
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
Genome Editing in Mammalian Cell Lines using CRISPR-Cas
In This Article
Summary
CRISPR-Cas is a powerful technology to engineer the complex genomes of plants and animals. Here, we detail a protocol to efficiently edit the human genome using different Cas endonucleases. We highlight important considerations and design parameters to optimize editing efficiency.
Abstract
The clustered regularly interspaced short palindromic repeats (CRISPR) system functions naturally in bacterial adaptive immunity, but has been successfully repurposed for genome engineering in many different living organisms. Most commonly, the wildtype CRISPR associated 9 (Cas9) or Cas12a endonuclease is used to cleave specific sites in the genome, after which the DNA double-stranded break is repaired via the non-homologous end joining (NHEJ) pathway or the homology-directed repair (HDR) pathway depending on whether a donor template is absent or present respectively. To date, CRISPR systems from different bacterial species have been shown to be capable of performing genome editing in mammalian cells. However, despite the apparent simplicity of the technology, multiple design parameters need to be considered, which often leave users perplexed about how best to carry out their genome editing experiments. Here, we describe a complete workflow from experimental design to identification of cell clones that carry desired DNA modifications, with the goal of facilitating successful execution of genome editing experiments in mammalian cell lines. We highlight key considerations for users to take note of, including the choice of CRISPR system, the spacer length, and the design of a single-stranded oligodeoxynucleotide (ssODN) donor template. We envision that this workflow will be useful for gene knockout studies, disease modeling efforts, or the generation of reporter cell lines.
Introduction
The ability to engineer the genome of any living organism has many biomedical and biotechnological applications, such as the correction of disease-causing mutations, construction of accurate cellular models for disease studies, or generation of agricultural crops with desirable traits. Since the turn of the century, various technologies have been developed for genome engineering in mammalian cells, including meganucleases1,2,3, zinc finger nucleases4,5, or transcription activator-like effector nucleases (TALENs)6,7,8,9. However, these earlier technologies are either difficult to program or tedious to assemble, thereby hampering their widespread adoption in research and the industry.
In recent years, the clustered regularly interspaced short palindromic repeats (CRISPR)- CRISPR-associated (Cas) system has emerged as a powerful new genome engineering technology10,11. Originally an adaptive immune system in bacteria, it has been successfully deployed for genome modification in plants and animals, including humans. A primary reason why CRISPR-Cas has gained so much popularity in such a short time is that the element that brings the key Cas endonuclease, such as Cas9 or Cas12a (also known as Cpf1), to the correct location in the genome is simply a short piece of chimeric single guide RNA (sgRNA), which is straightforward to design and cheap to synthesize. After being recruited to the target site, the Cas enzyme functions as a pair of molecular scissors and cleaves the bound DNA with its RuvC, HNH, or Nuc domains12,13,14. The resulting double stranded break (DSB) is subsequently repaired by the cells via either the non-homologous end joining (NHEJ) or homology-directed repair (HDR) pathway. In the absence of a repair template, the DSB is repaired by the error-prone NHEJ pathway, which can give rise to pseudo-random insertion or deletion of nucleotides (indels) at the cut site, potentially causing frameshift mutations in protein-coding genes. However, in the presence of a donor template that contains the desired DNA changes, the DSB is repaired by the high fidelity HDR pathway. Common types of donor templates include single-stranded oligonucleotides (ssODNs) and plasmids. The former is typically used if the intended DNA changes are small (for example, alteration of a single base pair), while the latter is typically used if one wishes to insert a relatively long sequence (for example, the coding sequence of a green fluorescent protein or GFP) into the target locus.
The endonuclease activity of the Cas protein requires the presence of a protospacer adjacent motif (PAM) at the target site15. The PAM of Cas9 is at the 3’ end of the protospacer, while the PAM of Cas12a (also called Cpf1) is at the 5’ end instead16. The Cas-guide RNA complex is unable to introduce a DSB if the PAM is absent17. Hence, the PAM places a constraint on the genomic locations where a particular Cas nuclease is able to cleave. Fortunately, Cas nucleases from different bacterial species typically exhibit different PAM requirements. Hence, by integrating various CRISPR-Cas systems into our engineering toolbox, we can expand the range of sites that may be targeted in a genome. Moreover, a natural Cas enzyme can be engineered or evolved to recognize alternative PAM sequences, further widening the scope of genomic targets accessible to manipulation18,19,20.
Although multiple CRISPR-Cas systems are available for genome engineering purposes, most users of the technology have relied mainly on the Cas9 nuclease from Streptococcus pyogenes (SpCas9) for multiple reasons. First, it requires a relatively simply NGG PAM, unlike many other Cas proteins that can only cleave in the presence of more complex PAMs. Second, it is the first Cas endonuclease to be successfully deployed in human cells21,22,23,24. Third, SpCas9 is by far the best characterized enzyme to date. If a researcher wishes to use another Cas nuclease, he or she would often be unclear about how best to design the experiment and how well other enzymes will perform in different biological contexts compared to SpCas9.
To provide clarity to the relative performance of different CRISPR-Cas systems, we have recently performed a systematic comparison of five Cas endonucleases – SpCas9, the Cas9 enzyme from Staphylococcus aureus (SaCas9), the Cas9 enzyme from Neisseria meningitidis (NmCas9), the Cas12a enzyme from Acidaminococcus sp. BV3L6 (AsCas12a), and the Cas12a enzyme from Lachnospiraceae bacterium ND2006 (LbCas12a)25. For a fair comparison, we evaluated the various Cas nucleases using the same set of target sites and other experimental conditions. The study also delineated design parameters for each CRISPR-Cas system, which would serve as a useful reference for users of the technology. Here, to better enable researchers to make use of the CRISPR-Cas system, we provide a step-by-step protocol for optimal genome engineering with different Cas9 and Cas12a enzymes (see Figure 1). The protocol not only includes experimental details but also important design considerations to maximize the likelihood of a successful genome engineering outcome in mammalian cells.

Figure 1: An overview of the workflow to generate genome edited human cell lines. Please click here to view a larger version of this figure.
{kind=link}
Protocol
1. Design of sgRNAs
- Select an appropriate CRISPR-Cas system.
- First, inspect the target region for the PAM sequences of all Cas9 and Cas12a nucleases that have been shown to be functional in mammalian cells16,21-32. Five frequently used enzymes are given in Table 1 together with their respective PAMs.
NOTE: Besides the endonucleases listed in Table 1, there are other less commonly used Cas enzymes that have been successfully deployed in mammalian cells as well, such as a Cas9 nuclease from Streptococcus thermophilus (St1Cas9) that recognizes the PAM NNAGAAW. If the desired target site does not contain a known PAM, then one would not be able to use the CRISPR-Cas system for genome engineering. - Second, consider any known properties of the target genomic locus or gene. Some properties to take into consideration include gene expression levels or chromatin accessibility and whether there are other closely related sequences as well.
NOTE: Certain enzymes are better suited for particular biological contexts. For example, to edit a repetitive genomic locus or a gene with several other close paralogs, it is recommended to use either AsCas12a (due to its lower tolerance for mismatches between the sgRNA and the target DNA than SpCas9 and LbCas12a) or SaCas9 (due to its requirement for longer spacers, which provides higher targeting specificity)25.
