
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Analyse morphologique et compositionnelle des pièges extracellulaires neutrophiles induits par des stimuli microbiens et chimiques
Dans cet article
Résumé
Un protocole pour l’induction et l’analyse de pièges extracellulaires (TNE) in vitro est présenté. La quantification de l’ADN, de la cathélicidine (LL37) et de l’activité enzymatique a fourni des données qui montrent la variabilité de la composition et de la morphologie des TNE induites par des stimuli microbiens et chimiques dans des conditions contrôlées similaires.
Résumé
Les neutrophiles fonctionnent comme la première ligne de défense cellulaire dans une réponse immunitaire innée en utilisant divers mécanismes, tels que la formation de pièges extracellulaires neutrophiles (TNE). Cette étude analyse les changements morphologiques et compositionnels des TNE induits par des stimuli microbiens et chimiques à l’aide de méthodologies in vitro normalisées pour l’induction et la caractérisation des TNE avec des cellules humaines. Les procédures décrites ici permettent d’analyser la morphologie (lytique ou non lytique) et la composition des NET (structures ADN-protéines et activité enzymatique), ainsi que l’effet des facteurs solubles ou du contact cellulaire sur ces caractéristiques. De plus, les techniques décrites ici pourraient être modifiées pour évaluer l’effet des facteurs solubles exogènes ou du contact cellulaire sur la composition des TNE.
Les techniques appliquées comprennent la purification de cellules polymorphonucléaires du sang périphérique humain à l’aide d’un double gradient de densité (1,079-1,098 g / mL), garantissant une pureté et une viabilité optimales (≥ 95%), comme démontré par la coloration de Wright, l’exclusion du bleu de trypan et la cytométrie en flux, y compris l’analyse FSC versus SSC et la coloration 7AAD. La formation de TNE est induite par des stimuli microbiens (Pseudomonas aeruginosa, Staphylococcus aureus et Candida albicans) et chimiques (acétate de myristate de phorbol, HOCl), et les TNE sont caractérisées par une coloration de l’ADN-DAPI, une immunocoloration pour le peptide antimicrobien cathélicidine (LL37) et une quantification de l’activité enzymatique (élastase neutrophile, cathepsine G et myéloperoxydase). Les images sont acquises par microscopie à fluorescence et analysées avec ImageJ.
Introduction
Les neutrophiles sont les leucocytes les plus abondants dans la circulation sanguine, jouant un rôle essentiel lors de la clairance des agents pathogènes par plusieurs mécanismes, dont la libération de grandes structures chromatiniques composées d’ADN et de plusieurs protéines antibactériennes nucléaires, cytoplasmiques et granulaires 1,2. L’antécédent direct décrivant ce rôle antimicrobien des neutrophiles a été fait par Takei et al.3 en 1996. Ces auteurs ont rapporté une nouvelle forme de mort différente de l’apoptose et de la nécroptose chez les neutrophiles, ont montré des changements morphologiques présentant une rupture nucléaire, suivie d’un débordement du nucléoplasme dans le cytoplasme, et une augmentation de la perméabilité de la membrane à partir de 3 h d’incubation avec l’acétate de myristate de phorbol (PMA)2,3. Cependant, ce n’est qu’en 2004 que le terme « pièges extracellulaires (TNE) » a été utilisé4.
La formation de TNE a été observée dans diverses conditions, telles que les infections bactériennes, fongiques5, virales6 et parasitaires, pour neutraliser, tuer et prévenir la dissémination microbienne7. D’autres études montrent qu’il peut également se produire dans des conditions non pathogènes par des stimuli stériles, tels que des cytokines, des cristaux d’acide urique monosodique ou de cholestérol, des auto-anticorps, des complexes immunitaires et des plaquettes activées7. Le lipopolysaccharide (LPS), l’interleukine-8 (IL-8) et la PMA ont été parmi les premiers stimuli in vitro décrits comme des inducteurs NET, et l’implication in vivo des TNE dans les processus pathogènes a été démontrée dans deux modèles d’inflammation aiguë : la dysenterie expérimentale et l’appendicite humaine spontanée4. L’ADN est une composante essentielle de la TNE. Sa structure et sa composition appropriées sont nécessaires à la séquestration et à la destruction des micro-organismes en délivrant une forte concentration locale de molécules antimicrobiennes vers les microbes capturés, comme le démontre un bref traitement par désoxyribonucléase (DNase) qui désintègre les TNE et leurs propriétés microbicides4. Outre l’ADN, les TNE comprennent des protéines attachées telles que les histones, l’élastase neutrophile (NE), la cathepsine G (CG), la protéinase 3, la lactoferrine, la gélatinase, la myéloperoxydase (MPO) et les peptides antimicrobiens (AMP) tels que le peptide cationique pro-inflammatoire cathélicidine LL-37, entre autres, 8,9. Ces agrégats peuvent former des fils plus grands avec des diamètres allant jusqu’à 50 nm. Ces facteurs peuvent perturber les facteurs de virulence microbienne ou l’intégrité de la membrane cellulaire pathogène; de plus, les AMPs peuvent stabiliser l’ADN dérivé de NET contre la dégradation par les nucléases bactériennes10.
Les mécanismes spécifiques régulant la formation des TNE n’ont pas encore été complètement clarifiés. La voie la mieux caractérisée menant à la libération de NET est la signalisation ERK, qui conduit à l’activation de la NADPH oxydase et à la production d’espèces réactives de l’oxygène (ROS), ainsi qu’à une augmentation du calcium intracellulaire qui déclenche l’activation de la voie MPO. Cela transforme à son tour le peroxyde d’hydrogène en acide hypochloreux, activant NE par oxydation11,12. L’entérite nécrotique est responsable de la dégradation des filaments d’actine du cytosquelette pour bloquer la phagocytose et de leur translocation vers le noyau pour traitement par clivage protéolytique et désamination par PAD4 qui entraînent la désensibilisation des fibres de chromatine, qui s’associent aux protéines granulaires et cytoplasmiques, et sont ensuite libérées extracellulairement7. Ces protéases comprennent celles libérées par le complexe azurosome des granules azurophiles et d’autres protéases telles que la cathepsine G13.
Selon les changements morphologiques des neutrophiles, les TNE sont classées en deux types : la formation de TNE suicidaires ou lytiques conduisant à la mort cellulaire4, et la formation de TNE vitale ou non lytique produite par des cellules viables médiées par une libération vésiculeuse d’ADN nucléaire ou mitochondrial, avec un reste d’un cytoplaste anucléé ayant une capacité phagocytaire14,15. Généralement, les TNE composées d’ADN mitochondrial présentent une morphologie allongéede fibres 14, tandis que celles structurées d’ADN nucléaire ont une apparence nuageuse3. Cependant, on ne sait pas comment le neutrophile choisit son origine ADN. Contrairement aux études précédentes qui décrivaient les voies canoniques des TNE comme nécessitant plusieurs heures, la voie vitale est rapidement activée en seulement 5 à 60 minutes15.
Malgré ces avancées, la composition de l’EVF varie en fonction du stimulus; par exemple, différentes souches mucoïdes et non mucoïdes de P. aeruginosa induisent la formation de TNE contenant 33 protéines communes et jusqu’à 50 protéines variables7. Ainsi, il est nécessaire d’homogénéiser les techniques qui permettent la génération de conclusions objectives dans les groupes de recherche. Cet article décrit un protocole avec diverses techniques qui permettent de comparer et d’évaluer la composition, la structure et la morphologie des TNE induites par différents micro-organismes: Staphylococcus aureus (bactérie à Gram positif), Pseudomonas aeruginosa (bactérie à Gram négatif) et Candida albicans (champignon), ainsi que des stimuli chimiques (PMA, HOCl) dans les neutrophiles humains d’individus sains. Les résultats représentatifs démontrent l’hétérogénéité des TNE en fonction de leur stimulus inducteur dans des conditions in vitro comparables, caractérisées par la coloration de l’ADN-DAPI, l’immunomarquage pour LL37 et la quantification de l’activité enzymatique (NE, CG et MPO).
Protocole
Les échantillons de sang ont été prélevés sous forme de dons de participants cliniquement sains après consentement éclairé. Toutes les expériences ont été réalisées avec l’autorisation du Comité d’éthique de la recherche humaine de la Faculté des sciences biochimiques de l’Université autonome Benito Juárez d’Oaxaca.
REMARQUE : Les critères d’inclusion dans l’étude étaient le sexe et l’âge indistincts, et cliniquement sains selon les réponses des participants à un questionnaire avant le prélèvement d’un échantillon de sang. Une analyse hématologique a été effectuée pour déterminer le nombre de cellules et exclure les infections ou l’anémie, ainsi que le test de la protéine C-réactive pour exclure l’inflammation chez le donneur.
1. Prélèvement de sang périphérique et obtention de l’emballage érythrocytaire et leucocytaire
- Prélever 10 mL de sang périphérique par ponction veineuse dans des tubes contenant 1,8 mg/mL de K2· L’EDTA comme anticoagulant (voir le tableau des matières) provenant de personnes cliniquement saines après avoir obtenu un consentement éclairé. Ensuite, effectuez une biométrie sanguine standard et un test de protéine C-réactive pour exclure une infection ou une inflammation, garantissant ainsi la qualité de l’échantillon.
- Centrifuger l’échantillon de sang périphérique à 82 x g pendant 15 min pour éliminer le plasma riche en plaquettes, suivi d’une deuxième centrifugation à 630 x g pendant 5 min. Jeter le plasma restant pour obtenir l’emballage érythrocytaire et leucocytes.
- Diluez-le dans un rapport 1:1 (v/v) avec 1x solution saline tamponnée au phosphate (DPBS) de Dulbecco.
2. Purification des neutrophiles polymorphonucléaires (PMN) à l’aide d’un gradient de densité double
REMARQUE: Effectuer la purification des neutrophiles immédiatement après le prélèvement du sang, car ils ont une durée de vie in vitro limitée d’environ 8 heures.
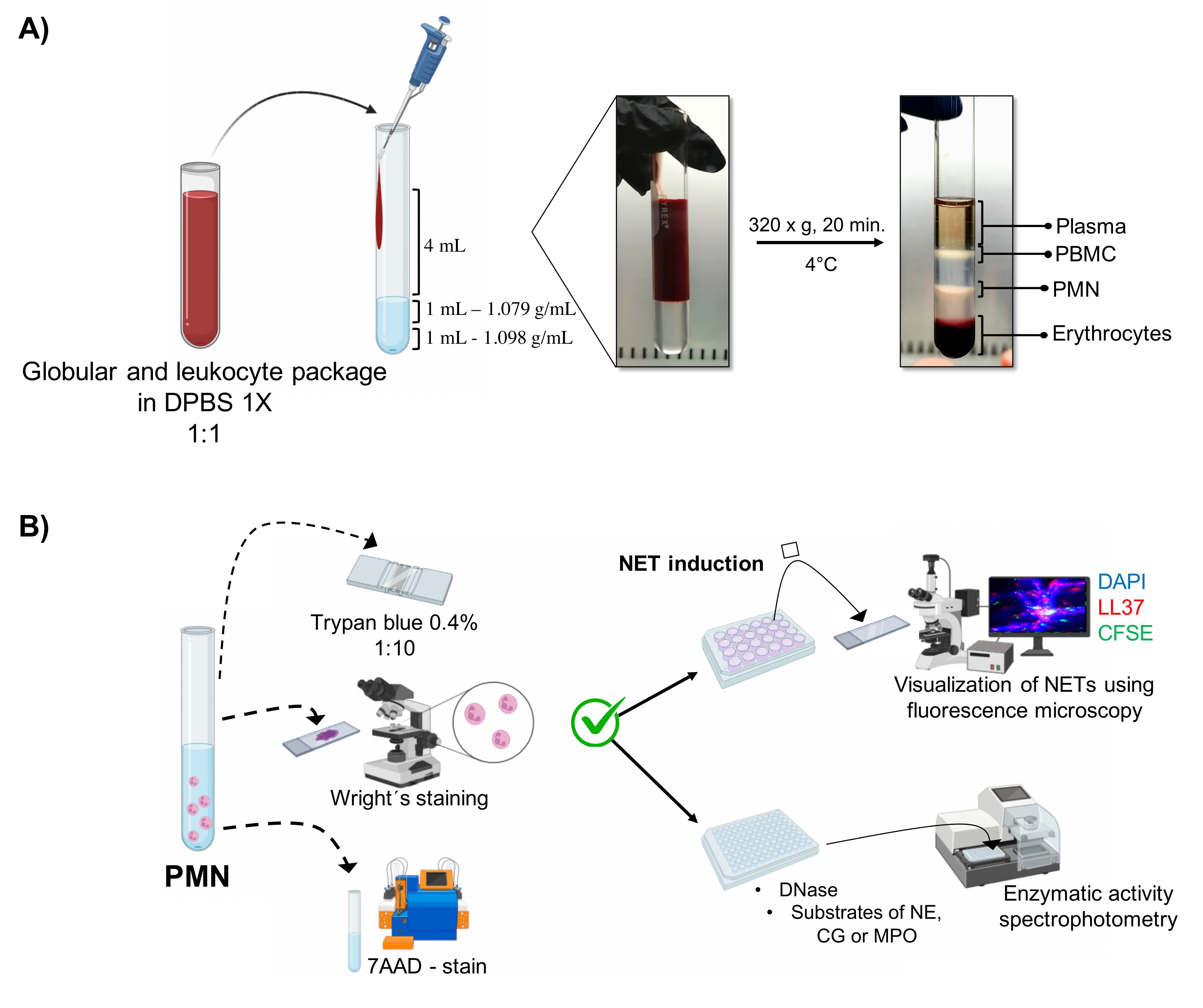
- Déposer les éléments suivants dans un tube de verre stérile de 10 mL (voir le tableau des matières) dans l’ordre : 1 mL de solution de densité de 1,098 g/mL, 1 mL de solution de densité de 1,079 g/mL (voir le tableau des matières), puis 4 mL de l’emballage érythrocytaire et leucocytaire dilué. Verser sur les murs sans rompre la tension superficielle entre les couches pour éviter qu’elles ne se mélangent.
- Centrifuger à 320 x g pendant 20 min à 4 °C, en évitant l’accélération/décélération afin que les forces élevées de la centrifugeuse ne perturbent pas la pente.
- Aspirer la phase qui correspond aux granulocytes (figure 1A) par pipetage, et transférer dans un autre tube de verre stérile de 10 mL. Laver avec 4 mL de 1x DPBS à 300 x g pendant 10 min à 4 °C.
- Jeter le surnageant et traiter les cellules avec un choc osmotique pour éliminer les érythrocytes restants. Ajouter 4 mL de solution saline à 0,2 % pendant 2 min à 4 °C et centrifuger à 300 x g pendant 10 min à 4 °C. Jetez le surnageant. Ajouter ensuite 4 mL de la solution isotonique (solution saline à 0,65 %) pendant 5 min à 4 °C pour rétablir l’intégrité de la membrane, et centrifuger à 300 x g pendant 10 min à 4 °C.
NOTE: La solution saline à 0,2% est un milieu hypotonique, avec une concentration de soluté inférieure à celle du milieu intracellulaire RBC. Le contact avec le milieu hypotonique permet à l’eau de diffuser dans les globules rouges, entraînant leur gonflement et leur hémolyse. Cette élimination des globules rouges du surnageant a été confirmée par une observation microscopique. - Retirez le surnageant. Remettez les cellules en suspension dans 4 mL de 1x DPBS pour éliminer les débris cellulaires, puis centrifugez à 300 x g pendant 10 min à 4 °C. Enfin, remettez en suspension la pastille cellulaire dans 2 ml de tampon de solution saline équilibrée (HBSS) de Hank froid.
3. Morphologie et viabilité des neutrophiles (figure 1B)
- Test d’exclusion du bleu trypan
- Diluer 5 μL de la suspension cellulaire dans 20 μL de bleu de trypan à 0,4 % (rapport 1:5). Compter les cellules dans une chambre de Neubauer et déterminer la viabilité cellulaire à l’aide d’un test d’exclusion. Considérez les cellules qui maintiennent l’intégrité de leur membrane sans perméabiliser le colorant comme viables.
- Monter 5 μL de la suspension cellulaire sur une glissière; sécher et tacher avec la teinture de Wright pendant 15 s. Fixer immédiatement l’échantillon avec un tampon phosphate pH 6,4 pendant 30 s. Laver avec suffisamment d’eau distillée et observer la morphologie au microscope optique (100x).
- 7AAD-coloration et analyse de cytométrie en flux
- Ajouter 1 x 105 cellules dans des tubes de cytométrie en flux et colorer avec 1 μL de 7AAD dans 100 μL de tampon FACS (1x DPBS, 0,1% d’azoture de sodium et 10% de plasma autologue décomplété) pendant 15 min à 4 °C dans l’obscurité.
- Laver avec 500 μL de tampon FACS à 300 x g pendant 10 min. Fixer les cellules avec 500 μL de paraformaldéhyde à 2% et conserver à 4 °C jusqu’à leur analyse dans le cytomètre en flux.
- Pour un contrôle des cellules mortes, fixer 1 x 105 cellules avec 200 μL de paraformaldéhyde à 4% pendant 30 min, et laver avec 500 μL de 1x PBS à 300 x g pendant 10 min à 4 °C. Retirez le surnageant et jetez-le. Ajouter ensuite 200 μL de Triton X-100 à 0,1 % pendant 1 h à 4 °C. Laver avec 500 μL de 1x PBS et teindre avec 7AAD comme à l’étape 3.2.1.
- À l’aide d’un cytomètre en flux (voir le tableau des matériaux), effectuer une analyse FSC versus SSC pour analyser la pureté cellulaire et SSC versus coloration 7AAD pour analyser la viabilité cellulaire. Lire 3 x 104 événements dans 100 μL de volume d’absorption à débit moyen (1 000 cellules/s) dans les milieux polymorphonucléaires (FSC, 400-490 et SSC, 300-320).
- Analyser les données saisies dans le logiciel de cytomètre en flux (voir le tableau des matériaux) et déterminer le pourcentage de pureté et de cellules positives pour 7AAD dans la population polymorphonucléaire, présenté au moyen de diagrammes de points et d’histogrammes.
4. Coloration des micro-organismes par le CFSE
- Ajouter 1 x 108 bactéries ou 1 x 106 pseudohyphes fongiques dans des microtubes de 1,5 mL et colorer avec 200 μL de carboxyfluorescéine succinimidylester (CFSE) dissous dans 1x PBS. Mélanger pendant quelques secondes et incuber à 37 °C pendant 10 min dans l’obscurité.
- Arrêter la réaction en ajoutant 500 μL de plasma décomplété, et centrifuger à 620 x g pendant 10 min pour les pseudohyphes ou à 1 800 x g pendant 10 min pour les bactéries.
- Jeter les surnageants et laver les granulés avec 1 mL de 1x PBS par centrifugation comme à l’étape 4.2. Enfin, remettre les micro-organismes en suspension dans 250 μL de 1x PBS.
- Préparer 50 μL d’aliquotes dans des microtubes de 1,5 mL avec 2 x 107 bactéries (MOI : 100) ou 2 x 105 pseudohyphes (MOI:1) pour l’induction NET.
5. Induction NET
- Placer des lamelles de verre stérile de 10 mm x 10 mm dans une plaque de 24 puits et couvrir de 10 μL de poly-L-lysine à 0,001 % pendant 1 h à température ambiante. Laver deux fois avec 100 μL de 1x PBS, sécher à l’air et irradier à la lumière UV pendant 15 min.
- Remplacer la solution HBSS de la suspension de neutrophiles à l’étape 2.5 par un milieu RPMI 1640 complété par du plasma autologue à 10%. Dans la plaque de 24 puits (étape 5.1), ajouter 350 μL de cette suspension cellulaire, pour une concentration finale de 2 x 105 neutrophiles/puits.
- Laisser les cellules adhérer au fond des puits en incubant pendant 20 min à 37 °C avec 5% de CO2.
- Ajouter les stimuli pour induire la formation de TNE dans 50 μL : la bactérie S. aureus (ATCC 25923), la bactérie à Gram négatif P. aeruginosa (ATCC 10145) à MOI 100 et les pseudohyphes de C. albicans (ATCC 10231) à MOI:1; stimuli biochimiques-PMA (200 nM) et HOCl (4,5 mM), et contrôle avec stimulus absent (50 μL de HBSS).
- Obtenir un volume final de 400 μL par puits. Mélanger sur une plaque agitatrice à 140 tr/min pendant 30 s et incuber pendant 4 h à 37 °C et 5% de CO2.
6. Visualisation des TNE par microscopie à fluorescence
- Immunomarquage de l’ADN et du LL37
- Après induction NET, retirer les surnageants des puits en les pipetant soigneusement, et fixer les cellules avec 300 μL de paraformaldéhyde à 4% pendant 30 min.
- Laver les cellules avec 200 μL de 1x PBS sans centrifugation, et ajouter 200 μL de tampon bloquant (10% de plasma décomplété dans 1x PBS) pendant 30 min.
- Pour la coloration LL-37, perméabiliser les cellules avec 200 μL de Triton X-100 à 0,2% dans 1x PBS pendant 10 minutes pour permettre à l’anticorps de pénétrer dans les cellules. Lavez 2x soigneusement avec 1x PBS pour enlever l’excès de détergent.
- Montez les lames de couverture sur des lames de verre (quatre lamelles de couverture sur chaque glissière). Colorer les cellules avec 2 μL de DAPI (voir Tableau des matériaux), sceller les lamelles de couverture et stocker à -20 °C jusqu’à leur analyse par microscopie confocale à fluorescence.
- Acquisition et analyse d’images fluorescentes
- Prenez des images NET pour quantifier leurs composants et utilisez les filtres correspondants dans le microscope confocale à fluorescence (voir Tableau des matériaux) pour acquérir les images avec le logiciel de l’ordinateur.
REMARQUE: Considérez que l’ADN est coloré avec DAPI (couleur bleue), montrant une excitation à 360 nm et une émission à 460 nm. Les micro-organismes sont colorés avec CFSE (couleur verte), qui a une excitation de 492 nm et une émission de 521 nm. Le peptide LL37 est marqué avec l’anticorps anti-LL37 Alexa Fluor 594 (couleur rouge), qui a une excitation de 594 nm et une émission de 614 nm. - Calibrer le microscope. Placez la diapositive et effectuez la mise au point à l’aide du contraste d’interférence différentiel (DIC) avec la lumière normale allumée. Choisissez En direct pour projeter l’image sur le moniteur.
- Éteignez la lumière et sélectionnez le canal correspondant au fluorochrome. Par exemple, sélectionnez filtre 365 nm/bleu pour DAPI, 43 HE DsRed pour Alexa 594 ou 38 HE GFP pour CFSE.
- Ajustez les paramètres avec l’anticorps témoin isotype pour LL37 et les cellules non colorées pour DAPI et CFSE. Réglez le même temps d’exposition, la même tension, le même contraste et les mêmes réglages d’objectif pour capturer toutes les images dans les mêmes conditions.
REMARQUE: Dans cette étude, le temps d’exposition, la tension et le contraste ont été fixés à 1,0 ms, 4,0 V et 0,0, respectivement, avec un objectif 40x. Ces valeurs peuvent être ajustées pour faciliter la meilleure capture d’image pour les échantillons. - Sélectionnez Accrochage pour capturer l’image. Enregistrer cinq images (quatre extrêmes et le centre) par puits, et de la colocalisation (fusion) de l’ADN / LL37 / CFSE.
- Définissez les trois classes de pixels comme arrière-plan avec les images indépendantes de chaque couleur et analysez la valeur du signal gris moyen par zone avec le logiciel Image J.
- Prenez des images NET pour quantifier leurs composants et utilisez les filtres correspondants dans le microscope confocale à fluorescence (voir Tableau des matériaux) pour acquérir les images avec le logiciel de l’ordinateur.
7. Quantification de l’activité enzymatique
- Dans une plaque de 96 puits, ajouter 90 μL de suspension cellulaire dans HBSS contenant 1 x 10 5 neutrophiles pour l’induction NET, et incuber pendant 20 min à 37 °C et5 % de CO2.
- Immédiatement, ajouter 10 μL des stimuli correspondants (concentration comme à l’étape 5.4) et incuber pendant 4 h à 37 °C avec 5% de CO2.
- Jetez les surnageants et lavez les cellules avec 100 μL de HBSS. Traiter avec 1 U/mL de DNase pendant 10 min à 37 °C pour favoriser la libération des structures ADN-protéine, et centrifuger à 1 800 x g pendant 10 min.
- Récupérer les surnageants et évaluer l’activité enzymatique du surnageant à l’aide de réactions colorimétriques décrites précédemment par White et coll.17.
- Déterminer l’activité enzymatique maximale de NE, CG et MPO dans les neutrophiles dans les mêmes conditions expérimentales sans ajouter de stimuli pour l’induction NET. Ensuite, congeler l’échantillon cellulaire à -70 °C et décongeler à 37 °C au bain-marie, générant un choc thermique pour favoriser la libération de protéines intracellulaires par lyse cellulaire. Centrifuger à 1 800 x g pendant 10 min et récupérer les surnageants.
- Ajouter 50 μL du surnageant à chaque puits dans des plaques de 96 puits, puis ajouter 50 μL de chaque substrat comme indiqué à l’étape 7.7.
- Ajouter 0,5 M de N-méthoxysuccinyl-Ala-Ala-Pro-Val-p-nitro aniline comme substrat pour NE, et 1 mM de N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide pour CG. Incuber pendant 3 h à température ambiante. Pour le MPO, ajouter 1,6 mM de 3,3',5'-tétraméthylbenzidine (TMB) et incuber pendant 30 min à température ambiante.
- Après incubation, ajouter 50 μL de la solution stop (0,5 MH2SO4)pour le MPO et mesurer l’absorbance à 405 nm pour l’entérite ne nétiques et CG et à 450 nm pour le MPO, à l’aide d’un spectrophotomètre.
- Comparer les valeurs obtenues avec les courbes d’étalonnage correspondantes et montrer les résultats de chaque condition par rapport à l’activité enzymatique maximale (100%).
8. Analyse statistique
- Analyser les données de mesure en triple exemplaire pour chaque expérience indépendante (n = 10) et effectuer une ANOVA pour l’analyse statistique en comparant les groupes avec un niveau de confiance de 95%.

Figure 1 : Purification PMN et protocole d’induction NET. (A) Le plasma a été retiré du sang périphérique pour obtenir l’emballage érythrocytaire et leucocytaire et dilué 1:1 (v/v) avec 1x DPBS. Ensuite, 4 mL de la dilution ont été ajoutés le long de la paroi au tube à gradient de double densité, et centrifugés à 320 x g pendant 20 min à 4 °C, obtenant la séparation des différentes couches cellulaires et récupérant celle correspondant à PMN. (B) Les cellules purifiées ont été comptées et leur morphologie a été analysée par coloration de Wright. La viabilité a été déterminée par exclusion du bleu de trypan et coloration 7AAD par cytométrie en flux. Une fois la pureté et la viabilité optimales des neutrophiles vérifiées, la formation de TNE a été induite par l’ajout de microbes (S. aureus, P. aeruginosa et C. albicans) ou de produits chimiques (PMA, HOCl) dans des plaques à 24 puits pour analyse par microscopie à fluorescence avec ADN-DAPI, anti-LL37 Alexa Fluor 594 et coloration micro-organisme-CFSE. Pour la quantification enzymatique, les TNE ont été induites dans des plaques de 96 puits pendant 3 h et traitées avec de la DNase, suivies de l’ajout de substrats pour chaque enzyme : NE, CG et MPO; Les changements de couleur ont été quantifiés par spectrophotométrie. DPBS = solution saline tamponnée au phosphate de Dulbecco; PBMC = cellules mononucléées du sang périphérique; PMN = neutrophiles polymorphonucléaires; NE = élastase neutrophile; CG = cathepsine G; MPO = Myéloperoxydase; PMA = acétate de myristate de Phorbol; HOCl = Acide hypochloreux. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Résultats
Pureté et viabilité des neutrophiles
Les phases cellulaires dynamiques sont visualisées dans le tube à partir de la purification à double gradient de densité. Au sein de ces couches, la couche correspondant aux granulocytes est supérieure à la couche de densité de 1,079 g/mL, ce qui se distingue des phases des mononucléocytes du sang périphérique (PBMC) et des érythrocytes (Figure 1A). La morphologie des cellules purifiées a été vérifiée avec la colorati...
Discussion
Une population très pure de neutrophiles viables doit être obtenue pour induire la libération de TNE, car ces cellules ont une durée de vie ex vivo limitée de 8 h en moyenne, période au cours de laquelle toutes les expériences doivent être effectuées. Pour cela, la méthodologie idéale est le gradient de double densité pour optimiser le temps de purification en isolant les cellules non activées plus sensibles à la stimulation exogène, contrairement au gradient de Ficoll-Histopaque ou aux techniques...
Déclarations de divulgation
Les auteurs déclarent n’avoir aucun conflit d’intérêts.
Remerciements
Ce travail a été soutenu par une subvention de science fondamentale (#285480) de CONACyT et par le Département de recherche en immunologie clinique de la Faculté des sciences biochimiques de l’Universidad Autónoma 'Benito Juárez' de Oaxaca. A.A.A, S.A.S.L et W.J.R.R. ont des bourses doctorales de CONACyT numéros #799779, #660793 et #827788, respectivement.
matériels
| Name | Company | Catalog Number | Comments |
| 24 Well plate for cell culture | Corning | 3526 | |
| 7-aminoactinomycin D (7-AAD) | BD Pharmingen | 51-668981E | |
| 96 Well plate for cell culture | Costar | 3596 | Flat bottom |
| Agitator | CRM Globe | CRM-OS1 | |
| Antibody LL37 | Santa Cruz Biotechnology | sc-166770 | |
| Blood collection tubes | BD VACUTAINER | 368171 | K2 EDTA 7.2 mg |
| Carboxyfluorescein succinimidyl ester (CFSE) | Sigma-Aldrich | 21878 | |
| Centrifuge | Hettich | 1406-01 | |
| Coverslip | Madesa | M03-CUB-22X22 | 22 mm x 22 mm |
| Dulbecco´s phosphate-buffered saline (DPBS) | Caisson | 1201022 | |
| Falcon tubes 50 mL | CORNING | 430829 | |
| Flow Cytometry Tubes | Miltenyi Biotec | 5 mL - Without caps | |
| FlowJo Software | BD Biosciences | Analyze flow cytometry data | |
| Fluorescence microscope | DM 2000 | LEICA | |
| Fluoroshield with DAPI | Sigma-Aldrich | F6057 | |
| Incubator | NUAIRE | UN-4750 | |
| MACSQuant Analyzer | Miltenyi Biotec | Flow cytometer | |
| Microplate reader photometer | Clarkson Laboratory - CL | ||
| Microtubes 1.5 mL | Zhejiang Runlab Tech | 35200N | wire snap |
| Minitab Software | Minitab | Statistical analysis | |
| Needles | BD VACUTAINER | 301746 | Diameter 1.34 mm |
| Optical microscope | VELAB | VE-B50 | |
| Percoll | GE Healthcare | 17-0891-01 | Solution for density gradient |
| Phosphate Buffered Saline (10x) | Caisson | PBL07-500ML | |
| Pyrex culture tubes | CORNING | CLS982025 | N°9820 |
| RPMI 1640 1x | Corning | 10-104-CV | contains Glutagro |
| Slides | Madesa | PDI257550 | 22 mm x 75 mm |
| Trypan Blue solution 0.4% | SIGMA | T8154-100ML |
Références
- De Buhr, N., Maren, K. B. How neutrophil extracellular traps become visible. Journal of Immunology Research. 2016, 4604713 (2016).
- Karlsson, A., Nixon, J. B., McPhail, L. C. Phorbol myristate acetate induces neutrophil NADPH-oxidase activity by two separate signal transduction pathways: dependent or independent of phosphatidylinositol 3-kinase. Journal of Leukocyte Biology. 67 (3), 396-404 (2000).
- Takei, H., Araki, A., Watanabe, H., Ichinose, A., Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. Journal of Leukocyte Biology. 59 (2), 229-240 (1996).
- Brinkmann, V., et al. Neutrophil extracellular traps kill bacteria. Science. 303 (5663), 1532-1535 (2004).
- Kenny, E. F., et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife. 6, 24437 (2017).
- Schultz, B. M., Acevedo, O. A., Kalergis, A. M., Bueno, S. M. Role of extracellular trap release during bacterial and viral infection. Frontiers in Microbiology. 13, 798853 (2022).
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nature Reviews Immunology. 18 (2), 134-147 (2018).
- Delgado-Rizo, V., et al. Neutrophil extracellular traps and its implications in inflammation: An overview. Frontiers in Immunology. 8, 81 (2017).
- Petretto, A., et al. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS One. 14 (7), 0218946 (2019).
- Neumann, A., et al. Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular traps against degradation by bacterial nucleases. Journal of Innate Immunity. 6 (6), 860-868 (2014).
- Hakkim, A., et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nature Chemical Biology. 7 (2), 75-77 (2011).
- Sabbatini, M., Magnelli, V., Renò, F. NETosis in wound healing: When enough Is enough. Cells. 10 (3), 494 (2021).
- Metzler, K. D., Goosmann, C., Lubojemska, A., Zychlinsky, A., Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Reports. 8 (3), 883-896 (2014).
- Clark, S. R., et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature Medicine. 13 (4), 463-469 (2007).
- Pilsczek, F. H., et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. Journal of Immunology. 185 (12), 7413-7425 (2010).
- Sosa, S. A., et al. Structural differences of neutrophil extracellular traps induced by biochemical and microbiologic stimuli under healthy and autoimmune milieus. Immunologic Research. 69 (3), 264-274 (2021).
- White, P. C., et al. Characterization, quantification, and visualization of neutrophil extracellular traps. Methods in Molecular Biology. 1537, 481-497 (2017).
- Boeltz, S., et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death and Differentiation. 26 (3), 395-408 (2019).
- Yousefi, S., Mihalache, C., Kozlowski, E., Schmid, I., Simon, H. U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death and Differentiation. 16 (11), 1438-1444 (2009).
- Brinkmann, V., Zychlinsky, A. Neutrophil extracellular traps: is immunity the second function of chromatin. The Journal of Cell Biology. 198 (5), 773-783 (2012).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.