
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Morphologische und kompositorische Analyse von neutrophilen extrazellulären Fallen, die durch mikrobielle und chemische Reize induziert werden
In diesem Artikel
Zusammenfassung
Hier wird ein Protokoll für die Induktion und Analyse von in vitro neutrophilen extrazellulären Fallen (NETs) vorgestellt. Die Quantifizierung von DNA, Cathelicidin (LL37) und Enzymaktivität ergab Daten, die die Variabilität in der Zusammensetzung und Morphologie von NETs zeigen, die durch mikrobielle und chemische Stimuli unter ähnlichen kontrollierten Bedingungen induziert werden.
Zusammenfassung
Neutrophile fungieren als erste Linie der zellulären Abwehr in einer angeborenen Immunantwort, indem sie verschiedene Mechanismen einsetzen, wie z.B. die Bildung von neutrophilen extrazellulären Fallen (NETs). Diese Studie analysiert die morphologischen und kompositorischen Veränderungen in NETs, die durch mikrobielle und chemische Reize induziert werden, unter Verwendung standardisierter In-vitro-Methoden für die NET-Induktion und Charakterisierung mit menschlichen Zellen. Die hier beschriebenen Verfahren ermöglichen die Analyse der NET-Morphologie (lytisch oder nicht-lytisch) und der Zusammensetzung (DNA-Proteinstrukturen und enzymatische Aktivität) sowie die Wirkung löslicher Faktoren oder zellulärer Kontakte auf solche Eigenschaften. Zusätzlich könnten die hier beschriebenen Techniken modifiziert werden, um die Wirkung von exogenen löslichen Faktoren oder zellulärem Kontakt auf die NET-Zusammensetzung zu bewerten.
Die angewandten Techniken umfassen die Reinigung von polymorphkernigen Zellen aus menschlichem peripherem Blut unter Verwendung eines doppelten Dichtegradienten (1,079-1,098 g/ml), der eine optimale Reinheit und Lebensfähigkeit (≥ 95%) garantiert, wie die Wright-Färbung, der Trypanblauausschluss und die Durchflusszytometrie, einschließlich FSC- versus SSC-Analyse und 7AAD-Färbung, zeigen. Die NET-Bildung wird mit mikrobiellen (Pseudomonas aeruginosa, Staphylococcus aureus und Candida albicans) und chemischen (Phorbolmyristatacetat, HOCl) Stimuli induziert, und die NETs sind durch DNA-DAPI-Färbung, Immunfärbung für das antimikrobielle Peptid Cathelicidin (LL37) und Quantifizierung der enzymatischen Aktivität (neutrophile Elastase, Cathepsin G und Myeloperoxidase) gekennzeichnet. Die Bilder werden mittels Fluoreszenzmikroskopie aufgenommen und mit ImageJ analysiert.
Einleitung
Neutrophile sind die am häufigsten vorkommenden Leukozyten im Blutkreislauf und spielen eine wesentliche Rolle bei der Beseitigung von Krankheitserregern durch verschiedene Mechanismen, einschließlich der Freisetzung großer Chromatinstrukturen, die aus DNA und mehreren nuklearen, zytoplasmatischen und granulären antibakteriellen Proteinen bestehen 1,2. Der direkte Vorläufer, der diese antimikrobielle Rolle von Neutrophilen beschreibt, wurde 1996 von Takei et al.3 erstellt. Diese Autoren berichteten über eine neue Form des Todes, die sich von Apoptose und Nekroptose bei Neutrophilen unterschied, morphologische Veränderungen zeigten, die einen Kernbruch zeigten, gefolgt von einem Austreten aus dem Nukleoplasma in das Zytoplasma und einer Erhöhung der Membranpermeabilität nach 3 Stunden Inkubation mit Phorbolmyristatacetat (PMA)2,3. Es dauerte jedoch bis 2004, bis der Begriff "neutrophile extrazelluläre Fallen (NETs)" verwendet wurde4.
Die NET-Bildung wurde unter verschiedenen Bedingungen beobachtet, wie z. B. bakteriellen,Pilz-5-,Virus-6- und parasitären Infektionen, um die mikrobielle Verbreitung zu neutralisieren, abzutöten und zu verhindern7. Andere Studien zeigen, dass es auch unter nicht-pathogenen Zuständen durch sterile Reize wie Zytokine, Mononatriumharnsäure- oder Cholesterinkristalle, Autoantikörper, Immunkomplexe und aktivierte Blutplättchen auftretenkann 7. Lipopolysaccharid (LPS), Interleukin-8 (IL-8) und PMA gehörten zu den ersten In-vitro-Stimuli, die als NET-Induktoren beschrieben wurden, und die In-vivo-Beteiligung von NET an pathogenen Prozessen wurde in zwei Modellen akuter Entzündung nachgewiesen: experimentelle Dysenterie und spontane menschliche Appendizitis4. DNA ist eine wesentliche NET-Komponente. Seine geeignete Struktur und Zusammensetzung sind für die Sequestrierung und Abtötung von Mikroorganismen notwendig, indem eine hohe lokale Konzentration antimikrobieller Moleküle an die gefangenen Mikroben abgegeben wird, wie eine kurze Desoxyribonuklease (DNase)-Behandlung zeigt, die NETs und ihre mikrobiziden Eigenschaften zersetzt4. NETs umfassen neben DNA u.a. angehängte Proteine wie Histone, neutrophile Elastase (NE), Cathepsin G (CG), Proteinase 3, Lactoferrin, Gelatinase, Myeloperoxidase (MPO) und antimikrobielle Peptide (AMPs) wie das kationische proinflammatorische Peptid Cathelicidin LL-37 u.a. 8,9. Solche Aggregate können größere Fäden mit Durchmessern von bis zu 50 nm bilden. Diese Faktoren können die mikrobiellen Virulenzfaktoren oder die Integrität der Zellmembran des Erregers stören. zusätzlich können die AMPs die NET-abgeleitete DNA gegen den Abbau durch bakterielle Nukleasenstabilisieren 10.
Die spezifischen Mechanismen, die die NET-Bildung regulieren, sind noch nicht vollständig geklärt. Der am besten charakterisierte Weg, der zur NET-Freisetzung führt, ist der ERK-Signalweg, der zur Aktivierung der NADPH-Oxidase und zur Produktion reaktiver Sauerstoffspezies (ROS) sowie zu erhöhtem intrazellulärem Kalzium führt, das die Aktivierung des MPO-Signalwegs auslöst. Dies wiederum wandelt Wasserstoffperoxid in hypochlorige Säure um und aktiviert NE durch Oxidation11,12. NE ist verantwortlich für den Abbau der Aktinfilamente des Zytoskeletts, um die Phagozytose zu blockieren, und für die Translokation in den Zellkern zur Verarbeitung durch proteolytische Spaltung und Desaminierung durch PAD4, die die Desensibilisierung von Chromatinfasern vorantreiben, die sich mit granulierten und zytoplasmatischen Proteinen verbinden und dann extrazellulär freigesetzt werden7. Zu diesen Proteasen gehören diejenigen, die aus dem Azurosomenkomplex des azurophilen Granulats und anderen Proteasen wie Cathepsin G13 freigesetzt werden.
Abhängig von den morphologischen Veränderungen der Neutrophilen werden NETs in zwei Typen eingeteilt: suizidale oder lytische NET-Bildung, die zum Zelltod führt4, und vitale oder nicht-lytische NET-Bildung, die von lebensfähigen Zellen erzeugt wird, die durch eine vesikuläre Freisetzung von nukleärer oder mitochondrialer DNA vermittelt wird, mit einem Überrest eines kernhaltigen Zytoplasten mit phagozytärer Fähigkeit14,15. Im Allgemeinen weisen NETs, die aus mitochondrialer DNA bestehen, eine länglicheFaser-14-Morphologie auf, während solche, die aus Kern-DNA strukturiert sind, ein wolkenartiges Aussehenaufweisen 3. Es ist jedoch nicht bekannt, wie die Neutrophile ihren DNA-Ursprung wählt. Im Gegensatz zu früheren Studien, die die kanonischen Signalwege von NETs als mehrere Stunden dauernd beschrieben, wird der Vitalweg in nur 5-60 min15 schnell aktiviert.
Trotz dieser Fortschritte variiert die NET-Zusammensetzung je nach Stimulus; Zum Beispiel induzieren verschiedene schleimige und nicht-mukoide Stämme von P. aeruginosa die Bildung von NETs, die 33 gemeinsame Proteine und bis zu 50 variable Proteine enthalten7. Daher ist es notwendig, Techniken zu homogenisieren, die die Generierung objektiver Schlussfolgerungen in Forschungsgruppen ermöglichen. Dieser Artikel beschreibt ein Protokoll mit verschiedenen Techniken, die den Vergleich und die Bewertung der Zusammensetzung, Struktur und Morphologie von NETs ermöglichen, die mit verschiedenen Mikroorganismen induziert werden: Staphylococcus aureus (grampositives Bakterium), Pseudomonas aeruginosa (gramnegatives Bakterium) und Candida albicans (Pilz) sowie chemische Stimuli (PMA, HOCl) in menschlichen Neutrophilen von gesunden Personen. Die repräsentativen Ergebnisse zeigen die Heterogenität von NETs in Abhängigkeit von ihrem induzierenden Stimulus unter vergleichbaren in vitro Bedingungen, gekennzeichnet durch DNA-DAPI-Färbung, Immunfärbung für LL37 und Quantifizierung der enzymatischen Aktivität (NE, CG und MPO).
Access restricted. Please log in or start a trial to view this content.
Protokoll
Die Blutproben wurden als Spenden von klinisch gesunden Teilnehmern nach Einverständniserklärung entnommen. Alle Experimente wurden mit Genehmigung der Ethikkommission für Humanforschung der Fakultät für Biochemische Wissenschaften der Universidad Autónoma "Benito Juárez" von Oaxaca durchgeführt.
HINWEIS: Die Einschlusskriterien in der Studie waren undeutliches Geschlecht und Alter und klinisch gesund gemäß den Antworten der Teilnehmer auf einen Fragebogen vor der Entnahme einer Blutprobe. Eine hämatologische Analyse wurde durchgeführt, um die Zellzahl zu bestimmen und Infektionen oder Anämie auszuschließen, sowie der C-reaktive Proteintest, um eine Entzündung beim Spender auszuschließen.
1. Periphere Blutentnahme und Gewinnung des Erythrozyten- und Leukozytenpakets
- 10 ml peripheres Blut werden durch Venenpunktion in Röhrchen mit 1,8 mg/ml K2 entnommen. EDTA als Antikoagulans (siehe Materialtabelle) von klinisch gesunden Personen nach Einholung der Einwilligung nach Einverständniserklärung. Führen Sie dann eine Standardblutbiometrie und einen C-reaktiven Proteintest durch, um Infektionen oder Entzündungen auszuschließen und die Qualität der Probe sicherzustellen.
- Zentrifugieren Sie die periphere Blutprobe bei 82 x g für 15 min, um das plättchenreiche Plasma zu entfernen, gefolgt von einer zweiten Zentrifugation bei 630 x g für 5 min. Verwerfen Sie das verbleibende Plasma, um das Erythrozyten- und Leukozytenpaket zu erhalten.
- Verdünnen Sie es im Verhältnis 1:1 (v/v) mit 1x Dulbecco's phosphatgepufferter Kochsalzlösung (DPBS).
2. Reinigung von polymorphkernigen neutrophilen (PMN) unter Verwendung eines Doppeldichtegradienten
HINWEIS: Führen Sie die Neutrophilenreinigung unmittelbar nach der Blutentnahme durch, da sie eine begrenzte In-vitro-Lebensdauer von etwa 8 h haben.
- In einem sterilen 10-ml-Glasröhrchen (siehe Materialtabelle) wird der Reihe nach abgeschieden: 1 ml Lösung mit 1,098 g/ml Dichte, 1 ml Lösung mit 1,079 g/ml Dichte (siehe Materialtabelle) und dann 4 ml des verdünnten Erythrozyten- und Leukozytenpakets. Gießen Sie über die Wände, ohne die Oberflächenspannung zwischen den Schichten zu unterbrechen, um zu verhindern, dass sie sich vermischen.
- Zentrifuge bei 320 x g für 20 min bei 4 °C, wobei Beschleunigung/Verzögerung vermieden wird, damit die hohen Kräfte der Zentrifuge den Gradienten nicht stören.
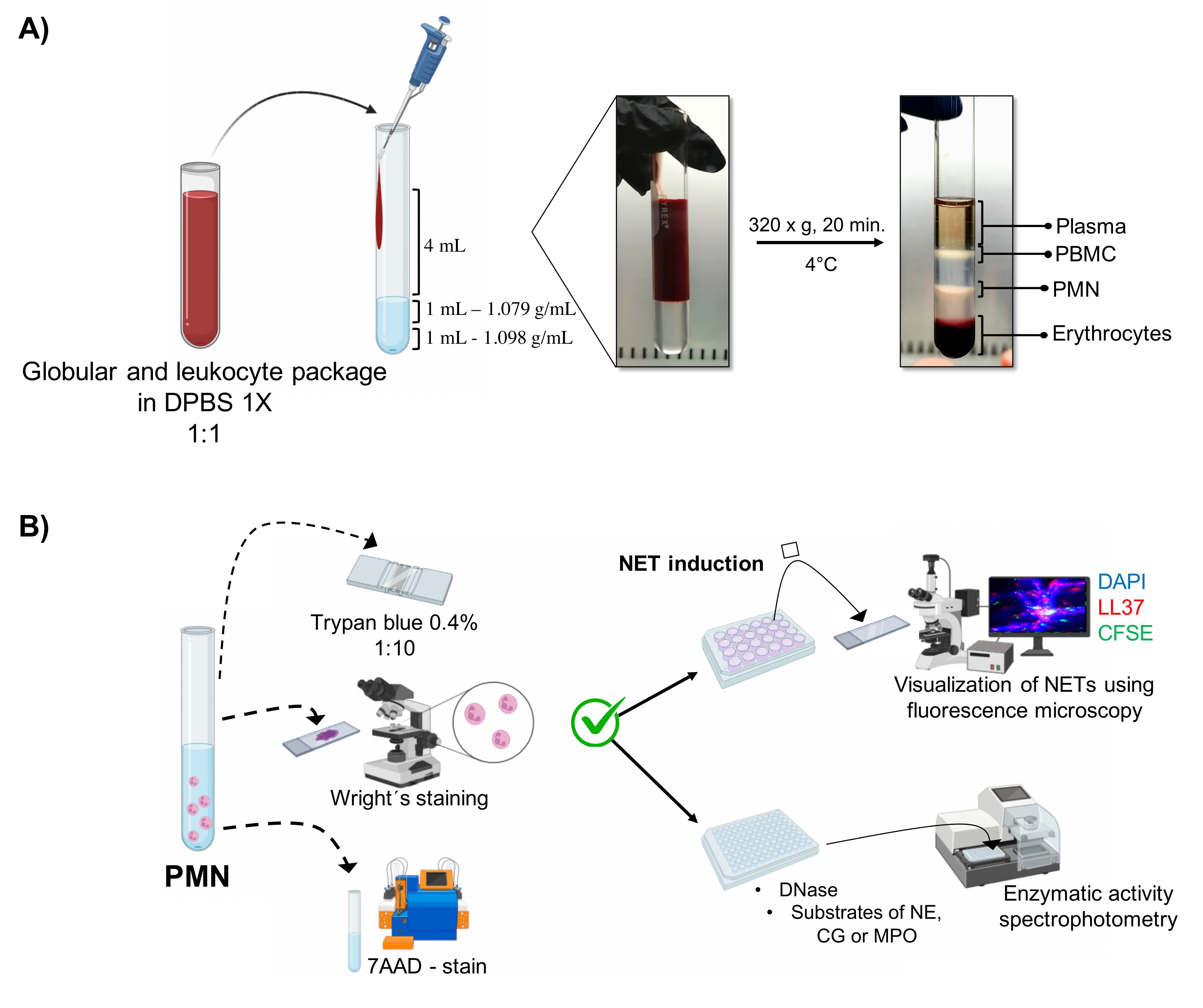
- Die Phase, die den Granulozyten entspricht (Abbildung 1A), wird durch Pipettieren abgesaugt und in ein anderes steriles 10-ml-Glasröhrchen überführt. Mit 4 mL 1x DPBS bei 300 x g für 10 min bei 4 °C waschen.
- Verwerfen Sie den Überstand und behandeln Sie die Zellen mit einem osmotischen Schock, um die verbleibenden Erythrozyten zu entfernen. 4 ml 0,2%ige Kochsalzlösung für 2 min bei 4 °C zugeben und bei 300 x g für 10 min bei 4 °C zentrifugieren. Verwerfen Sie den Überstand. Dann 4 ml der isotonischen Lösung (0,65% Kochsalzlösung) für 5 min bei 4 °C hinzufügen, um die Integrität der Membran wiederherzustellen, und bei 300 x g für 10 min bei 4 °C zentrifugieren.
HINWEIS: Die 0,2%ige Kochsalzlösung ist ein hypotones Medium mit einer niedrigeren Konzentration des gelösten Stoffes im Vergleich zu der des intrazellulären Erythrozytenmediums. Durch den Kontakt mit dem hypotonischen Medium kann Wasser in die Erythrozyten diffundieren, was zu Schwellungen und Hämolyse führt. Diese Entfernung von Erythrozyten aus dem Überstand wurde durch mikroskopische Beobachtungen bestätigt. - Entfernen Sie den Überstand. Resuspendieren Sie die Zellen in 4 ml 1x DPBS, um Zelltrümmer zu entfernen, und zentrifugieren Sie dann bei 300 x g für 10 min bei 4 °C. Zum Schluss resuspendieren Sie das Zellpellet in 2 ml kaltem Hank's Balanced Salt Solution (HBSS) -Puffer.
3. Morphologie und Lebensfähigkeit der neutrophilen Granulozyten (Abbildung 1B)
- Ausschlusstest mit Trypanblau
- Verdünnen Sie 5 μL der Zellsuspension in 20 μL 0,4% Trypanblau (Verhältnis 1:5). Zählen Sie die Zellen in einer Neubauer-Kammer und bestimmen Sie die Zelllebensfähigkeit mit einem Ausschlusstest. Betrachten Sie die Zellen, die die Integrität ihrer Membran erhalten, ohne den Farbstoff zu permeabilisieren, als lebensfähig.
- Montieren Sie 5 μL der Zellsuspension auf einem Objektträger; trocknen und mit Wright-Fleck für 15 s flecken. Die Probe wird sofort mit einem Phosphatpuffer pH 6,4 für 30 s fixiert. Mit ausreichend destilliertem Wasser waschen und die Morphologie unter einem Lichtmikroskop (100x) beobachten.
- 7AAD-Färbung und Durchflusszytometrie-Analyse
- Geben Sie 1 x 105 Zellen in Durchflusszytometrieröhrchen und färben Sie mit 1 μL 7AAD in 100 μL FACS-Puffer (1x DPBS, 0,1% Natriumazid und 10% autologes dekomplementiertes Plasma) für 15 min bei 4 °C im Dunkeln.
- Mit 500 μL FACS-Puffer bei 300 x g 10 min waschen. Die Zellen werden mit 500 μL 2%igem Paraformaldehyd fixiert und bis zur Analyse im Durchflusszytometer bei 4 °C gelagert.
- Für eine Totzellkontrolle 1 x 105 Zellen mit 200 μL 4% Paraformaldehyd für 30 min fixieren und mit 500 μL 1x PBS bei 300 x g für 10 min bei 4 °C waschen. Ziehen Sie den Überstand ab und entsorgen Sie ihn. Dann fügen Sie 200 μL 0,1% Triton X-100 für 1 h bei 4 °C hinzu. Mit 500 μL 1x PBS waschen und mit 7AAD wie in Schritt 3.2.1 färben.
- Führen Sie mit einem Durchflusszytometer (siehe Materialtabelle) eine FSC- versus SSC-Analyse durch, um die Zellreinheit zu analysieren, und eine SSC- versus 7AAD-Färbung, um die Zelllebensfähigkeit zu analysieren. Lesen Sie 3 x 104 Ereignisse in 100 μL Aufnahmevolumen bei mittlerem Durchfluss (1.000 Zellen/s) in den polymorphkernigen Einstellungen (FSC, 400-490 und SSC, 300-320).
- Analysieren Sie die erfassten Daten in der Durchflusszytometer-Software (siehe Materialtabelle) und bestimmen Sie den Prozentsatz an Reinheit und positiven Zellen für 7AAD in der polymorphkernigen Population, dargestellt durch Punktdiagramme und Histogramme.
4. CFSE-Färbung von Mikroorganismen
- Fügen Sie 1 x 108 Bakterien oder 1 x 106 Pilzpseudohyphen in 1,5 ml Mikroröhrchen hinzu und färben Sie sie mit 200 μL 5 μM Carboxyfluorescein-Succinimidylester (CFSE), gelöst in 1x PBS. Einige Sekunden mixen und bei 37 °C 10 min im Dunkeln inkubieren.
- Stoppen Sie die Reaktion durch Zugabe von 500 μL dekomplementiertem Plasma und zentrifugieren Sie bei 620 x g für 10 min für Pseudohyphen oder bei 1.800 x g für 10 min für Bakterien.
- Die Überstände werden verworfen und die Pellets mit 1 ml 1x PBS unter Zentrifugation wie in Schritt 4.2 gewaschen. Zum Schluss resuspendieren Sie die Mikroorganismen in 250 μL 1x PBS.
- Bereiten Sie 50 μL Aliquots in Mikroröhrchen von 1,5 ml mit 2 x 107 Bakterien (MOI: 100) oder 2 x 105 Pseudohyphen (MOI: 1) für die NET-Induktion vor.
5. NET-Induktion
- 10 mm x 10 mm große sterile Glasdeckgläser in eine 24-Well-Platte geben und 1 Stunde bei Raumtemperatur mit 10 μL 0,001% Poly-L-Lysin abdecken. Zweimal mit 100 μL 1x PBS waschen, an der Luft trocknen und 15 min mit UV-Licht bestrahlen.
- Ersetzen Sie die HBSS-Lösung der neutrophilen Suspension in Schritt 2.5 durch RPMI 1640-Medium, das mit 10%igem Eigenblutplasma ergänzt ist. Zur 24-Well-Platte (Schritt 5.1) werden 350 μL dieser Zellsuspension hinzugefügt, um eine Endkonzentration von 2 x 105 Neutrophilen/Vertiefung zu erhalten.
- Lassen Sie die Zellen am Boden der Vertiefungen haften, indem Sie 20 Minuten bei 37 °C mit 5% CO2 inkubieren.
- Fügen Sie die Stimuli hinzu, um die NET-Bildung in 50 μL zu induzieren: mikrobielles Stimuli-Gram-positives Bakterium S. aureus (ATCC 25923), gramnegatives Bakterium P. aeruginosa (ATCC 10145) bei MOI 100 und Pseudohyphen von C. albicans (ATCC 10231) bei MOI:1; biochemische Stimuli-PMA (200 nM) und HOCl (4,5 mM) und Kontrolle mit fehlendem Stimulus (50 μL HBSS).
- Erhalten Sie ein Endvolumen von 400 μL pro Vertiefung. Auf einem Plattenschüttler bei 140 U/min für 30 s mischen und 4 h bei 37 °C und 5 %CO2 inkubieren.
6. Visualisierung von NETs durch Fluoreszenzmikroskopie
- DNA- und LL37-Immunfärbung
- Nach der NET-Induktion die Überstände durch vorsichtiges Pipettieren aus den Vertiefungen entfernen und die Zellen 30 min lang mit 300 μL 4% Paraformaldehyd fixieren.
- Waschen Sie die Zellen mit 200 μL 1x PBS ohne Zentrifugieren und fügen Sie 200 μL Blockierpuffer (10% dekomplementiertes Plasma in 1x PBS) für 30 min hinzu.
- Für die LL-37-Färbung permeabilisieren Sie die Zellen mit 200 μL 0,2% Triton X-100 in 1x PBS für 10 min, damit der Antikörper in die Zellen eindringen kann. 2x vorsichtig mit 1x PBS waschen, um das überschüssige Reinigungsmittel zu entfernen.
- Montieren Sie die Deckgläser auf Glasobjektträger (vier Deckgläser auf jedem Objektträger). DNA färbt die Zellen mit 2 μL DAPI (siehe Materialtabelle), versiegelt die Deckgläser und lagert sie bei -20 °C bis zu ihrer Analyse durch konfokale Fluoreszenzmikroskopie.
- Aufnahme und Analyse von Fluoreszenzbildern
- Nehmen Sie NET-Bilder auf, um ihre Komponenten zu quantifizieren, und verwenden Sie die entsprechenden Filter im konfokalen Fluoreszenzmikroskop (siehe Materialtabelle), um die Bilder mit der Computersoftware zu erfassen.
HINWEIS: Beachten Sie, dass die DNA mit DAPI (blaue Farbe) gefärbt ist, was eine Anregung bei 360 nm und eine Emission bei 460 nm zeigt. Die Mikroorganismen sind mit CFSE (grüne Farbe) gefärbt, das eine Anregung von 492 nm und eine Emission von 521 nm aufweist. Das LL37-Peptid ist mit einem Anti-LL37 Alexa Fluor 594-Antikörper (rote Farbe) markiert, der eine Anregung von 594 nm und eine Emission von 614 nm aufweist. - Kalibrieren Sie das Mikroskop. Platzieren Sie das Dia und fokussieren Sie mit differenziellem Interferenzkontrast (DIC) bei eingeschaltetem normalem Licht. Wählen Sie "Live ", um das Bild auf den Monitor zu projizieren.
- Schalten Sie das Licht aus und wählen Sie den entsprechenden Fluorochrom-Kanal. Wählen Sie z. B. Filter 365 nm/blau für DAPI, 43 HE DsRed für Alexa 594 oder 38 HE GFP für CFSE.
- Passen Sie die Einstellungen mit dem Isotyp-Kontrollantikörper für LL37 und ungefärbten Zellen für DAPI und CFSE an. Stellen Sie die gleichen Belichtungszeit-, Spannungs-, Kontrast- und Objektiveinstellungen ein, um alle Bilder unter den gleichen Bedingungen aufzunehmen.
HINWEIS: In dieser Studie wurden Belichtungszeit, Spannung und Kontrast auf 1,0 ms, 4,0 V bzw. 0,0 mit einem 40-fachen Objektiv eingestellt. Diese Werte können angepasst werden, um die beste Bildaufnahme für die Proben zu ermöglichen. - Wählen Sie Ausrichten , um das Bild aufzunehmen. Speichern Sie fünf Bilder (vier Extreme und das Zentrum) pro Vertiefung und von der Kolokalisation (Merge) von DNA/LL37/CFSE.
- Definieren Sie die drei Pixelklassen als Hintergrund mit den unabhängigen Bildern jeder Farbe und analysieren Sie den mittleren Grausignalwert pro Bereich mit der Image J-Software.
- Nehmen Sie NET-Bilder auf, um ihre Komponenten zu quantifizieren, und verwenden Sie die entsprechenden Filter im konfokalen Fluoreszenzmikroskop (siehe Materialtabelle), um die Bilder mit der Computersoftware zu erfassen.
7. Quantifizierung der enzymatischen Aktivität
- In eine 96-Well-Platte werden 90 μl Zellsuspension in HBSS gegeben, die 1 x 10 5 Neutrophile für die NET-Induktion enthält, und 20 Minuten lang bei 37 °C und5 %CO2 inkubiert.
- Sofort werden 10 μL der entsprechenden Stimuli (Konzentration wie in Schritt 5.4) zugegeben und 4 h bei 37 °C mit 5 %CO2 inkubiert.
- Verwerfen Sie die Überstände und waschen Sie die Zellen mit 100 μL HBSS. Mit 1 U/ml DNase für 10 min bei 37 °C behandeln, um die Freisetzung von DNA-Proteinstrukturen zu begünstigen, und 10 min bei 1.800 x g zentrifugieren.
- Gewinnen Sie die Überstände zurück und bewerten Sie die Enzymaktivität im Überstand unter Verwendung kolorimetrischer Reaktionen, wie zuvor von White et al.17 beschrieben.
- Bestimmen Sie die maximale Enzymaktivität von NE, CG und MPO in Neutrophilen unter den gleichen experimentellen Bedingungen, ohne irgendwelche Stimuli für die NET-Induktion hinzuzufügen. Anschließend wird die Zellprobe bei -70 °C eingefroren und bei 37 °C in einem Wasserbad aufgetaut, wodurch ein Temperaturschock erzeugt wird, um die Freisetzung von Intrazellproteinen durch Zelllyse zu begünstigen. Zentrifugieren Sie bei 1.800 x g für 10 min und gewinnen Sie die Überstände zurück.
- In 96-Well-Platten werden 50 μl des Überstands in jede Vertiefung gegeben und dann 50 μl jedes Substrats zugegeben, wie in Schritt 7.7 angegeben.
- Man fügt 0,5 m N-Methoxysuccinyl-Ala-Pro-Val-p-nitro-Anilin als Substrat für NE und 1 mM N-Succinyl-Ala-Ala-Pro-Phe-p-nitroanilid für CG hinzu. 3 h bei Raumtemperatur inkubieren. Für MPO 1,6 mM 3,3', 5,5'-Tetramethylbenzidin (TMB) zugeben und 30 min bei Raumtemperatur inkubieren.
- Nach der Inkubation werden 50 μl der Stopplösung (0,5 M H2SO4) für MPO hinzugefügt und die Extinktion bei 405 nm für NE und CG und 450 nm für MPO mit einem Spektralphotometer gemessen.
- Vergleichen Sie die erhaltenen Werte mit den entsprechenden Kalibrierkurven und zeigen Sie die Ergebnisse jeder Bedingung in Bezug auf die maximale Enzymaktivität (100%).
8. Statistische Auswertung
- Analysieren Sie die Messdaten in dreifacher Ausfertigung für jedes unabhängige Experiment (n = 10) und führen Sie eine ANOVA für die statistische Analyse durch, indem Sie Gruppen mit einem Konfidenzniveau von 95 % vergleichen.

Abbildung 1: PMN-Aufreinigung und NET-Induktionsprotokoll. (A) Plasma wurde aus dem peripheren Blut entnommen, um das Erythrozyten- und Leukozytenpaket zu erhalten, und 1:1 (v/v) mit 1x DPBS verdünnt. Dann wurden 4 ml der Verdünnung entlang der Wand in das Doppeldichtegradientenröhrchen gegeben und bei 320 x g für 20 Minuten bei 4 °C zentrifugiert, um die Trennung verschiedener Zellschichten zu erhalten und die PMN entsprechende Zellschicht zurückzugewinnen. (B) Die gereinigten Zellen wurden gezählt und ihre Morphologie wurde durch Wright-Färbung analysiert. Die Lebensfähigkeit wurde durch Trypanblauausschluss und 7AAD-Färbung mittels Durchflusszytometrie bestimmt. Sobald die optimale Reinheit und Lebensfähigkeit von Neutrophilen überprüft wurde, wurde die NET-Bildung durch Zugabe von Mikroben (S. aureus, P. aeruginosa und C. albicans) oder Chemikalien (PMA, HOCl) in 24-Well-Platten zur Analyse durch Fluoreszenzmikroskopie mit DAPI-DNA, Anti-LL37 Alexa Fluor 594 und Mikroorganismen-CFSE-Färbung induziert. Zur Enzymquantifizierung wurden NETs in 96-Well-Platten für 3 h induziert und mit DNase behandelt, gefolgt von der Zugabe von Substraten für jedes Enzym: NE, CG und MPO; Farbveränderungen wurden durch Spektrophotometrie quantifiziert. DPBS = Dulbecco's phosphatgepufferte Kochsalzlösung; PBMC = Mononukleäre Zellen des peripheren Blutes; PMN = Polymorphkernige Neutrophile; NE = Neutrophile Elastase; CG = Cathepsin G; MPO = Myeloperoxidase; PMA = Phorbolmyristatacetat; HOCl = Hypochlorige Säure. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Reinheit und Lebensfähigkeit von Neutrophilen
Die dynamischen zellulären Phasen werden in der Röhre aus der Double-Density-Gradientenreinigung visualisiert. Innerhalb dieser Schichten befindet sich die Schicht, die den Granulozyten entspricht, oberhalb der Dichteschicht von 1,079 g/ml, die von den Phasen der peripheren Blutmononukleozyten (PBMCs) und Erythrozyten unterschieden wird (Abbildung 1A). Die Morphologie der gereinigten Zellen wurde mit Wright-Färbung verifiz...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Um die Freisetzung von NETs zu induzieren, muss eine hochreine Population lebensfähiger Neutrophilen erhalten werden, da diese Zellen eine begrenzte Ex-vivo-Lebensdauer von durchschnittlich 8 Stunden haben, ein Zeitraum, in dem alle Experimente durchgeführt werden müssen. Zu diesem Zweck ist die ideale Methodik der Double-Density-Gradient zur Optimierung der Reinigungszeit durch Isolierung nicht aktivierter Zellen, die im Gegensatz zu Ficoll-Histopaque-Gradienten- oder Dextran-Sedimentationstechniken besser a...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren erklären, dass sie keine Interessenkonflikte haben.
Danksagungen
Diese Arbeit wurde durch ein Basic Science Grant (#285480) von CONACyT und von der Abteilung für klinische Immunologieforschung der Fakultät für Biochemische Wissenschaften der Universidad Autónoma 'Benito Juárez' de Oaxaca unterstützt. A.A.A, S.A.S.L. und W.J.R.R. haben Doktorandenstipendien der CONACyT-Nummern #799779, #660793 bzw. #827788.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| 24 Well plate for cell culture | Corning | 3526 | |
| 7-aminoactinomycin D (7-AAD) | BD Pharmingen | 51-668981E | |
| 96 Well plate for cell culture | Costar | 3596 | Flat bottom |
| Agitator | CRM Globe | CRM-OS1 | |
| Antibody LL37 | Santa Cruz Biotechnology | sc-166770 | |
| Blood collection tubes | BD VACUTAINER | 368171 | K2 EDTA 7.2 mg |
| Carboxyfluorescein succinimidyl ester (CFSE) | Sigma-Aldrich | 21878 | |
| Centrifuge | Hettich | 1406-01 | |
| Coverslip | Madesa | M03-CUB-22X22 | 22 mm x 22 mm |
| Dulbecco´s phosphate-buffered saline (DPBS) | Caisson | 1201022 | |
| Falcon tubes 50 mL | CORNING | 430829 | |
| Flow Cytometry Tubes | Miltenyi Biotec | 5 mL - Without caps | |
| FlowJo Software | BD Biosciences | Analyze flow cytometry data | |
| Fluorescence microscope | DM 2000 | LEICA | |
| Fluoroshield with DAPI | Sigma-Aldrich | F6057 | |
| Incubator | NUAIRE | UN-4750 | |
| MACSQuant Analyzer | Miltenyi Biotec | Flow cytometer | |
| Microplate reader photometer | Clarkson Laboratory - CL | ||
| Microtubes 1.5 mL | Zhejiang Runlab Tech | 35200N | wire snap |
| Minitab Software | Minitab | Statistical analysis | |
| Needles | BD VACUTAINER | 301746 | Diameter 1.34 mm |
| Optical microscope | VELAB | VE-B50 | |
| Percoll | GE Healthcare | 17-0891-01 | Solution for density gradient |
| Phosphate Buffered Saline (10x) | Caisson | PBL07-500ML | |
| Pyrex culture tubes | CORNING | CLS982025 | N°9820 |
| RPMI 1640 1x | Corning | 10-104-CV | contains Glutagro |
| Slides | Madesa | PDI257550 | 22 mm x 75 mm |
| Trypan Blue solution 0.4% | SIGMA | T8154-100ML |
Referenzen
- De Buhr, N., Maren, K. B. How neutrophil extracellular traps become visible. Journal of Immunology Research. 2016, 4604713(2016).
- Karlsson, A., Nixon, J. B., McPhail, L. C. Phorbol myristate acetate induces neutrophil NADPH-oxidase activity by two separate signal transduction pathways: dependent or independent of phosphatidylinositol 3-kinase. Journal of Leukocyte Biology. 67 (3), 396-404 (2000).
- Takei, H., Araki, A., Watanabe, H., Ichinose, A., Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. Journal of Leukocyte Biology. 59 (2), 229-240 (1996).
- Brinkmann, V., et al. Neutrophil extracellular traps kill bacteria. Science. 303 (5663), 1532-1535 (2004).
- Kenny, E. F., et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife. 6, 24437(2017).
- Schultz, B. M., Acevedo, O. A., Kalergis, A. M., Bueno, S. M. Role of extracellular trap release during bacterial and viral infection. Frontiers in Microbiology. 13, 798853(2022).
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nature Reviews Immunology. 18 (2), 134-147 (2018).
- Delgado-Rizo, V., et al. Neutrophil extracellular traps and its implications in inflammation: An overview. Frontiers in Immunology. 8, 81(2017).
- Petretto, A., et al. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS One. 14 (7), 0218946(2019).
- Neumann, A., et al. Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular traps against degradation by bacterial nucleases. Journal of Innate Immunity. 6 (6), 860-868 (2014).
- Hakkim, A., et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nature Chemical Biology. 7 (2), 75-77 (2011).
- Sabbatini, M., Magnelli, V., Renò, F. NETosis in wound healing: When enough Is enough. Cells. 10 (3), 494(2021).
- Metzler, K. D., Goosmann, C., Lubojemska, A., Zychlinsky, A., Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Reports. 8 (3), 883-896 (2014).
- Clark, S. R., et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature Medicine. 13 (4), 463-469 (2007).
- Pilsczek, F. H., et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. Journal of Immunology. 185 (12), 7413-7425 (2010).
- Sosa, S. A., et al. Structural differences of neutrophil extracellular traps induced by biochemical and microbiologic stimuli under healthy and autoimmune milieus. Immunologic Research. 69 (3), 264-274 (2021).
- White, P. C., et al. Characterization, quantification, and visualization of neutrophil extracellular traps. Methods in Molecular Biology. 1537, 481-497 (2017).
- Boeltz, S., et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death and Differentiation. 26 (3), 395-408 (2019).
- Yousefi, S., Mihalache, C., Kozlowski, E., Schmid, I., Simon, H. U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death and Differentiation. 16 (11), 1438-1444 (2009).
- Brinkmann, V., Zychlinsky, A. Neutrophil extracellular traps: is immunity the second function of chromatin. The Journal of Cell Biology. 198 (5), 773-783 (2012).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten