
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Modification du génome dans des lignées de cellules mammifères à l’aide de CRISPR-Cas
Dans cet article
Résumé
SAE-CRISPR est une technologie puissante à l’ingénieur les génomes complexes de plantes et d’animaux. Ici, nous détaillons un protocole pour modifier efficacement le génome humain endonucléases de Cas différents. Nous mettons en évidence les considérations importantes et des paramètres de conception pour optimiser l’efficacité édition.
Résumé
Le système régulièrement dois‑je courts palindromes répétitions (CRISPR) en cluster fonctionne naturellement dans l’immunité adaptative bactérienne, mais a été réorienté avec succès pour le génie du génome chez de nombreux organismes de vie différents. Le plus souvent, le type sauvage CRISPR associés 9 (Cas9) ou endonucléase de Cas12a est utilisé en conjonction avec des sites spécifiques dans le génome, après quoi la rupture d’ADN double brin est réparée par l’intermédiaire de la fin non homologue, rejoindre la voie (NHEJ) ou le (réparation axés sur l’homologie Voie HDR) selon si un modèle de donneur est absent ou présentent respectivement. A ce jour, systèmes CRISPR provenant de différentes espèces bactériennes ont démontré être capable d’effectuer le montage de génome dans les cellules de mammifères. Toutefois, malgré l’apparente simplicité de la technologie, plusieurs paramètres de conception doivent être prises en considération qui laissent souvent perplexes sur la façon de mieux pour réaliser leur génome édition expériences les utilisateurs. Nous décrivons ici un workflow complet de conception expérimentale à l’identification des clones de cellules qui transportent les adaptations ADN désirées, dans le but de faciliter l’exécution réussie du génome édition des expériences dans des lignées cellulaires de mammifères. Nous mettons en évidence les considérations clés pour les utilisateurs de prendre note du, y compris le choix du système CRISPR, la longueur de l’entretoise et la conception d’un modèle de donateurs monocaténaire ODN (ssODN). Nous prévoyons que ce flux de travail vous sera utile pour les études de gènes knockout, modélisation des efforts, la maladie ou la génération de reporter des lignées cellulaires.
Introduction
La capacité à l’ingénieur le génome de tout organisme vivant a beaucoup d’applications biomédicales et biotechnologiques, telles que la correction des pathogènes mutations, construction de modèles cellulaires précis pour les études de la maladie, ou génération d’agricoles cultures avec traits désirables. Depuis le tournant du siècle, plusieurs technologies ont été développées pour le génie du génome dans les cellules de mammifères, y compris les méganucléases1,2,3, zinc finger nucleases4,5, ou transcription activator comme effecteur nucléases (TAPS)6,7,8,9. Cependant, ces technologies antérieures sont difficiles à programmer ou fastidieux à monter, ce qui entrave leur adoption généralisée dans la recherche et l’industrie.
Ces dernières années, le cluster régulièrement ponctuées de courtes répétitions palindromiques (CRISPR) - système associées à CRISPR (Cas) a émergé comme un puissant génome nouvel ingénierie technologie10,11. Initialement un système immunitaire adaptatif chez les bactéries, il a été correctement déployé pour la modification du génome chez les plantes et les animaux, y compris les humains. Des principales raisons pourquoi CRISPR-Cas a gagné en popularité autant en si peu de temps qui est l’élément qui apporte l’endonucléase de Cas clé, tels que Cas9 ou Cas12a (également connu comme Cpf1), à l’emplacement correct dans le génome est simplement un petit morceau de guide unique chimérique RN A (sgRNA), qui est simple de conception et pas cher à synthétiser. Après recrutement vers le site cible, l’enzyme Cas fonctionne comme une paire de ciseaux moléculaires et fend l’ADN lié avec son RuvC, HNH ou Nuc domaines12,13,14. La rupture de brin double (DSB) qui en résulte est réparée par la suite par les cellules via la fin non homologue rejoindre (NHEJ) ou voie de réparation axés sur l’homologie (HDR). En l’absence d’un modèle de la réparation, l’ORD est réparé par la voie NHEJ erreurs, qui peut donner lieu à pseudo-aléatoire d’insertion ou de suppression de nucléotides (indels) sur le site de la coupe, causant potentiellement déphasage des mutations dans les gènes codant pour des protéines. Toutefois, en présence d’un modèle de donateurs qui contient les modifications souhaitées de l’ADN, l’ORD est réparé par la voie du HDR haute fidélité. Types communs de modèles de donateurs comprennent des oligonucléotides monocaténaires (ssODNs) et plasmides. Le premier est généralement utilisé si les changements prévus de l’ADN sont de petite taille (par exemple, modification d’un seul nucléotide), alors que ce dernier est généralement utilisé si l'on veut introduire une séquence relativement longue (par exemple, la séquence codante d’une protéine fluorescente verte ou GFP) dans le locus de la cible.
L’activité endonucléasique de la protéine Cas nécessite la présence d’un motif adjacent de protospacer (PAM) à la cible site15. Le PAM de Cas9 est à l’extrémité 3' de la protospacer, tandis que le PAM de Cas12a (également appelé Cpf1) est à l’extrémité 5' à la place16. Le Cas-guide RNA complexe ne peut pas introduire un ORD si la PAM est absente17. Par conséquent, la PAM place une contrainte sur les génomiques emplacements où une nucléase de Cas particulier est capable de cliver. Heureusement, nucléases Cas de différentes espèces de bactéries présentent généralement des exigences différentes de PAM. Par conséquent, en intégrant différents systèmes CRISPR-Cas dans notre boîte à outils technique, nous pouvons élargir l’éventail des sites qui peuvent être ciblés dans un génome. En outre, une enzyme naturelle de Cas peut être conçue ou évoluée pour reconnaître les alternatives des séquences de PAM, encore élargir la portée de génomiques cibles accessibles aux manipulations18,19,20.
Bien que plusieurs systèmes CRISPR-Cas soient disponibles pour fins d’ingénierie du génome, la plupart des utilisateurs de la technologie sont appuient principalement sur la nucléase Cas9 de Streptococcus pyogenes (SpCas9) pour de multiples raisons. Tout d’abord, il faut un PAM relativement simplement NGG, contrairement à plusieurs autres protéines de Cas qui peuvent s’attacher uniquement en présence de SDS plus complexes. Deuxièmement, il est le premier endonucléase de Cas qui seront déployés avec succès dans les cellules humaines21,22,23,24. En troisième lieu, le SpCas9 est de loin la meilleure enzyme caractérisé à ce jour. Si un chercheur souhaite utiliser la nucléase un autre Cas, il ou elle serait souvent peu claire sur la meilleure façon de concevoir l’expérience et comment bien les autres enzymes seront produira dans différents contextes biologiques par rapport à SpCas9.
Pour fournir la clarté à la performance relative des différents systèmes CRISPR-Cas, nous avons récemment effectué une comparaison systématique des cinq endonucléases de Cas – SpCas9, l’enzyme Cas9 Staphylococcus aureus (SaCas9), l’enzyme Cas9 de Neisseria meningitidis (NmCas9), l’enzyme Cas12a de Acidaminococcus SP. BV3L6 (AsCas12a) et l’enzyme Cas12a de bactérie Lachnospiraceae ND2006 (LbCas12a)25. Pour une comparaison équitable, nous avons évalué les nucléases de Cas différents, en utilisant le même ensemble de sites cibles et autres conditions expérimentales. Les paramètres de conception d’étude aussi délimité pour chaque système de SAE-CRISPR, qui servirait de référence utile pour les utilisateurs de la technologie. Ici, pour mieux permettre aux chercheurs de faire usage de la SAE-CRISPR système, nous fournissons un protocole étape par étape pour le génie de génome optimale avec des enzymes de Cas9 et Cas12a différentes (voir Figure 1). Le protocole inclut non seulement les détails expérimentaux, mais les considérations de conception aussi important pour maximiser la probabilité d’un résultat technique génome réussie dans les cellules de mammifères.

Figure 1 : Une vue d’ensemble du flux de travail pour générer du génome édité des lignées cellulaires humaines. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
Protocole
1. conception de sgRNAs
- Sélectionnez un système CRISPR-Cas approprié.
- Tout d’abord, examinez la région cible pour les séquences de PAM de tous les nucléases Cas9 et Cas12a qui sont sont révélées être fonctionnel dans les cellules de mammifères16,21-32. Cinq enzymes fréquemment utilisés sont donnés dans le tableau 1 ainsi que de leurs respectifs PAMs.
Remarque : outre les endonucléases énumérés au tableau 1, il existe d’autres enzymes Cas moins fréquemment utilisés qui ont été déployées avec succès dans les cellules de mammifères, tels qu’une nucléase Cas9 de Streptococcus thermophilus (St1Cas9) qui reconnaît le PAM NNAGAAW. Si le site cible ne contient-elle pas un PAM connu, on ne serait pas en mesure d’utiliser le système CRISPR-Cas pour le génie du génome. - Deuxièmement, examiner toutes les propriétés connues du locus génomiques de cible ou de gène. Certaines propriétés de prendre en considération incluent les niveaux d’expression de gène ou l’accessibilité de la chromatine et si il y a d’autres étroitement liés aussi bien des séquences.
Remarque : Certaines enzymes conviennent mieux aux contextes biologiques particuliers. Par exemple, pour modifier un locus génomique répétitif ou un gène avec plusieurs autres paralogues étroites, il est recommandé d’utiliser soit AsCas12a (en raison de sa faible tolérance pour les incompatibilités entre le sgRNA et l’ADN cible que SpCas9 et LbCas12a) ou SaCas9 (dû à sa exigence d’entretoises plus longues, qui fournit la plus grande spécificité de ciblage)25.
- Tout d’abord, examinez la région cible pour les séquences de PAM de tous les nucléases Cas9 et Cas12a qui sont sont révélées être fonctionnel dans les cellules de mammifères16,21-32. Cinq enzymes fréquemment utilisés sont donnés dans le tableau 1 ainsi que de leurs respectifs PAMs.
| Endonucléase de cas | PAM | Longueur de la cale d’espacement optimal |
| SpCas9 | NGG | 17-22 nt inclus |
| SaCas9 | NNGRRT | ≥ 21 nt |
| NmCas9 | NNNNGATT | ≥ 19 nt |
| AsCas12a et LbCas12a | TTTV | ≥ 19 nt |
Tableau 1 : certains couramment enzymes Cas avec leurs apparentées PAMs et les longueurs optimales sgRNA. N = aucune nucléotides (A, T, G ou C) ; R = A ou G ; V = A, C ou G.
- Sélectionnez une séquence de l’espaceur approprié. Identifier une séquence aussi unique que possible afin de minimiser le risque d’événements hors-cible clivage, soit en examinant le génome cible avec BLAST33 , soit en utilisant plusieurs outils en ligne librement disponibles, tels que : (a) programme de laboratoire de Feng Zhang34 (http://crispr.mit.edu/) ; (b) CHOPCHOP35 (http://chopchop.cbu.uib.no/) ; (c) E-CRISP36 (http://www.e-crisp.org/E-CRISP/) ; (d) CRISPOR37 (http://crispor.tefor.net/) ; (e) Cas-OFFinder38 (http://www.rgenome.net/cas-offinder/).
Remarque : La durée optimale de l’espacement peut varier de 17 à 25 nucléotides (nt) inclusives, selon les Cas, enzyme est utilisée (voir tableau 1). Pour Cas9, l’entretoise est en amont de la PAM, tandis que pour Cas12a, l’entretoise est en aval de la PAM. en outre, l’efficacité HDR chute rapidement avec l’augmentation de distance du site coupe. Par conséquent, pour montage d’ADN précis, positionner la sgRNA aussi près que possible sur le site de la modification prévue. - Une synthèse des oligonucléotides d’ADN avec les surplombs appropriés pour le plasmide CRISPR qui est utilisé.
- Ajouter un nucléotide G devant l’entretoise si la première position du guide n’est pas un G. Déterminez la séquence inverse et complémentaire de l’entretoise. Ajouter dans les surplombs nécessaires pour des fins de clonage.
NOTE : À titre d’illustration, pour les plasmides utilisé dans notre évaluation étude25, les oligonucléotides synthétisés sont indiquées dans le tableau 2. Utilisez l’exemple donné dans la Figure 2 comme un guide, si nécessaire. Plusieurs plasmides CRISPR sont disponibles sur le marché (p. ex., Addgene). Certains des plus populaires plasmides sont donnés dans la Table des matières.
- Ajouter un nucléotide G devant l’entretoise si la première position du guide n’est pas un G. Déterminez la séquence inverse et complémentaire de l’entretoise. Ajouter dans les surplombs nécessaires pour des fins de clonage.
| Plasmide CRISPR | Séquence |
| pSpCas9 et pSaCas9 | Sens : 5' - NNNNNNNNNNNNNNNNNNNNN CACC (G) - 3' Antisens : 3' - (C) NNNNNNNNNNNNNNNNNNNNNCAAA - 5' |
| pNmCas9 | Sens : 5' - NNNNNNNNNNNNNNNNNNNNN CACC (G) - 3' Antisens : 3' - (C) NNNNNNNNNNNNNNNNNNNNNCAAC - 5' |
| pAsCas12a et pLbCas12a | Sens : 5' - AGATNNNNNNNNNNNNNNNNNNNNN - 3' Antisens : 3' - NNNNNNNNNNNNNNNNNNNNNAAAA - 5' |
Tableau 2 : oligonucléotides requis pour le clonage sgRNA séquences dans des plasmides CRISPR utilisés dans une évaluation récente étudient25. Les surplombs sont indiqués en italique.

Figure 2 : Un exemple illustrant comment sélectionner des sites cibles et concevoir des oligonucléotides de clonage dans des plasmides CRISPR. Le locus génomiques cible ici est 45 d’exon du gène humain CACNA1D. Les PAMs pour SpCas9 et SaCas9 sont respectivement NGG et NNGRRT et sont surlignées en rouge, tandis que la PAM pour AsCas12a et LbCas12a est TTTN et est surligné en vert. La barre horizontale rouge indique le protospacer pour SpCas9 et SaCas9, tandis que la barre verte horizontale indique le protospacer pour les deux enzymes Cas12a. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
2. clonage d’oligonucléotides dans un vecteur colonne vertébrale
- Phosphoryler et recuire des oligonucléotides sens et antisens.
- Si les oligonucléotides sont lyophilisés, les remettre en suspension à une concentration de 100 µM en acide de tris-EDTA (EDTA) (TE mettre en mémoire tampon, voir Table des matières) ou FD2O.
- Préparer un mélange de réaction de 10µl contenant 1 µL d’oligonucléotides sens, 1 µL d’oligonucléotides antisens, 1 µL de tampon de ligase T4 ADN (10 x), 1 µL de T4 polynucléotide kinase (PNK) et 6 µL de ddH2O. Mix bien en pipettant également et placer le mélange réactionnel dans un cy thermique Cler en utilisant les paramètres suivants : 37 ° C pendant 30 min, 95 ° C pendant 5 min et rampe jusqu'à 25 ° C à 6 ° C/min.
- Diluer la réaction mix 1/100 FD2O (p. ex., mélange de réaction de 2 µL + 198 µL ddH2O).

Figure 3 : Un exemple d’un plasmide CRISPR. (un) A carte indiquant les différentes caractéristiques importantes du plasmide. Ici, le promoteur de l’EF-1 a conduit l’expression de Cas9, tandis que le promoteur U6 entraîne l’expression de la sgRNA. Amp(R) indique un gène de résistance à l’ampicilline dans le plasmide. (b) la séquence de la « site clonage BbsI-BplI » dans le plasmide. La séquence de reconnaissance de BbsI est GAAGAC et est indiquée en rouge, tandis que la séquence de reconnaissance du BplI est GAG-N5- CTC et est indiquée en vert. (c) des amorces qui peut être utilisé pour la PCR sur colonie pour vérifier si la séquence de sgRNA a été clonée avec succès dans le plasmide. L’apprêt hU6_forward est indiqué par une flèche mauve sur la carte plasmidique, tandis que l’amorce universelle de M13R(-20) est indiquée par une flèche rose sur la carte plasmidique. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
- Digérer le plasmide CRISPR avec une enzyme de restriction appropriée.
NOTE : Clonage de sgRNAs généralement dépendent de Golden Gate assembly avec type IIs enzymes de restriction. Différentes enzymes peuvent être utilisés pour différents plasmides CRISPR. Pour pSpCas9, utilisez BbsI ou BplI (voir Figure 3). Pour pSaCas9, pNmCas9, pAsCas12a et pLbCas12a, utilisent BsmBI.- Préparer un mélange de réaction de 20 µL contenant 1 µg de vecteur plasmidique circulaire, 2 µL de tampon (10 x), de 1 µL d’enzyme de restriction (par exemple, BbsI, BplI ou BsmBI) et FD2O pour un volume final de 20 µL. Incubez la réaction à 37 ° C pendant 2,5 heures.
- Ajouter 1 µL de la phosphatase alcaline (SAP) de crevette à la réaction et incuber à 37 ° C pendant 30 min.
- Étancher la réaction en ajoutant 5 µL de 6 x chargement ADN teindre (voir Table des matières), mélange bien et résoudre la réaction sur un gel d’agarose 0,8 % avec 1 x tampon tris-acétate-EDTA (TAE). Ensuite, la bande de fréquences correcte de l’accise et procéder au gel purifier le vecteur linéarisé.
- Ligaturer les oligonucléotides recuits dans le plasmide CRISPR digéré.
- Préparer un mélange de réaction 10 µL : 50 ng de vecteur linéarisé, 1 µL d’oligonucléotides recuits dilués, 1 µL de tampon de ligase d’ADN T4 (10 x), de 1 µL de T4 DNA ligase et FD2O pour un volume final de 10 µL (voir Table des matières). Incubez la réaction à 16 ° C durant la nuit ou à la température ambiante pendant 2 h.
Remarque : Pour accélérer le processus de la ligature, utilisez concentré T4 DNA ligase et incuber le mélange de réaction à température ambiante pendant 15 min (voir Table des matières).
- Préparer un mélange de réaction 10 µL : 50 ng de vecteur linéarisé, 1 µL d’oligonucléotides recuits dilués, 1 µL de tampon de ligase d’ADN T4 (10 x), de 1 µL de T4 DNA ligase et FD2O pour un volume final de 10 µL (voir Table des matières). Incubez la réaction à 16 ° C durant la nuit ou à la température ambiante pendant 2 h.
- Transformer chimiquement compétent e. coli de produits ligaturés les cellules (voir supplémentaire 1 fichier). Étaler les cellules bactériennes transformées sur une gélose LB avec de l’ampicilline 100 µg/mL.
- Effectuer colonie chaîne par polymérase (PCR) pour identifier les bactéries avec insert.
- Préparer deux séries de tubes de la bande PCR stériles. La valeur 1, ajouter 4,7 µL de ddH2O, tandis que dans l’ensemble 2, ajouter 50 µL de bouillon LB avec l’antibiotique approprié (par exemple, 100 µg/mL ampicilline).
- Avec un embout de la pipette stérile, prélever une colonie au large de la plaque, il saisissez brièvement dans un tube à la valeur 1 et laisser le pourboire dans un tube Set 2. Répétez pour quelques colonies, en veillant à utiliser des tubes PCR différents chaque fois.
Remarque : En général, quatre colonies de dépistage est suffisante. Toutefois, cela peut varier selon l’efficacité de clonage. - Ajouter les réactifs suivants dans chaque tube Set 1 (pour un 10 µL PCR) : 5 µL de 2 x PCR mélange principal (avec colorant de chargement, voir la Table des matières), 0,15 µL d’oligonucléotide antisens ou de sens (100 µM), 0,15 µL d’apprêt (100 µM) ciblant le plasmide CRISPR en aval soit un flux de la cassette de sgRNA respectivement (voir Figure 3).
Remarque : Le produit PCR devrait idéalement donner une taille de produit d’environ 150 bp ou plus grands, positives afin que les bandes ne se trompent pas comme dimères d’amorce. - Exécuter les PCR dans un thermocycleur en utilisant les paramètres suivants : 95 ° C pendant 3 min, 95 ° C pendant 30 s (étape 2), 60 ° C pendant 30 s (étape 3), 72 ° C pendant 30 s (étape 4), répétez les étapes 2\u20124 pour un autre 34 cycles, 72 ° C pendant 5 min et de tenir à 4 ° C.
Remarque : La température de recuit de 60 ° C devrez peut-être être optimisé pour les amorces. Le temps de l’élongation de 30 s peut également varier selon la taille attendue d’amplicon PCR et l’ADN polymérase utilisée. - Résoudre les réactions sur un gel d’agarose à 1 % à l’aide de 1 x TAE tampon.
- Ensemencer une colonie qui donne une bande positive pour l’ACP en transférant 50 µL de sa culture de tube Set 2 correspondant dans un plus grand tube conique contenant 5 mL LB avec un antibiotique approprié. Laissez la culture se développer pendant la nuit dans un 37 ° C incubateur-agitateur.
- Isoler les plasmides la culture durant la nuit en faisant un miniprep kit (voir Table des matières) et des séquences à l’aide de l’amorce PCR de colonie qui n’est pas le sens ou antisens oligonucleotide (hU6_forward ou M13R(-20) à la Figure 3).
Remarque : Si nécessaire, effectuez un maxiprep du plasmide CRISPR séquence-vérifié pour obtenir une plus grande quantité pour des expériences en aval.
3. conception et synthèse des modèles de réparation
Remarque : Pour la mécanique de précision de génome, un modèle précisant les modifications souhaitées d’ADN doit être fournie ainsi que les réactifs CRISPR. Des petites modifications de l’ADN tels que l’altération d’un seul nucléotide, ssODN modèles de donateurs sont plus approprié (voir section 3.1). Pour les plus grandes modifications ADN telles que l’insertion d’un GFP balise 5' ou 3' d’un gène codant pour des protéines particulière, plasmide donneur modèles sont plus approprié (voir la section 3.2).
- Concevoir et synthétiser un modèle de donneur de ssODN (voir Figure 4).
- Déterminer que le bon chapelet dont le modèle devrait suivre la séquence.
Remarque : Cas12a montre une préférence pour les ssODNs de la séquence du brin non ciblés, tandis que Cas9 montre une préférence pour les ssODNs du brin cible des séquences plutôt25 (voir Figure 4a). - S’assurer que la séquence réparée n’est pas encore targetable par la nucléase de Cas sélectionnée. Par exemple, muter la PAM de telle sorte qu’il n’y a aucun changement d’acides aminés ou d’éliminer la PAM depuis le modèle donneur si il n’a aucune conséquence fonctionnelle. Utilisez l’exemple donné dans la Figure 4b comme guide, si nécessaire.
- Décider si un modèle symétrique ou asymétrique des donateurs est souhaité. Pour les donateurs symétriques, assurez-vous que chaque bras d’homologie flanquant le site de modification de l’ADN est au moins 17 nt long25. Pour les modèles de donateurs asymétrique, utilisez plus long bras 5′ des changements ADN désirées (voir Figure 4a). Ce qui est important, s’assurer que le bras court est autour de 37 nt de longueur, tandis que l’autre branche de l’homologie est environ 77 nt en longueur25,,39.
- Synthétiser le modèle conçu comme un morceau d’ADN simple brin.
Remarque : SsODNs asymétriques peut, mais ne sont pas toujours, présentent une plus grande efficacité HDR que ssODNs symétrique. En général, un donateur asymétrique généralement effectue au moins ainsi qu’un donneur symétrique, lorsque conçu correctement. Toutefois, le modèle asymétrique coûte beaucoup plus parce qu’elle est plus longue et nécessite donc la purification (PAGE) l’électrophorèse sur gel de polyacrylamide ou une procédure spéciale de synthèse. Débouchures de routine gène habituellement s’appuient sur la voie de réparation NHEJ et ne nécessitent pas un modèle de réparation. Toutefois, si l’efficacité de l’élimination directe est faible, concevoir un modèle de donneur de ssODN qui contient une mutation de changement de phase et est au moins 120 nt en longueur25,40.
- Déterminer que le bon chapelet dont le modèle devrait suivre la séquence.

Figure 4 : Conception des modèles de donneur ssODN. (a) schéma illustrant les diverses conceptions possibles. Les rectangles rouges horizontales indiquent le brin non ciblés (NT), tandis que les rectangles bleus indiquent le brin de cible (T). En outre, les petits rectangles verts indiquent les modifications de l’ADN désirées (tels que les changements de nucléotide). Lorsqu’on utilise un ssODN symétrique, la longueur minimale de chaque bras de l’homologie devrait être au moins 17 nt (mais peut être plus long). Pour ssODNs asymétrique, le ssODN de T 37/77 semble être optimale pour HDR induite par le SpCas9, alors que le ssODN NT de 77/37 semble être optimale pour HDR induite par le Cas12a. L = bras gauche homologie ; R = le bras droit d’homologie. (b) un exemple pour montrer comment créer des modèles de ssODN. Ici, le locus génomiques de la cible est exon 45 du gène humain CACNA1D. La PAM pour Cas9 est rose et soulignés, tandis que la PAM pour Cas12a est brune et soulignés. Vise à créer une mutation faux-sens (surligné en vert) en convertissant AGU (encodage sérine) à AGG (encodage arginine). Pour éviter de re-ciblage par Cas12a, le PAM TTTC est mutée au CTA. Notez qu’il n’y a aucun changement dans les acides aminés (UAU et UAC les deux codent pour la tyrosine). Pour mieux prévenir re-ciblage de Cas9, un codon AGU est remplacé par un codon de l’UCC (gras), les deux qui codent pour la sérine. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
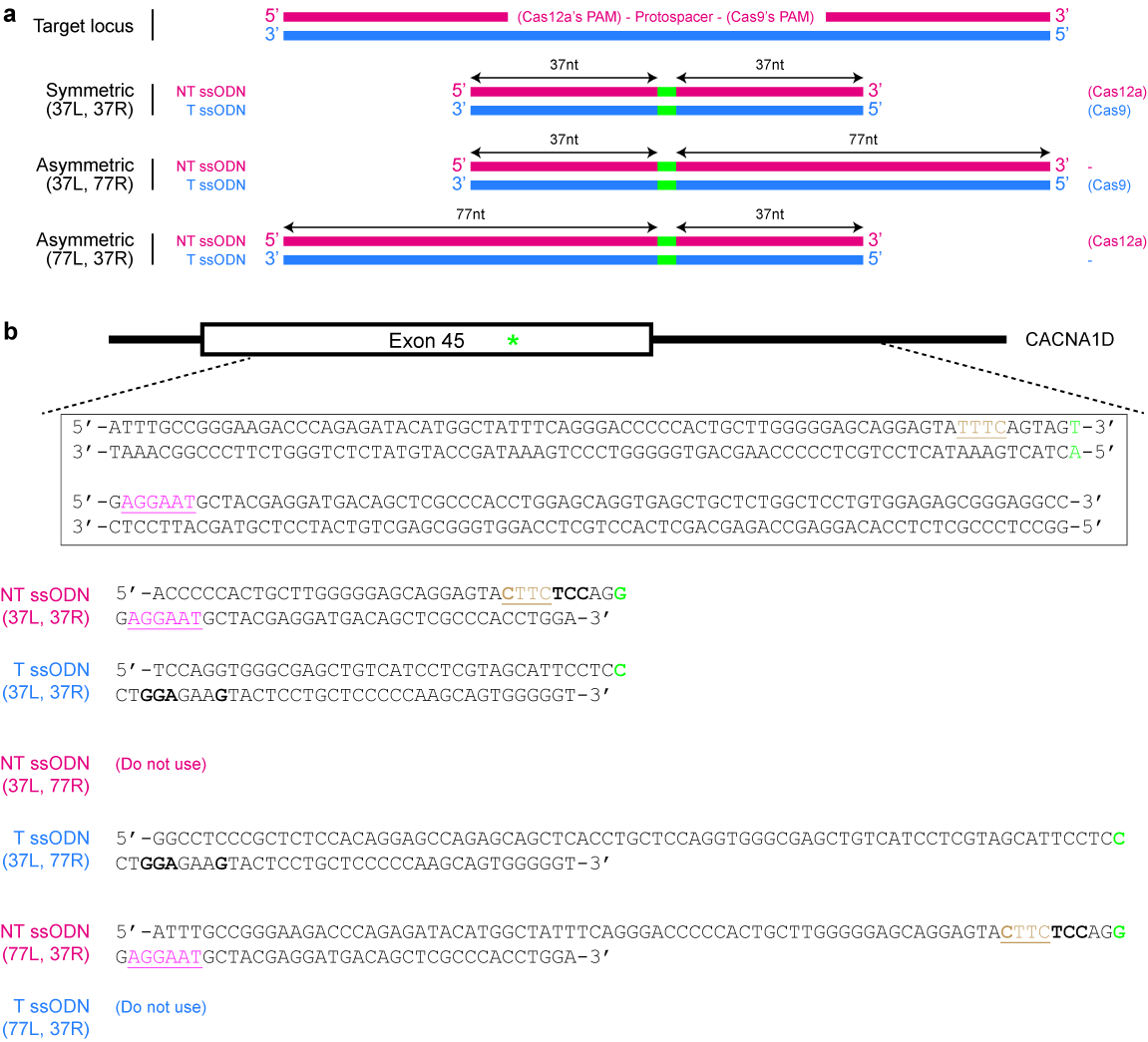{kind=link}
- Concevoir et cloner un modèle de donneur de plasmide approprié. Par exemple, il peut contenir une séquence GFP flanquée de longs bras qui sont homologues au locus génomiques de la cible (voir Figure 5).
- S’assurer que la séquence modifiée n’est pas encore targetable par la nucléase de Cas sélectionnée. Par exemple, le protospacer peut être divisée par la balise (GFP) insérée. Par ailleurs, la PAM peut être mutée ou retirée du modèle donateur d’une manière qui n’affecte pas la fonction du gène.
- Amplifier les bras d’homologie de l’ADN génomique à l’aide de la PCR. La longueur de chaque bras de l’homologie est typiquement de 1000 à 1500 bp.
Remarque : Afin de faciliter le clonage, veillez à ce que l’amorce vers l’avant pour le bras gauche d’homologie et l’amorce inverse pour l’homologie droite bras chacun a au moins 20 nt qui se chevauchent de séquence avec une épine dorsale vector sélectionnés. En outre, s’assurer que l’apprêt inverse pour le bras gauche d’homologie et l’amorce vers l’avant pour le bras droit d’homologie ont quelques séquences qui se chevauchent avec la balise de l’épitope ainsi. - Cloner les deux branches de l’homologie et la balise (GFP) dans la colonne vertébrale de vecteur à l’aide de l’Assemblée de Gibson41 (voir la Table des matières). Vérifier le plasmide par Sanger sequencing utilisant avant et marche arrière amorces qui sont en amont et en aval du modèle donneur respectivement.
NOTE : Sanger séquençage est largement et à un prix avantageux disponible comme un service commercial. Envoyer une partie aliquote du plasmide avec les amorces de séquençage à un fournisseur de services. - Linéariser le modèle donateur avec une enzyme de restriction qui coupe le plasmide seulement une fois en amont du bras gauche une homologie ou en aval du bras droit d’homologie.
NOTE : Récemment, un double-coupe des donateurs, qui sont flanqué par les séquences sgRNA-PAM et sont libéré du plasmide après clivage par la nucléase de Cas correspondante, s’est avéré augmenter l’efficacité HDR42. Lorsque les séquences sgRNA-PAM sont insérés en amont et en aval de l’homologie de gauche et de droite d’armes respectivement (par exemple en assemblée de Gibson), la longueur de bras d’homologie peut être ramenée à 300 bp et il n’y a aucun besoin de linéariser le plasmide.

Figure 5 : Conception et clonage d’un modèle de donneur de plasmide. (un) le but dans cet exemple précis est de fusionner P2A-GFP à l’extrémité C-terminale de la protéine de l’ACCB. Le rectangle horizontal bleu indique le bras gauche d’homologie, tandis que le rectangle horizontal rouge indique le bras droit d’homologie. Lettres majuscules indiquent les séquences codant pour des protéines, tandis que les lettres minuscules indiquent des séquences non codantes. Les PAMs pour SpCas9 et Cas12a sont en italiques et soulignées. (b), un donneur de plasmide modèle qui peut être utilisé de façon endogène tag P2A-GFP à l’extrémité C-terminale de l’ACCB. Les séquences d’amorces fourni permet de cloner le plasmide par Assemblée de Gibson. Les conditions de la PCR sont comme suit : 98 ° C pendant 3 min, 98 ° C pendant 30 s (étape 2), 63 ° C pendant 30 s (étape 3), 72 ° C pendant 1 min (étape 4), répétez les étapes 2\u20124 pour un autre 34 cycles, 72 ° C pendant 3 min et maintenez à 4 ° C. Lettres noires correspondent à des séquences de vecteur, lettres bleues correspondent au bras gauche une homologie, lettres vertes correspondent aux P2A-GFP et lettres rouges correspondent au bras droit d’homologie. Notez qu’une fois la séquence codant pour P2A-GFP est réussi à intégrer dans le locus cible, re-ciblage de SpCas9 sera impossible, depuis seulement 9 nt de sa protospacer (GTGCACCAG) est laissés intacts. En outre, afin d’éviter de re-ciblage de Cas12a, trois basepairs immédiatement en aval du STOP codon (en gras) sont supprimés de la séquence de plasmide. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
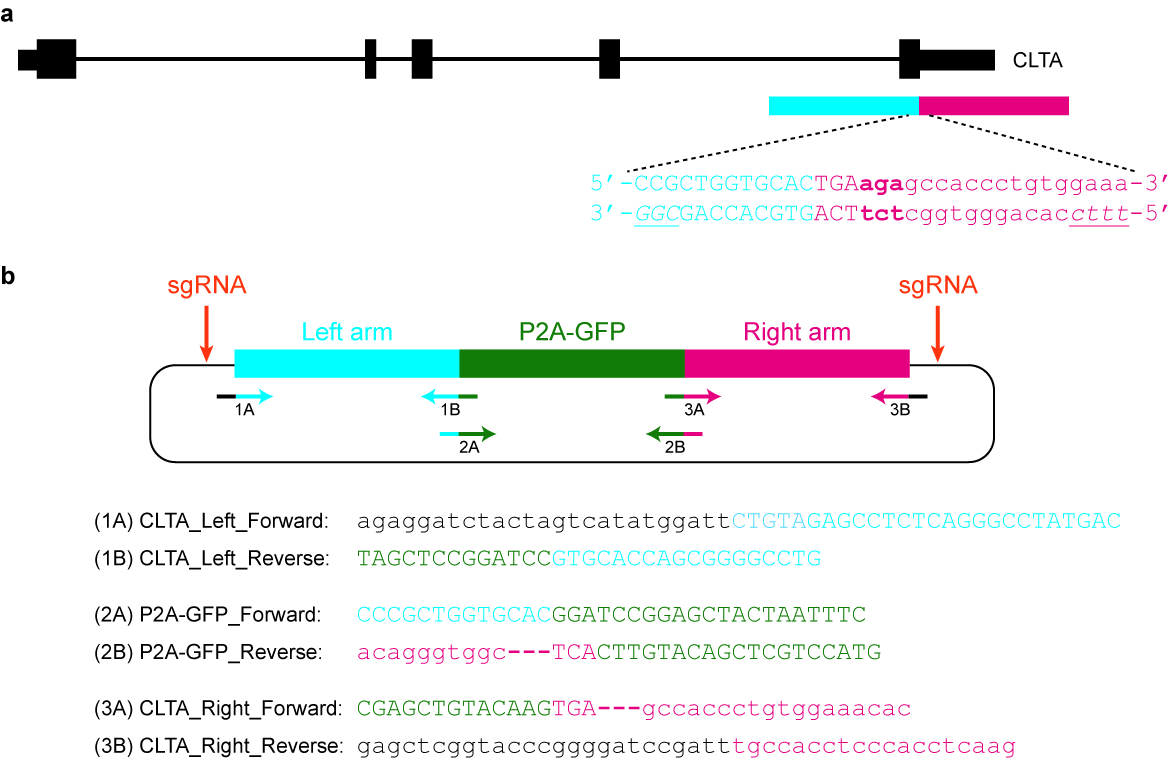{kind=link}
4. la transfection de cellules
NOTE : Les parties restantes du protocole sont rédigés avec cellules HEK293T à l’esprit. Le milieu de culture utilisé est composé de Dulbecco Eagle modifié (DMEM) additionné de 4,5 g/L de glucose 10 % sérum fœtal (SVF), 2 mM de L-glutamine et 0,1 % pénicilline/streptomycine. Différentes étapes du protocole devrez peut-être être modifié selon la ligne de cellule réelle utilisée. Tous les travaux de culture cellulaire se fait dans une enceinte de sécurité biologique de classe II pour assurer un environnement de travail stérile.
- Semences 1,8 x 105 cellules dans une plaque de culture de tissus de 24 puits un jour avant la transfection.
- Dissocier les cellules en aspirant les médias et en ajoutant ensuite 150 µL de 0,25 % trypsine-EDTA / puits. Incuber les cellules à 37 ° C pendant 2 min.
- Neutraliser la trypsine en ajoutant 150 µL (ou 1 x volume) des milieux de culture cellulaire. Transférer la suspension de cellules dans un tube conique. Tournez en bas des cellules dans une centrifugeuse de paillasse supérieure à 1000 x g pendant 5 min.
- Aspirer le surnageant et remettre en suspension avec 5 mL de milieux de culture cellulaire. Dans un tube à centrifuger distincte, aliquote de 10 µL de solution de bleu de trypan de 0,4 %. Puis, ajouter dans les 10 µL resuspendues cellules de l’étape 4.1.2 et bien mélanger.
- Distribuer 10 µL du mélange (cellules + bleu trypan) dans un hémocytomètre. Passez à compter les cellules manuellement ou en utilisant un compteur de cellules automatisées.
- Cellules de5 graines 1,8 x 10 dans un puits d’une plaque de culture de tissus de 24 puits.
- Préparer un mélange de transfection contenant soit 500 ng de plasmide CRISPR (pour NHEJ-mediated édition) ou 300 ng de plasmide CRISPR et 300 ng de modèle de donateurs (pour HDR-mediated modification), conformément aux instructions fournies avec le réactif de transfection (voir Table des matières). Incuber à température ambiante pendant toute la durée de temps recommandé (généralement autour de 10\u201220 min).
- Ajouter le mélange de transfection pour les cellules de façon goutte à goutte, et agiter doucement la plaque après.
- Incuber à 37 ° C dans un 5 % humidifié incubateur à air CO2 pendant 24 h (pour les expériences NHEJ) ou 72 h (pour les expériences axées sur le HDR).
5. fluorescence activé la cellule triant (FACS) des cellules transfectées
- Dissocier les cellules en aspirant les médias et en ajoutant ensuite 150 µL de 0,25 % de trypsine-EDTA / puits. Incuber les cellules à 37 ° C pendant 2 min.
- Neutraliser la trypsine en ajoutant 150 µL (ou 1 x volume) des milieux de culture cellulaire. Transférer la suspension de cellules dans un tube à centrifuger. Tournez en bas les cellules dans un microcentrifuge à 235 x g pendant 5 min.
- Aspirer le surnageant et remettre en suspension les cellules avec 2 % sérum fœtal (SVF) dans une solution saline tamponnée au phosphate (PBS). Filtrer les cellules grâce à un maillage de 30 µm ou filtre dans un tube de 5 mL FACS de cellules.
- Préparer un autre tube à centrifuger avec environ 100 µL milieux de culture ou 2 % FBS dans du PBS pour la collection de cellules.
- Sur le cytomètre en flux, la porte des cellules avec des cellules non transfectées comme témoin négatif. Trier et collecter les cellules transfectées, selon laquelle fluorescence marqueur est présent sur le plasmide CRISPR utilisé. Par exemple, si le plasmide contient un gène de mCherry, trier des cellules positives DP.
NOTE : Les plasmides différents CRISPR auront différents marqueurs sélectionnables. L’ensemble des plasmides (pSpCas9, pSaCas9, pNmCas9, pAsCpf1 et pLbCpf1) utilisés dans cette étude d’évaluation portent soit la protéine fluorescente orange (OFP), soit le gène mCherry.
6. l’expansion des clones individuels
- Centrifuger les cellules triées dans une centrifugeuse de paillasse supérieure à la vitesse maximale (18 000 x g) pendant 5 min. aspirer le surnageant et resuspendre le culot avec les milieux de culture 300 µL. 200 cellules µL dans une plaque de culture de tissus de 24 puits de graines et les laisser récupérer pendant quelques jours dans un incubateur à 37 ° C. Gardez le reste des 100 cellules µL au chapitre 7.
- Une fois que les cellules commencent à devenir confluentes, leur passage selon les étapes 4.1.1\u20124.1.3. Les cellules faiblement dans un plat de vitroplants de 100 mm à laisser suffisamment d’espace pour les colonies individuelles à cultiver des semences. Incuber à 37 ° C dans un 5 % humidifié incubateur à air CO2 .
NOTE : Essayez diverses dilutions. Cellules isolées ont besoin de suffisamment d’espace pour grandir en tant que colonies individuelles. Cependant, ils également ne peut pas être trop clairsemées, comme certaines lignées cellulaires ne se développent pas bien quand le nombre de cellules est trop peu nombreux. - Une fois que les colonies commencent à se former, cueillez-les au microscope (avec un grossissement de 4 x) et placer chaque clone dans un puits individuel d’une plaque de 24 puits contenant des milieux de culture cellulaire. Incuber à 37 ° C dans un 5 % humidifié incubateur à air CO2 jusqu'à ce que les cellules deviennent confluentes.
NOTE : Une alternative à des dilutions sériées et cueillette de colonie consiste à utiliser la cytométrie en flux pour trier des cellules individuelles dans une plaque de 96 puits. Toutefois, cela peut ne pas fonctionner pour certaines lignées de cellules qui ne se développent pas bien lorsque qu’une cellule est présente.
7. évaluation de l’efficacité d’édition
- Extraire l’ADN génomique par centrifugation les cellules restantes de 100 µL triés (de l’étape 6.1) dans une centrifugeuse de paillasse supérieure à la vitesse maximale (18 000 x g) pendant 5 min. aspirer le surnageant et continuer à isoler l’ADN génomique à l’aide d’un kit d’extraction (voir Table de Matériaux).
- Effectuer l’endonucléase T7 j’ai (T7EI) dosage de clivage (voir Figure 6).
- Mettre en place un 50µl PCR contenant 10 µL de tampon de réaction PCR (x 5), 1 µL du mélange dNTP (10 mM), 2,5 µL d’apprêt avant défini par l’utilisateur (10 µM), 2,5 µL d’apprêt inverse définie par l’utilisateur (10 µM), 0,5 µL de l’ADN polymérase, 2\u20125 µL de matrice d’ADN génomique (en fonction de combien de cellules ont été triées), puis haut jusqu'à 50 µL avec FD2O (voir Table des matières).
Remarque : Les amorces sont conçues pour flanquer le locus génomiques cible et au rendement qu'un produit PCR d’autour de 400\u2012700 BP. habituellement une amorce est placé plus près vers le site coupé de l’enzyme de Cas que l’autre amorce, afin que le résultat du test T7EI est deux bandes distinctes sur un ag s’est posée de gel (voir Figure 6). - Exécutez le PCR dans un thermocycleur avec les paramètres suivants : 98 ° C pendant 3 min, 98 ° C pendant 30 s (étape 2), 63 ° C pendant 30 s (étape 3), 72 ° C pendant 30 s (étape 4), répétez les étapes 2\u20124 pour un autre 34 cycles, 72 ° C pendant 2 min et de tenir à 4 ° C.
- Résoudre la réaction sur un gel d’agarose à 2 % à l’aide de 1 x TAE tampon.
- Le produit PCR du gel avec un scalpel propre, forte de l’accise et purifier l’ADN en utilisant un gel extraction kit, conformément aux instructions du fabricant. Mesurer la concentration du produit PCR à l’aide d’un spectrophotomètre à une longueur d’onde d’absorbance de nm 260 (voir Table des matières).
- Préparer un mélange d’essai contenant 200 ng d’ADN, 2 µL de réaction T7EI mettre en mémoire tampon (10 x) et surmonté jusqu'à 19 µL avec FD2O (voir Table des matières).
- Re-recuire le produit PCR dans un thermocycleur en utilisant les paramètres suivants : 95 ° C pendant 5 min, rampe jusqu'à 25 ° C à 6 ° C/min, puis maintenez à 4 ° C.
- Ajouter 5 U T7EI du produit de PCR re-recuit, mélanger bien de pipetage et incuber à 37 ° C pendant 50 min.
- Résoudre l’ADN digéré T7EI sur un gel d’agarose de 2,5 % à l’aide de 1 x TAE tampon.
- Le gel d’image, quantifier les intensités de bande à l’aide de ImageJ et calculer le taux de formation d’indel selon la formule suivante :

où a représente l’intensité du produit PCR intact et b et c correspondent à l’intensité de la produits de clivage43.- Afin de quantifier l’intensité d’une bande de ImageJ, tout d’abord dessiner une boîte rectangulaire autour de la bande, comme à proximité de sa frontière que possible. Ensuite, cliquez sur analyser , puis Définissez les mesures. Vérifiez que les options de la zone grise valeur moyenneet densité intégrée sont cochées. Fermez la fenêtre de paramètres en cliquant sur OK. En troisième lieu, cliquez sur analyser , puis sur mesure. La valeur moyenne ou RawIntDen est utilisé comme l’intensité de la bande.
- Mettre en place un 50µl PCR contenant 10 µL de tampon de réaction PCR (x 5), 1 µL du mélange dNTP (10 mM), 2,5 µL d’apprêt avant défini par l’utilisateur (10 µM), 2,5 µL d’apprêt inverse définie par l’utilisateur (10 µM), 0,5 µL de l’ADN polymérase, 2\u20125 µL de matrice d’ADN génomique (en fonction de combien de cellules ont été triées), puis haut jusqu'à 50 µL avec FD2O (voir Table des matières).

Figure 6 : Vérification des cellules pour génome réussi édition résultats. (un) A schéma illustrant deux couramment utilisé des essais, à savoir le clivage de l’incompatibilité doser avec l’endonucléase de T7 j’enzyme (T7EI) et séquençage de la génération suivant (NGS) ou séquençage ciblé amplicon. Les rectangles bleus horizontales indiquent que l’ADN et les cercles jaunes indiquent modifications induites par le système CRISPR-Cas. Amorces pour le dosage de la T7E1 sont signalées en vert, tandis que les amorces pour générer des amplicons pour NGS sont notées en rouge. (b), à la conception d’amorce de séquences pour le dosage de clivage T7EI et end. Ici, le locus génomiques de la cible est exon 45 du gène humain CACNA1D. Le site de la modification envisagée est indiqué par un astérisque. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
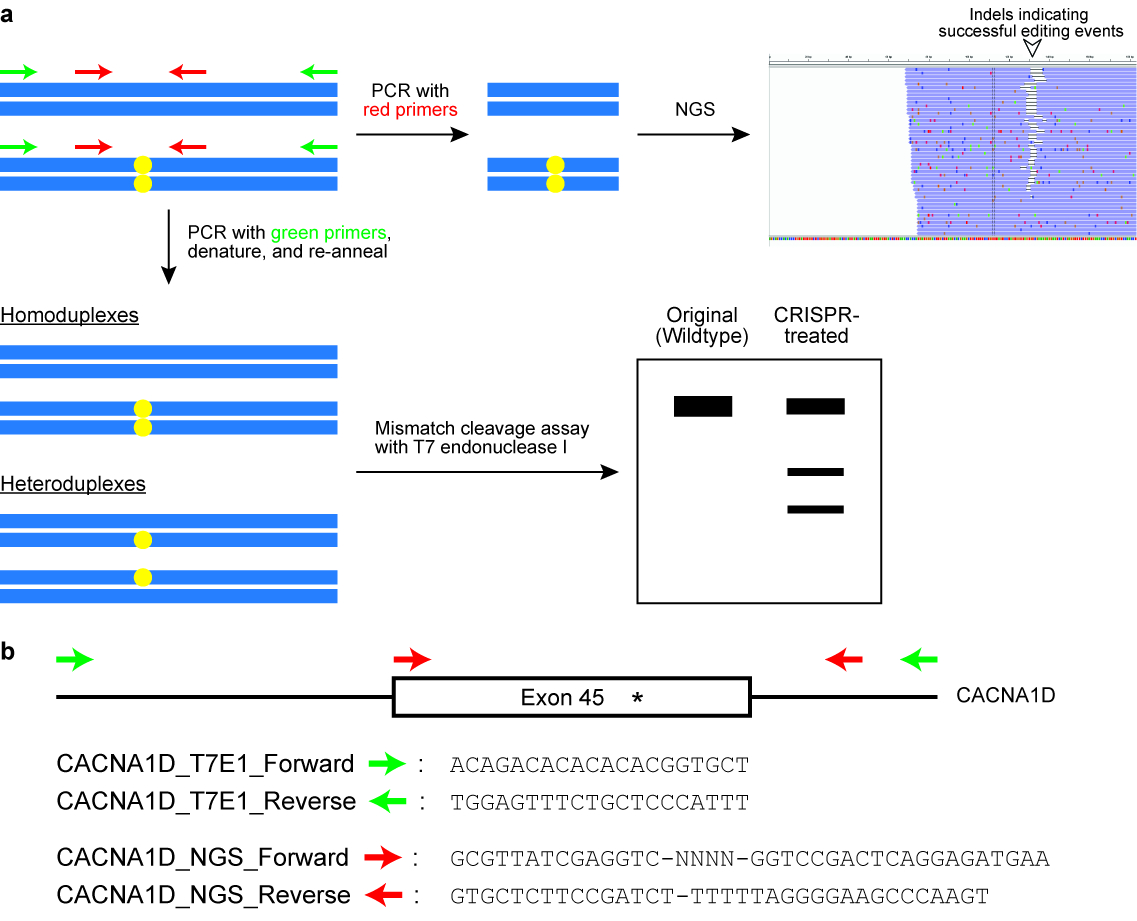{kind=link}
- Effectuer le séquençage de l’amplicon ciblés (voir Figure 6).
- Concevoir des amorces PCR pour amplifier le locus génomiques de la cible. Positionnez l’un des amorces d’être inférieur à 100 bp mais plus de 20 bp de la protospacer.
Remarque : En général, la taille totale du produit PCR est conçue pour être autour 150\u2012300 bp (voir Figure 6). - Ajoutez des séquences supplémentaires pour les amorces comme suit : (a) 5' \u2012GCGTTATCGAGGTC - NNNN-[Forward Primer] – 3' ; (b) 5' - GTGCTCTTCCGATCT-[Reverse Primer] – 3 '.
- Mettre en place un mélange de réaction PCR µL 50 contenant 10 µL de tampon de réaction PCR (x 5), 1 µL de dNTP (10 mM), 5 µL de primer une (10 µM), 5 µL d’apprêt b (10 µM), 0,5 polymérase ADN µL, matrice d’ADN génomique µL (en fonction de combien de cellules ont été triées) 2\u20125 , puis albums jusqu'à 50 µL avec FD20.
- Exécutez le PCR dans un thermocycleur avec les paramètres suivants : 98 ° C pendant 3 min, 98 ° C pendant 30 s (étape 2), 63 ° C pendant 30 s (étape 3), 72 ° C pendant 15 s (étape 4), répétez les étapes 2\u20124 pour un autre 34 cycles, 72 ° C pendant 2 min et de tenir à 4 ° C.
- Résoudre la réaction sur gel d’agarose à 2 % et de purifier le produit de la PCR en utilisant un gel extraction kit conformément aux instructions du fabricant. Quantifier l’ADN à l’aide d’un spectrophotomètre à une longueur d’onde d’absorbance de nm 260 (voir la Table des matières).
- Synthétiser ce qui suit ronde 2 amorces PCR : (c) 5' \u2012 AATGATACGGCGACCACCGAGATCTACACCCTACACGAGCGTTATCGAGGTC – 3'; (d) 5' \u2012CAAGCAGAAGACGGCATACGAGAT-[barcode] - GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3'
- Mettre en place une réaction de PCR de 20 µL mix contenant 4 µL de tampon de réaction PCR (x 5), 0,4 µL de dNTP (10 mM), 2 µL de primer c (10 µM), 2 µL de primer d (10 µM), 0.2 µL de l’ADN polymérase, 2 µL de matrice d’ADN (de l’étape 7.3.5 dilué au 1/100) et 9,4 µL de ddH2O.
Remarque : Le facteur de dilution pour la matrice d’ADN peut varier selon sa concentration initiale. Si la concentration est environ 20\u201240 ng/µL, utiliser un facteur de dilution au 1/100. En outre, choisir un code à barres différente pour chaque échantillon de laboratoire, si la même amorce un et b apprêt sont utilisés à l’étape 7.3.3. - Exécutez le PCR dans un thermocycleur avec les paramètres suivants : 98 ° C pendant 3 min, 98 ° C pendant 30 s (étape 2), 65 ° C pendant 30 s (étape 3), 72 ° C pendant 30 s (étape 4), répétez les étapes 2\u20124 pour un autre 14 cycles, 72 ° C pendant 2 min et 4 ° C tenir.
- Résoudre 5 µL de chaque réaction sur un gel d’agarose à 2 % pour déterminer le succès de la moissonneuse-batteuse PCRs. tous les échantillons de l’ensemble (en supposant qu’un code-barres différent a été utilisé pour chaque échantillon) et nettoyer l’ADN regroupée à l’aide d’un kit de purification de PCR selon du fabricant instructions. Si certains des RFT présentent plus d’une bande (indiquant la présence de produits non spécifiques), effectuer une étape d’extraction de gel supplémentaire.
- La bibliothèque sur un instrument de séquençage à haut débit de séquence (voir Table des matières) conformément aux instructions du fabricant à produire des lectures de bp 151 paires. L’amorce de séquençage de lecture 1 est conçu sur mesure et doit être fourni séparément. Sa séquence est la suivante : Read1_seq : 5' – CCACCGAGATCTACACCCTACACGAGCGTTATCGAGGTC – 3'. L’amorce de séquençage de lecture 2 amorce de séquençage index standard et sont sont fournis dans la cartouche de réactif.
Remarque : Le dosage de T7EI et le séquençage de l’amplicon ciblées sont utilisées pour vérifier l’efficacité de la modification du génome. Cependant, d’autres expériences peuvent être réalisés pour évaluer l’efficacité de l’édition, selon le type de modifications de l’ADN introduit. Par exemple, si un site de restriction est créé sur le site cible, un test de polymorphisme de longueur de fragment restriction est possible. Il est similaire à l’essai de T7EI sauf qu’une endonucléase de restriction sert à digérer le produit PCR à la place.
- Concevoir des amorces PCR pour amplifier le locus génomiques de la cible. Positionnez l’un des amorces d’être inférieur à 100 bp mais plus de 20 bp de la protospacer.
8. sélection des clones individuels
- De l’étape 6.3, fractionner les cellules une fois qu’ils commencent à devenir confluentes. Pour chaque clone individuel, recueillir les cellules restantes et extraire l’ADN génomique d’après étape 7.1.
- Effectuer l’essai de T7EI pour tous les clones individuels conformément à l’article 7.2, à l’exception d’une modification. Amplifier le locus génomiques cible des cellules de type sauvage et à l’étape 7.2.5, au lieu d’utiliser 200 ng de test ADN seulement, mélanger 100 ng de test ADN avec 100 ng de type sauvage de l’ADN.
NOTE : La raison de l’étape modifiée est que certains clones peuvent avoir subi la conversion réussie bialléliques et sont homozygotes mutants. Dans de tels cas, il n’y aura aucune bande de clivage dans l’essai de T7EI si l’ADN de type sauvage se mélange pas. - Séquence du site cible chez les clones qui présentent des bandes de clivage dans le dosage de la T7EI.
- Amplifier le locus génomique modifié selon les étapes 7.2.1–7.2.4.
- Mettre en place la réaction suivante de clonage : 4 µL du produit de PCR, 1 µL de solution de sel, 1 µL de vecteur TOPO (voir la Table des matières).
- Mélanger doucement de pipetage et incuber à température ambiante pendant au moins 5 min.
- Transformer chimiquement compétent e. coli de 3 µL du mélange de la réaction des cellules (par exemple les TOP10 ou Stbl3) (voir fichier complémentaire 1). Étaler les cellules bactériennes transformées sur une gélose LB avec 50 kanamycine μg/mL.
- Le lendemain, ensemencer au moins 10 colonies dans des milieux liquides LB contenant 50 kanamycine μg/mL.
- Lorsque les cultures bactériennes sont turbide, isolez les plasmides en utilisant un miniprep kit (voir Table des matières) et des séquences à l’aide de la norme M13 forward ou M13 reverse primer.
- Effectuer un transfert western (également connu sous le nom d’immunoblot) afin de déterminer l’absence ou la présence de la protéine ciblée (si le génome édition expérience consiste à assommer un gène codant pour des protéines par l’intermédiaire de mutations de déphasage). Voir fiche complémentaire 1.
Remarque : D’autres expériences peuvent être effectuées pour identifier le clone portant les modifications génomiques désirées. Par exemple, une analyse phénotypique peut être effectuée si assommant un gène particulier est connu pour causer certains changements dans le comportement cellulaire.
Résultats
Pour effectuer un génome édition experiment, un plasmide CRISPR exprimant un sgRNA ciblant que le locus d’intérêt doit être cloné. Tout d’abord, le plasmide est digéré par une enzyme de restriction (généralement de type IIs enzyme) de linéariser il. Il est recommandé de régler le produit digéré sur un gel d’agarose de 1 % aux côtés d’un plasmide non digéré de distinguer une digestion complète ou partielle. Comme les plasmides non digérés sont surenroulés, ils ont tendance à courir plus vit...
Discussion
Le système CRISPR-Cas est une puissante technologie révolutionnaire à l’ingénieur les génomes et les transcriptions des plantes et des animaux. De nombreuses espèces bactériennes ont été trouvés pour contenir les systèmes CRISPR-Cas, qui peuvent éventuellement être adaptées pour le génome et transcriptome fins44d’ingénierie. Bien que l’endonucléase Cas9 de Streptococcus pyogenes (SpCas9) a été la première enzyme qui seront déployés avec succès dans les cellule...
Déclarations de divulgation
Les auteurs n’ont pas de concurrence des intérêts financiers.
Remerciements
M.H.T. est pris en charge par un organisme de délivrance de bureau conseil commun Science Technology and Research (1431AFG103), un Conseil National de la recherche médicale accorde (OFIRG/0017/2016), accorde à la National Research Foundation (NRF2013-THE001-046 et NRF2013-THE001-093), un Subvention du ministère de l’éducation niveau 1 (RG50/17 (S)), une startup subvention de Nanyang Technological University et les fonds pour le concours International génétiquement ingénierie Machine (iGEM) de Nanyang Technological University.
matériels
| Name | Company | Catalog Number | Comments |
| T4 Polynucleotide Kinase (PNK) | NEB | M0201 | |
| Shrimp Alkaline Phosphatase (rSAP) | NEB | M0371 | |
| Tris-Acetate-EDTA (TAE) Buffer, 50X | 1st Base | BUF-3000-50X4L | Dilute to 1X before use. The 1X solution contains 40 mM Tris, 20 mM acetic acid, and 1 mM EDTA. |
| Tris-EDTA (TE) Buffer, 10X | 1st Base | BUF-3020-10X4L | Dilute to 1X before use. The 1X solution contains 10 mM Tris (pH 8.0) and 1 mM EDTA. |
| BbsI | NEB | R0539 | |
| BsmBI | NEB | R0580 | |
| T4 DNA Ligase | NEB | M0202 | 400,000 units/ml |
| Quick Ligation Kit | NEB | M2200 | An alternative to T4 DNA Ligase. |
| Rapid DNA Ligation Kit | Thermo Scientific | K1423 | An alternative to T4 DNA Ligase. |
| Zero Blunt TOPO PCR Cloning Kit | Thermo Scientific | 451245 | The salt solution comes with the TOPO vector in the kit. |
| NEBuilder HiFi DNA Assembly Master Mix | NEB | E2621L | Kit for Gibson assembly. |
| One Shot Stbl3 Chemically Competent E.Coli | Thermo Scientific | C737303 | |
| LB Broth (Lennox), powder | Sigma Aldrich | L3022 | Reconstitute in ddH20, and autoclave before use |
| LB Broth with Agar (Lennox), powder | Sigma Aldrich | L2897 | Reconstitute in ddH20, and autoclave before use |
| SOC media | - | - | 2.5 mM KCl, 10 mM MgCl2, 20 mM glucose in 1 L of LB Broth |
| Ampicillin (Sodium), USP Grade | Gold Biotechnology | A-301 | |
| REDiant 2X PCR Mastermix | 1st Base | BIO-5185 | |
| Agarose | 1st Base | BIO-1000 | |
| T7 Endonuclease I | NEB | M0302 | |
| Plasmid DNA Extraction Miniprep Kit | Favorgen | FAPDE 300 | |
| Dulbecco's Modified Eagle Medium (DMEM), High Glucose | Hyclone | SH30081.01 | 4.5 g/L Glucose, no L-glutamine, HEPES and Sodium Pyruvate |
| L-Glutamine, 200mM | Gibco | 25030 | |
| Penicillin-Streptomycin, 10, 000U/mL | Gibco | 15140 | |
| 0.25% Trypsin-EDTA, 1X | Gibco | 25200 | |
| Fetal Bovine Serum | Hyclone | SV30160 | FBS is heat inactivated before use at 56 oC for 30 min |
| Phosphate Buffered Saline, 1X | Gibco | 20012 | |
| jetPRIME transfection reagent | Polyplus Transfection | 114-75 | |
| QuickExtract DNA Extraction Solution, 1.0 | Epicentre | LUCG-QE09050 | |
| ISOLATE II Genomic DNA Kit | Bioline | BIO-52067 | An alternative to QuickExtract |
| Q5 High-Fidelity DNA Polymerase | NEB | M0491 | |
| Deoxynucleotide (dNTP) Solution Mix | NEB | N0447 | |
| 6X DNA Loading Dye | Thermo Scientific | R0611 | 10 mM Tris-HCl (pH 7.6) 0.03% bromophenol blue, 0.03% xylene cyanol FF, 60% glycerol, 60 mM EDTA |
| Protease Inhibitor Cocktail, Set3 | Merck | 539134 | |
| Nitrocellulose membrane, 0.2µm | Bio-Rad | 1620112 | |
| Tris-glycine-SDS buffer, 10X | Bio-Rad | 1610772 | Dilute to 1X before use. The 1x solution contains 25 mM Tris, 192 mM glycine, and 0.1% SDS. |
| Tris-glycine buffer, 10X | 1st base | BUF-2020 | Dilute to 1X before use. The 1x solution contains 25 mM Tris and 192 mM glycine. |
| Ponceau S solution | Sigma Aldrich | P7170 | |
| TBS, 20X | 1st base | BUF-3030 | Dilute to 1X before use. The 1x solution contains 25 mM Tris-HCl (pH 7.5) and 150 mM NaCl. |
| Tween 20 | Sigma Aldrich | P9416 | |
| Skim Milk for immunoassay | Nacalai Tesque | 31149-75 | |
| WesternBright Sirius-femtogram HRP | Advansta | K12043 | |
| Antibody for β-actin (C4) | Santa Cruz Biotechnology | sc-47778 | Lot number: C0916 |
| MiSeq system | Illumina | SY-410-1003 | |
| NanoDrop spectrophotometer | Thermo Scientific | ND-2000 | |
| Qubit fluorometer | Thermo Scientific | Q33226 | |
| EVOS FL Cell Imaging System | Thermo Scientific | AMF4300 | |
| CRISPR plasmid: pSpCas9(BB)-2A-GFP (PX458) | Addgene | 48138 | Single vector system: The gRNA is expressed from the same plasmid. |
| CRISPR plasmid: pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA | Addgene | 61591 | Single vector system: The gRNA is expressed from the same plasmid. |
| CRISPR plasmid: xCas9 3.7 | Addgene | 108379 | Dual vector system: The gRNA is expressed from a different plasmid. |
| CRISPR plasmid: pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | 42230 | Single vector system: The gRNA is expressed from the same plasmid. |
| CRISPR plasmid: hCas9 | Addgene | 41815 | Dual vector system: The gRNA is expressed from a different plasmid. |
| CRISPR plasmid: eSpCas9(1.1) | Addgene | 71814 | Single vector system: The gRNA is expressed from the same plasmid. |
| CRISPR plasmid: VP12 (SpCas9-HF1) | Addgene | 72247 | Dual vector system: The gRNA is expressed from a different plasmid. |
Références
- Epinat, J. C., et al. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Research. 31 (11), 2952-2962 (2003).
- Arnould, S., et al. Engineered I-CreI derivatives cleaving sequences from the human XPC gene can induce highly efficient gene correction in mammalian cells. Journal of Molecular Biology. 371 (1), 49-65 (2007).
- Chapdelaine, P., Pichavant, C., Rousseau, J., Paques, F., Tremblay, J. P. Meganucleases can restore the reading frame of a mutated dystrophin. Gene Therapy. 17 (7), 846-858 (2010).
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics. 188 (4), 773-782 (2011).
- Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S., Gregory, P. D. Genome editing with engineered zinc finger nucleases. Nature Reviews Genetics. 11 (9), 636-646 (2010).
- Miller, J. C., et al. A TALE nuclease architecture for efficient genome editing. Nature Biotechnology. 29 (2), 143-148 (2011).
- Zhang, F., et al. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nature Biotechnology. 29 (2), 149-153 (2011).
- Boch, J., et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 326 (5959), 1509-1512 (2009).
- Moscou, M. J., Bogdanove, A. J. A simple cipher governs DNA recognition by TAL effectors. Science. 326 (5959), 1501 (2009).
- Hsu, P. D., Lander, E. S., Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 157 (6), 1262-1278 (2014).
- Sander, J. D., Joung, J. K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nature Biotechnology. 32 (4), 347-355 (2014).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nishimasu, H., et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 156 (5), 935-949 (2014).
- Yamano, T., et al. Crystal Structure of Cpf1 in Complex with Guide RNA and Target DNA. Cell. 165 (4), 949-962 (2016).
- Swarts, D. C., Mosterd, C., van Passel, M. W., Brouns, S. J. CRISPR interference directs strand specific spacer acquisition. PLoS One. 7 (4), e35888 (2012).
- Zetsche, B., et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 163 (3), 759-771 (2015).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 507 (7490), 62-67 (2014).
- Hu, J. H., et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 556 (7699), 57-63 (2018).
- Kleinstiver, B. P., et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nature Biotechnology. 33 (12), 1293-1298 (2015).
- Kleinstiver, B. P., et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 523 (7561), 481-485 (2015).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Jinek, M., et al. RNA-programmed genome editing in human cells. Elife. 2, e00471 (2013).
- Cho, S. W., Kim, S., Kim, J. M., Kim, J. S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nature Biotechnology. 31 (3), 230-232 (2013).
- Wang, Y., et al. Systematic evaluation of CRISPR-Cas systems reveals design principles for genome editing in human cells. Genome Biology. 19 (1), 62 (2018).
- Ran, F. A., et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 520 (7546), 186-191 (2015).
- Hou, Z., et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proceedings of the National Academy of Sciences U S A. 110 (39), 15644-15649 (2013).
- Kim, E., et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nature Communications. 8, 14500 (2017).
- Edraki, A., et al. A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Molecular Cell. , (2018).
- Chatterjee, P., Jakimo, N., Jacobson, J. M. Minimal PAM specificity of a highly similar SpCas9 ortholog. Science Advances. 4 (10), (2018).
- Muller, M., et al. Streptococcus thermophilus CRISPR-Cas9 Systems Enable Specific Editing of the Human Genome. Mol Therapy. 24 (3), 636-644 (2016).
- Esvelt, K. M., et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nature Methods. 10 (11), 1116-1121 (2013).
- Boratyn, G. M., et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Research. 41 (Web Server issue), W29-W33 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M., Valen, E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Research. 42 (Web Server issue), W401-W407 (2014).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Haeussler, M., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Bae, S., Park, J., Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 30 (10), 1473-1475 (2014).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Richardson, C. D., Ray, G. J., Bray, N. L., Corn, J. E. Non-homologous DNA increases gene disruption efficiency by altering DNA repair outcomes. Nature Communications. 7, 12463 (2016).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. 6 (5), 343-345 (2009).
- Zhang, J. P., et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biology. 18 (1), 35 (2017).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Shmakov, S., et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nature Reviews Microbiology. 15 (3), 169-182 (2017).
- Moreno-Mateos, M. A., et al. CRISPR-Cpf1 mediates efficient homology-directed repair and temperature-controlled genome editing. Nature Communications. 8 (1), 2024 (2017).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 3, e04766 (2014).
- Yang, L., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Watanabe, K., et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nature Biotechnology. 25 (6), 681-686 (2007).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.