- First, inspect the target region for the PAM sequences of all Cas9 and Cas12a nucleases that have been shown to be functional in mammalian cells16,21-32. Five frequently used enzymes are given in Table 1 together with their respective PAMs.
| Cas Endonuclease | PAM | Optimal spacer length |
| SpCas9 | NGG | 17-22 nt inclusive |
| SaCas9 | NNGRRT | ≥ 21 nt |
| NmCas9 | NNNNGATT | ≥ 19 nt |
| AsCas12a and LbCas12a | TTTV | ≥ 19 nt |
Table 1: Some commonly used Cas enzymes with their cognate PAMs and optimal sgRNA lengths. N = Any nucleotide (A, T, G, or C); R = A or G; V = A, C, or G.
- Select a suitable spacer sequence. Identify as unique a sequence as possible to minimize the risk of off-target cleavage events, either by examining the target genome with BLAST33 or by using several freely available online tools, such as: (a) Program from Feng Zhang’s laboratory34 (http://crispr.mit.edu/); (b) CHOPCHOP35 (http://chopchop.cbu.uib.no/); (c) E-CRISP36 (http://www.e-crisp.org/E-CRISP/); (d) CRISPOR37 (http://crispor.tefor.net/); (e) Cas-OFFinder38 (http://www.rgenome.net/cas-offinder/).
NOTE: The optimal length of the spacer can vary from 17 to 25 nucleotides (nt) inclusive, depending on which Cas enzyme is used (see Table 1). For Cas9, the spacer is upstream of the PAM, while for Cas12a, the spacer is downstream of the PAM. Additionally, HDR efficiency drops rapidly with increasing distance from the cut site. Hence, for precise DNA editing, position the sgRNA as close as possible to the intended modification site. - Synthesize DNA oligonucleotides with the appropriate overhangs for the CRISPR plasmid that is being used.
- Add a G nucleotide in front of the spacer if the first position of the guide is not a G. Determine the reverse complementary sequence of the spacer. Add in the necessary overhangs for cloning purposes.
NOTE: By way of illustration, for the plasmids used in our evaluation study25, the oligonucleotides to be synthesized are shown in Table 2. Use the example given in Figure 2 as a guide, if necessary. Many CRISPR plasmids are available from commercial sources (e.g., Addgene). Some of the more popular plasmids are given in the Table of Materials.
- Add a G nucleotide in front of the spacer if the first position of the guide is not a G. Determine the reverse complementary sequence of the spacer. Add in the necessary overhangs for cloning purposes.
| CRISPR Plasmid | Sequence |
| pSpCas9 and pSaCas9 | Sense: 5' - CACC(G)NNNNNNNNNNNNNNNNNNNNN - 3' Antisense: 3' - (C)NNNNNNNNNNNNNNNNNNNNNCAAA - 5' |
| pNmCas9 | Sense: 5' - CACC(G)NNNNNNNNNNNNNNNNNNNNN - 3' Antisense: 3' - (C)NNNNNNNNNNNNNNNNNNNNNCAAC - 5' |
| pAsCas12a and pLbCas12a | Sense: 5' - AGATNNNNNNNNNNNNNNNNNNNNN - 3' Antisense: 3' - NNNNNNNNNNNNNNNNNNNNNAAAA - 5' |
Table 2: Oligonucleotides required for cloning sgRNA sequences into CRISPR plasmids used in a recent evaluation study25. The overhangs are italicized.

Figure 2: An example illustrating how to select target sites and design oligonucleotides for cloning into CRISPR plasmids. The target genomic locus here is exon 45 of the human CACNA1D gene. The PAMs for SpCas9 and SaCas9 are NGG and NNGRRT respectively and are highlighted in red, while the PAM for AsCas12a and LbCas12a is TTTN and is highlighted in green. The red horizontal bar indicates the protospacer for SpCas9 and SaCas9, while the green horizontal bar indicates the protospacer for the two Cas12a enzymes. Please click here to view a larger version of this figure.
{kind=link}
2. Cloning of oligonucleotides into a backbone vector
- Phosphorylate and anneal sense and antisense oligonucleotides.
- If the oligonucleotides are lyophilized, resuspend them to a 100 µM concentration in tris- ethylenediaminetetraacetic acid (EDTA) (TE buffer, see Table of Materials) or ddH2O.
- Prepare a 10 µL reaction mix containing 1 µL of sense oligonucleotide, 1 µL of antisense oligonucleotide, 1 µL of T4 DNA ligase buffer (10x), 1 µL of T4 polynucleotide kinase (PNK), and 6 µL of ddH2O. Mix well by pipetting and place the reaction mix in a thermal cycler using the following parameters: 37 °C for 30 min, 95 °C for 5 min, and ramp down to 25 °C at 6 °C/min.
- Dilute the reaction mix 1:100 in ddH2O (e.g., 2 µL reaction mix + 198 µL ddH2O).

Figure 3: An example of a CRISPR plasmid. (a) A map indicating different important features of the plasmid. Here, the EF-1a promoter drives the expression of Cas9, while the U6 promoter drives the expression of the sgRNA. Amp(R) indicates an ampicillin-resistance gene in the plasmid. (b) The sequence of the “BbsI-BplI cloning site” in the plasmid. The recognition sequence of BbsI is GAAGAC and is indicated in red, while the recognition sequence of BplI is GAG-N5-CTC and is indicated in green. (c) Primers that can be used for colony PCR to check whether the sgRNA sequence has been successfully cloned into the plasmid. The hU6_forward primer is indicated by a purple arrow on the plasmid map, while the universal M13R(-20) primer is indicated by a pink arrow on the plasmid map. Please click here to view a larger version of this figure.
{kind=link}
- Digest the CRISPR plasmid with an appropriate restriction enzyme.
NOTE: Cloning of sgRNAs typically rely on Golden Gate assembly with type IIs restriction enzymes. Different enzymes may be used for different CRISPR plasmids. For pSpCas9, use BbsI or BplI (see Figure 3). For pSaCas9, pNmCas9, pAsCas12a, and pLbCas12a, use BsmBI.- Prepare a 20 µL reaction mix containing 1 µg of circular plasmid vector, 2 µL of buffer (10x), 1 µL of restriction enzyme (e.g., BbsI, BplI, or BsmBI), and ddH2O to a final volume of 20 µL. Incubate the reaction at 37 °C for 2.5 h.
- Add 1 µL of shrimp alkaline phosphatase (SAP) to the reaction, and incubate at 37 °C for another 30 min.
- Quench the reaction by adding 5 µL of 6x DNA loading dye (see Table of Materials), mix well, and resolve the reaction on a 0.8% agarose gel with 1x tris-acetate-EDTA (TAE) buffer. Then, excise the correct band and proceed to gel purify the linearized vector.
- Ligate the annealed oligonucleotides into the digested CRISPR plasmid.
- Prepare a 10 µL reaction mix: 50 ng of linearized vector, 1 µL of diluted annealed oligonucleotides, 1 µL of T4 DNA ligase buffer (10x), 1 µL of T4 DNA ligase, and ddH2O to a final volume of 10 µL (see Table of Materials). Incubate the reaction at 16 °C overnight or at room temperature for 2 h.
NOTE: To speed up the ligation process, use concentrated T4 DNA ligase and incubate the reaction mix at room temperature for 15 min (see Table of Materials).
- Prepare a 10 µL reaction mix: 50 ng of linearized vector, 1 µL of diluted annealed oligonucleotides, 1 µL of T4 DNA ligase buffer (10x), 1 µL of T4 DNA ligase, and ddH2O to a final volume of 10 µL (see Table of Materials). Incubate the reaction at 16 °C overnight or at room temperature for 2 h.
- Transform ligated products into chemically competent E. coli cells (see Supplementary File 1). Spread the transformed bacterial cells on an LB agar plate with 100 µg/mL ampicillin.
- Perform colony polymerase chain reaction (PCR) to identify the bacteria with insert.
- Prepare two sets of sterile PCR strip tubes. In Set 1, add 4.7 µL of ddH2O, while in Set 2, add 50 µL of LB broth with appropriate antibiotic (e.g., 100 µg/mL ampicillin).
- With a sterile pipette tip, pick a colony off the plate, swipe it briefly in a Set 1 tube, and leave the tip in a Set 2 tube. Repeat for a few colonies, making sure to use different PCR tubes each time.
NOTE: Typically, screening four colonies is sufficient. However, this may vary depending on the cloning efficiency. - Add the following reagents to each Set 1 tube (for a 10 µL PCR): 5 µL of 2x PCR master mix (with loading dye, see Table of Materials), 0.15 µL of sense or antisense oligonucleotide (100 µM), 0.15 µL of primer (100 µM) targeting the CRISPR plasmid downstream or upstream of the sgRNA cassette respectively (see Figure 3).
NOTE: The PCR product should ideally yield a product size of approximately 150 bp or larger, so that any positive bands are not mistaken as primer dimers. - Run the PCRs in a thermal cycler using the following parameters: 95 °C for 3 min, 95 °C for 30 s (step 2), 60 °C for 30 s (step 3), 72 °C for 30 s (step 4), repeat steps 2‒4 for another 34 cycles, 72 °C for 5 min, and hold at 4 °C.
NOTE: The annealing temperature of 60 °C may need to be optimized for the primers designed. The elongation time of 30 s can also vary depending on the expected PCR amplicon size and the DNA polymerase used. - Resolve the reactions on a 1% agarose gel using 1x TAE buffer.
- Inoculate a colony that yields a positive band in the PCR by transferring 50 µL of its culture from the corresponding Set 2 tube into a larger conical tube containing 5 mL LB with an appropriate antibiotic. Let the culture grow overnight in a 37 °C incubator-shaker.
- Isolate plasmids from the overnight culture using a miniprep kit (see Table of Materials) and sequence the sample using the colony PCR primer that is not the sense or antisense oligonucleotide (hU6_forward or M13R(-20) in Figure 3).
NOTE: If necessary, perform a maxiprep of the sequence-verified CRISPR plasmid to obtain a larger amount for downstream experiments.
3. Design and synthesis of repair templates
NOTE: For precision genome engineering, a template specifying the desired DNA modifications needs to be provided together with the CRISPR reagents. For small DNA edits such as alteration of a single nucleotide, ssODN donor templates are most suitable (see section 3.1). For larger DNA edits such as insertion of a GFP tag 5’ or 3’ of a particular protein-coding gene, plasmid donor templates are most suitable (see section 3.2).
- Design and synthesize a ssODN donor template (see Figure 4).
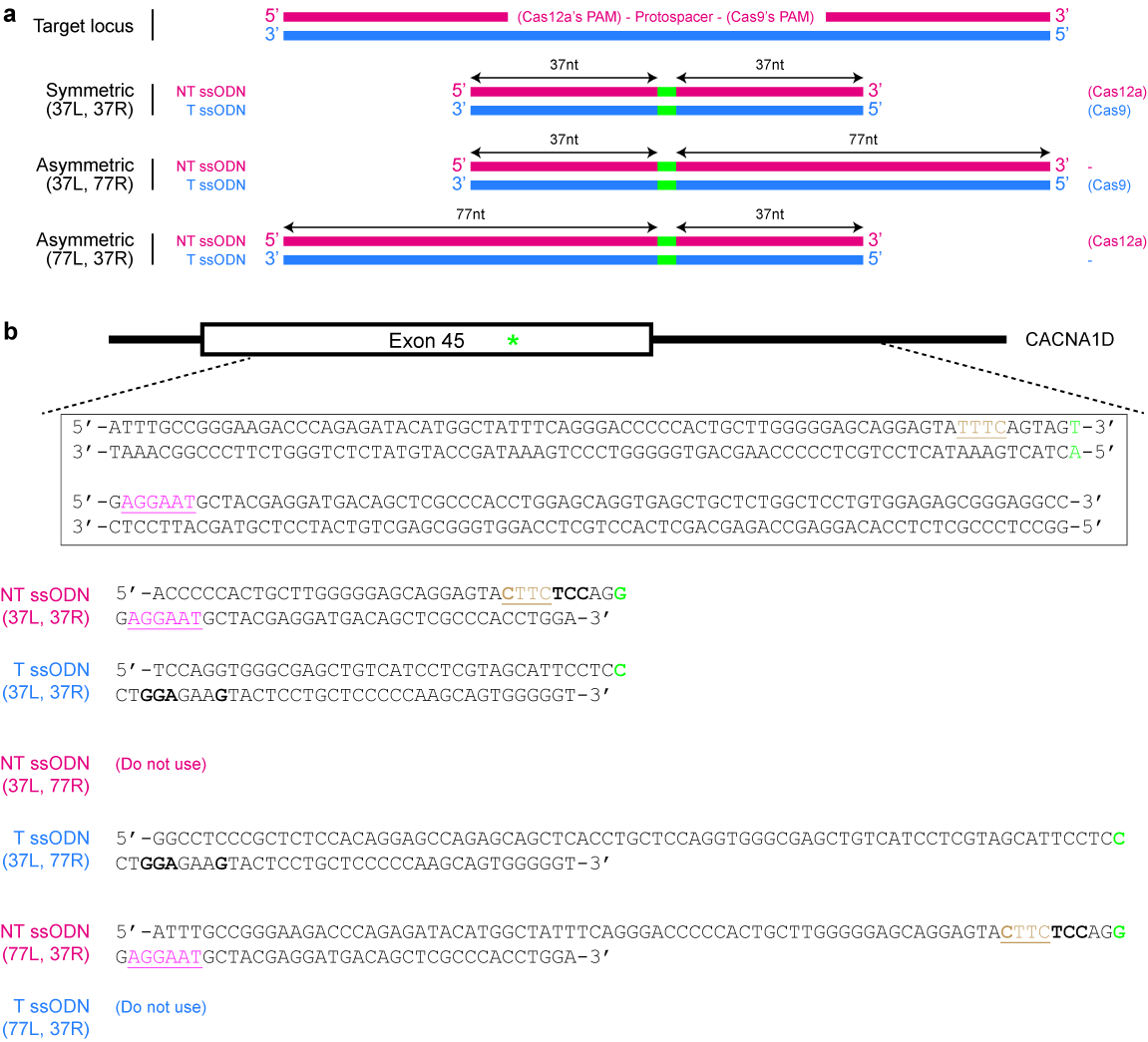
- Determine the correct strand whose sequence the template should follow.
NOTE: Cas12a exhibits a preference for ssODNs of the non-target strand sequence, while Cas9 exhibits a preference for ssODNs of the target strand sequence instead25 (see Figure 4a). - Ensure that the repaired sequence is not targetable by the selected Cas nuclease again. For example, mutate the PAM in such a way that there is no amino acid change or eliminate the PAM from the donor template if it has no functional consequence. Use the example given in Figure 4b as a guide, if necessary.
- Decide if a symmetric or asymmetric donor template is desired. For symmetric donors, ensure that each homology arm flanking the DNA modification site is at least 17 nt long25. For asymmetric donor templates, use longer arms 5′ of the desired DNA changes (see Figure 4a). Importantly, ensure that the shorter arm is around 37 nt in length, while the other homology arm is around 77 nt in length25,39.
- Synthesize the designed template as a piece of single-stranded DNA.
NOTE: Asymmetric ssODNs can, but do not always, exhibit higher HDR efficiency than symmetric ssODNs. In general, an asymmetric donor typically performs at least as well as a symmetric donor, when designed correctly. However, the asymmetric template costs much more because it is longer and hence requires polyacrylamide gel electrophoresis (PAGE) purification or a special synthesis procedure. Routine gene knockouts usually rely on the NHEJ repair pathway and do not require a repair template. However, if the knockout efficiency is low, design a ssODN donor template that contains a frameshift mutation and is at least 120 nt in length25,40.
- Determine the correct strand whose sequence the template should follow.

Figure 4: Design of ssODN donor templates. (a) Schematic illustrating various possible designs. The red horizontal rectangles indicate the non-target (NT) strand, while the blue rectangles indicate the target (T) strand. In addition, the small green rectangles indicate the desired DNA modifications (such as single nucleotide changes). When a symmetric ssODN is used, the minimum length of each homology arm should be at least 17 nt (but can be longer). For asymmetric ssODNs, the 37/77 T ssODN appears to be optimal for SpCas9-induced HDR, while the 77/37 NT ssODN appears to be optimal for Cas12a-induced HDR. L = left homology arm; R = right homology arm. (b) A specific example to demonstrate how to design ssODN templates. Here, the target genomic locus is exon 45 of the human CACNA1D gene. The PAM for Cas9 is pink and underlined, while the PAM for Cas12a is brown and underlined. The goal is to create a missense mutation (highlighted in green) by converting AGU (encoding serine) to AGG (encoding arginine). To prevent re-targeting by Cas12a, the TTTC PAM is mutated to CTTC. Note that there is no change in amino acid (UAU and UAC both code for tyrosine). To further prevent re-targeting by Cas9, an AGU codon is replaced with a UCC codon (bold), both of which code for serine. Please click here to view a larger version of this figure.
{kind=link}
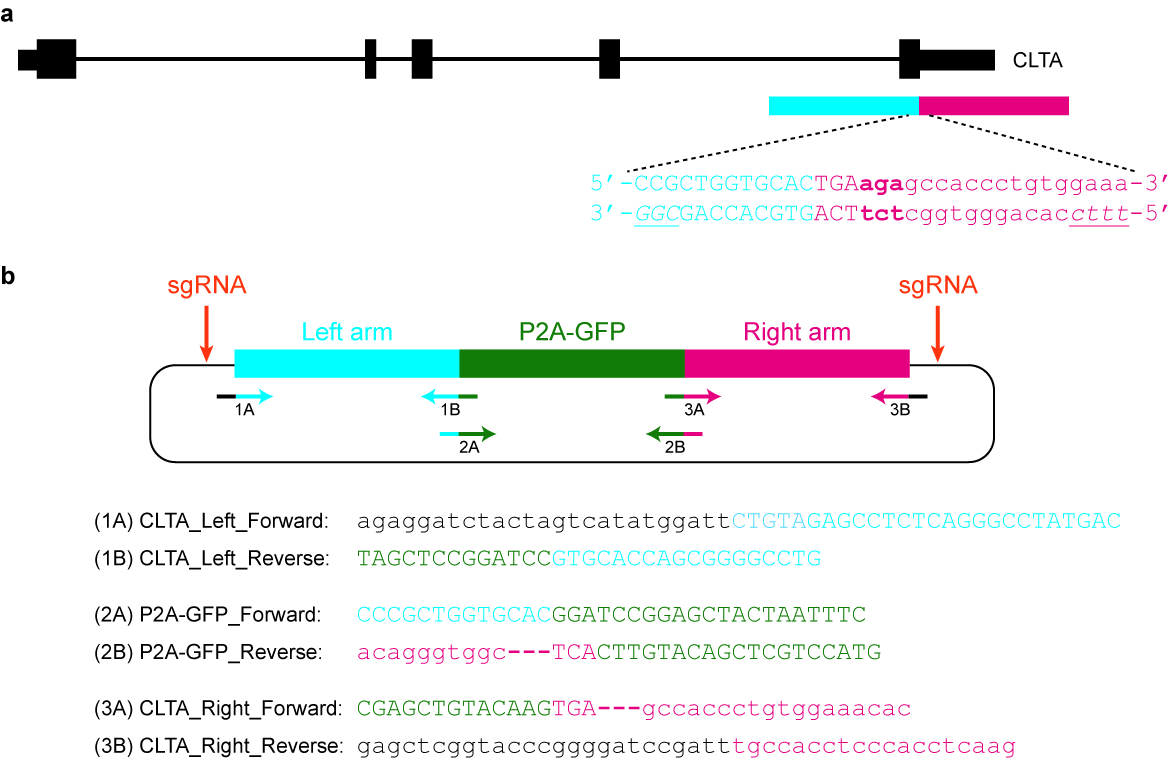
- Design and clone an appropriate plasmid donor template. For example, it may contain a GFP sequence flanked by long arms that are homologous to the target genomic locus (see Figure 5).
- Ensure that the modified sequence is not targetable by the selected Cas nuclease again. For example, the protospacer may be split by the inserted (GFP) tag. Alternatively, the PAM may be mutated or removed from the donor template in such a way that does not affect gene function.
- Amplify the homology arms from genomic DNA using PCR. The length of each homology arm is typically 1000 to 1500 bp.
NOTE: To facilitate cloning, ensure that the forward primer for the left homology arm and the reverse primer for the right homology arm each has at least 20 nt overlapping sequence with a selected vector backbone. Additionally, ensure that the reverse primer for the left homology arm and the forward primer for the right homology arm has some overlapping sequences with the epitope tag as well. - Clone the two homology arms and the (GFP) tag into the vector backbone using Gibson assembly41 (see the Table of Materials). Verify the plasmid by Sanger sequencing using forward and reverse primers that are upstream and downstream of the donor template respectively.
NOTE: Sanger sequencing is widely and cheaply available as a commercial service. Send an aliquot of the plasmid together with the sequencing primers to a service provider. - Linearize the donor template with a restriction enzyme that cuts the plasmid only once either upstream of the left homology arm or downstream of the right homology arm.
NOTE: Recently, a double-cut donor, which is flanked by the sgRNA-PAM sequences and is released from the plasmid after cleavage by the corresponding Cas nuclease, has been shown to increase HDR efficiency42. When the sgRNA-PAM sequences are inserted upstream and downstream of the left and right homology arms respectively (for example by Gibson assembly), the homology arm length may be reduced to 300 bp and there is no need to linearize the plasmid.

Figure 5: Design and cloning of a plasmid donor template. (a) The goal in this specific example is to fuse P2A-GFP to the C-terminus of the CLTA protein. The blue horizontal rectangle indicates the left homology arm, while the red horizontal rectangle indicates the right homology arm. Capital letters indicate protein-coding sequences, while lowercase letters indicate non-coding sequences. The PAMs for SpCas9 and Cas12a are italicized and underlined. (b) A plasmid donor template that can be used to endogenously tag P2A-GFP at the C-terminus of CLTA. The provided primer sequences can be used to clone the plasmid by Gibson assembly. The PCR conditions are as follows: 98 °C for 3 min, 98 °C for 30 s (step 2), 63 °C for 30 s (step 3), 72 °C for 1 min (step 4), repeat steps 2‒4 for another 34 cycles, 72 °C for 3 min, and hold at 4 °C. Black letters correspond to vector sequences, blue letters correspond to the left homology arm, green letters correspond to P2A-GFP, and red letters correspond to the right homology arm. Note that once the sequence encoding P2A-GFP is successfully integrated into the target locus, re-targeting by SpCas9 will not be possible, since only 9 nt of its protospacer (GTGCACCAG) will be left intact. Moreover, in order to prevent re-targeting by Cas12a, three basepairs immediately downstream of the STOP codon (in bold) are deleted from the plasmid sequence. Please click here to view a larger version of this figure.
{kind=link}
4. Cell transfection
NOTE: The remaining parts of the protocol are written with HEK293T cells in mind. The culture medium used consists of Dulbecco Modified Eagle Medium (DMEM) supplemented with 4.5 g/L glucose, 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 0.1% penicillin/streptomycin. Different steps of the protocol may have to be modified according to the actual cell line used. All cell culture work is done in a Class II Biosafety Cabinet to ensure a sterile work environment.
- Seed 1.8 x 105 cells in a 24-well tissue culture plate one day prior to transfection.
- Dissociate the cells by aspirating the media and then adding 150 µL 0.25% Trypsin-EDTA per well. Incubate the cells at 37 °C for 2 min.
- Neutralize the trypsin by adding 150 µL (or 1x volume) of cell culture media. Transfer the cell suspension to a conical tube. Spin down the cells in a bench top centrifuge at 1000 x g for 5 min.
- Aspirate the supernatant, and resuspend with 5 mL of cell culture media. In a separate centrifuge tube, aliquot 10 µL of 0.4% trypan blue solution. Then, add in 10 µL resuspended cells from step 4.1.2 and mix thoroughly.
- Pipette 10 µL of the mixture (cells + trypan blue) into a hemocytometer. Proceed to count the cells manually or by using an automated cell counter.
- Seed 1.8 x 105 cells into one well of a 24-well tissue culture plate.
- Prepare a transfection mix containing either 500 ng of CRISPR plasmid (for NHEJ-mediated editing) or 300 ng of CRISPR plasmid and 300 ng of donor template (for HDR-mediated editing), according to the instructions provided with the transfection reagent (see Table of Materials). Incubate at room temperature for the recommended time duration (typically around 10‒20 min).
- Add the transfection mix to the cells in a dropwise fashion, and gently swirl the plate after.
- Incubate at 37 °C in a humidified 5% CO2 air incubator for 24 h (for NHEJ-based experiments) or 72 h (for HDR-based experiments).
5. Fluorescence activated cell sorting (FACS) of transfected cells
- Dissociate the cells by aspirating the media and then adding 150 µL of 0.25% trypsin-EDTA per well. Incubate the cells at 37 °C for 2 min.
- Neutralize the trypsin by adding 150 µL (or 1x volume) of cell culture media. Transfer the cell suspension to a centrifuge tube. Spin down the cells in a microcentrifuge at 235 x g for 5 min.
- Aspirate the supernatant and resuspend the cells with 2% fetal bovine serum (FBS) in phosphate-buffered saline (PBS). Filter the cells through a 30 µm mesh or cell strainer in a 5 mL FACS tube.
- Prepare another centrifuge tube with approximately 100 µL culture media or 2% FBS in PBS for collection of cells.
- On the flow cytometer, gate the cells with non-transfected cells as a negative control. Sort and collect the transfected cells, according to which fluorescence marker is present on the CRISPR plasmid used. For example, if the plasmid carries a mCherry gene, sort for RFP-positive cells.
NOTE: Different CRISPR plasmids will have different selectable markers. The set of plasmids (pSpCas9, pSaCas9, pNmCas9, pAsCpf1, and pLbCpf1) used in this evaluation study carry either the orange fluorescent protein (OFP) or the mCherry gene.
6. Expansion of individual clones
- Centrifuge the sorted cells in a bench top centrifuge at maximum speed (18,000 x g) for 5 min. Aspirate the supernatant and resuspend the pellet with 300 µL culture media. Seed 200 µL cells in a 24-well tissue culture plate and let them recover for a few days in a 37 °C incubator. Keep the remaining 100 µL cells for section 7.
- Once the cells start becoming confluent, passage them according to steps 4.1.1‒4.1.3. Seed the cells sparsely in a 100 mm tissue culture dish to allow sufficient space for individual colonies to grow. Incubate at 37 °C in a humidified 5% CO2 air incubator.
NOTE: Try various dilutions. Single cells need sufficient space to grow as individual colonies. However, they also cannot be overly sparse, as some cell lines do not grow well when the number of cells are too few. - Once the colonies are starting to form, pick them under the microscope (with a 4x magnification) and place each clone into an individual well of a 24-well plate that contains cell culture media. Incubate at 37 °C in a humidified 5% CO2 air incubator until the cells are becoming confluent.
NOTE: An alternative to serial dilutions and colony picking is to use flow cytometry to sort for single cells into a 96-well plate. However, this may not work for some cell lines that do not grow well when only one cell is present.
7. Evaluation of editing efficiency
- Extract genomic DNA by centrifuging the remaining 100 µL sorted cells (from step 6.1) in a bench top centrifuge at maximum speed (18,000 x g) for 5 min. Aspirate the supernatant and proceed to isolate genomic DNA using an extraction kit (see Table of Materials).
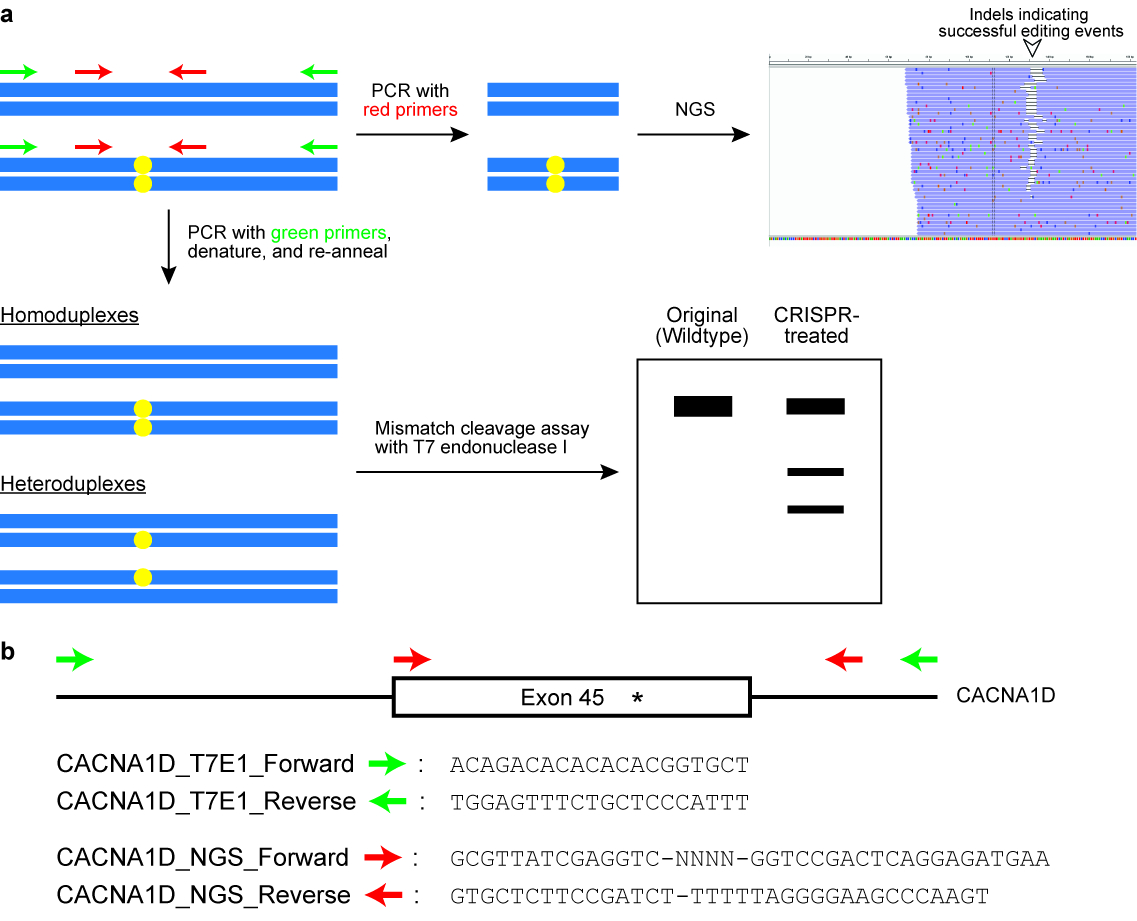
- Perform the T7 endonuclease I (T7EI) cleavage assay (see Figure 6).
- Set up a 50 µL PCR containing 10 µL of PCR reaction buffer (5x), 1 µL of dNTP mix (10 mM), 2.5 µL of user-defined forward primer (10 µM), 2.5 µL of user-defined reverse primer (10 µM), 0.5 µL of DNA polymerase, 2‒5 µL of genomic DNA template (depending on how many cells have been sorted), then top up to 50 µL with ddH2O (see Table of Materials).
NOTE: The primers are designed to flank the target genomic locus and yield a PCR product of around 400‒700 bp. Usually one primer is positioned closer to the cut site of the Cas enzyme than the other primer, so that the result of the T7EI assay is two distinct bands on an agarose gel (see Figure 6). - Run the PCR in a thermal cycler with the following parameters: 98 °C for 3 min, 98 °C for 30 s (step 2), 63 °C for 30 s (step 3), 72 °C for 30 s (step 4), repeat steps 2‒4 for another 34 cycles, 72 °C for 2 min, and hold at 4 °C.
- Resolve the reaction on a 2% agarose gel using 1x TAE buffer.
- Excise the PCR product from the gel with a clean, sharp scalpel and purify the DNA using a gel extraction kit, according to manufacturer’s instructions. Measure the concentration of the PCR product using a spectrophotometer at a wavelength of 260 nm absorbance (see Table of Materials).
- Prepare an assay mix containing 200 ng of DNA, 2 µL of T7EI reaction buffer (10x), and topped up to 19 µL with ddH2O (see Table of Materials).
- Re-anneal the PCR product in a thermal cycler using the following parameters: 95 °C for 5 min, ramp down to 25 °C at 6 °C/min, then hold at 4 °C.
- Add 5 U T7EI to the re-annealed PCR product, mix well by pipetting, and incubate at 37 °C for 50 min.
- Resolve the T7EI-digested DNA on a 2.5% agarose gel using 1x TAE buffer.
- Image the gel, quantify the band intensities using ImageJ, and calculate rate of indel formation using the following formula:

where a represents the intensity of the intact PCR product and b and c correspond to the intensities of the cleavage products43.- To quantify the intensity of a band in ImageJ, first draw a rectangular box around the band as close to its boundary as possible. Second, click on Analyze and then Set Measurements. Ensure that the area, mean gray value, and integrated density options are checked. Exit the settings window by clicking ok. Third, click on Analyze and then Measure. The mean or RawIntDen value is used as the band intensity.
- Set up a 50 µL PCR containing 10 µL of PCR reaction buffer (5x), 1 µL of dNTP mix (10 mM), 2.5 µL of user-defined forward primer (10 µM), 2.5 µL of user-defined reverse primer (10 µM), 0.5 µL of DNA polymerase, 2‒5 µL of genomic DNA template (depending on how many cells have been sorted), then top up to 50 µL with ddH2O (see Table of Materials).

Figure 6: Checking cells for successful genome editing outcomes. (a) A schematic illustrating two commonly used assays, namely the mismatch cleavage assay with the T7 endonuclease I (T7EI) enzyme and next generation sequencing (NGS) or targeted amplicon sequencing. The blue horizontal rectangles indicate DNA and the yellow circles indicate modifications induced by the CRISPR-Cas system. Primers for the T7E1 assay are denoted in green, while primers for generating amplicons for NGS are denoted in red. (b) Design of primer sequences for the T7EI cleavage assay and for NGS. Here, the target genomic locus is exon 45 of the human CACNA1D gene. The intended modification site is indicated by an asterisk. Please click here to view a larger version of this figure.
{kind=link}
- Perform targeted amplicon sequencing (see Figure 6).
- Design PCR primers to amplify the target genomic locus. Position one of the primers to be less than 100 bp but more than 20 bp from the protospacer.
NOTE: Typically, the total PCR product size is designed to be around 150‒300 bp (see Figure 6). - Append additional sequences to the primers as follows: (a) 5’‒GCGTTATCGAGGTC-NNNN-[Forward Primer] – 3’; (b) 5’ - GTGCTCTTCCGATCT-[Reverse Primer]–3’.
- Set up a 50 µL PCR reaction mix containing 10 µL of PCR reaction buffer (5x), 1 µL of dNTP (10 mM), 5 µL of primer a (10 µM), 5 µL of primer b (10 µM), 0.5 µL DNA polymerase, 2‒5 µL genomic DNA template (depending on how many cells have been sorted), then top up to 50 µL with ddH20.
- Run the PCR in a thermal cycler with the following parameters: 98 °C for 3 min, 98 °C for 30 s (step 2), 63 °C for 30 s (step 3), 72 °C for 15 s (step 4), repeat steps 2‒4 for another 34 cycles, 72 °C for 2 min, and hold at 4 °C.
- Resolve the reaction on 2% agarose gel and purify the PCR product using a gel extraction kit according to manufacturer’s instructions. Quantify the DNA using a spectrophotometer at a wavelength of 260 nm absorbance (see the Table of Materials).
- Synthesize the following round 2 PCR primers: (c) 5’‒ AATGATACGGCGACCACCGAGATCTACACCCTACACGAGCGTTATCGAGGTC–3’; (d) 5’‒CAAGCAGAAGACGGCATACGAGAT-[barcode]-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT–3’
- Set up a 20 µL PCR reaction mix containing 4 µL of PCR reaction buffer (5x), 0.4 µL of dNTP (10 mM), 2 µL of primer c (10 µM), 2 µL of primer d (10 µM), 0.2 µL of DNA polymerase, 2 µL of DNA template (from step 7.3.5, diluted 1:100), and 9.4 µL of ddH2O.
NOTE: The dilution factor for the DNA template may vary depending on its original concentration. If the concentration is around 20‒40 ng/µL, use a 1:100 dilution factor. In addition, pick a different barcode for each experimental sample, if the same primer a and primer b are used in step 7.3.3. - Run the PCR in a thermal cycler with the following parameters: 98 °C for 3 min, 98 °C for 30 s (step 2), 65 °C for 30 s (step 3), 72 °C for 30 s (step 4), repeat steps 2‒4 for another 14 cycles, 72 °C for 2 min, and 4 °C hold.
- Resolve 5 µL of each reaction on a 2% agarose gel to determine the success of the PCRs. Combine all the samples together (assuming that a different barcode has been used for each sample) and clean up the pooled DNA using a PCR purification kit according to manufacturer’s instructions. If some of the PCRs exhibit more than one band (indicating the presence of non-specific products), perform an additional gel extraction step.
- Sequence the library on a high throughput sequencing instrument (see Table of Materials) according to manufacturer’s instructions to produce paired 151 bp reads. The read 1 sequencing primer is custom designed and has to be provided separately. Its sequence is as follows: Read1_seq: 5’–CCACCGAGATCTACACCCTACACGAGCGTTATCGAGGTC–3’. The read 2 sequencing primer and index sequencing primer are standard and are provided in the reagent cartridge.
NOTE: The T7EI assay and targeted amplicon sequencing are commonly used to check the efficiency of genome editing. However, other experiments may be performed to evaluate editing efficiency, depending on the type of DNA modifications introduced. For example, if a restriction site is created at the target site, a restriction fragment length polymorphism (RFLP) assay can be performed. It is similar to the T7EI assay except that a restriction endonuclease is used to digest the PCR product instead.
- Design PCR primers to amplify the target genomic locus. Position one of the primers to be less than 100 bp but more than 20 bp from the protospacer.
8. Screening of individual clones
- From step 6.3, split the cells once they start getting confluent. For each individual clone, collect the remaining cells and extract genomic DNA according to step 7.1.
- Perform the T7EI assay for all the individual clones according to section 7.2, except for one modification. Amplify the target genomic locus from wildtype cells and in step 7.2.5, instead of using 200 ng of test DNA only, mix 100 ng of test DNA with 100 ng of wildtype DNA.
NOTE: The reason for the modified step is that some clones may have undergone successful biallelic conversion and are homozygous mutants. In such cases, there will be no cleavage bands in the T7EI assay if the wildtype DNA is not mixed in. - Sequence the target site in the clones that exhibit cleavage bands in the T7EI assay.
- Amplify the modified genomic locus according to steps 7.2.1–7.2.4.
- Set up the following cloning reaction: 4 µL of PCR product, 1 µL of salt solution, 1 µL of TOPO vector (see the Table of Materials).
- Mix gently by pipetting and incubate at room temperature for at least 5 min.
- Transform 3 µL of the reaction mix into chemically competent E. coli cells (such as TOP10 or Stbl3) (see Supplementary File 1). Spread the transformed bacterial cells on an LB agar plate with 50 μg/mL kanamycin.
- The next day, inoculate at least 10 colonies in LB liquid media containing 50 μg/mL kanamycin.
- When the bacterial cultures are turbid, isolate the plasmids using a miniprep kit (see Table of Materials) and sequence them using the standard M13 forward or M13 reverse primer.
- Perform a western blot (also known as immunoblot) to determine the absence or presence of the targeted protein (if the genome editing experiment involves knocking out a protein-coding gene via frameshift mutations). See Supplementary File 1.
NOTE: Other experiments can be performed to identify the clone carrying the desired genomic modifications. For example, a phenotypic assay can be performed if knocking out a particular gene is known to cause certain changes in cellular behavior.
Results
To perform a genome editing experiment, a CRISPR plasmid expressing a sgRNA targeting the locus-of-interest needs to be cloned. First, the plasmid is digested with a restriction enzyme (typically a type IIs enzyme) to linearize it. It is recommended to resolve the digested product on a 1% agarose gel alongside an undigested plasmid to distinguish between a complete and partial digestion. As undigested plasmids are supercoiled, they tend to run faster than their linearized counterparts (see Figure 7
Discussion
The CRISPR-Cas system is a powerful, revolutionary technology to engineer the genomes and transcriptomes of plants and animals. Numerous bacterial species have been found to contain CRISPR-Cas systems, which may potentially be adapted for genome and transcriptome engineering purposes44. Although the Cas9 endonuclease from Streptococcus pyogenes (SpCas9) was the first enzyme to be deployed successfully in human cells21,22,
Disclosures
The authors do not have competing financial interests.
Acknowledgements
M.H.T. is supported by an Agency for Science Technology and Research’s Joint Council Office grant (1431AFG103), a National Medical Research Council grant (OFIRG/0017/2016), National Research Foundation grants (NRF2013-THE001-046 and NRF2013-THE001-093), a Ministry of Education Tier 1 grant (RG50/17 (S)), a startup grant from Nanyang Technological University, and funds for the International Genetically Engineering Machine (iGEM) competition from Nanyang Technological University.
Materials
| Name | Company | Catalog Number | Comments |
| T4 Polynucleotide Kinase (PNK) | NEB | M0201 | |
| Shrimp Alkaline Phosphatase (rSAP) | NEB | M0371 | |
| Tris-Acetate-EDTA (TAE) Buffer, 50X | 1st Base | BUF-3000-50X4L | Dilute to 1X before use. The 1X solution contains 40 mM Tris, 20 mM acetic acid, and 1 mM EDTA. |
| Tris-EDTA (TE) Buffer, 10X | 1st Base | BUF-3020-10X4L | Dilute to 1X before use. The 1X solution contains 10 mM Tris (pH 8.0) and 1 mM EDTA. |
| BbsI | NEB | R0539 | |
| BsmBI | NEB | R0580 | |
| T4 DNA Ligase | NEB | M0202 | 400,000 units/ml |
| Quick Ligation Kit | NEB | M2200 | An alternative to T4 DNA Ligase. |
| Rapid DNA Ligation Kit | Thermo Scientific | K1423 | An alternative to T4 DNA Ligase. |
| Zero Blunt TOPO PCR Cloning Kit | Thermo Scientific | 451245 | The salt solution comes with the TOPO vector in the kit. |
| NEBuilder HiFi DNA Assembly Master Mix | NEB | E2621L | Kit for Gibson assembly. |
| One Shot Stbl3 Chemically Competent E.Coli | Thermo Scientific | C737303 | |
| LB Broth (Lennox), powder | Sigma Aldrich | L3022 | Reconstitute in ddH20, and autoclave before use |
| LB Broth with Agar (Lennox), powder | Sigma Aldrich | L2897 | Reconstitute in ddH20, and autoclave before use |
| SOC media | - | - | 2.5 mM KCl, 10 mM MgCl2, 20 mM glucose in 1 L of LB Broth |
| Ampicillin (Sodium), USP Grade | Gold Biotechnology | A-301 | |
| REDiant 2X PCR Mastermix | 1st Base | BIO-5185 | |
| Agarose | 1st Base | BIO-1000 | |
| T7 Endonuclease I | NEB | M0302 | |
| Plasmid DNA Extraction Miniprep Kit | Favorgen | FAPDE 300 | |
| Dulbecco's Modified Eagle Medium (DMEM), High Glucose | Hyclone | SH30081.01 | 4.5 g/L Glucose, no L-glutamine, HEPES and Sodium Pyruvate |
| L-Glutamine, 200mM | Gibco | 25030 | |
| Penicillin-Streptomycin, 10, 000U/mL | Gibco | 15140 | |
| 0.25% Trypsin-EDTA, 1X | Gibco | 25200 | |
| Fetal Bovine Serum | Hyclone | SV30160 | FBS is heat inactivated before use at 56 oC for 30 min |
| Phosphate Buffered Saline, 1X | Gibco | 20012 | |
| jetPRIME transfection reagent | Polyplus Transfection | 114-75 | |
| QuickExtract DNA Extraction Solution, 1.0 | Epicentre | LUCG-QE09050 | |
| ISOLATE II Genomic DNA Kit | Bioline | BIO-52067 | An alternative to QuickExtract |
| Q5 High-Fidelity DNA Polymerase | NEB | M0491 | |
| Deoxynucleotide (dNTP) Solution Mix | NEB | N0447 | |
| 6X DNA Loading Dye | Thermo Scientific | R0611 | 10 mM Tris-HCl (pH 7.6) 0.03% bromophenol blue, 0.03% xylene cyanol FF, 60% glycerol, 60 mM EDTA |
| Protease Inhibitor Cocktail, Set3 | Merck | 539134 | |
| Nitrocellulose membrane, 0.2µm | Bio-Rad | 1620112 | |
| Tris-glycine-SDS buffer, 10X | Bio-Rad | 1610772 | Dilute to 1X before use. The 1x solution contains 25 mM Tris, 192 mM glycine, and 0.1% SDS. |
| Tris-glycine buffer, 10X | 1st base | BUF-2020 | Dilute to 1X before use. The 1x solution contains 25 mM Tris and 192 mM glycine. |
| Ponceau S solution | Sigma Aldrich | P7170 | |
| TBS, 20X | 1st base | BUF-3030 | Dilute to 1X before use. The 1x solution contains 25 mM Tris-HCl (pH 7.5) and 150 mM NaCl. |
| Tween 20 | Sigma Aldrich | P9416 | |
| Skim Milk for immunoassay | Nacalai Tesque | 31149-75 | |
| WesternBright Sirius-femtogram HRP | Advansta | K12043 | |
| Antibody for β-actin (C4) | Santa Cruz Biotechnology | sc-47778 | Lot number: C0916 |
| MiSeq system | Illumina | SY-410-1003 | |
| NanoDrop spectrophotometer | Thermo Scientific | ND-2000 | |
| Qubit fluorometer | Thermo Scientific | Q33226 | |
| EVOS FL Cell Imaging System | Thermo Scientific | AMF4300 | |
| CRISPR plasmid: pSpCas9(BB)-2A-GFP (PX458) | Addgene | 48138 | Single vector system: The gRNA is expressed from the same plasmid. |
| CRISPR plasmid: pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA | Addgene | 61591 | Single vector system: The gRNA is expressed from the same plasmid. |
| CRISPR plasmid: xCas9 3.7 | Addgene | 108379 | Dual vector system: The gRNA is expressed from a different plasmid. |
| CRISPR plasmid: pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | 42230 | Single vector system: The gRNA is expressed from the same plasmid. |
| CRISPR plasmid: hCas9 | Addgene | 41815 | Dual vector system: The gRNA is expressed from a different plasmid. |
| CRISPR plasmid: eSpCas9(1.1) | Addgene | 71814 | Single vector system: The gRNA is expressed from the same plasmid. |
| CRISPR plasmid: VP12 (SpCas9-HF1) | Addgene | 72247 | Dual vector system: The gRNA is expressed from a different plasmid. |
References
- Epinat, J. C., et al. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Research. 31 (11), 2952-2962 (2003).
- Arnould, S., et al. Engineered I-CreI derivatives cleaving sequences from the human XPC gene can induce highly efficient gene correction in mammalian cells. Journal of Molecular Biology. 371 (1), 49-65 (2007).
- Chapdelaine, P., Pichavant, C., Rousseau, J., Paques, F., Tremblay, J. P. Meganucleases can restore the reading frame of a mutated dystrophin. Gene Therapy. 17 (7), 846-858 (2010).
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics. 188 (4), 773-782 (2011).
- Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S., Gregory, P. D. Genome editing with engineered zinc finger nucleases. Nature Reviews Genetics. 11 (9), 636-646 (2010).
- Miller, J. C., et al. A TALE nuclease architecture for efficient genome editing. Nature Biotechnology. 29 (2), 143-148 (2011).
- Zhang, F., et al. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nature Biotechnology. 29 (2), 149-153 (2011).
- Boch, J., et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 326 (5959), 1509-1512 (2009).
- Moscou, M. J., Bogdanove, A. J. A simple cipher governs DNA recognition by TAL effectors. Science. 326 (5959), 1501 (2009).
- Hsu, P. D., Lander, E. S., Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 157 (6), 1262-1278 (2014).
- Sander, J. D., Joung, J. K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nature Biotechnology. 32 (4), 347-355 (2014).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nishimasu, H., et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 156 (5), 935-949 (2014).
- Yamano, T., et al. Crystal Structure of Cpf1 in Complex with Guide RNA and Target DNA. Cell. 165 (4), 949-962 (2016).
- Swarts, D. C., Mosterd, C., van Passel, M. W., Brouns, S. J. CRISPR interference directs strand specific spacer acquisition. PLoS One. 7 (4), e35888 (2012).
- Zetsche, B., et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 163 (3), 759-771 (2015).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 507 (7490), 62-67 (2014).
- Hu, J. H., et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 556 (7699), 57-63 (2018).
- Kleinstiver, B. P., et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nature Biotechnology. 33 (12), 1293-1298 (2015).
- Kleinstiver, B. P., et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 523 (7561), 481-485 (2015).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Jinek, M., et al. RNA-programmed genome editing in human cells. Elife. 2, e00471 (2013).
- Cho, S. W., Kim, S., Kim, J. M., Kim, J. S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nature Biotechnology. 31 (3), 230-232 (2013).
- Wang, Y., et al. Systematic evaluation of CRISPR-Cas systems reveals design principles for genome editing in human cells. Genome Biology. 19 (1), 62 (2018).
- Ran, F. A., et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 520 (7546), 186-191 (2015).
- Hou, Z., et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proceedings of the National Academy of Sciences U S A. 110 (39), 15644-15649 (2013).
- Kim, E., et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nature Communications. 8, 14500 (2017).
- Edraki, A., et al. A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Molecular Cell. , (2018).
- Chatterjee, P., Jakimo, N., Jacobson, J. M. Minimal PAM specificity of a highly similar SpCas9 ortholog. Science Advances. 4 (10), (2018).
- Muller, M., et al. Streptococcus thermophilus CRISPR-Cas9 Systems Enable Specific Editing of the Human Genome. Mol Therapy. 24 (3), 636-644 (2016).
- Esvelt, K. M., et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nature Methods. 10 (11), 1116-1121 (2013).
- Boratyn, G. M., et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Research. 41 (Web Server issue), W29-W33 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M., Valen, E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Research. 42 (Web Server issue), W401-W407 (2014).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Haeussler, M., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Bae, S., Park, J., Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 30 (10), 1473-1475 (2014).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Richardson, C. D., Ray, G. J., Bray, N. L., Corn, J. E. Non-homologous DNA increases gene disruption efficiency by altering DNA repair outcomes. Nature Communications. 7, 12463 (2016).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. 6 (5), 343-345 (2009).
- Zhang, J. P., et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biology. 18 (1), 35 (2017).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Shmakov, S., et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nature Reviews Microbiology. 15 (3), 169-182 (2017).
- Moreno-Mateos, M. A., et al. CRISPR-Cpf1 mediates efficient homology-directed repair and temperature-controlled genome editing. Nature Communications. 8 (1), 2024 (2017).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 3, e04766 (2014).
- Yang, L., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Watanabe, K., et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nature Biotechnology. 25 (6), 681-686 (2007).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved