
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Análise morfológica e composicional de armadilhas extracelulares de neutrófilos induzidas por estímulos microbianos e químicos
Neste Artigo
Resumo
Apresenta-se aqui um protocolo para a indução e análise de armadilhas extracelulares (TNEs) de neutrófilos in vitro . A quantificação do DNA, da catelicidina (LL37) e da atividade enzimática produziu dados que mostram a variabilidade na composição e morfologia dos TNEs induzidos por estímulos microbianos e químicos em condições controladas semelhantes.
Resumo
Os neutrófilos funcionam como a primeira linha de defesa celular em uma resposta imune inata, empregando diversos mecanismos, como a formação de armadilhas extracelulares de neutrófilos (TNEs). Este estudo analisa as alterações morfológicas e composicionais em TNEs induzidas por estímulos microbianos e químicos utilizando metodologias padronizadas in vitro para indução e caracterização da TNE com células humanas. Os procedimentos aqui descritos permitem a análise da morfologia da NET (lítica ou não lítica) e da composição (estruturas DNA-proteína e atividade enzimática), e o efeito de fatores solúveis ou contato celular sobre tais características. Além disso, as técnicas aqui descritas poderiam ser modificadas para avaliar o efeito de fatores solúveis exógenos ou de contato celular na composição da TNE.
As técnicas aplicadas incluem a purificação de células polimorfonucleares do sangue periférico humano usando um gradiente de densidade dupla (1,079-1,098 g/mL), garantindo pureza e viabilidade ideais (≥ 95%), conforme demonstrado pela coloração de Wright, exclusão de azul de tripano e citometria de fluxo, incluindo análise FSC versus SSC e coloração 7AAD. A formação de NET é induzida com estímulos microbianos (Pseudomonas aeruginosa, Staphylococcus aureus e Candida albicans) e químicos (acetato de miristato de phorbol, HOCl), e os TNEs são caracterizados por coloração DNA-DAPI, imunocoloração para o peptídeo antimicrobiano catelicidina (LL37) e quantificação da atividade enzimática (elastase de neutrófilos, catepsina G e mieloperoxidase). As imagens são adquiridas por microscopia de fluorescência e analisadas com ImageJ.
Introdução
Os neutrófilos são os leucócitos mais abundantes na corrente sanguínea, desempenhando um papel essencial durante a depuração de agentes patogênicos por diversos mecanismos, incluindo a liberação de grandes estruturas cromatina compostas de DNA e várias proteínas antibacterianas nucleares, citoplasmáticas e granulares 1,2. O antecedente direto descrevendo esse papel antimicrobiano dos neutrófilos foi feito por Takei et al.3 em 1996. Esses autores relataram uma nova forma de morte diferente da apoptose e necroptose em neutrófilos, mostraram alterações morfológicas exibindo ruptura nuclear, seguidas de derramamento do nucleoplasma para o citoplasma, e um aumento na permeabilidade da membrana a partir de 3 h de incubação com acetato de miristato de forbol (PMA)2,3. No entanto, foi somente em 2004 que o termo "armadilhas extracelulares de neutrófilos (TNEs)" foi utilizado4.
A formação de NET tem sido observada em diversas condições, como infecções bacterianas, fúngicas5, virais6 e parasitárias, para neutralizar, matar e prevenir a disseminação microbiana7. Outros estudos mostram que também pode ocorrer em condições não patogênicas por estímulos estéreis, como citocinas, ácido úrico monossódico ou cristais de colesterol, autoanticorpos, imunocomplexos e plaquetas ativadas7. Lipopolissacarídeo (LPS), interleucina-8 (IL-8) e PMA estavam entre os primeiros estímulos in vitro descritos como indutores de NET, e o envolvimento in vivo da TNE em processos patogênicos foi demonstrado em dois modelos de inflamação aguda: disenteria experimental e apendicite humana espontânea4. O DNA é um componente essencial da NET. Sua estrutura e composição adequadas são necessárias para o sequestro e morte de microrganismos, fornecendo uma alta concentração local de moléculas antimicrobianas em direção aos micróbios capturados, como demonstrado por um breve tratamento com desoxirribonuclease (DNase) que desintegra TNEs e suas propriedades microbicidas4. Além do DNA, os TNEs compreendem proteínas aderentes, como histonas, elastase de neutrófilos (NE), catepsina G (GC), proteinase 3, lactoferrina, gelatinase, mieloperoxidase (MPO) e peptídeos antimicrobianos (AMPs), como o peptídeo pró-inflamatório catiônico catelicidina LL-37, entre outros 8,9. Tais agregados podem formar roscas maiores com diâmetros de até 50 nm. Esses fatores podem perturbar os fatores de virulência microbiana ou a integridade da membrana celular do patógeno; além disso, os AMPs podem estabilizar o DNA derivado da TNE contra a degradação por nucleases bacterianas10.
Os mecanismos específicos que regulam a formação da NET ainda não foram completamente esclarecidos. A via mais bem caracterizada que leva à liberação de NET é através da sinalização ERK, que leva à ativação da NADPH oxidase e à produção de espécies reativas de oxigênio (ROS), bem como ao aumento do cálcio intracelular que desencadeia a ativação da via MPO. Este, por sua vez, transforma o peróxido de hidrogênio em ácido hipocloroso, ativando o NE por oxidação11,12. O NE é responsável por degradar os filamentos de actina do citoesqueleto para bloquear a fagocitose e translocá-los para o núcleo para processamento por clivagem proteolítica e desaminação por PAD4 que impulsionam a dessensibilização das fibras cromatinas, que se associam a proteínas granulográficas e citoplasmáticas, e são então liberadas extracelularmente7. Essas proteases incluem aquelas liberadas do complexo azurossomo dos grânulos azurófilos e outras proteases, como a catepsina G13.
Dependendo das alterações morfológicas nos neutrófilos, os TNEs são classificados em dois tipos: formação de TNE suicida ou lítica levando à morte celular4 e formação de TNE vital ou não lítica produzida por células viáveis mediadas por uma liberação vesicular de DNA nuclear ou mitocondrial, com remanescente de citoplasto anucleado com capacidade fagocítica14,15. Geralmente, TNEs compostos de DNA mitocondrial apresentam morfologia de fibra alongada14, enquanto aqueles estruturados de DNA nuclear têm aparência de nuvem3. No entanto, não se sabe como o neutrófilo escolhe sua origem no DNA. Ao contrário de estudos anteriores que descreveram as vias canônicas dos TNEs como exigindo várias horas, a via vital é rapidamente ativada em apenas 5-60 min15.
Apesar desses avanços, a composição da TNE varia de acordo com o estímulo; por exemplo, diferentes cepas mucoides e não mucoides de P. aeruginosa induzem a formação de TNEs contendo 33 proteínas comuns e até 50 proteínas variáveis7. Assim, faz-se necessário homogeneizar técnicas que permitam a geração de conclusões objetivas em grupos de pesquisa. Este trabalho descreve um protocolo com várias técnicas que permitem comparar e avaliar a composição, estrutura e morfologia de TNEs induzidos com diferentes microrganismos: Staphylococcus aureus (bactéria gram-positiva), Pseudomonas aeruginosa (bactéria gram-negativa) e Candida albicans (fungo), bem como estímulos químicos (PMA, HOCl) em neutrófilos humanos de indivíduos saudáveis. Os resultados representativos demonstram a heterogeneidade dos TNEs dependendo de seu estímulo indutor em condições in vitro comparáveis, caracterizadas por coloração DNA-DAPI, imunocoloração para LL37 e quantificação da atividade enzimática (NE, GC e MPO).
Protocolo
As amostras de sangue foram obtidas como doações de participantes clinicamente saudáveis após consentimento informado. Todos os experimentos foram realizados com a permissão do Comitê de Ética em Pesquisa com Seres Humanos da Faculdade de Ciências Bioquímicas da Universidad Autónoma 'Benito Juárez' de Oaxaca.
NOTA: Os critérios de inclusão no estudo foram sexo e idade indistintos e clinicamente saudáveis de acordo com as respostas dos participantes a um questionário antes de uma amostra de sangue. Uma análise hematológica foi realizada para determinar a contagem de células e descartar infecções ou anemia, bem como o teste de proteína C-reativa para descartar inflamação no doador.
1. Coleta de sangue periférico e obtenção da embalagem de eritrócitos e leucócitos
- Coletar 10 mL de sangue periférico por punção venosa em tubos com 1,8 mg/mL de K2· EDTA como anticoagulante (ver Tabela de Materiais) de indivíduos clinicamente saudáveis após a obtenção do consentimento informado. Em seguida, realize a biometria sanguínea padrão e o teste de proteína C-reativa para descartar infecção ou inflamação, garantindo a qualidade da amostra.
- Centrifugar a amostra de sangue periférico a 82 x g durante 15 min para remover o plasma rico em plaquetas, seguido de uma segunda centrifugação a 630 x g durante 5 min. Descarte o plasma restante para obter a embalagem de eritrócitos e leucócitos.
- Diluí-lo na proporção de 1:1 (v/v) com 1x solução salina tamponada com fosfato (DPBS) da Dulbecco.
2. Purificação de neutrófilos polimorfonucleares (PMN) usando um gradiente de dupla densidade
NOTA: Realizar a purificação de neutrófilos imediatamente após a coleta do sangue, porque eles têm uma vida útil in vitro limitada de cerca de 8 h.
- Depositar o seguinte num tubo de vidro estéril de 10 ml (ver Tabela de Materiais) por ordem: 1 ml de solução de densidade de 1,098 g/ml, 1 ml de solução de densidade de 1,079 g/ml (ver Tabela de Materiais) e, em seguida, 4 ml da embalagem diluída de eritrócitos e leucócitos. Despeje sobre as paredes sem quebrar a tensão superficial entre as camadas para evitar que elas se misturem.
- Centrífuga a 320 x g durante 20 min a 4 °C, evitando a aceleração/desaceleração de modo a que as altas forças da centrífuga não perturbem o gradiente.
- Aspirar a fase que corresponde aos granulócitos (Figura 1A) por pipetagem e transferir para outro tubo de vidro estéril de 10 mL. Lavar com 4 ml de 1x DPBS a 300 x g durante 10 min a 4 °C.
- Descarte o sobrenadante e trate as células com choque osmótico para remover os eritrócitos restantes. Adicionar 4 mL de solução salina a 0,2% por 2 min a 4 °C e centrifugar a 300 x g por 10 min a 4 °C. Descarte o sobrenadante. Em seguida, adicione 4 mL da solução isotônica (solução salina a 0,65%) por 5 min a 4 °C para restaurar a integridade da membrana e centrifugar a 300 x g por 10 min a 4 °C.
NOTA: A solução salina a 0,2% é um meio hipotônico, com menor concentração de soluto em relação à do meio intracelular eritrocitária. O contato com o meio hipotônico permite que a água se difunda no eritrócito, levando ao seu inchaço e hemólise. Esta remoção de hemácias do sobrenadante foi confirmada por observação microscópica. - Remova o sobrenadante. Ressuspeite as células em 4 mL de 1x DPBS para remover detritos celulares e, em seguida, centrifugar a 300 x g por 10 min a 4 °C. Finalmente, ressuspenda o pellet celular em 2 mL de tampão de solução salina balanceada de Hank (HBSS) frio.
3. Morfologia e viabilidade dos neutrófilos (Figura 1B)
- Teste de exclusão azul de tripano
- Diluir 5 μL da suspensão celular em 20 μL de azul de tripano a 0,4% (proporção de 1:5). Conte as células em uma câmara de Neubauer e determine a viabilidade celular usando um teste de exclusão. Considere as células que mantêm a integridade de sua membrana sem permeabilizar o corante como viáveis.
- Montagem de 5 μL da suspensão celular numa lâmina; seco e manchado com a mancha de Wright por 15 s. Fixar imediatamente a amostra com tampão fosfato pH 6,4 durante 30 s. Lave com água destilada suficiente e observe a morfologia sob um microscópio óptico (100x).
- Análise de coloração 7AAD e citometria de fluxo
- Adicione 1 x 105 células aos tubos de citometria de fluxo e core com 1 μL de 7AAD em 100 μL de tampão FACS (1x DPBS, azida de sódio a 0,1% e plasma descomplementado autólogo a 10%) por 15 min a 4 °C no escuro.
- Lavar com 500 μL de tampão FACS a 300 x g durante 10 min. Fixar as células com 500 μL de paraformaldeído a 2% e armazenar a 4 °C até à sua análise no citómetro de fluxo.
- Para um controle de células mortas, fixar 1 x 105 células com 200 μL de paraformaldeído a 4% por 30 min e lavar com 500 μL de 1x PBS a 300 x g por 10 min a 4 °C. Retire o sobrenadante e descarregue. Em seguida, adicione 200 μL de Triton X-100 a 0,1% por 1 h a 4 °C. Lave com 500 μL de 1x PBS e deixe com 7AAD como na etapa 3.2.1.
- Usando um citômetro de fluxo (ver Tabela de Materiais), realize a análise FSC versus SSC para analisar a pureza celular e a coloração SSC versus 7AAD para analisar a viabilidade celular. Leia 3 x 104 eventos em 100 μL de volume de absorção em fluxo médio (1.000 células/s) nas configurações polimorfonucleares (FSC, 400-490 e SSC, 300-320).
- Analisar os dados capturados no software do citômetro de fluxo (ver Tabela de Materiais) e determinar a porcentagem de pureza e células positivas para 7DAA na população polimorfonuclear, apresentada através de gráficos de pontos e histogramas.
4. Coloração CFSE de microrganismos
- Adicionar 1 x 108 bactérias ou 1 x 106 pseudo-hifas fúngicas em microtubos de 1,5 mL e corar com 200 μL de 5 μM de éster succinimidílico de carboxifluoresceína (CFSE) dissolvido em 1x PBS. Misturar durante alguns segundos e incubar a 37 °C durante 10 minutos no escuro.
- Pare a reação adicionando 500 μL de plasma descomplementado e centrifugar a 620 x g por 10 min para pseudo-hifas ou a 1.800 x g por 10 min para bactérias.
- Rejeitar os sobrenadantes e lavar os pellets com 1 ml de 1x PBS com centrifugação como no passo 4.2. Finalmente, ressuspenda os microrganismos em 250 μL de 1x PBS.
- Preparar alíquotas de 50 μL em microtubos de 1,5 mL com 2 x 107 bactérias (MOI: 100) ou 2 x 105 pseudo-hifas (MOI:1) para indução NET.
5. Indução NET
- Coloque as tampas de vidro estéril de 10 mm x 10 mm em uma placa de 24 poços e cubra com 10 μL de poli-L-lisina a 0,001% por 1 h à temperatura ambiente. Lave duas vezes com 100 μL de 1x PBS, seque ao ar e irradie com luz UV por 15 min.
- Substitua a solução HBSS da suspensão de neutrófilos no passo 2.5 por um meio RPMI 1640 suplementado com plasma autólogo a 10%. À placa de 24 poços (passo 5.1), adicionar 350 μL desta suspensão celular, para uma concentração final de 2 x 105 neutrófilos/poço.
- Permitir que as células adiram ao fundo dos poços incubando por 20 min a 37 °C com 5% de CO2.
- Adicione os estímulos para induzir a formação de NET em 50 μL: estímulos microbianos-gram-positivos bactéria S. aureus (ATCC 25923), bactéria gram-negativa P. aeruginosa (ATCC 10145) no MOI 100 e pseudo-hifas de C. albicans (ATCC 10231) no MOI:1; estímulos bioquímicos-PMA (200 nM) e HOCl (4,5 mM), e controle com estímulo ausente (50 μL de HBSS).
- Obter um volume final de 400 μL por poço. Misturar num agitador de placas a 140 rpm durante 30 s e incubar durante 4 h a 37 °C e 5% de CO2.
6. Visualização de TNEs por microscopia de fluorescência
- DNA e imunocoloração LL37
- Após a indução NET, remova os sobrenadantes dos poços pipetando cuidadosamente e fixe as células com 300 μL de paraformaldeído a 4% por 30 min.
- Lave as células com 200 μL de 1x PBS sem centrifugação e adicione 200 μL de tampão de bloqueio (10% de plasma descomplementado em 1x PBS) por 30 min.
- Para a coloração LL-37, permeabilizar as células com 200 μL de Triton X-100 a 0,2% em PBS 1x por 10 min para permitir que o anticorpo entre nas células. Lave 2x cuidadosamente com 1x PBS para remover o excesso de detergente.
- Monte as tampas em lâminas de vidro (quatro folhas de cobertura em cada lâmina). O DNA cora as células com 2 μL de DAPI (ver Tabela de Materiais), sela as folhas de cobertura e armazena a -20 °C até sua análise por microscopia de fluorescência confocal.
- Aquisição e análise de imagens fluorescentes
- Pegue imagens NET para quantificar seus componentes e use os filtros correspondentes no microscópio de fluorescência confocal (consulte Tabela de materiais) para adquirir as imagens com o software do computador.
NOTA: Considere que o DNA é corado com DAPI (cor azul), mostrando excitação a 360 nm e emissão a 460 nm. Os microrganismos são corados com CFSE (cor verde), que tem uma excitação de 492 nm e uma emissão de 521 nm. O peptídeo LL37 é marcado com anticorpo anti-LL37 Alexa Fluor 594 (cor vermelha), que tem uma excitação de 594 nm e uma emissão de 614 nm. - Calibre o microscópio. Coloque o slide e o foco usando contraste de interferência diferencial (DIC) com a luz normal acesa. Escolha Live para projetar a imagem no monitor.
- Desligue a luz e selecione o canal correspondente do fluorocromo. Por exemplo, selecione filtro 365 nm/azul para DAPI, 43 HE DsRed para Alexa 594 ou GFP 38 HE para CFSE.
- Ajuste as configurações com o anticorpo de controle de isotipo para LL37 e células não coradas para DAPI e CFSE. Defina as mesmas configurações de tempo de exposição, tensão, contraste e lente para capturar todas as imagens sob as mesmas condições.
NOTA: Neste estudo, o tempo de exposição, a tensão e o contraste foram fixados em 1,0 ms, 4,0 V e 0,0, respectivamente, com objetivo de 40x. Esses valores podem ser ajustados para facilitar a melhor captura de imagem para as amostras. - Selecione Ajustar para capturar a imagem. Salve cinco imagens (quatro extremos e o centro) por poço, e da colocalização (fusão) de DNA/LL37/CFSE.
- Defina as três classes de pixels como plano de fundo com as imagens independentes de cada cor e analise o valor do Sinal Cinza Médio por área com o software Image J.
- Pegue imagens NET para quantificar seus componentes e use os filtros correspondentes no microscópio de fluorescência confocal (consulte Tabela de materiais) para adquirir as imagens com o software do computador.
7. Quantificação da atividade enzimática
- Em uma placa de 96 poços, adicionar 90 μL de suspensão celular em HBSS contendo 1 x 10 5 neutrófilos para indução NET e incubar por 20 min a 37 °C e5 % de CO2.
- Imediatamente adicionar 10 μL dos estímulos correspondentes (concentração como na etapa 5.4) e incubar por 4 h a 37 °C com 5% de CO2.
- Descarte os sobrenadantes e lave as células com 100 μL de HBSS. Tratar com 1 U/mL de DNase por 10 min a 37 °C para favorecer a liberação de estruturas DNA-proteína e centrifugar a 1.800 x g por 10 min.
- Recuperar os sobrenadantes e avaliar a atividade enzimática no sobrenadante utilizando reações colorimétricas conforme descrito anteriormente por White et al.17.
- Determinar a atividade enzimática máxima de NE, CG e MPO em neutrófilos sob as mesmas condições experimentais sem adicionar nenhum estímulo para indução NET. Em seguida, congelar a amostra celular a -70 °C e descongelar a 37 °C em banho-maria, gerando um choque de temperatura para favorecer a liberação de proteínas intracelulares por lise celular. Centrifugar a 1.800 x g por 10 min e recuperar os sobrenadantes.
- Adicionar 50 μL do sobrenadante a cada poço em placas de 96 poços e, em seguida, adicionar 50 μL de cada substrato, conforme indicado na etapa 7.7.
- Adicionar 0,5 M de N-metoxisuccinil-Ala-Ala-Pro-Val-p-nitro anilina como substrato para NE, e 1 mM de N-succinil-Ala-Ala-Pro-Phe-p-nitroanilide para CG. Incubar durante 3 h à temperatura ambiente. No caso da OSM, adicionar 1,6 mM de 3,3', 5,5'-tetrametilbenzidina (TMB) e incubar durante 30 min à temperatura ambiente.
- Após incubação, adicionar 50 μL da solução de parada (0,5 M H2SO4) para MPO e medir a absorbância a 405 nm para NE e CG e 450 nm para MPO, usando um espectrofotômetro.
- Comparar os valores obtidos com as curvas de calibração correspondentes e mostrar os resultados de cada condição em relação à atividade enzimática máxima (100%).
8. Análise estatística
- Analise os dados de medição em triplicado para cada experimento independente (n = 10) e realize uma ANOVA para análise estatística, comparando grupos com um nível de confiança de 95%.

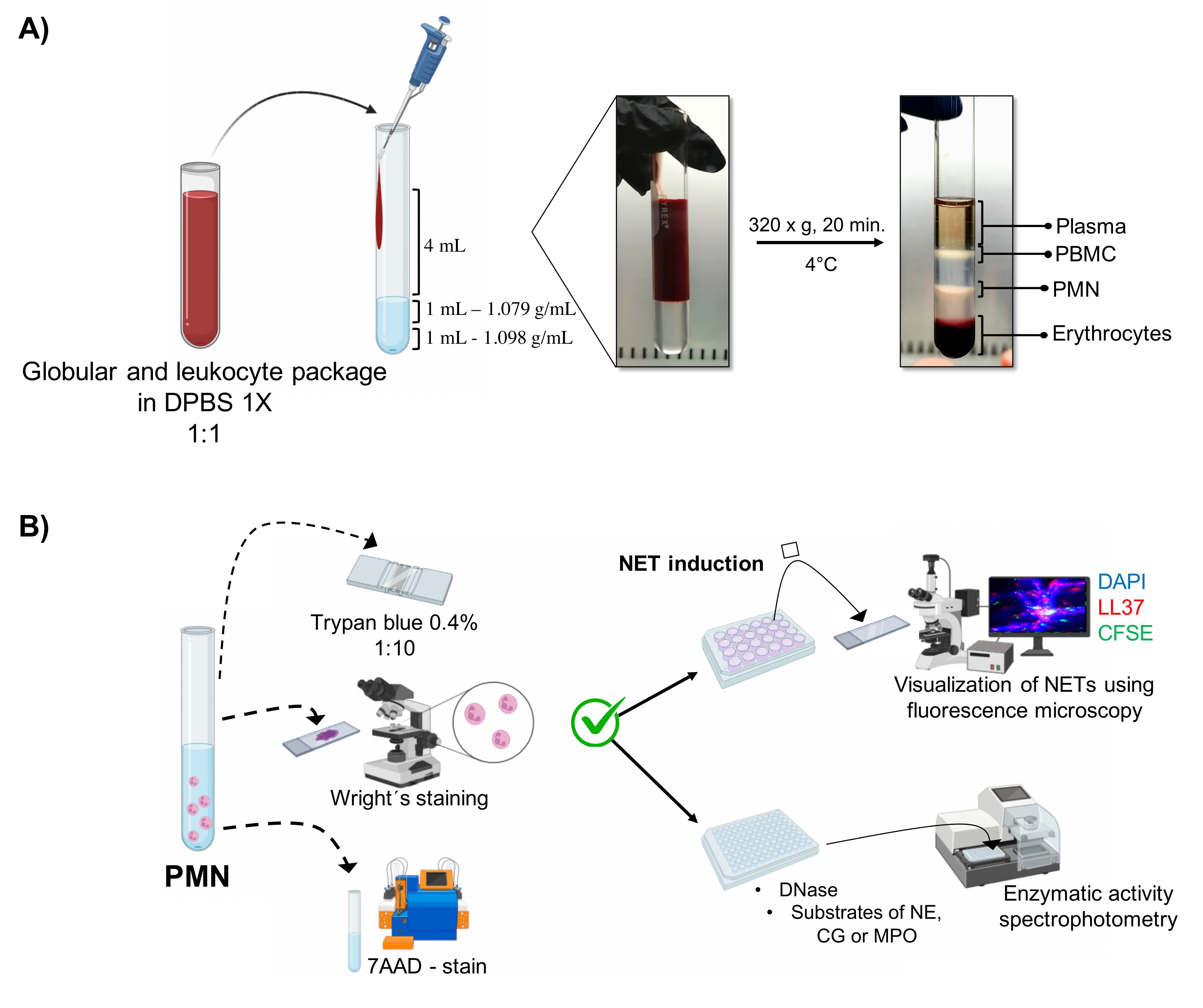
Figura 1: Purificação de PMN e protocolo de indução NET. (A) O plasma foi retirado do sangue periférico para obtenção da embalagem de eritrócitos e leucócitos e diluído 1:1 (v/v) com 1x DPBS. Em seguida, 4 mL da diluição foram adicionados ao longo da parede ao tubo de gradiente de dupla densidade, e centrifugados a 320 x g por 20 min a 4 °C, obtendo-se a separação das diferentes camadas celulares e recuperando-se a correspondente ao PMN. (B) As células purificadas foram contadas e sua morfologia foi analisada pela coloração de Wright. A viabilidade foi determinada pela exclusão do azul de tripano e coloração da DAAA 7 por citometria de fluxo. Uma vez verificada a pureza e a viabilidade ideais dos neutrófilos, a formação de NET foi induzida pela adição de micróbios (S. aureus, P. aeruginosa e C. albicans) ou produtos químicos (PMA, HOCl) em placas de 24 poços para análise por microscopia de fluorescência com DAPI-DNA, anti-LL37 Alexa Fluor 594 e coloração de microrganismos-CFSE. Para quantificação enzimática, os TNEs foram induzidos em placas de 96 poços por 3 h e tratados com DNase, seguidos da adição de substratos para cada enzima: NE, GC e MPO; as alterações de cor foram quantificadas por espectrofotometria. DPBS = solução salina tamponada com fosfato de Dulbecco; PBMC = Células mononucleares do sangue periférico; PMN = neutrófilos polimorfonucleares; NE = Elastase de neutrófilos; GC = Catepsina G; MPO = Mieloperoxidase; PMA = Acetato de miristato de Phorbol; HOCl = Ácido hipocloroso. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
Resultados
Pureza e viabilidade dos neutrófilos
As fases celulares dinâmicas são visualizadas no tubo a partir da purificação do gradiente de dupla densidade. Dentro dessas camadas, a camada correspondente aos granulócitos está acima da camada de densidade de 1,079 g/mL, distinta das fases de mononucleócitos do sangue periférico (PBMCs) e eritrócitos (Figura 1A). A morfologia das células purificadas foi verificada com a coloração de Wright por meio da observação de c?...
Discussão
Uma população altamente pura de neutrófilos viáveis deve ser obtida para induzir a liberação de TNEs, uma vez que essas células têm uma vida útil ex vivo limitada de 8 h em média, um período dentro do qual todos os experimentos devem ser realizados. Para tanto, a metodologia ideal é o gradiente de dupla densidade para otimizar o tempo de purificação, isolando células não ativadas mais responsivas à estimulação exógena, em contraste com o gradiente Ficoll-Histopaque ou as técnicas de sediment...
Divulgações
Os autores declaram que não têm conflitos de interesse.
Agradecimentos
Este trabalho foi apoiado por uma bolsa de ciência básica (#285480) do CONACyT e pelo Departamento de Pesquisa em Imunologia Clínica da Faculdade de Ciências Bioquímicas da Universidad Autónoma 'Benito Juárez' de Oaxaca. A.A.A, S.A.S.L e W.J.R.R. têm bolsas de doutorado dos números CONACyT #799779, #660793 e #827788, respectivamente.
Materiais
| Name | Company | Catalog Number | Comments |
| 24 Well plate for cell culture | Corning | 3526 | |
| 7-aminoactinomycin D (7-AAD) | BD Pharmingen | 51-668981E | |
| 96 Well plate for cell culture | Costar | 3596 | Flat bottom |
| Agitator | CRM Globe | CRM-OS1 | |
| Antibody LL37 | Santa Cruz Biotechnology | sc-166770 | |
| Blood collection tubes | BD VACUTAINER | 368171 | K2 EDTA 7.2 mg |
| Carboxyfluorescein succinimidyl ester (CFSE) | Sigma-Aldrich | 21878 | |
| Centrifuge | Hettich | 1406-01 | |
| Coverslip | Madesa | M03-CUB-22X22 | 22 mm x 22 mm |
| Dulbecco´s phosphate-buffered saline (DPBS) | Caisson | 1201022 | |
| Falcon tubes 50 mL | CORNING | 430829 | |
| Flow Cytometry Tubes | Miltenyi Biotec | 5 mL - Without caps | |
| FlowJo Software | BD Biosciences | Analyze flow cytometry data | |
| Fluorescence microscope | DM 2000 | LEICA | |
| Fluoroshield with DAPI | Sigma-Aldrich | F6057 | |
| Incubator | NUAIRE | UN-4750 | |
| MACSQuant Analyzer | Miltenyi Biotec | Flow cytometer | |
| Microplate reader photometer | Clarkson Laboratory - CL | ||
| Microtubes 1.5 mL | Zhejiang Runlab Tech | 35200N | wire snap |
| Minitab Software | Minitab | Statistical analysis | |
| Needles | BD VACUTAINER | 301746 | Diameter 1.34 mm |
| Optical microscope | VELAB | VE-B50 | |
| Percoll | GE Healthcare | 17-0891-01 | Solution for density gradient |
| Phosphate Buffered Saline (10x) | Caisson | PBL07-500ML | |
| Pyrex culture tubes | CORNING | CLS982025 | N°9820 |
| RPMI 1640 1x | Corning | 10-104-CV | contains Glutagro |
| Slides | Madesa | PDI257550 | 22 mm x 75 mm |
| Trypan Blue solution 0.4% | SIGMA | T8154-100ML |
Referências
- De Buhr, N., Maren, K. B. How neutrophil extracellular traps become visible. Journal of Immunology Research. 2016, 4604713 (2016).
- Karlsson, A., Nixon, J. B., McPhail, L. C. Phorbol myristate acetate induces neutrophil NADPH-oxidase activity by two separate signal transduction pathways: dependent or independent of phosphatidylinositol 3-kinase. Journal of Leukocyte Biology. 67 (3), 396-404 (2000).
- Takei, H., Araki, A., Watanabe, H., Ichinose, A., Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. Journal of Leukocyte Biology. 59 (2), 229-240 (1996).
- Brinkmann, V., et al. Neutrophil extracellular traps kill bacteria. Science. 303 (5663), 1532-1535 (2004).
- Kenny, E. F., et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife. 6, 24437 (2017).
- Schultz, B. M., Acevedo, O. A., Kalergis, A. M., Bueno, S. M. Role of extracellular trap release during bacterial and viral infection. Frontiers in Microbiology. 13, 798853 (2022).
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nature Reviews Immunology. 18 (2), 134-147 (2018).
- Delgado-Rizo, V., et al. Neutrophil extracellular traps and its implications in inflammation: An overview. Frontiers in Immunology. 8, 81 (2017).
- Petretto, A., et al. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS One. 14 (7), 0218946 (2019).
- Neumann, A., et al. Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular traps against degradation by bacterial nucleases. Journal of Innate Immunity. 6 (6), 860-868 (2014).
- Hakkim, A., et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nature Chemical Biology. 7 (2), 75-77 (2011).
- Sabbatini, M., Magnelli, V., Renò, F. NETosis in wound healing: When enough Is enough. Cells. 10 (3), 494 (2021).
- Metzler, K. D., Goosmann, C., Lubojemska, A., Zychlinsky, A., Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Reports. 8 (3), 883-896 (2014).
- Clark, S. R., et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature Medicine. 13 (4), 463-469 (2007).
- Pilsczek, F. H., et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. Journal of Immunology. 185 (12), 7413-7425 (2010).
- Sosa, S. A., et al. Structural differences of neutrophil extracellular traps induced by biochemical and microbiologic stimuli under healthy and autoimmune milieus. Immunologic Research. 69 (3), 264-274 (2021).
- White, P. C., et al. Characterization, quantification, and visualization of neutrophil extracellular traps. Methods in Molecular Biology. 1537, 481-497 (2017).
- Boeltz, S., et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death and Differentiation. 26 (3), 395-408 (2019).
- Yousefi, S., Mihalache, C., Kozlowski, E., Schmid, I., Simon, H. U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death and Differentiation. 16 (11), 1438-1444 (2009).
- Brinkmann, V., Zychlinsky, A. Neutrophil extracellular traps: is immunity the second function of chromatin. The Journal of Cell Biology. 198 (5), 773-783 (2012).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados