costar | 3596 | Flat bottom |
| Agitator | CRM Globe | CRM-OS1 | |
| Antibody LL37 | Santa Cruz Biotechnology | sc-166770 | |
| Blood collection tubes | BD VACUTAINER | 368171 | K2 EDTA 7.2 mg |
| Carboxyfluorescein succinimidyl ester (CFSE) | Sigma-Aldrich | 21878 | |
| Centrifuge | Hettich | 1406-01 | |
| Coverslip | Madesa | M03-CUB-22X22 | 22 mm x 22 mm |
| Dulbecco´s phosphate-buffered saline (DPBS) | Caisson | 1201022 | |
| Falcon tubes 50 mL | CORNING | 430829 | |
| Flow Cytometry Tubes | Miltenyi Biotec | 5 mL - Without caps | |
| FlowJo Software | BD Biosciences | Analyze flow cytometry data | |
| Fluorescence microscope | DM 2000 | LEICA | |
| Fluoroshield with DAPI | Sigma-Aldrich | F6057 | |
| Incubator | NUAIRE | UN-4750 | |
| MACSQuant Analyzer | Miltenyi Biotec | Flow cytometer | |
| Microplate reader photometer | Clarkson Laboratory - CL | ||
| Microtubes 1.5 mL | Zhejiang Runlab Tech | 35200N | wire snap |
| Minitab Software | Minitab | Statistical analysis | |
| Needles | BD VACUTAINER | 301746 | Diameter 1.34 mm |
| Optical microscope | VELAB | VE-B50 | |
| Percoll | GE Healthcare | 17-0891-01 | Solution for density gradient |
| Phosphate Buffered Saline (10x) | Caisson | PBL07-500ML | |
| Pyrex culture tubes | CORNING | CLS982025 | N°9820 |
| RPMI 1640 1x | Corning | 10-104-CV | contains Glutagro |
| Slides | Madesa | PDI257550 | 22 mm x 75 mm |
| Trypan Blue solution 0.4% | SIGMA | T8154-100ML |
Riferimenti
- De Buhr, N., Maren, K. B. How neutrophil extracellular traps become visible. Journal of Immunology Research. 2016, 4604713 (2016).
- Karlsson, A., Nixon, J. B., McPhail, L. C. Phorbol myristate acetate induces neutrophil NADPH-oxidase activity by two separate signal transduction pathways: dependent or independent of phosphatidylinositol 3-kinase. Journal of Leukocyte Biology. 67 (3), 396-404 (2000).
- Takei, H., Araki, A., Watanabe, H., Ichinose, A., Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. Journal of Leukocyte Biology. 59 (2), 229-240 (1996).
- Brinkmann, V., et al. Neutrophil extracellular traps kill bacteria. Science. 303 (5663), 1532-1535 (2004).
- Kenny, E. F., et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife. 6, 24437 (2017).
- Schultz, B. M., Acevedo, O. A., Kalergis, A. M., Bueno, S. M. Role of extracellular trap release during bacterial and viral infection. Frontiers in Microbiology. 13, 798853 (2022).
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nature Reviews Immunology. 18 (2), 134-147 (2018).
- Delgado-Rizo, V., et al. Neutrophil extracellular traps and its implications in inflammation: An overview. Frontiers in Immunology. 8, 81 (2017).
- Petretto, A., et al. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS One. 14 (7), 0218946 (2019).
- Neumann, A., et al. Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular traps against degradation by bacterial nucleases. Journal of Innate Immunity. 6 (6), 860-868 (2014).
- Hakkim, A., et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nature Chemical Biology. 7 (2), 75-77 (2011).
- Sabbatini, M., Magnelli, V., Renò, F. NETosis in wound healing: When enough Is enough. Cells. 10 (3), 494 (2021).
- Metzler, K. D., Goosmann, C., Lubojemska, A., Zychlinsky, A., Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Reports. 8 (3), 883-896 (2014).
- Clark, S. R., et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature Medicine. 13 (4), 463-469 (2007).
- Pilsczek, F. H., et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. Journal of Immunology. 185 (12), 7413-7425 (2010).
- Sosa, S. A., et al. Structural differences of neutrophil extracellular traps induced by biochemical and microbiologic stimuli under healthy and autoimmune milieus. Immunologic Research. 69 (3), 264-274 (2021).
- White, P. C., et al. Characterization, quantification, and visualization of neutrophil extracellular traps. Methods in Molecular Biology. 1537, 481-497 (2017).
- Boeltz, S., et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death and Differentiation. 26 (3), 395-408 (2019).
- Yousefi, S., Mihalache, C., Kozlowski, E., Schmid, I., Simon, H. U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death and Differentiation. 16 (11), 1438-1444 (2009).
- Brinkmann, V., Zychlinsky, A. Neutrophil extracellular traps: is immunity the second function of chromatin. The Journal of Cell Biology. 198 (5), 773-783 (2012).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati