
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Edição do genoma em linhas de células de mamíferos usando CRISPR-Cas
Neste Artigo
Resumo
CRISPR-Cas é uma tecnologia poderosa para projetar os genomas complexos de plantas e animais. Aqui, detalhamos um protocolo eficiente editar o genoma humano usando diferentes endonucleases de Cas. Destacamos importantes considerações e parâmetros de projeto para otimizar a eficiência de edição.
Resumo
O sistema regularmente intercaladas curta palíndromo repetições (CRISPR) em cluster funciona naturalmente na imunidade adaptativa bacteriana, mas tem com êxito foi realocado para engenharia do genoma em muitos organismos vivos diferentes. Mais comumente, a sua CRISPR associado 9 (Cas9) ou Cas12a endonuclease é usado para decompor a locais específicos no genoma, após o qual o intervalo de dupla-hélice do DNA é reparado através de fim não-homóloga aderir a caminho (NHEJ) ou o reparo de homologia-dirigido ( Caminho HDR) dependendo se é um modelo de doador ausentes ou apresentam respectivamente. Até à data, sistemas CRISPR de diferentes espécies bacterianas foram mostrados para ser capaz de realizar a edição de genoma em células de mamíferos. No entanto, apesar da aparente simplicidade da tecnologia, vários parâmetros de projeto precisam ser considerados, que muitas vezes deixam perplexos sobre como melhor realizar seu genoma edição de experiências de usuários. Aqui, descrevemos um fluxo de trabalho completo de delineamento experimental para identificação de clones de células que levam a modificações de DNA desejadas, com o objetivo de facilitar a execução bem-sucedida do genoma edição experimentos em linhas de células de mamíferos. Destacamos as considerações-chave para que os usuários anotem, incluindo a escolha do sistema CRISPR, o comprimento do espaçador e o design de um modelo de doador single-stranded oligodeoxynucleotide (ssODN). Vislumbramos que este fluxo de trabalho será útil para estudos de gene nocaute, doença modelagem esforços, ou linhas de célula a geração de repórter.
Introdução
A capacidade para projetar o genoma de qualquer organismo vivo tem muitas aplicações biomédicas e biotecnológicas, como a correção da doença-causando mutações, construção de modelos de celulares precisos para estudos de doença, ou geração de produtos agrícolas culturas com características desejáveis. Desde a virada do século, diversas tecnologias foram desenvolvidas para a engenharia do genoma em células de mamíferos, incluindo meganucleases1,2,3, zinco dedo nucleases4,5, ou transcrição como ativador effector nucleases (TALENs)6,7,8,9. No entanto, estas tecnologias anteriores são difíceis de programa ou tedioso para montar, prejudicando, assim, sua adoção generalizada na investigação e a indústria.
Nos últimos anos, o cluster regularmente intercaladas curtas palíndromos repetições (CRISPR) - sistema CRISPR-associado (Cas) tem emergido como um poderoso novo genoma engenharia tecnologia10,11. Originalmente um sistema imune adaptativo em bactérias, tem sido com sucesso implantado para a modificação do genoma em plantas e animais, incluindo seres humanos. Uma razão principal por que CRISPR-Cas ganhou tanta popularidade em tão pouco tempo é que o elemento que traz a chave endonuclease de Cas, tais como Cas9 ou Cas12a (também conhecido como Cpf1), para o local correto no genoma é simplesmente um pedaço curto de quimérico único guia RN Um (sgRNA), que é simples para o projeto e barato para sintetizar. Depois de ser recrutado para o site de destino, a enzima Cas funciona como um par de tesouras moleculares e cliva o DNA acoplado com sua RuvC, HNH ou Nuc domínios12,13,14. A resultante quebra encalhada dobro (DSB) é posteriormente reparada pelas células através de fim não-homóloga (NHEJ) juntar-se ou no caminho de reparação homologia-dirigido (HDR). Na ausência de um modelo de reparação, o ORL é reparado por via NHEJ propenso a erro, que pode dar origem a pseudo-aleatório inserção ou supressão de nucleotídeos (puntuais) no local do corte, potencialmente causando frameshift mutações nos genes codificantes de proteínas. No entanto, na presença de um modelo de doador que contém as alterações de DNA desejadas, o ORL é reparado por via HDR de alta fidelidade. Tipos comuns de modelos de doadores incluem oligonucleotides single-stranded (ssODNs) e plasmídeos. O primeiro costuma ser usado se as alterações de DNA pretendidas são pequenas (por exemplo, a alteração de um único par de base), enquanto o último é geralmente usado se deseja inserir uma sequência relativamente longa (por exemplo, a sequência de codificação de uma proteína verde fluorescente ou GFP) para o local de destino.
A atividade de endonuclease da proteína Cas requer a presença de um motivo protospacer adjacentes (PAM) para o local de destino15. O PAM de Cas9 é na extremidade 3' da protospacer, enquanto o PAM de Cas12a (também chamado Cpf1) é, ao invés, na extremidade 5'16. O Cas-guia complexo RNA é incapaz de apresentar um ORL se o PAM estiver ausente17. Daí, o PAM coloca uma restrição na genômicas locais onde um determinado nuclease Cas é capaz de decompor. Felizmente, nucleases de Cas de diferentes espécies bacterianas geralmente apresentam exigências diferentes do PAM. Portanto, integrando vários sistemas CRISPR-Cas em nossa caixa de ferramentas de engenharia, podemos expandir a gama de sites que podem ser alvo de um genoma. Além disso, uma enzima natural do Cas pode ser projetada ou evoluiu para reconhecer sequências de PAM alternativas, ainda mais, alargar o âmbito da genômicos alvos acessíveis para manipulação de19,18,20.
Embora vários sistemas CRISPR-Cas estão disponíveis para fins de engenharia do genoma, a maioria dos usuários da tecnologia dependem principalmente da nuclease Cas9 de Streptococcus pyogenes (SpCas9) por várias razões. Em primeiro lugar, requer um PAM relativamente simplesmente NGG, ao contrário de muitas outras proteínas de Cas que podem cleave apenas na presença de PAMs mais complexas. Em segundo lugar, é a primeira endonuclease Cas seja implantado com sucesso em células humanas21,22,23,24. Em terceiro lugar, SpCas9 é, de longe, a melhor enzima caracterizada até à data. Se um pesquisador deseja usar outro nuclease Cas, ele ou ela muitas vezes seria incerto sobre qual a melhor forma de projetar o experimento e bem como outras enzimas irão realizar em diferentes contextos biológicos em relação ao SpCas9.
Para fornecer clareza para o desempenho relativo dos diferentes sistemas de CRISPR-Cas, recentemente Efetuamos uma comparação sistemática de cinco endonucleases de Cas-SpCas9, a enzima Cas9 de Staphylococcus aureus (SaCas9), a enzima Cas9 de Neisseria meningitidis (NmCas9), a enzima Cas12a de SP Acidaminococcus BV3L6 (AsCas12a) e a enzima Cas12a de bactéria Lachnospiraceae ND2006 (LbCas12a)25. Para uma comparação justa, avaliamos as nucleases de Cas diferentes usando o mesmo conjunto de sites de destino e outras condições experimentais. Os parâmetros de projeto de estudo também delineado para cada sistema CRISPR-Cas, que serviu como uma referência útil para os usuários da tecnologia. Aqui, para melhor permitir aos investigadores fazer uso das CRISPR-Cas sistema, nós fornecemos um protocolo passo a passo para engenharia de genoma ideal com diferentes enzimas Cas9 e Cas12a (ver Figura 1). O protocolo inclui não apenas detalhes experimentais, mas considerações de design também é importante para maximizar a probabilidade de um resultado de sucesso do genoma engenharia em células de mamíferos.

Figura 1 : Uma visão geral do fluxo de trabalho para gerar o genoma edição linha celular humana. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Protocolo
1. design de sgRNAs
- Selecione um sistema adequado de CRISPR-Cas.
- Primeiro, inspecione a região de destino para as sequências de PAM de todas as nucleases Cas9 e Cas12a que foram mostradas para ser funcional em células de mamíferos16,21-32. Cinco enzimas usadas com frequência são descritas na tabela 1 , juntamente com seus respectivas PAMs.
Nota: além das endonucleases listados na tabela 1, há outras enzimas Cas menos comumente usadas que foram implantadas com sucesso em células de mamíferos, tais como uma nuclease Cas9 de Streptococcus thermophilus (St1Cas9) que reconhece o PAM NNAGAAW. Se o site de destino desejado não contiver uma PAM conhecida, então um não seria capaz de usar o sistema CRISPR-Cas para engenharia de genoma. - Em segundo lugar, considere qualquer Propriedades conhecidas do locus genômico alvo ou gene. Algumas propriedades de tomar em consideração incluem os níveis de expressão do gene ou acessibilidade da cromatina e se existem outros estreitamente relacionados com sequências também.
Nota: Certas enzimas são mais adequadas para contextos biológicos particulares. Por exemplo, para editar um locus genômico repetitivo ou um gene com vários outros paralogs próximos, recomenda-se usar qualquer AsCas12a (devido a sua baixa tolerância para incompatibilidades entre o sgRNA e o ADN do alvo do que SpCas9 e LbCas12a) ou SaCas9 (devido a sua requisito para espaçadores mais longos, que fornece maior especificidade direcionamento)25.
- Primeiro, inspecione a região de destino para as sequências de PAM de todas as nucleases Cas9 e Cas12a que foram mostradas para ser funcional em células de mamíferos16,21-32. Cinco enzimas usadas com frequência são descritas na tabela 1 , juntamente com seus respectivas PAMs.
| Endonuclease de CAS | PAM | Comprimento do espaçador ideal |
| SpCas9 | NGG | 17-22 nt inclusiva |
| SaCas9 | NNGRRT | ≥ 21 nt |
| NmCas9 | NNNNGATT | ≥ 19 nt |
| AsCas12a e LbCas12a | TTTV | ≥ 19 nt |
Tabela 1: algumas usadas enzimas Cas com seus cognatas PAMs e comprimentos ideal sgRNA. N = qualquer nucleotídeo (A, T, G ou C); R = A ou G; V = A, C, ou G.
- Selecione uma sequência de espaçador adequado. Identificar uma sequência tão única quanto possível para minimizar o risco de eventos fora do alvo clivagem, examinando o genoma alvo com explosão33 ou usando várias ferramentas online disponíveis gratuitamente, tais como: (a) programa de laboratório de Feng Zhang34 (http://crispr.mit.edu/); (b) CHOPCHOP35 (http://chopchop.cbu.uib.no/); (c) E CRISP-36 (http://www.e-crisp.org/E-CRISP/); (d) CRISPOR37 (http://crispor.tefor.net/); (e) Cas-OFFinder38 (http://www.rgenome.net/cas-offinder/).
Nota: O comprimento ideal do espaçador pode variar de 17 a 25 nucleotídeos (nt) inclusivos, dependendo de qual Cas enzima é usada (ver tabela 1). Para Cas9, o espaçador é a montante do PAM, enquanto que para Cas12a, o espaçador é a jusante do Pam, adicionalmente, eficiência HDR cai rapidamente com o aumento da distância do local do corte. Portanto, para edição precisa de DNA, posicione o sgRNA tão próximo quanto possível ao site da modificação pretendida. - Sintetiza oligonucleotídeos de DNA com as saliências apropriados para o plasmídeo CRISPR que está sendo usado.
- Adicione um nucleotídeo G na frente do espaçador, se a primeira posição do guia não é um G. Determine a sequência inversa complementar do espaçador. Adicione as saliências necessárias para fins de clonagem.
Nota: A título de ilustração, para o plasmídeo utilizado em nosso estudo de avaliação25, os oligonucleotides de ser sintetizada são mostrados na tabela 2. Use o exemplo dado na Figura 2 como um guia, se necessário. Muitos plasmídeos CRISPR estão disponíveis a partir de fontes comerciais (por exemplo, Addgene). Alguns dos Plasmideos mais populares são dadas na Tabela de materiais.
- Adicione um nucleotídeo G na frente do espaçador, se a primeira posição do guia não é um G. Determine a sequência inversa complementar do espaçador. Adicione as saliências necessárias para fins de clonagem.
| Plasmídeo CRISPR | Sequência de |
| pSpCas9 e pSaCas9 | Sentido: 5' - NNNNNNNNNNNNNNNNNNNNN CACC (G) - 3' Antisentido: 3 - (C) NNNNNNNNNNNNNNNNNNNNNCAAA - 5' ' |
| pNmCas9 | Sentido: 5' - NNNNNNNNNNNNNNNNNNNNN CACC (G) - 3' Antisentido: 3 - (C) NNNNNNNNNNNNNNNNNNNNNCAAC - 5' ' |
| pAsCas12a e pLbCas12a | Sentido: 5' - AGATNNNNNNNNNNNNNNNNNNNNN - 3' Antisentido: 3 - NNNNNNNNNNNNNNNNNNNNNAAAA - 5' ' |
Tabela 2: Oligonucleotides, necessários para clonagem sgRNA sequências em plasmídeos CRISPR usados em uma avaliação recente estudo25. As saliências são em itálico.

Figura 2 : Um exemplo ilustrando como selecionar sites de destino e projetar oligonucleotides para clonagem em plasmídeos CRISPR. O locus genômico de alvo aqui é 45 de exon do gene humano CACNA1D. As PAMs para SpCas9 e SaCas9 são NGG e NNGRRT, respectivamente e são destacadas em vermelho, enquanto o PAM para AsCas12a e LbCas12a é TTTN e é realçado em verde. A barra horizontal vermelha indica a protospacer para SpCas9 e SaCas9, enquanto a barra horizontal verde indica a protospacer para as duas enzimas Cas12a. Clique aqui para ver uma versão maior desta figura.
{kind=link}
2. clonagem de oligonucleotídeos em um vetor de coluna vertebral
- Fosforilar e recoze sentido e antisense oligonucleotides.
- Se os oligonucleotides são liofilizados, resuspenda-los a uma concentração de 100 µM em ácido tris-ácido etilenodiaminotetracético (EDTA) (TE de buffer, consulte Tabela de materiais) ou ddH2O.
- Preparar uma mistura de reação de 10 µ l contendo 1 µ l do oligonucleotide do sentido, 1 µ l do oligonucleotide antisentido, 1 µ l de debuffer T4 DNA ligase (10x), 1 µ l da quinase de polinucleotido T4 (PNK) e 6 µ l de DDQ2O. Mix bem pipetando e coloque a mistura de reação em uma térmica cy Cler, usando os seguintes parâmetros: 37 ° C por 30 min, 95 ° C por 5 min e rampa até 25 ° C a 6 ° C/min.
- Dilua a reação mistura 1: 100 em ddH2O (por exemplo, mistura de reação de 2 µ l + µ l 198 ddH2O).

Figura 3 : Um exemplo de um plasmídeo CRISPR. (um) A mapa indicando diferentes características importantes do plasmídeo. Aqui, o promotor de EF-1a conduz a expressão de Cas9, enquanto o promotor U6 conduz a expressão da sgRNA. Amp(R) indica um gene de resistência a ampicilina no plasmídeo. (b) a sequência do "BbsI-BplI clonagem site" no plasmídeo. A sequência de reconhecimento de BbsI é GAAGAC e é indicada em vermelho, enquanto a sequência de reconhecimento de BplI é mordaça-N5- CTC e é indicada em verde. (c) Primers que pode ser usado para PCR de colônia para verificar se a sequência de sgRNA foi clonada com sucesso para o plasmídeo. A cartilha do hU6_forward é indicada por uma seta roxa no mapa do plasmídeo, enquanto o primer universal de M13R(-20) é indicado por uma seta rosa no mapa do plasmídeo. Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Digeri o plasmídeo CRISPR com uma enzima de restrição apropriada.
Nota: Clonagem de sgRNAs normalmente dependem Golden Gate assembly com tipo de enzimas de restrição do IIs. Enzimas diferentes podem ser utilizadas para diferentes plasmídeos CRISPR. Para pSpCas9, use BbsI ou BplI (ver Figura 3). Para pSaCas9, pNmCas9, pAsCas12a e pLbCas12a, use BsmBI.- Preparar uma mistura de reação de 20 µ l contendo 1 µ g de vetor do plasmídeo circular, 2 µ l de tampão (10x), 1 µ l de enzima de restrição (por exemplo, BbsI, BplI ou BsmBI) e ddH2O até um volume final de 20 µ l. Incubar a reação a 37 ° C para 2,5 h.
- Adicionar 1 µ l de fosfatase alcalina do camarão (SAP) para a reação e incubar a 37 ° C por mais 30 minutos.
- Saciar a reação pela adição de 5 µ l de 6 x carregamento de DNA tingir (ver Tabela de materiais), misture bem e resolver a reação em um gel de agarose 0,8% com tampão tris-Acetato-EDTA (TAE) 1x. Em seguida, excisar a banda correcta e proceder ao gel purificar o vetor tornado linear.
- Ligam os oligonucleotides recozidos para o plasmídeo CRISPR digerido.
- Preparar uma mistura de reação de 10 µ l: 50 ng do vetor tornado linear, 1 µ l de oligonucleotides recozidos diluídos, 1 µ l de tampão de T4 DNA ligase (10x), 1 μL de T4 DNA ligase e ddH2O até um volume final de 10 µ l (veja a Tabela de materiais). Incube a reação a 16 ° C durante a noite ou em temperatura ambiente por 2 h.
Nota: Para acelerar o processo de ligadura, usar concentrado T4 DNA ligase e incubar a mistura de reação em temperatura ambiente por 15 min (ver Tabela de materiais).
- Preparar uma mistura de reação de 10 µ l: 50 ng do vetor tornado linear, 1 µ l de oligonucleotides recozidos diluídos, 1 µ l de tampão de T4 DNA ligase (10x), 1 μL de T4 DNA ligase e ddH2O até um volume final de 10 µ l (veja a Tabela de materiais). Incube a reação a 16 ° C durante a noite ou em temperatura ambiente por 2 h.
- Transformar produtos ligados quimicamente competentes de Escherichia coli eritrócitos (ver arquivo complementar 1). Espalhe as células bacterianas transformadas em uma placa de ágar LB com ampicilina 100 de µ g/mL.
- Execute colônia reação em cadeia da polimerase (PCR) para identificar as bactérias com inserção.
- Prepare-se dois conjuntos de tubos estéreis de tira PCR. Em Set 1, adicionar 4,7 µ l de DDQ2O, enquanto em Set 2, adicionar 50 µ l de caldo LB com antibiótico apropriado (por exemplo, ampicilina 100 de µ g/mL).
- Com uma ponta de pipeta estéril, escolhe uma colônia no prato, passá-lo brevemente em um tubo de Set 1 e deixe a ponta em um tubo de Set 2. Repita para algumas colônias, certificando-se de usar tubos PCR diferentes cada vez.
Nota: Normalmente, quatro colônias de triagem é suficiente. No entanto, pode variar dependendo da eficiência de clonagem. - Adicione os seguintes reagentes a cada tubo de 1 conjunto (para um 10 µ l de PCR): 5 µ l de mistura de mestre 2 x PCR (com tintura de carregamento, consulte Tabela de materiais), 0,15 µ l do sentido ou antisentido oligonucleotide (100 µM), 0,15 µ l de primer (100 µM) visando o plasmídeo CRISPR rio abaixo ou acima fluxo da fita sgRNA respectivamente (ver Figura 3).
Nota: O produto do PCR idealmente deve render um tamanho de produto de aproximadamente 150 bp ou bandas maiores, assim que qualquer positivas não estão erradas como dímeros da primeira demão. - Executar o PCRs em um termociclador usando os seguintes parâmetros: 95 ° C por 3 min, 95 ° C por 30 s (etapa 2), 60 ° C por 30 s (etapa 3), 72 ° C por 30 s (etapa 4), repita as etapas 2\u20124 por mais 34 ciclos, 72 ° C por 5 min e segurar a 4 ° C.
Nota: A temperatura do recozimento de 60 ° C pode precisar ser otimizado para os primers desenhados. O tempo de alongamento de 30 s também podem variar dependendo o tamanho esperado do amplicon PCR e o polymerase do DNA usado. - Resolva as reações em um gel de agarose a 1% usando 1 x TAE buffer.
- Inocule uma colônia que produz uma banda positiva na PCR Transferindo 50 µ l de sua cultura do tubo correspondente Set 2 em um tubo cônico maior contendo 5 mL LB com um antibiótico apropriado. Deixe a cultura crescer durante a noite em um 37 ° C incubadora-agitador.
- Isole o plasmídeo da noite cultura usando um miniprep kit (veja a Tabela de materiais) e sequenciar a amostra usando o primer PCR de colônia que não é o sentido ou antisentido oligonucleotide (hU6_forward ou M13R(-20) na Figura 3).
Nota: Se necessário, execute um maxiprep do plasmídeo CRISPR sequência verificada para obter uma maior quantidade de experiências a jusante.
3. design e síntese de modelos de reparação
Nota: Para a engenharia de precisão do genoma, um modelo especificando as modificações de DNA desejadas precisa ser fornecido juntamente com os reagentes CRISPR. Para pequenas edições de ADN como a alteração de um único nucleotídeo, modelos de doador de ssODN são mais adequado (ver secção 3.1). Para edições de DNA maiores tais como a inserção de um GFP marca 5' ou 3' de um determinado gene codificantes de proteínas, modelos de doador do plasmídeo são mais adequado (ver secção 3.2).
- Projetar e sintetizar um modelo de doador ssODN (ver Figura 4).
- Determine que a correta vertente cuja sequência deve seguir o modelo.
Nota: Cas12a exibe uma preferência para a ssODNs das sequências não-alvo, enquanto Cas9 exibe uma preferência para ssODNs da strand, alvo de sequências em vez disso25 (consulte a Figura 4um). - Assegurar que a sequência reparada não targetable pela nuclease Cas selecionada novamente. Por exemplo, mutação do PAM de tal forma que não há alteração de aminoácidos ou eliminar o PAM do modelo doador se não tem nenhuma consequência funcional. Use o exemplo dado na Figura 4b como um guia, se necessário.
- Decida se um modelo de doador simétrica ou assimétrica é desejado. Para doadores simétricos, certifique-se de que cada braço de homologia flanqueando o site de modificação do DNA tem pelo menos 17 nt longa25. Para modelos de doador assimétrica, use mais 5 ' braços das alterações de DNA desejadas (consulte a Figura 4um). Importante, certifique-se que o braço mais curto é cerca de 37 nt de comprimento, enquanto o outro braço de homologia é em torno de 77 nt em comprimento25,39.
- Sintetiza o modelo concebido como uma peça de ADN single-stranded.
Nota: SsODNs assimétricas pode, mas não sempre, apresentam maior eficiência HDR que ssODNs simétrica. Em geral, um doador assimétrico normalmente executa pelo menos assim como um doador simétrico, quando projetado corretamente. No entanto, o modelo assimétrico custa muito mais porque é mais longo e, portanto, requer purificação de eletroforese (página) do gel de polyacrylamide ou um procedimento especial de síntese. Nocautes de rotina gene geralmente dependem da via de reparo NHEJ e não necessitam de um modelo de reparação. No entanto, se a eficiência de nocaute é baixa, projetar um modelo de doador de ssODN que contém uma mutação frameshift e é pelo menos 120 nt em comprimento25,40.
- Determine que a correta vertente cuja sequência deve seguir o modelo.

Figura 4 : Design de modelos de doador ssODN. (a) esquema ilustrando vários projetos possíveis. Os retângulos horizontais vermelhos indicam a vertente não-alvo (NT), enquanto os retângulos azuis indicam a vertente de alvo (T). Além disso, os pequenos retângulos verdes indicam as modificações de DNA desejadas (por exemplo, alterações de nucleotídeo único). Quando um ssODN simétrico é usado, o comprimento mínimo de cada braço de homologia deve ser de pelo menos 17 nt (mas pode ser mais longo). Para ssODNs assimétrica, o ssODN de T 37/77 parece ser ideal para HDR induzida por SpCas9, enquanto o 77/37 NT ssODN parece ser ideal para HDR induzida por Cas12a. L = braço esquerdo homologia; R = o braço direito de homologia. (b) um exemplo específico para demonstrar como criar modelos de ssODN. Aqui, o locus genômico alvo é exon 45 do gene humano CACNA1D. O PAM para Cas9 é rosa e sublinhado, enquanto que o PAM para Cas12a é marrom e sublinhado. O objetivo é criar uma mutação missense (realçado em verde) através da conversão de Araújo (codificação serina) AGG (codificação de arginina). Para evitar re-direcionamento por Cas12a, o TTTC PAM é uma mutação de CTTC. Observe que não há alteração no aminoácido (UAU e UAC ambos codificam tirosina). Para aumentar a prevenção de re-direcionamento por Cas9, um codão AGU é substituído com um códon UCC (negrito), ambos do qual código para serina. Clique aqui para ver uma versão maior desta figura.
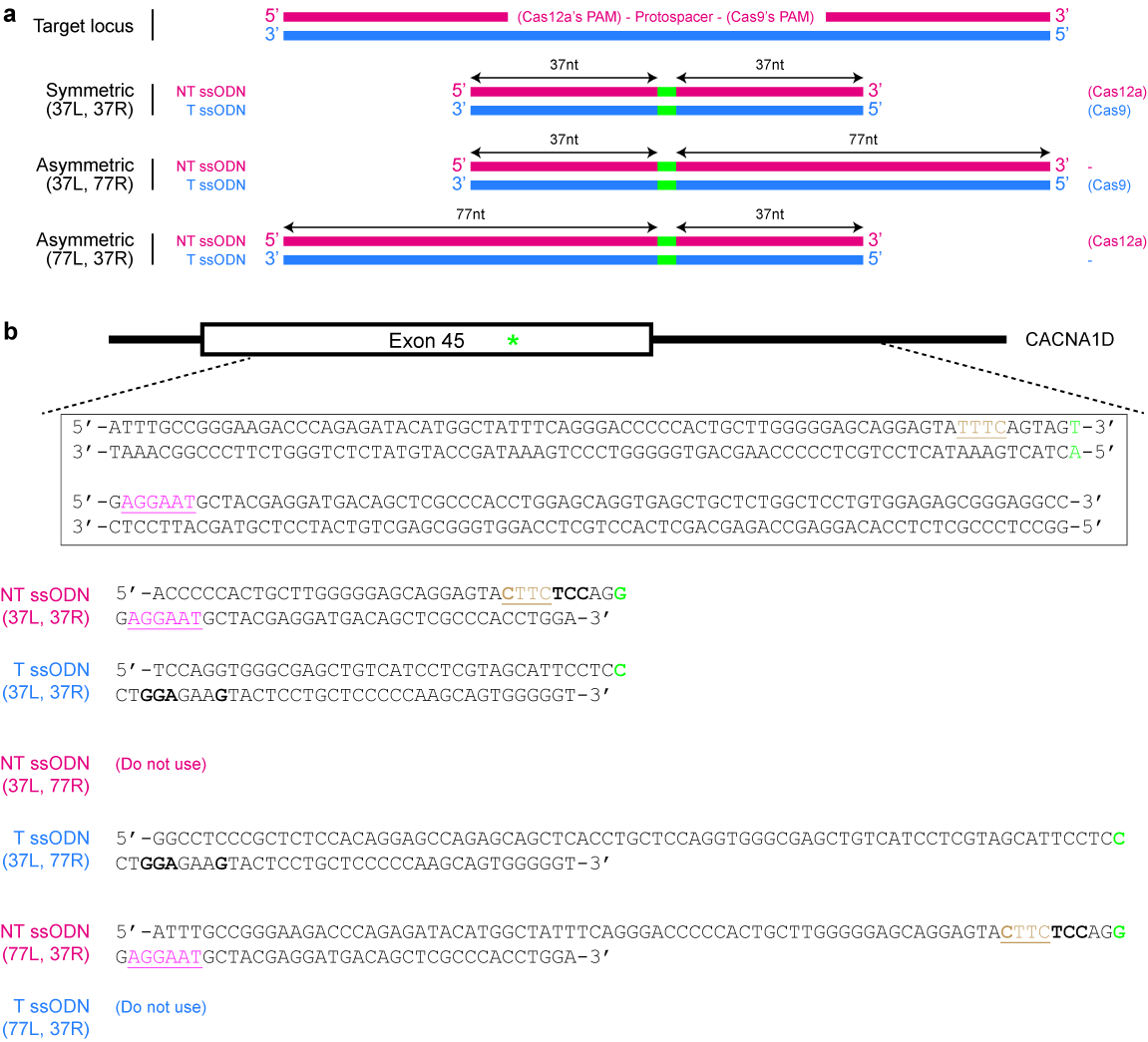{kind=link}
- Design e clonar um modelo de doador apropriado do plasmídeo. Por exemplo, pode conter uma sequência GFP, ladeada por braços longos que são homólogos para o locus genômico de destino (consulte a Figura 5).
- Assegurar que a sequência modificada não targetable pela nuclease Cas selecionada novamente. Por exemplo, o protospacer pode ser dividido pela tag (GFP) inserida. Alternativamente, o PAM pode ser uma mutação ou removido do modelo de forma a que não afeta a função do gene doador.
- Amplifica os braços de homologia do DNA genômico usando PCR. O comprimento de cada braço de homologia é tipicamente bp 1000 a 1500.
Nota: Para facilitar a clonagem, certifique-se que o primer para a frente para o braço esquerdo de homologia e o primer reverso para a homologia certo braço cada uma tem pelo menos 20 nt sobrepostas a sequência com um backbone de vetor selecionado. Além disso, certifique-se que o primer reverso para o braço esquerdo de homologia e o primer para a frente para o braço direito de homologia tem algumas sequências de sobreposição com o epítopo tag também. - Clonar os dois braços de homologia e a tag (GFP) na espinha dorsal de vetor usando Gibson montagem41 (ver a Tabela de materiais). Verifique se o plasmídeo por Sanger sequenciamento utilizando-se para a frente e reverter as primeiras demão que são respectivamente o montante e a jusante do modelo doador.
Nota: Sanger sequenciamento é amplamente e mais barato disponível como um serviço comercial. Envie uma aliquota do plasmídeo juntamente com os primers de sequenciamento para um provedor de serviço. - Linearizar o modelo doador com uma enzima de restrição que corta o plasmídeo somente uma vez ou montante do braço esquerdo homologia ou a jusante do braço direito de homologia.
Nota: Recentemente, um doador de duplo corte, que é ladeado por sequências de sgRNA-PAM e é libertado o plasmídeo após clivagem pela correspondente nuclease Cas, foi mostrado para aumentar a eficiência HDR42. Quando as sequências de sgRNA-PAM são inseridas a montante e a jusante de homologia a esquerda e direita respectivamente armas (por exemplo, montagem de Gibson), o comprimento do braço de homologia pode ser reduzido para 300 bp e não há nenhuma necessidade para linearizar o plasmídeo.

Figura 5 : Design e clonagem de um modelo de doador plasmídeo. (um) o objetivo neste exemplo específico é fundir P2A-GFP para o C-terminal da proteína CLTA. O retângulo horizontal azul indica o braço esquerdo de homologia, enquanto o retângulo horizontal vermelho indica o braço direito de homologia. Letras maiusculas indicam sequências codificantes de proteínas, enquanto letras minúsculas indicam sequências não-codificantes. As PAMs para SpCas9 e Cas12a estão em itálico e sublinhadas. (b) um doador do plasmídeo modelo que pode ser usado a tag endogenamente P2A-GFP no C-terminal da CLTA. As sequências de cartilha fornecido podem ser usadas para clonar o plasmídeo por montagem Gibson. As condições PCR são as seguintes: 98 ° C por 3 min, 98 ° C por 30 s (etapa 2), 63 ° C por 30 s (etapa 3), 72 ° C por 1 min (etapa 4), repita as etapas 2\u20124 por mais 34 ciclos, 72 ° C por 3 min e manter a 4 ° C. Letras pretas correspondem às sequências de vetor, letras azuis corresponde ao braço esquerdo homologia, letras verdes correspondem a P2A-GFP e letras vermelhas correspondem ao braço direito de homologia. Note que uma vez a sequência de codificação P2A-GFP é integrada com sucesso para o locus de alvo, re-direcionamento por SpCas9 não será possível, desde apenas 9 nt de sua protospacer (GTGCACCAG) será deixado intacto. Além disso, para evitar re-direcionamento por Cas12a, três basepairs imediatamente a jusante da parada de codon (em negrito) são excluídos da sequência de plasmídeo. Clique aqui para ver uma versão maior desta figura.
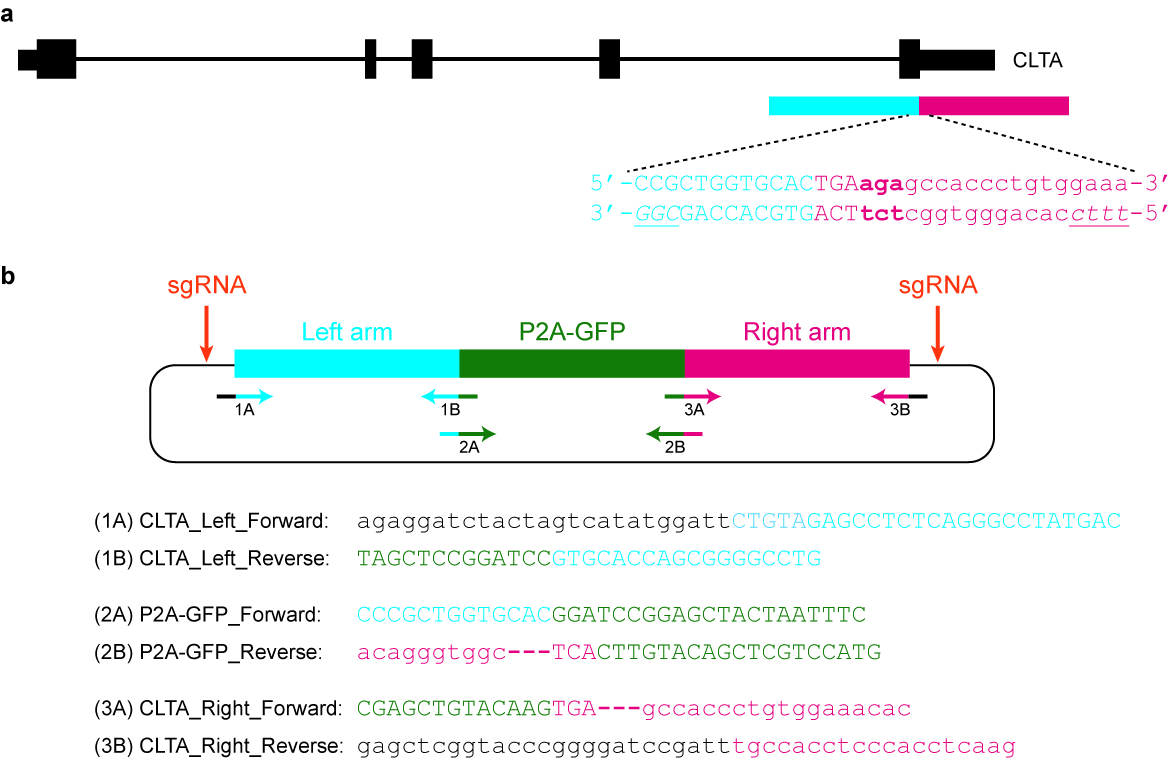{kind=link}
4. transfecção celular
Nota: As partes restantes do protocolo são escritas com células HEK293T em mente. O meio de cultura utilizado consiste de Dulbecco modificado águia médio (DMEM) suplementado com 0,1% penicilina/estreptomicina, 10% de soro fetal bovino (FBS), 2 mM L-glutamina e glicose de 4,5 g/L. Diferentes etapas do protocolo podem ter que ser modificado de acordo com a linha de células reais usada. Todo trabalho de cultura de células é feito em um gabinete de segurança biológica classe II para garantir um ambiente de trabalho estéril.
- Semente de 1,8 x 105 células em uma placa de cultura de tecido de 24-bem um dia antes do transfection.
- Dissociar as células por aspiração a mídia e adicionando 150 µ l 0,25% Trypsin-EDTA por bem. Incube as células a 37 ° C por 2 min.
- Neutralize a tripsina adicionando 150 µ l (ou 1 x volume) de meios de cultura de células. Transfira a suspensão de células para um tubo cónico. Gire para baixo as células em uma centrífuga de bancada superior a 1000 x g por 5 min.
- Aspire o sobrenadante e ressuspender com 5 mL de meio de cultura celular. Num tubo de centrífuga separado, alíquota 10 µ l de solução de azul de Tripan 0,4%. Em seguida, adicione 10 células µ l resuspended do passo 4.1.2 e misture bem.
- Pipete 10 µ l da mistura (células + trypan azul) para um hemocytometer. Prossiga para contar as células manualmente ou usando um contador automatizado de células.
- Semente de 1,8 x 105 células em um poço de uma placa de cultura de tecidos de 24-bem.
- Preparar uma mistura de transfeccao contendo qualquer 500 ng do plasmídeo CRISPR (para edição de mediadas por NHEJ) ou 300 ng de CRISPR plasmídeo e 300 ng do modelo do doador (para HDR-mediada edição), de acordo com as instruções fornecidas com o reagente de transfeccao (veja Tabela de materiais). Incube a temperatura ambiente para a duração do tempo recomendado (tipicamente em torno de 10\u201220 min).
- Adicione a mistura de Transfeccao para as células em forma de gota a gota, e agite suavemente a placa depois.
- Incubar a 37 ° C em um umidificado 5% CO2 incubadora de ar para 24 h (para experimentos baseados em NHEJ) ou 72 h (para experimentos baseados em HDR).
5. fluorescência ativado celular classificação (FACS) de células transfectadas
- Dissocia as células por aspiração a mídia e adicionando 150 µ l de 0,25% do trypsin-EDTA por bem. Incube as células a 37 ° C por 2 min.
- Neutralize a tripsina adicionando 150 µ l (ou 1 x volume) de meios de cultura de células. Transfira a suspensão de células para um tubo de centrífuga. Gire para baixo as células numa microcentrifuga a 235 x g por 5 min.
- Aspirar o sobrenadante e ressuspender as células com 2% de soro fetal bovino (FBS) em tampão fosfato salino (PBS). As células através de uma malha de 30 µm de filtro ou filtro em um tubo de 5 mL FACS da pilha.
- Preparar um outro tubo de centrífuga com aproximadamente 100 µ l meios de cultura ou 2% FBS em PBS para a coleção de células.
- Sobre o citômetro de fluxo, portão as células com células não transfectadas como controlo negativo. Classificar e coletar as células transfectadas, de acordo com o qual fluorescência marcador está presente sobre o plasmídeo CRISPR usado. Por exemplo, se o plasmídeo carrega um gene mCherry, classificar para células de RFP-positivo.
Nota: Plasmídeos CRISPR diferentes terão diferentes marcadores selecionáveis. O conjunto de plasmídeos (pSpCas9, pSaCas9, pNmCas9, pAsCpf1 e pLbCpf1) utilizado neste estudo de avaliação carregam a proteína fluorescente laranja (Pee) ou o gene mCherry.
6. expansão de clones individuais
- Centrifugar as células classificadas em uma centrífuga de bancada superior à velocidade máxima (18.000 x g) 5 min. Aspire o sobrenadante e ressuspender o precipitado com 300 µ l meios de cultura. 200 µ l as células em uma placa de cultura de tecido de 24-poço de sementes e deixá-los a recuperar por alguns dias em uma incubadora de 37 ° C. Manter as restante 100 células µ l para a seção 7.
- Uma vez que as células começam a se tornar confluentes, passagem-los de acordo com passos 4.1.1\u20124.1.3. Semente escassamente as células numa placa de cultura de tecido de 100 mm para permitir espaço suficiente para as colônias individuais crescer. Incubar a 37 ° C em um 5% umidificado incubadora de CO2 ar.
Nota: Tente várias diluições. Células single precisam de espaço suficiente para crescer como colônias individuais. No entanto, eles também não podem ser excessivamente esparsos, como algumas linhas de célula não cresce bem quando o número de células é muito poucos. - Uma vez que as colônias estão começando a se formar, buscá-las sob o microscópio (com uma ampliação de 4x) e coloque cada clone em um poço individual de uma placa de 24 que contém células de meios de cultura. Incubar a 37 ° C em um umidificado 5% CO2 incubadora de ar até as células estão se tornando confluentes.
Nota: Uma alternativa para diluições em série e colônia picking é usar citometria de fluxo para classificar para células individuais em uma placa de 96 poços. No entanto, isso pode não funcionar para algumas linhas de célula que não crescem bem quando apenas uma célula está presente.
7. avaliação de eficiência de edição
- Extrair DNA genômico por centrifugação as células restantes 100 µ l classificados (da etapa 6.1) em uma centrífuga de bancada superior à velocidade máxima (18.000 x g) 5 min. Aspire o sobrenadante e prossiga para isolar o DNA genômico, usando um kit de extração (ver tabela de de Materiais).
- Executar a endonuclease T7 eu (T7EI) ensaio de clivagem (ver Figura 6).
- Configurar um 50 µ l contendo 10 µ l de tampão de reação de PCR (5x), 1 µ l de dNTP mix (10 mM), 2,5 µ l de definido pelo usuário para a frente da primeira demão (10 µM), 2,5 µ l de primer reverso definido pelo usuário (10 µM), 0,5 µ l de DNA polimerase, 2\u20125 µ l do modelo de DNA genômico (dependendo de quantas células têm de PCR sido classificada), então superior até 50 µ l com O ddH2(ver Tabela de materiais).
Nota: Os primers são projetados para flanquear o alvo genômico locus e o rendimento de que um produto PCR de em torno de 400\u2012700 BP normalmente uma primeira demão está posicionado mais perto para o local de corte da enzima Cas do que o outro primer, para que o resultado do ensaio T7EI é duas bandas distintas em um ag surgiu o gel (veja a Figura 6). - Executar o PCR em um termociclador com os seguintes parâmetros: 98 ° C por 3 min, 98 ° C por 30 s (etapa 2), 63 ° C por 30 s (etapa 3), 72 ° C por 30 s (etapa 4), repita as etapas 2\u20124 por mais 34 ciclos, 72 ° C por 2 min e segurar a 4 ° C.
- Resolva a reação em um gel de agarose 2% usando 1 x TAE buffer.
- Impostos especiais de consumo do produto PCR do gel com um bisturi limpo e afiado e purificar o DNA usando um kit de extração do gel, de acordo com as instruções do fabricante. Medir a concentração do produto do PCR usando um espectrofotômetro no comprimento de onda de 260 absorvância nm (ver Tabela de materiais).
- Preparar uma mistura de ensaio contendo 200 ng de DNA, 2 µ l de reação T7EI buffer (10x) e liderou até 19 µ l com O ddH2(ver Tabela de materiais).
- Re-recozer o produto do PCR em um termociclador usando os seguintes parâmetros: 95 ° C por 5 min, rampa até 25 ° C a 6 ° C/min, em seguida, mantenha a 4 ° C.
- Adicionar 5 U T7EI para o produto do PCR re-recozido, misture bem por pipetagem e incubar a 37 ° C, durante 50 min.
- Resolva o DNA T7EI-digerido em um gel de agarose de 2,5% usando 1 x TAE buffer.
- O gel de imagem, quantificar as intensidades de banda usando o ImageJ e calcular a taxa de formação de indel usando a seguinte fórmula:

onde a representa a intensidade do produto PCR intacto e b e c correspondem as intensidades dos produtos de clivagem43.- Para quantificar a intensidade de uma banda no ImageJ, primeiro desenhe uma caixa retangular em torno da banda como perto de seu limite possível. Em segundo lugar, clique em analisar e, em seguida, Definir as medições. Certifique-se de que as opções de área, quer dizer valor cinzae densidade integrada são verificadas. Sai da janela de configurações clicando okey. Em terceiro lugar, clique em Analyze e então medida. A média ou valor de RawIntDen é usado como a intensidade da banda.
- Configurar um 50 µ l contendo 10 µ l de tampão de reação de PCR (5x), 1 µ l de dNTP mix (10 mM), 2,5 µ l de definido pelo usuário para a frente da primeira demão (10 µM), 2,5 µ l de primer reverso definido pelo usuário (10 µM), 0,5 µ l de DNA polimerase, 2\u20125 µ l do modelo de DNA genômico (dependendo de quantas células têm de PCR sido classificada), então superior até 50 µ l com O ddH2(ver Tabela de materiais).

Figura 6 : Verificação de células para genoma bem sucedida edição resultados. (um) A esquema ilustrando dois comumente usados ensaios, nomeadamente a clivagem de incompatibilidade do ensaio com a endonuclease T7 enzima (T7EI) e sequenciamento de próxima geração (NGS) ou alvo amplicon sequenciamento. Os retângulos horizontais azuis indicam que DNA e círculos amarelos indicam modificações induzidas pelo sistema CRISPR-Cas. Primers para o ensaio de T7E1 são indicados em verde, enquanto primers para gerar amplicons para NGS são indicados em vermelho. (b) Design de primer sequências para o ensaio de clivagem T7EI e para NGS. Aqui, o locus genômico alvo é exon 45 do gene humano CACNA1D. O site da modificação pretendida é indicado por um asterisco. Clique aqui para ver uma versão maior desta figura.
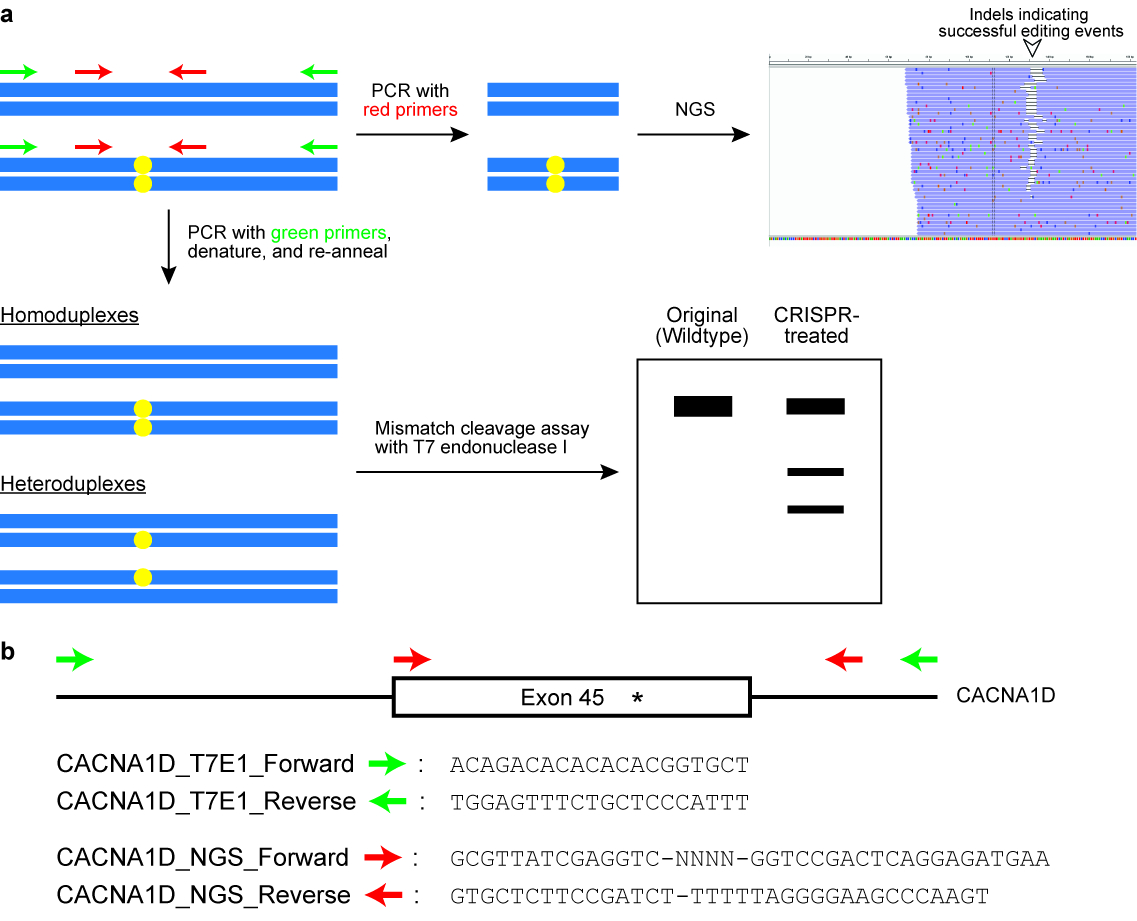{kind=link}
- Realizar o sequenciamento amplicon alvo (ver Figura 6).
- Projeto do PCR primers para amplificar o locus genômico do alvo. Posicione um dos primers para ser menos de 100 bp mas mais de 20 bp do protospacer.
Nota: Normalmente, o tamanho total do produto do PCR é projetado para ser em torno de 150\u2012300 bp (ver Figura 6). - Acrescentar sequências adicionais para as primeiras demão da seguinte forma: (a) 5' \u2012GCGTTATCGAGGTC - NNNN-[frente Primer] – 3'; (b) 5 com a sua cara ' - GTGCTCTTCCGATCT-[reverso Primer] – 3 '.
- Configurar um 50 mistura de reação de PCR µ l contendo 10 µ l de tampão de reação de PCR (5x), 1 µ l de dNTP (10 mM), 5 µ l de primer um (10 µM), 5 µ l de primer b (10 µM), 0,5 DNA-polimerase µ l, 2\u20125 modelo µ l genoma de DNA (dependendo de quantas células têm sido classificadas) , em seguida, cubra a 50 µ l com DDQ20.
- Executar o PCR em um termociclador com os seguintes parâmetros: 98 ° C por 3 min, 98 ° C por 30 s (etapa 2), 63 ° C por 30 s (etapa 3), 72 ° C por 15 s (etapa 4), repita as etapas 2\u20124 por mais 34 ciclos, 72 ° C por 2 min e segurar a 4 ° C.
- Resolver a reação em gel de agarose 2% e purificar o produto do PCR usando um kit de extração do gel de acordo com as instruções do fabricante. Quantificar o DNA usando um espectrofotômetro no comprimento de onda de 260 absorvância nm (ver a Tabela de materiais).
- Sintetizar o seguinte rodada 2 iniciadores de PCR: (c) 5' \u2012 3 – AATGATACGGCGACCACCGAGATCTACACCCTACACGAGCGTTATCGAGGTC'; (d) 5' \u2012CAAGCAGAAGACGGCATACGAGAT-[código de barras] - GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3'
- Configurar uma reação de PCR de 20 µ l de mistura contendo 4 μL de tampão de reação de PCR (5x), 0,4 µ l de dNTP (10 mM), 2 µ l de cartilha c (10 µM), 2 µ l de cartilha d (10 µM), 0,2 µ l de DNA polimerase, 2 µ l do molde de ADN (da etapa 7.3.5 diluído 1: 100) e 9,4 µ l de DDQ2O.
Nota: O fator de diluição para o modelo de ADN pode variar dependendo de sua concentração original. Se a concentração é em torno de 20\u201240 ng / µ l, use um fator de diluição de 1: 100. Além disso, escolher um código de barras diferente para cada amostra experimental, se o mesmo primer um e b primeira demão são utilizados na etapa 7.3.3. - Executar o PCR em um termociclador com os seguintes parâmetros: 98 ° C por 3 min, 98 ° C por 30 s (etapa 2), 65 ° C por 30 s (etapa 3), 72 ° C por 30 s (etapa 4), repita as etapas 2\u20124 por mais 14 ciclos, 72 ° C por 2 min e 4 ° C segurar.
- Resolver a 5 µ l de cada reação em um gel de agarose 2% para determinar o sucesso do PCRs. Combine todas as amostras juntas (supondo que foi usado um código de barras diferente para cada amostra) e limpar o DNA agrupado usando um kit de purificação de PCR de acordo com do fabricante instruções. Se alguns dos PCRs apresentam mais de uma banda (indicando a presença de produtos não específicos), execute uma etapa de extração do gel de adicionais.
- A biblioteca em um instrumento de sequenciamento de alta taxa de transferência de sequências (ver Tabela de materiais) de acordo com as instruções do fabricante para produzir emparelhados 151 leituras de bp. O primer de sequenciamento de leitura 1 é projetado e tem de ser fornecido separadamente. Sua sequência é a seguinte: Read1_seq: 5'-CCACCGAGATCTACACCCTACACGAGCGTTATCGAGGTC-3'. A primeira demão de sequenciamento de leitura 2 e primeira demão de sequenciamento de índice são padrão e são fornecidos no cartucho de reagente.
Nota: O ensaio de T7EI e sequenciamento de amplicons alvo são comumente usados para verificar a eficiência do genoma de edição. No entanto, outros experimentos podem ser realizados para avaliar a eficiência de edição, dependendo do tipo de modificações do DNA introduzido. Por exemplo, se um sítio de restrição é criado no local de destino, um ensaio de polimorfismo (RFLP) de comprimento de fragmento de restrição pode ser executado. É semelhante para o ensaio de T7EI, exceto que uma endonuclease de restrição é usada para digerir o produto do PCR, em vez disso.
- Projeto do PCR primers para amplificar o locus genômico do alvo. Posicione um dos primers para ser menos de 100 bp mas mais de 20 bp do protospacer.
8. seleção de clones individuais
- Da etapa 6.3, dividi as células, uma vez que eles começam a ficar confluente. Para cada clone individual, coletar as células restantes e extrair DNA genômico de acordo com o passo 7.1.
- Realize o ensaio de T7EI para todos os clones individuais de acordo com a seção 7.2, com exceção de uma modificação. Amplificar o locus genômico alvo de sua células e na etapa 7.2.5, em vez de usar 200 ng do teste de DNA só, mistura 100 ng de DNA de teste com 100 ng de DNA de sua.
Nota: A razão para o passo modificado é que alguns clones podem ter sofrido biallelic bem sucedida conversão e são homozigotos mutantes. Em tais casos, não haverá nenhum bandas de clivagem no ensaio de T7EI se o DNA de sua não é misturado em. - O local de destino em clones que apresentam bandas de clivagem no ensaio de T7EI de sequência.
- Amplifica o locus genômico modificado de acordo com passos 7.2.1–7.2.4.
- Configurar a seguinte reação de clonagem: 4 µ l do produto do PCR, 1 µ l da solução de sal, 1 µ l de vetor TOPO (ver a Tabela de materiais).
- Misture suavemente pipetando e incubar a temperatura ambiente pelo menos 5 min.
- Transformar a 3 µ l da reação mistura quimicamente competentes de Escherichia coli células (tais como TOP10 ou Stbl3) (ver arquivo complementar 1). Espalhou-se a células bacterianas transformadas em uma placa de ágar LB com 50 canamicina μg/mL.
- No dia seguinte, inocule pelo menos 10 colônias em LB mídia líquida contendo 50 canamicina μg/mL.
- Quando as culturas bacterianas são turvas, isolar o plasmídeo usando um miniprep kit (veja a Tabela de materiais) e sequência-los usando o M13 padrão para a frente ou reverso primer M13.
- Realize um western blot (também conhecido como immunoblot) para determinar a ausência ou a presença da proteína alvo (se o genoma edição experimento envolve bater para fora um gene codificantes de proteínas através de mutações frameshift). Consulte o arquivo complementar 1.
Nota: Outras experiências podem ser executadas para identificar o clone carregando as modificações genômicas desejadas. Por exemplo, um ensaio fenotípico pode ser realizado se bater para fora de um determinado gene é conhecido por causar certas mudanças no comportamento celular.
Resultados
Para executar um genoma edição experimento, um plasmídeo CRISPR expressando uma sgRNA como alvo que o locus de interesse precisa ser clonado. Primeiro, o plasmídeo é digerido com uma enzima de restrição (normalmente um tipo de enzima do IIs) para linearizá-lo. É recomendável para resolver o produto digerido em um gel de agarose 1% ao lado de um plasmídeo não digerido para distinguir entre uma digestão completa e parcial. Como plasmídeo não digerido supercoiled, eles tendem a correr mais rápido do que suas...
Discussão
O sistema CRISPR-Cas é uma poderosa, revolucionária tecnologia para projetar os genomas e transcriptomes de plantas e animais. Numerosas espécies bacterianas foram encontradas para conter sistemas CRISPR-Cas, que potencialmente podem ser adaptados para o genoma e transcriptoma fins44de engenharia. Embora a endonuclease Cas9 de Streptococcus pyogenes (SpCas9) foi a primeira enzima a ser implantado com sucesso em células humanas21,22<...
Divulgações
Os autores não têm concorrentes interesses financeiros.
Agradecimentos
M.H.T. é suportado por uma agência de tecnologia da ciência e da pesquisa concessão de escritório comum do Conselho (1431AFG103), um Conselho Nacional de pesquisa médica grant (OFIRG/0017/2016), Fundação Nacional de pesquisa concede (NRF2013-THE001-046 e NRF2013-THE001-093), um Concessão do Ministério da educação Tier 1 (RG50/17 (S)), uma startup grant da Universidade Tecnológica de Nanyang e fundos para a competição internacional geneticamente engenharia máquina (Medren) da Universidade Tecnológica de Nanyang.
Materiais
| Name | Company | Catalog Number | Comments |
| T4 Polynucleotide Kinase (PNK) | NEB | M0201 | |
| Shrimp Alkaline Phosphatase (rSAP) | NEB | M0371 | |
| Tris-Acetate-EDTA (TAE) Buffer, 50X | 1st Base | BUF-3000-50X4L | Dilute to 1X before use. The 1X solution contains 40 mM Tris, 20 mM acetic acid, and 1 mM EDTA. |
| Tris-EDTA (TE) Buffer, 10X | 1st Base | BUF-3020-10X4L | Dilute to 1X before use. The 1X solution contains 10 mM Tris (pH 8.0) and 1 mM EDTA. |
| BbsI | NEB | R0539 | |
| BsmBI | NEB | R0580 | |
| T4 DNA Ligase | NEB | M0202 | 400,000 units/ml |
| Quick Ligation Kit | NEB | M2200 | An alternative to T4 DNA Ligase. |
| Rapid DNA Ligation Kit | Thermo Scientific | K1423 | An alternative to T4 DNA Ligase. |
| Zero Blunt TOPO PCR Cloning Kit | Thermo Scientific | 451245 | The salt solution comes with the TOPO vector in the kit. |
| NEBuilder HiFi DNA Assembly Master Mix | NEB | E2621L | Kit for Gibson assembly. |
| One Shot Stbl3 Chemically Competent E.Coli | Thermo Scientific | C737303 | |
| LB Broth (Lennox), powder | Sigma Aldrich | L3022 | Reconstitute in ddH20, and autoclave before use |
| LB Broth with Agar (Lennox), powder | Sigma Aldrich | L2897 | Reconstitute in ddH20, and autoclave before use |
| SOC media | - | - | 2.5 mM KCl, 10 mM MgCl2, 20 mM glucose in 1 L of LB Broth |
| Ampicillin (Sodium), USP Grade | Gold Biotechnology | A-301 | |
| REDiant 2X PCR Mastermix | 1st Base | BIO-5185 | |
| Agarose | 1st Base | BIO-1000 | |
| T7 Endonuclease I | NEB | M0302 | |
| Plasmid DNA Extraction Miniprep Kit | Favorgen | FAPDE 300 | |
| Dulbecco's Modified Eagle Medium (DMEM), High Glucose | Hyclone | SH30081.01 | 4.5 g/L Glucose, no L-glutamine, HEPES and Sodium Pyruvate |
| L-Glutamine, 200mM | Gibco | 25030 | |
| Penicillin-Streptomycin, 10, 000U/mL | Gibco | 15140 | |
| 0.25% Trypsin-EDTA, 1X | Gibco | 25200 | |
| Fetal Bovine Serum | Hyclone | SV30160 | FBS is heat inactivated before use at 56 oC for 30 min |
| Phosphate Buffered Saline, 1X | Gibco | 20012 | |
| jetPRIME transfection reagent | Polyplus Transfection | 114-75 | |
| QuickExtract DNA Extraction Solution, 1.0 | Epicentre | LUCG-QE09050 | |
| ISOLATE II Genomic DNA Kit | Bioline | BIO-52067 | An alternative to QuickExtract |
| Q5 High-Fidelity DNA Polymerase | NEB | M0491 | |
| Deoxynucleotide (dNTP) Solution Mix | NEB | N0447 | |
| 6X DNA Loading Dye | Thermo Scientific | R0611 | 10 mM Tris-HCl (pH 7.6) 0.03% bromophenol blue, 0.03% xylene cyanol FF, 60% glycerol, 60 mM EDTA |
| Protease Inhibitor Cocktail, Set3 | Merck | 539134 | |
| Nitrocellulose membrane, 0.2µm | Bio-Rad | 1620112 | |
| Tris-glycine-SDS buffer, 10X | Bio-Rad | 1610772 | Dilute to 1X before use. The 1x solution contains 25 mM Tris, 192 mM glycine, and 0.1% SDS. |
| Tris-glycine buffer, 10X | 1st base | BUF-2020 | Dilute to 1X before use. The 1x solution contains 25 mM Tris and 192 mM glycine. |
| Ponceau S solution | Sigma Aldrich | P7170 | |
| TBS, 20X | 1st base | BUF-3030 | Dilute to 1X before use. The 1x solution contains 25 mM Tris-HCl (pH 7.5) and 150 mM NaCl. |
| Tween 20 | Sigma Aldrich | P9416 | |
| Skim Milk for immunoassay | Nacalai Tesque | 31149-75 | |
| WesternBright Sirius-femtogram HRP | Advansta | K12043 | |
| Antibody for β-actin (C4) | Santa Cruz Biotechnology | sc-47778 | Lot number: C0916 |
| MiSeq system | Illumina | SY-410-1003 | |
| NanoDrop spectrophotometer | Thermo Scientific | ND-2000 | |
| Qubit fluorometer | Thermo Scientific | Q33226 | |
| EVOS FL Cell Imaging System | Thermo Scientific | AMF4300 | |
| CRISPR plasmid: pSpCas9(BB)-2A-GFP (PX458) | Addgene | 48138 | Single vector system: The gRNA is expressed from the same plasmid. |
| CRISPR plasmid: pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA | Addgene | 61591 | Single vector system: The gRNA is expressed from the same plasmid. |
| CRISPR plasmid: xCas9 3.7 | Addgene | 108379 | Dual vector system: The gRNA is expressed from a different plasmid. |
| CRISPR plasmid: pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | 42230 | Single vector system: The gRNA is expressed from the same plasmid. |
| CRISPR plasmid: hCas9 | Addgene | 41815 | Dual vector system: The gRNA is expressed from a different plasmid. |
| CRISPR plasmid: eSpCas9(1.1) | Addgene | 71814 | Single vector system: The gRNA is expressed from the same plasmid. |
| CRISPR plasmid: VP12 (SpCas9-HF1) | Addgene | 72247 | Dual vector system: The gRNA is expressed from a different plasmid. |
Referências
- Epinat, J. C., et al. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Research. 31 (11), 2952-2962 (2003).
- Arnould, S., et al. Engineered I-CreI derivatives cleaving sequences from the human XPC gene can induce highly efficient gene correction in mammalian cells. Journal of Molecular Biology. 371 (1), 49-65 (2007).
- Chapdelaine, P., Pichavant, C., Rousseau, J., Paques, F., Tremblay, J. P. Meganucleases can restore the reading frame of a mutated dystrophin. Gene Therapy. 17 (7), 846-858 (2010).
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics. 188 (4), 773-782 (2011).
- Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S., Gregory, P. D. Genome editing with engineered zinc finger nucleases. Nature Reviews Genetics. 11 (9), 636-646 (2010).
- Miller, J. C., et al. A TALE nuclease architecture for efficient genome editing. Nature Biotechnology. 29 (2), 143-148 (2011).
- Zhang, F., et al. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nature Biotechnology. 29 (2), 149-153 (2011).
- Boch, J., et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 326 (5959), 1509-1512 (2009).
- Moscou, M. J., Bogdanove, A. J. A simple cipher governs DNA recognition by TAL effectors. Science. 326 (5959), 1501 (2009).
- Hsu, P. D., Lander, E. S., Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 157 (6), 1262-1278 (2014).
- Sander, J. D., Joung, J. K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nature Biotechnology. 32 (4), 347-355 (2014).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nishimasu, H., et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 156 (5), 935-949 (2014).
- Yamano, T., et al. Crystal Structure of Cpf1 in Complex with Guide RNA and Target DNA. Cell. 165 (4), 949-962 (2016).
- Swarts, D. C., Mosterd, C., van Passel, M. W., Brouns, S. J. CRISPR interference directs strand specific spacer acquisition. PLoS One. 7 (4), e35888 (2012).
- Zetsche, B., et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 163 (3), 759-771 (2015).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 507 (7490), 62-67 (2014).
- Hu, J. H., et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 556 (7699), 57-63 (2018).
- Kleinstiver, B. P., et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nature Biotechnology. 33 (12), 1293-1298 (2015).
- Kleinstiver, B. P., et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 523 (7561), 481-485 (2015).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Jinek, M., et al. RNA-programmed genome editing in human cells. Elife. 2, e00471 (2013).
- Cho, S. W., Kim, S., Kim, J. M., Kim, J. S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nature Biotechnology. 31 (3), 230-232 (2013).
- Wang, Y., et al. Systematic evaluation of CRISPR-Cas systems reveals design principles for genome editing in human cells. Genome Biology. 19 (1), 62 (2018).
- Ran, F. A., et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 520 (7546), 186-191 (2015).
- Hou, Z., et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proceedings of the National Academy of Sciences U S A. 110 (39), 15644-15649 (2013).
- Kim, E., et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nature Communications. 8, 14500 (2017).
- Edraki, A., et al. A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Molecular Cell. , (2018).
- Chatterjee, P., Jakimo, N., Jacobson, J. M. Minimal PAM specificity of a highly similar SpCas9 ortholog. Science Advances. 4 (10), (2018).
- Muller, M., et al. Streptococcus thermophilus CRISPR-Cas9 Systems Enable Specific Editing of the Human Genome. Mol Therapy. 24 (3), 636-644 (2016).
- Esvelt, K. M., et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nature Methods. 10 (11), 1116-1121 (2013).
- Boratyn, G. M., et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Research. 41 (Web Server issue), W29-W33 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M., Valen, E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Research. 42 (Web Server issue), W401-W407 (2014).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Haeussler, M., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Bae, S., Park, J., Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 30 (10), 1473-1475 (2014).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Richardson, C. D., Ray, G. J., Bray, N. L., Corn, J. E. Non-homologous DNA increases gene disruption efficiency by altering DNA repair outcomes. Nature Communications. 7, 12463 (2016).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. 6 (5), 343-345 (2009).
- Zhang, J. P., et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biology. 18 (1), 35 (2017).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Shmakov, S., et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nature Reviews Microbiology. 15 (3), 169-182 (2017).
- Moreno-Mateos, M. A., et al. CRISPR-Cpf1 mediates efficient homology-directed repair and temperature-controlled genome editing. Nature Communications. 8 (1), 2024 (2017).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 3, e04766 (2014).
- Yang, L., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Watanabe, K., et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nature Biotechnology. 25 (6), 681-686 (2007).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados