
Method Article
Techniques d’immunohistochimie pour analyser la prolifération cellulaire et la neurogenèse chez le rat à l’aide de l’analogue de thymidine BrdU
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
Cet article présente quatre des techniques les plus courantes pour visualiser les cellules BrdU positives afin de mesurer la neurogenèse adulte chez le rat. Ce travail comprend des instructions pour la préparation des réactifs, l’administration d’analogues de thymidine, la perfusion transcardique, la préparation tissulaire, la réaction immunohistochimique de la peroxydase, l’immunofluorescence, l’amplification du signal, la contre-coloration, l’imagerie microscopique et l’analyse cellulaire.
Résumé
L’une des choses les plus importantes dans le domaine de la neurogenèse hippocampique adulte (AHN) est l’identification des cellules nouvellement générées. L’immunodétection des analogues de la thymidine (tels que la 5-Bromo-2'-désoxyuridine (BrdU)) est une technique standard utilisée pour visualiser ces cellules nouvellement générées. Par conséquent, BrdU est généralement injecté chez de petits animaux par voie intrapéritonéale, de sorte que l’analogue de la thymidine est incorporé dans les cellules en division pendant la synthèse de l’ADN. La détection est effectuée par analyse immunohistochimique des tranches de cerveau. Chaque groupe de recherche qui a utilisé cette technique peut comprendre qu’elle nécessite une attention particulière aux moindres détails pour obtenir une coloration réussie. Par exemple, une étape importante est la dénaturation de l’ADN avec HCl, qui lui permet d’atteindre le noyau cellulaire pour le colorer. Cependant, les rapports scientifiques existants décrivent très peu de ces étapes en détail. Par conséquent, la normalisation de la technique est difficile pour les nouveaux laboratoires, car il peut s’écouler plusieurs mois avant d’obtenir des résultats positifs et positifs. Le but de ce travail est de décrire et d’élaborer les étapes pour obtenir des résultats positifs et réussis de la technique d’immunomarquage en détail lors du travail avec l’analogue de thymidine BrdU. Le protocole comprend la préparation et la configuration du réactif, l’administration d’un analogue de thymidine chez un rongeur, la perfusion transcardique, la préparation tissulaire, la réaction immunohistochimique de la peroxydase, l’utilisation du complexe avidine-biotine, l’immunofluorescence, la contre-coloration, l’imagerie microscopique et l’analyse cellulaire.
Introduction
L’idée que de nouveaux neurones sont générés dans le cerveau humain adulte tout au long de la vie fascine la communauté scientifique depuis des décennies. La connaissance que le cerveau génère de nouveaux neurones tout au long de sa vie a été obtenue grâce à la détection de cellules sous la division 1,2. La détection de neurones nouvellement générés dans le cerveau adulte a d’abord été identifiée par injection intracrânienne de thymidine tritiée (thymidine-H3) chez le rat et détection de cellules dans le cycle cellulaire par autoradiogrammes 1,2. La division cellulaire de la glie et la présence de neuroblastes ont été rapportées, ce qui a été la première donnée prometteuse sur la neurogenèse postnatale1. Néanmoins, l’utilisation et la détection de thymidine-H3 impliquaient l’utilisation de la radioactivité, ce qui peut être nocif pour les personnes qui la gèrent. Le premier effort examinant la pertinence de l’immunohistochimie BrdU dans l’étude de la prolifération, de la migration et de l’origine des cellules dans le système nerveux est apparu en 1988 par Miller et Nowakowski3. En 1998, un article publié par Eriksson et ses collègues a montré que de nouveaux neurones étaient visualisés post-mortem dans le cerveau adulte humain de patients auxquels on avait injecté de la 5-Bromo-2′-désoxyuridine (BrdU)4. Ces patients ont reçu l’injection de BrdU (250 mg par voie intraveineuse) pour marquer la croissance des tumeurs4. Cette technique a été adoptée dans des modèles animaux. L’introduction de ces méthodes a marqué une étape importante pour le domaine car elle a permis la détection de cellules nouvellement générées sans l’utilisation de composés radioactifs. Cette procédure est devenue l’étalon-or pour mesurer la prolifération cellulaire dans les niches cérébrales adultes afin de promouvoir la poursuite de la recherche dans le domaine.
La limite de la technique analogue de la thymidine est qu’elle ne permet pas de déterminer l’identité cellulaire des cellules nouvellement générées. Cependant, l’immunohistochimie nous permet de réaliser une technique de double ou triple marquage de la même cellule, ce qui valide le devenir cellulaire des cellules nouvellement générées et même leurs stades de maturation, conduisant à une évolution ultérieure du domaine. Cette méthode a été caractérisée pour différencier les cellules nouvellement générées en cellules gliales, neurones indifférenciés ou une cellule granulaire complètement mature, et même pour déterminer si elles participent activement aux circuits. Une autre percée dans le domaine a été l’utilisation de modèles transgéniques pour identifier les cellules indifférenciées sous le domaine de la nestine. Les souris transgéniques nestine-GFP expriment une protéine fluorescente verte améliorée (GFP), qui est sous le contrôle du promoteur de la nestine. La nestine est un filament intermédiaire caractérisé par des cellules progénitrices5. Les souris transgéniques nestine-GFP ont permis d’établir les étapes précoces du développement impliquées dans la neurogenèse6. Cependant, une limitation importante est de pouvoir maintenir une colonie de souris transgéniques nestine-GFP dans des conditions spéciales dans une installation de laboratoire qui devient rentable pour certains groupes scientifiques, en particulier ceux des pays en développement.
Les techniques mentionnées ci-dessus ont des avantages et des inconvénients. Cependant, l’identification des cellules proliférantes par immunohistochimie (IHC) et la possibilité de réaliser une technique de double ou triple marquage par immunofluorescence pour identifier le stade de maturation cellulaire ou le destin cellulaire représentent le moyen le plus réalisable de mesurer la neurogenèse adulte, jusqu’à présent. Le processus d’identification par immunohistochimie consiste à marquer des protéines, un domaine protéique ou des nucléotides avec un anticorps spécifique qui permet leur reconnaissance appelé anticorps primaire. Ce dernier est reconnu par l’anticorps secondaire, qui est marqué d’un chromogène (p. ex. peroxydase de raifort) ou d’un fluorochrome (p. ex., FITC) couplé à l’anticorps secondaire. Les microscopes peuvent détecter à la fois les signaux des chromogènes et des fluorochromes. En utilisant IHC, il est possible d’identifier des protéines membranaires, des protéines du cytosquelette ou des composants nucléaires tels que BrdU. D’autre part, BrdU peut être trouvé dans le noyau cellulaire puisqu’il est incorporé dans l’ADN pendant la phase S par compétition. Par conséquent, une étape cruciale est la dénaturation de l’ADN avec HCl, qui ouvre les liaisons ADN pour permettre à l’anticorps BrdU d’accéder à BrdU dans l’ADN. Il est essentiel de savoir que le BrdU est présent en concentration saturée dans le sérum de souris et de rat pendant 15 et 60 min respectivement, après administration intrapéritonéale, puis chute rapidement à des niveaux indétectables à 60 et 120 min respectivement7.
Ici, nous décrivons quatre techniques IHC différentes mais étroitement liées: détection indirecte chromogène par réaction de peroxydase de raifort (HRP) avec DAB (3,3'-diaminobenzidine) sans amplification du signal (étape 4.1), amplification du complexe avidine-biotine (ABC) (étape 4.1), détection indirecte par immunofluorescence sans amplification du signal (étape 4.4) et amplification marquée streptavidine-biotine (LSAB) (étape 4.3). Chaque méthode présente des avantages et des inconvénients et pourrait être utile pour des besoins tissulaires spécifiques (voir le tableau 1). Nous avons décidé de suivre les méthodes indirectes de l’ICH en raison de leur prix abordable et de leur simplicité pour passer des méthodes de détection chromogènes aux méthodes de détection fluorescentes lors de l’utilisation d’anticorps primaires non conjugués. L’approche HRP est une méthode IHC couramment utilisée en raison de son prix abordable, de sa grande stabilité, de son taux de renouvellement élevé et de la disponibilité totale des substrats. Néanmoins, nous recommandons d’utiliser un contrôle positif pour confirmer que la méthode de coloration fonctionne correctement et l’utilisation d’un contrôle négatif pour tester efficacement la fonction des anticorps. Les immunocolorations multiples ou les méthodes IHC multiplex (voir étape 6) sont des outils puissants pour acquérir de grandes quantités de données à partir de la section tissulaire en une seule expérience. Cette technique est particulièrement importante lorsque la disponibilité des échantillons est limitée. Un autre avantage est la possibilité d’identifier simultanément des protéines spécifiques co-exprimées dans le même espace cellulaire tout en préservant l’intégrité des tissus. Le multiplex permet de colorer différents marqueurs exprimés au cours de stades prolifératifs spécifiques (par exemple, nestine, GFAP, DCX, Ki-67), ce qui nous permet d’atteindre une recherche plus détaillée sur la prolifération et la différenciation8. Il est crucial de choisir des anticorps compatibles avec la technique de fixation utilisée pour éviter la réactivité croisée. Nous recommandons de tester chaque nouvel anticorps (y compris BrdU) individuellement pour ajuster et affiner la méthode. Ensuite, introduisez la double coloration séquentielle et, enfin, commencez le processus d’immunomarquage simultané lorsque la méthode séquentielle est entièrement dominée. Il est crucial de choisir des anticorps secondaires appropriés pour cette méthode.
| Méthode | Méthode spécifique | Avantages | Inconvénients |
| Méthode de détection indirecte | Réaction de peroxydase avec DAB | 1. Sensibilité supérieure à la méthode de détection directe et de fluorescence indirecte. 2. Résistance plus élevée au photoblanchiment que les fluorochromes. 3. Coût inférieur à celui de la méthode de détection par fluorescence | 1. Difficile pour le multiplexage avec moins de colorants. 2. Compliqué pour les cibles co-exprimées dans le même espace cellulaire. 3. Plage dynamique réduite pour des cibles simultanées rares et abondantes sur le même tissu. |
| Fluorescence | 1. Le meilleur et le plus facile pour le multiplexage avec plus de colorants. 2. Idéal pour les cibles co-exprimées dans le même espace cellulaire. 3. Meilleure plage dynamique pour des cibles simultanées rares et très abondantes sur le même tissu. 4. Aucune étape supplémentaire. | 1. Sensibilité inférieure à celle de la réaction indirecte de peroxydase avec la méthode DAB. 2. Faible résistance au photoblanchiment au fil du temps. 3. Plus cher. | |
| Méthode d’amplification de signal | Complexe Avidine-Biotine (ABC) | 1. Sensibilité supérieure à celle de la méthode de détection directe et indirecte. 2. Réduire l’arrière-plan | 1. Étapes supplémentaires. 2. Plus cher qu’une amplification autrement. |
| Streptaviridine-biotine marquée (LSAB) | 1. Sensibilité supérieure à celle de la méthode de détection directe et indirecte. 2. Pénétration tissulaire plus importante que la méthode ABC. 3. Réduire l’arrière-plan | 1. Étapes supplémentaires. 2. Plus cher que la méthode ABC. | |
| Pas de méthode d’amplification supplémentaire | 1. Réduction des coûts. 2. Aucune étape supplémentaire. 3. Idéal pour les cibles abondantes élevées. | 1. Sensibilité plus faible : problématique sans cibles abondantes. |
Tableau 1 : Avantages/inconvénients des techniques IHC. Ce tableau montre les avantages/inconvénients des méthodes de détection indirecte : réaction de la peroxydase avec (3,3'-diaminobenzidine) DAB et fluorescence ; et méthodes d’amplification du signal : complexe avidine-biotine (ABC), streptavidine-biotine marquée (LSAB), et non méthode d’amplification supplémentaire.
Une image haute résolution est fondamentale pour effectuer une analyse appropriée et présenter les résultats. Il existe deux approches pour améliorer la résolution : 1) l’utilisation d’une meilleure conception de microscope (par exemple, confocale, multiphotonique) ou 2) l’inversion numérique du processus de flou pour améliorer les images à l’aide de la déconvolution9. Malheureusement, la microscopie confocale n’est pas abordable en raison des coûts élevés de l’équipement et de son entretien10. Un microscope à épifluorescence à grand champ et la déconvolution subséquente des images z-stack constituent une alternative appropriée et peu coûteuse à la microscopie confocale 8,9. Comme indiqué ci-dessus, l’objectif de la déconvolution est de restaurer le signal original qui a été dégradé par le système d’acquisition9, en réduisant le flou, la brume floue et la distorsion montrées dans l’image obtenue par un microscope épifluorescence ou confocale à l’aide d’algorithmes mathématiquesde suppression 10. L’image floue acquise peut être modélisée mathématiquement à la suite de la mise en relation des objets observés avec une fonction d’étalement de points 3D (PSF). La PSF est un diagramme de diffraction théorique des points de lumière émis par l’échantillon de tissu et collectés par le microscope. Le fichier PSF est créé avec les conditions spécifiques de chaque image, telles que l’espacement des cellules CCD de la caméra, l’indice de réfraction du support utilisé, l’ouverture numérique de la lentille de l’objectif, la longueur d’onde d’émission du fluorophore, la taille de l’image, le nombre d’images dans la méthode de traitement z-stack et l’espace entre elles (voir les spécifications techniques dans le tableau 2). En d’autres termes, le fichier PSF résume les effets de la configuration d’imagerie sur les observations au microscope9. Cependant, nous utilisons le plug-in 3D de diffraction PSF (https://imagej.net/Diffraction_PSF_3D) pour créer notre propre fichier PSF spécifique pour chaque image z-stack. Les images Z-stack sont une série de sections optiques numérisées à partir de profondeurs définies (axe z) au même emplacement XY de la diapositive. Un ordinateur compile les informations obtenues à partir du plan focal en réaffectant des signaux provenant d’objets situés dans d’autres plans focaux. Pour créer des images z-stack, il est nécessaire de prendre des images à partir de différentes couches focalisées des diapositives (par exemple, dix images différentes de la même zone XY toutes les profondeurs de 1 μm). Ensuite, nous utilisons un logiciel de microscopie fourni par le fabricant ou Fidji pour créer une z-stack ou une image 3D. Le résultat sera un fichier image à pile unique (par exemple, dix images avec des focus différents). Il existe plusieurs outils et solutions logicielles spécifiques aux clients, tels que les logiciels open source pour la microscopie de déconvolution. Nous montrerons les résultats du processus de déconvolution en utilisant DeconvolutionLab29 qui est un plugin Fiji11 (distribution d’ImageJ12). La déconvolution aidera à améliorer la résolution des micrographies finales (voir Figure 1B,C ). Pour plus d’informations et d’instructions, nous vous recommandons fortement de lire la référence13.

Figure 1 : Image représentative de la déconvolution 3D pour plusieurs canaux de couleur. (A) DG à faible grossissement. (B) Les images z-stack originales pour chaque canal et l’image fusionnée. (C) Images 3D déconvolutées z-stack pour chaque canal et l’image fusionnée. Ce cerveau provenait du rat qui faisait partie du groupe d’activité physique. La méthode d’amplification de la streptavidine-biotine marquée (LSAB) a été utilisée. Il a montré un anticorps conjugué à la streptavidine Cy3 pour indiquer BrdU (rouge), DAPI comme contre-coloration (bleu) et protéine acide fibrillaire gliale (GFAP) comme marqueur astroglial (vert). ML = couche moléculaire; GCL = couche cellulaire granulaire; SGZ = zone sous-granulaire. Veuillez cliquer ici pour voir une version agrandie de cette figure.
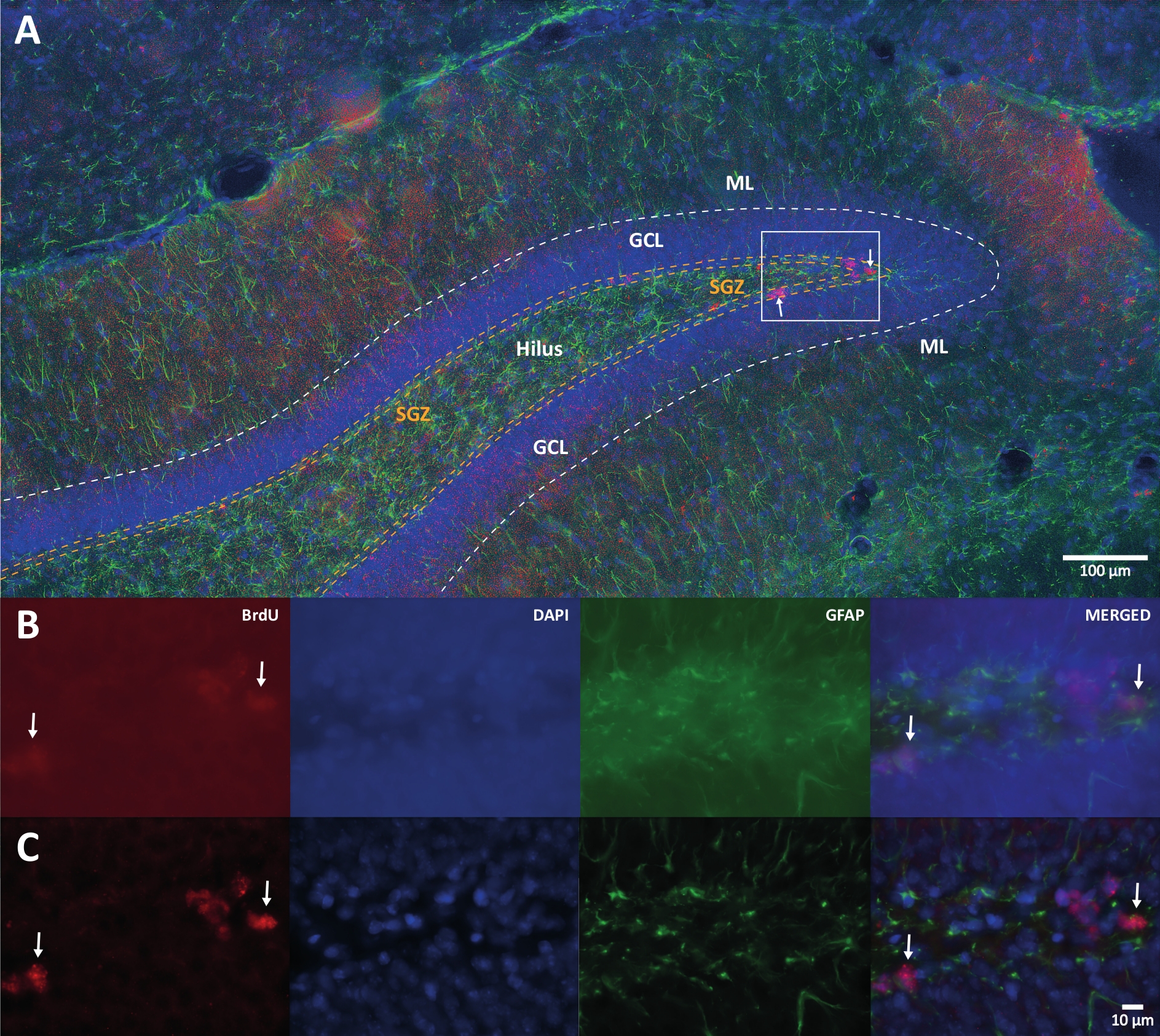{kind=link}
Le but de ce travail est de fournir une description détaillée des étapes pour obtenir des résultats positifs et positifs avec l’immunomarquage et d’énumérer les étapes couramment utilisées dans les études basées sur BrdU, sans l’utilisation d’un microscope confocal. La coloration BrdU est une technique qui nécessite plusieurs étapes qui doivent être soigneusement suivies pour obtenir une coloration réussie. La normalisation de ces techniques de coloration prend généralement des mois et exige beaucoup de temps et de ressources. Nous nous attendions à ce que cet article puisse fournir de l’information aux groupes qui débutent dans ce domaine en réduisant le temps et les erreurs.
Protocole
Toutes les procédures suivent le guide des National Institutes of Health pour le soin et l’utilisation des animaux de laboratoire (NIH Publications N° 8023, révisé en 1978) et les lois locales mexicaines pour minimiser le nombre d’animaux utilisés et leurs souffrances. Le comité d’éthique de l’Universidad Iberoamericana a approuvé les protocoles expérimentaux pour l’utilisation d’animaux dans cette étude.
1. Préparation et configuration du réactif
REMARQUE: La plupart des solutions peuvent être préparées plusieurs jours avant utilisation, sauf indication contraire.
- Solution BrdU
- Récupérez la solution BrdU du congélateur à -20 °C et laissez-la s’équilibrer à température ambiante (RT).
- Calculer la masse de BrdU nécessaire pour une dose de 50 mg/kg en fonction du poids corporel du rat. Calculer le volume de solution saline à 0,9 % (0,9 g de NaCl dans 100 mL deH2Ostérile) nécessaire pour obtenir une solution de travail de 20 mg/mL. Préparer un excédent pour fournir au moins 0,5 mL par rat par injection.
NOTE: La dose administrée aux animaux de laboratoire doit être sûre, avec un minimum d’effets secondaires et efficace. Il a été rapporté que la durée de la coloration avec 100 mg / kg BrdU ne l’emporte pas sur la toxicité potentiellement plus élevée par rapport à la dose de 50 mg / kg7. Aucune différence significative n’a été observée dans le nombre de cellules marquées BrdU/mm3 pour 50 et 100 mg/kg i.p. chez le rat7. Il est préférable d’injecter une petite dose pour minimiser la souffrance des animaux. - Peser la solution de BrdU et l’ajouter à la solution saline dans un tube conique et un vortex.
REMARQUE : Préchauffer la solution saline à 45 °C dans un bain-marie pour des volumes supérieurs à 1 mL. - Placer le tube dans un bain-marie à 50 °C pendant 10 à 10 minutes et vortex toutes les 2 à 12 minutes jusqu’à dissolution complète. Filtrer la solution avec un filtre à seringue pour injection stérile. Couvrir le tube avec du papier d’aluminium, le refroidir à température ambiante et l’utiliser immédiatement.
ATTENTION : La solution de BrdU est toxique et potentiellement cancérigène. Préparez-le dans la hotte. La solution BrdU doit être manipulée avec un équipement de protection (EPI) approprié. Il est recommandé de préparer la solution immédiatement avant utilisation. Cependant, la solution est stable pendant 24 h sous RT. Veuillez le protéger de la lumière.
- Pour préparer 1 L de solution saline tamponnée au phosphate 0,1 M (PBS) à pH 7,4, ajouter 240 mg de phosphate de potassium monobasique (KH2PO 4), 1,44 g de phosphate dibasique de sodium (Na2HPO4), 200 mg de chlorure de potassium (KCl) et 8 g de chlorure de sodium (NaCl) à 800 mL d’eau bidistillée (ddH2O) sous agitation constante. Ajuster le pH à 7,4 et ajouter leH2Obidistillé jusqu’à un volume total de 1 L. Conserver à 4 °C jusqu’à 1 semaine.
- Pour 100 mL de PBS+, ajouter 3 % (3 mL) de sérum de cheval normal et 0,3 % (300 μL) de Triton X-100 à 0,1 M PBS (pH 7,4). Conserver dans des aliquotes de 20 à 20 ml à -20 °C pendant une période allant jusqu’à 3 mois.
REMARQUE : Alternativement, TBS peut être utilisé à la place de PBS. Tout autre sérum différent des anticorps et des tissus expérimentaux de l’hôte convient. - Pour 100 mL de PBS++, ajouter 10 % (10 mL) de sérum de cheval normal et 0,3 % (300 μL) de Triton X-100 à 0,1 M PBS pH 7,4. Conserver dans des aliquotes de 20 à 20 ml à -20 °C pendant une période allant jusqu’à 3 mois.
- Pour 1 L de solution cryoprotectrice, mélanger 250 mL d’éthylène glycol et 250 mL de glycérol, remuer constamment jusqu’à consistance homogène. Porter lentement à 1 L avec PBS. Filtre avec papier filtre de grade 4 (20\u201225 μm). Conserver à 4 °C ou RT jusqu’à 1 an.
- Préparer 4% de paraformaldéhyde dans 0,1 M PBS (solution de PFA) comme suit. Pour 1 L de solution, ajouter lentement 40 g de poudre de paraformaldéhyde à 800 mL de 60\u201265 °C 0,1 M PBS sous agitation constante. Remuer jusqu’à ce que le paraformaldéhyde soit complètement dissous tout en contrôlant la température (60\u201265 °C). Si nécessaire, ajoutez quelques gouttes de 1 M de NaOH pour clarifier la solution. Lorsque la solution atteint la température ambiante, filtrer avec du papier filtre de grade 4 (20-25 μm).
ATTENTION : Le paraformaldéhyde est toxique et est suspecté d’être cancérigène, préparez-le dans la hotte. Conserver à 4 °C et de préférence utiliser dans un délai de 2 jours. La solution PFA prête à l’emploi est disponible dans le commerce. - Pour 1 L de tampon de citrate de sodium (SCB) de 10 mM à pH 6, ajouter 1,204 g de citrate de sodium (dihydraté) et 1,134 g d’acide citrique à 800 mL deH2Odouble distillé sous agitation constante. Ajuster le pH à 6,0 et ajouterddH2Ojusqu’à 1 L. Conserver à 4 °C jusqu’à 6 mois.
- Préparer 50 mL de HCl 2 N en ajoutant lentement 8,25 mL de HCl 12 N (solution mère concentrée) à 41,75 mL deddH2Osous agitation constante.
ATTENTION : Préparer dans la hotte. La solution doit être préparée immédiatement avant utilisation.
REMARQUE: 2 N HCl sera utilisé pour la dénaturation de l’ADN, une étape cruciale. Comme BrdU est incorporé dans l’ADN, HCl est utilisé pour ouvrir les liaisons ADN permettant aux anticorps BrdU d’accéder à BrdU dans l’ADN. - Préparer la solution de blocage de la peroxydase endogène comme suit. Préparer 100 mL de peroxyde d’hydrogène à 0,6 % en mélangeant 2 mL de peroxyde d’hydrogène à 30 % avec 98 mL deddH2Osous agitation constante.
NOTE: La solution doit être préparée immédiatement avant utilisation. Gardez-le dans l’obscurité car H 2 O2est sensible à la lumière. PBS ou TBS peuvent être utilisés à la place de l’eau. - Préparer la solution de complexe avidine-biotine (ABC) selon les instructions du fabricant. Pour 5 mL d’ABC dans 0,1 M de PBS, ajouter 2 gouttes (≈100 μL) de réactif A et mélanger, puis ajouter 2 gouttes (≈100 μL) de réactif B et mélanger.
NOTE: La solution doit être préparée et laissée rouler pendant 20 à 230 minutes avant utilisation. - Préparez le substrat de DAB (diaminobenzidine) peroxydase (HRP) à l’aide de la trousse en suivant les instructions du fabricant. À 5 mL deddH2O, ajouter 2 gouttes (≈ 84 μL) de réactif 1 et mélanger, ajouter 4 gouttes (≈ 100 μL) de réactif 2 et mélanger, puis ajouter 2 gouttes (≈ 80 μL) de réactif 3 et mélanger. Enfin, si désiré, ajouter 2 gouttes (≈ 80 μL) de réactif 4 (Nickel) et mélanger.
NOTE: La solution doit être préparée immédiatement avant utilisation.
ATTENTION : Le DAB est toxique et potentiellement cancérigène. Il doit être manipulé avec soin et jeté conformément à la réglementation sur les déchets dangereux de chaque établissement. Pour inactiver le DAB, ajoutez plusieurs gouttes d’eau de Javel (hypochlorite de sodium); La solution deviendra noire. - Préparer 100 mL de solution de violet de crésyle en ajoutant 100 mg d’acétate de violet de crésyle et 250 μL d’acide acétique à 80 mL deddH2Oà 55\u201260 °C. Régler le volume à 100 mL, filtrer et conserver à 4 °C dans un récipient de couleur foncée.
REMARQUE: L’utilisateur est encouragé à tester différentes concentrations de la solution de violet crésyle avant de l’utiliser sur des échantillons de tissus précieux. Le résultat peut être plus foncé pour la contre-coloration avec certains échantillons de tissus, ce qui peut diminuer la capacité de compter les cellules BrdU positives avec précision.
2. Administration d’un analogue de la thymidine BrdU
- Maîtriser l’animal de laboratoire (p. ex., le rat Wistar mâle de 90 jours pesant 350 g) en immobilisant la cavité abdominale inférieure.
- Administrer la solution BrdU (50 mg/kg) par voie intrapéritonéale (i.p.) à l’aide d’une aiguille de 23 G et d’une seringue de 1 mL.
REMARQUE: Ajustez le volume d’injection en fonction du poids de l’animal. Utilisez une aiguille de 23 à 23 g et une seringue de 1 à 20125 ml pour rats adultes. Le volume maximal tolérable d’injection intrapéritonéale chez le rat adulte est de 10 mL. Différentes voies peuvent être utilisées pour administrer la solution BrdU14. Par exemple, injection intrapéritonéale ou administration orale dans l’eau potable.
3. Préparation des tissus
REMARQUE: Les rats âgés de trois mois ont été autorisés ad libitum à l’activité physique (roue sans fin) pendant sept jours. Le jour 6, des rats ont reçu une injection de BrdU (étape 2) 3 fois à des intervalles de 12 heures. Effectuer les étapes décrites dans la section 3 8 h après la dernière injection de BrdU.
- Injectez du pentobarbital (50 mg/kg i.p.) et attendez quelques minutes jusqu’à ce que l’animal soit anesthésié en profondeur.
REMARQUE : Assurez-vous que l’animal est complètement anesthésié avant de continuer. Pincez soigneusement l’une des pattes ou la queue. Si l’animal réagit au stimulus, attendez quelques minutes de plus. Si l’animal ne réagit pas au pincement, passez à l’étape suivante. - Exposez le cœur en coupant la peau de la cavité abdominale sous le sternum, en démontant les côtes et en coupant le diaphragme.
- Fixation de perfusion transcardique
- Insérez une aiguille dans le ventricule gauche et faites une petite incision dans l’oreillette droite. À l’aide d’une pompe ou de la gravité, perfuser (débit 5\u20127 mL/min.) 0,1 M PBS jusqu’à ce que le sang total soit drainé et que la solution devienne claire.
- À l’aide d’une pompe ou de la gravité, effectuer une perfusion à froid (débit de 5 à 127 mL/min) avec une solution de PFA pour fixer le tissu jusqu’à ce que la queue devienne rigide.
REMARQUE: Habituellement, un rat de 300 g nécessite environ 100 à 100 à 150 mL de la solution de PFA. La fixation tissulaire est facultative. Ainsi, le cerveau peut être extrait pour être utilisé dans divers processus afin de minimiser l’utilisation d’animaux dans les expériences.
- Dissection et post-fixation
- Décapita l’animal et extraire doucement le cerveau du crâne. Immerger le cerveau dans un tube conique contenant une solution de PFA (~40 mL pour un rat de 250 mg) pendant 1 à 4 °C.
REMARQUE: Ne pas trop fixer (plus de 48 h), car cela peut épuiser la coloration tissulaire en raison de l’indisponibilité des antigènes. - Préparer 100 mL de solution de saccharose à 30 %, en ajoutant 30 g de saccharose à 70 mL de solution de PBS 0,1 M sous agitation constante. Ajouter la solution PBS 0,1 M à 100 mL. Immerger le cerveau dans un tube conique contenant une solution de saccharose à 30 % (35 mL) pendant environ 1 à 4 °C jusqu’à ce que le cerveau coule au fond du tube.
- Décapita l’animal et extraire doucement le cerveau du crâne. Immerger le cerveau dans un tube conique contenant une solution de PFA (~40 mL pour un rat de 250 mg) pendant 1 à 4 °C.
- Couper les sections coronales du cerveau
NOTE: L’utilisation d’un cryostat-microtome nécessite des conseils et une formation. Pour des instructions détaillées, voir la référence15.- Immergez tout le cerveau dans l’iso-pentane à -80 °C et maintenez-le à -80 °C pendant 10 min. Placez le cerveau dans une matrice d’encastrement sur une plaque cryostat-microtome.
REMARQUE : Dans certaines conditions, la congélation rapide du cerveau à -80 °C peut provoquer des fractures ou des dommages aux tissus. L’utilisateur doit être conscient de ce problème. Si c’est le cas, utilisez de l’isopentane à -20 °C pour congeler le cerveau. - À l’aide d’un cryostat-microtome (température de -25 à -20 °C), découpez des coupes coronales de 40 μm d’épaisseur. Transférer séquentiellement les sections dans une plaque de culture cellulaire de 24 puits avec solution de cryoprotection en suivant le guide de la figure 2. Conserver à -20 °C jusqu’à utilisation, jusqu’à quelques mois.
REMARQUE : Par la suite, traiter tous les tissus en sections sérielles flottantes de 40 μm dans des plaques à 12 puits avec des inserts de mailles en agitation douce et continue (10 tr/min). Il est possible de stocker des coupes cérébrales pendant des années dans de bonnes conditions.
- Immergez tout le cerveau dans l’iso-pentane à -80 °C et maintenez-le à -80 °C pendant 10 min. Placez le cerveau dans une matrice d’encastrement sur une plaque cryostat-microtome.

Figure 2 : Illustration schématique du transfert séquentiel de sections du cryostat-microtome dans une plaque de culture cellulaire à 24 puits avec solution de cryoprotection. Commencez au puits A1 et placez les tranches suivantes dans la rangée A; après A6-puits, passez à la rangée B suivante, ainsi de suite. En arrivant à la J6, retournez à l’A1 et continuez. Cet arrangement permet la quantification de la nième section (par exemple, sixième pour la neurogenèse, équivalent au contenu d’une colonne) d’une région entière du cerveau. Veuillez cliquer ici pour voir une version agrandie de cette figure.
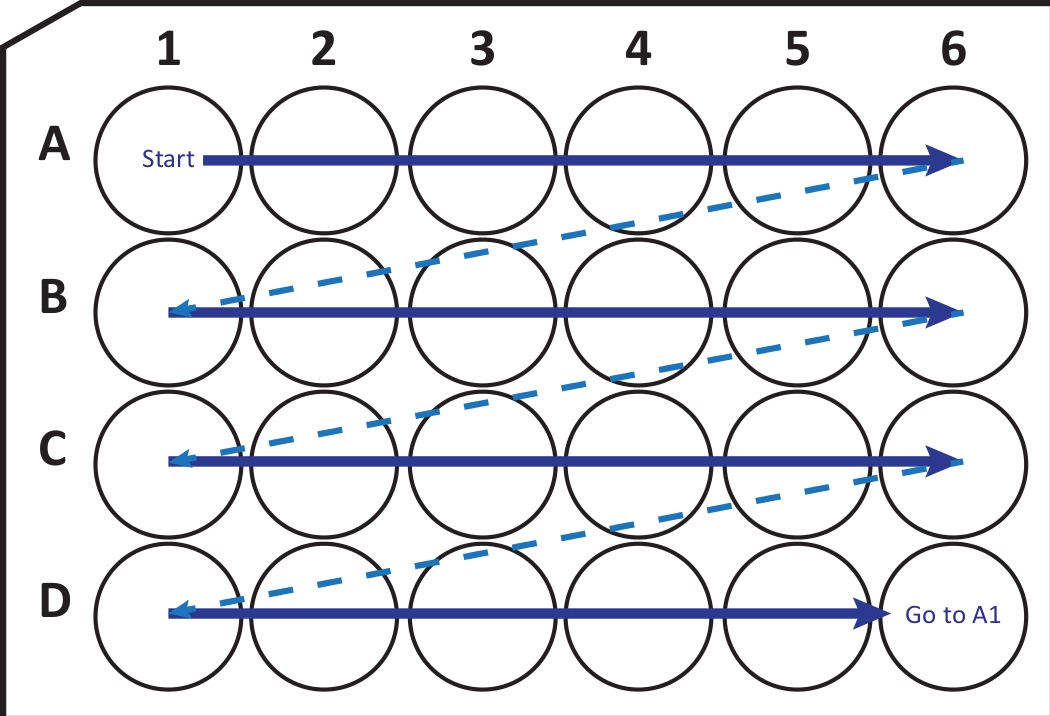{kind=link}
4. Immunomarquage
NOTE: Voir le tableau 1 pour le résumé des avantages et des inconvénients de chaque technique.
- Détection de BrdU par réaction de peroxydase avec DAB
REMARQUE : Effectuez les étapes 4.1.1 à 4.1.5 le jour 1.- Transvaser les tranches de la solution de cryoprotection vers 0,1 M de PBS à température ambiante. Rincer trois fois pendant 10 minutes chacune, avec 0,1 M PBS.
- Incuber des tranches pendant 30 min dans une solution de blocage de la peroxydase endogène pour inactiver la peroxydase endogène. Rincer 3 fois, 10 min chacune, avec 0,1 M PBS. Vous pouvez éventuellement effectuer le prélèvement d’antigènes (voir rubrique 5). Incuber les tranches pendant 20 min avec 2 N HCl à 37 °C. Rincer dans un tampon de borate 0,1 M (pH 8,5) pendant 10 min. Rincer 3 fois pendant 10 min chacune, avec du PBS glacé de 0,1 M.
- Incuber les tranches pendant 2 h à température ambiante avec PBS++ (solution bloquante). Incuber avec un anticorps primaire anti-BrdU (hôte souris) à une concentration de 1:250 dans PBS+ pendant une nuit à 4 °C.
- Le jour 2, rincer les tranches 3 fois pendant 10 minutes chacune avec 0,1 M de PBS.
- Incuber avec un anticorps secondaire conjugué à 1:250 HRP (anti-souris) dans PBS+ pendant 2\u20124 h à température ambiante. Rincer 3 fois pendant 10 min chacune avec 0,1 M PBS.
- Transférer les tranches dans la solution de substrat de la peroxydase DAB (HRP) et incuber pendant 2 à 2 min. Lorsque les tranches deviennent gris foncé, visualisez le tissu à l’aide d’une loupe ou d’un microscope. Si des cellules positives sont présentes, rincer 3 fois (pendant 15 minutes chacune) avec de l’eau du robinet (pour réduire le bruit de fond). Laver 3 fois pendant 10 min chacune avec 0,1 M PBS.
- Monter soigneusement les tranches sur des lames gélatinisées à l’aide d’une brosse douce, sécher à l’air pendant la nuit à température ambiante. Contre-teinture (voir rubrique 7.1), ajouter un support de montage permanent et placer des lamelles de couverture. Conserver à 4 °C jusqu’à 6 mois.
- Détection de BrdU par réaction peroxydase avec le complexe avidine-biotine-peroxydase
REMARQUE : Effectuez les étapes 4.2.1 à 4.2.5 le jour 1.- Transférer les tranches de la solution de cryoprotection à 0,1 M PBS pour les porter à température ambiante. Rincer 3 fois pendant 10 min chacune avec 0,1 M PBS.
- Incuber pendant 30 min avec une solution de blocage de la peroxydase endogène pour inactiver la peroxydase endogène. Rincer 3 fois pendant 10 min chacune dans 0,1 M PBS. Vous pouvez éventuellement effectuer le prélèvement d’antigènes (voir rubrique 5).
- Incuber pendant 20 min avec 2 N HCl à 37 °C. Rincer dans un tampon de borate 0,1 M (pH 8,5) pendant 10 min. Laver 3 fois pendant 10 minutes chacune avec 0,1 M de PBS glacé.
- Incuber pendant 2 h à température ambiante dans du PBS++ (solution bloquante).
- Incuber avec l’anticorps primaire anti-BrdU (hôte souris) 1:250 dans PBS+ pendant une nuit à 4 °C.
- Le jour 2, rincer 3 fois pendant 10 minutes chacune avec 0,1 M de PBS.
- Incuber avec un anticorps secondaire biotinylé 1:250 (anti-souris) dans PBS+ pendant 2\u20124 h à température ambiante. Rincer 3 fois pendant 10 min chacune avec 0,1 M PBS.
- Incuber dans la solution ABC pendant 1 h à température ambiante. Rincer 3 fois pendant 10 min chacune avec 0,1 M PBS.
- Transférer les tranches dans la solution de substrat de la DAB peroxydase (HRP) et incuber pendant 2 à 2 min. Lorsque les tranches deviennent gris foncé, visualisez le tissu à l’aide d’une loupe ou d’un microscope. Si des cellules positives sont présentes, rincer 3 fois (15 min chacune) avec de l’eau du robinet (pour réduire le bruit de fond) suivi de 3 fois avec 0,1 M PBS laver pendant 10 min chacun.
NOTE: La solution doit être préparée immédiatement avant utilisation. Il faut prendre soin d’éviter que les tranches de cerveau ne collent les unes aux autres en raison de taches brunes irrégulières dans les tissus. - Monter soigneusement les tranches sur des lames gélatinisées à l’aide d’une brosse douce, puis sécher à l’air libre pendant la nuit à température ambiante.
- Contre-teindre si nécessaire (voir étape 7.1), ajouter un support de montage permanent et placer des lamelles de couverture. Conserver à 4 °C jusqu’à 6 mois.
- Détection de BrdU par immunofluorescence à l’aide d’une amplification marquée de la streptavidine-biotine (LSAB)
Remarque : Effectuez les étapes 4.3.1 à 4.3.4 le jour 1.- Transvaser les tranches de la solution de cryoprotection vers 0,1 M de PBS à température ambiante. Rincer 3 fois pendant 10 min chacune avec 0,1 M PBS. Vous pouvez éventuellement effectuer le prélèvement d’antigènes (voir rubrique 5).
- Incuber pendant 20 min dans 2 N HCl à 37 °C. Rincer dans un tampon de borate 0,1 M (pH 8,5) pendant 10 min. Rincer 3 fois pendant 10 min chacune dans du PBS glacé de 0,1 M.
- Incuber pendant 2 h à température ambiante dans du PBS++ (solution bloquante). Incuber avec l’anticorps primaire anti-BrdU 1:250 (hôte souris) dans PBS+ pendant une nuit à 4 °C.
- Le jour 2, rincer 3 fois pendant 10 minutes chacune avec 0,1 M de PBS.
- Incuber avec un anticorps secondaire biotinylé 1:250 (anti-souris) dans PBS+ pendant 2\u20124 h à température ambiante. Rincer 3 fois pendant 10 min chacune avec 0,1 M PBS. Incuber avec 1:250 de streptavidine (Cy3) conjuguée au fluorochrome dans du PBS (ne pas utiliser de sérum) pendant 1 à 1 h à température ambiante. Rincer 3 fois pendant 10 min chacune avec 0,1 M PBS.
REMARQUE: Le sérum peut contenir de la biotine et ne doit pas être ajouté aux diluants. Utilisez plutôt du PBS contenant 0,3 % de Triton X-100. - Monter soigneusement les tranches sur des lames gélatinisées à l’aide d’une brosse douce, sécher à l’air pendant la nuit à température ambiante ou monter immédiatement avec un support de montage approprié. Contre-teinture (voir étape 7.2), ajouter un support de montage permanent et placer des lamelles de couverture. Conserver à 4 °C jusqu’à 6 mois.
- Détection du BrdU par immunofluorescence indirecte
REMARQUE : effectuez les étapes 4.4.1 à 4.4.4 le jour 1.- Transvaser les tranches de la solution de cryoprotection à 0,1 M PBS jusqu’à ce qu’elle atteigne la température ambiante. Rincer 3 fois pendant 10 min chacune avec 0,1 M PBS. Effectuer le prélèvement d’antigènes si nécessaire (facultatif, voir rubrique 5).
- Incuber pendant 20 min dans 2 N HCl à 37 °C. Rincer dans un tampon de borate 0,1 M (pH 8,5) pendant 10 min. Rincer 3 fois pendant 10 min chacune avec 0,1 M de PBS glacé. Incuber pendant 2 h à température ambiante avec PBS++ (solution bloquante). Incuber avec l’anticorps primaire anti-BrdU 1:250 (hôte souris) dans PBS+ pendant une nuit à 4 °C.
- Le jour 2, rincer 3 fois pendant 10 minutes chacune avec 0,1 M de PBS.
- Incuber avec un anticorps secondaire conjugué au fluorochrome 1:250 (anti-souris) dans PBS+ pendant 2\u20124 h à température ambiante. Rincer 3 fois pendant 10 min chacune avec 0,1 M PBS.
- Monter soigneusement les tranches sur des lames gélatinisées à l’aide d’une brosse douce, sécher à l’air pendant la nuit à température ambiante ou monter immédiatement avec un support de montage approprié. Contre-teinture (voir étape 7.2), ajouter un support de montage permanent et placer des lamelles de couverture. Conserver à 4 °C jusqu’à 6 mois.
5. Récupération d’antigènes (facultatif)
NOTE: La récupération d’antigènes est une étape facultative destinée à corriger la perte d’antigénicité causée par une fixation qui modifie la structure tertiaire et quaternaire de nombreux antigènes, les rendant indétectables par les anticorps. Cette étape peut être ajoutée au protocole d’origine.
- Au micro-ondes ou au bain-marie, préchauffer une solution tampon de citrate de sodium (SCB) de 10 mM pH 6 à 90\u201295 °C (selon l’altitude, la solution commence à bouillir autour de cette température). Remplir 80 % d’un tube conique de 50 ml (40 ml) avec un SCB préchauffé. Transférer les tranches sur des inserts de mailles dans le tube conique avec SCB. Couvrir le tube avec un bouchon à vis percé de trous faits avec une aiguille de 18\u201220 G.
- Conserver les tranches pendant 30 min dans le SCB à 80\u201285 °C en alternant les cycles de réchauffement au micro-ondes au niveau de puissance minimal. Si nécessaire, remplissez le tube conique avec SCB. Transférer les tranches immédiatement après avec les inserts de maille dans du PBS glacé de 0,1 M et rincer 3 fois pendant 10 minutes chacune.
6. Immunocolorations multiples (facultatif)
REMARQUE : Voir la section d’introduction pour connaître la raison d’être de cette étape.
- Immunocolorations multiples simultanées
- Préparer un cocktail avec les anticorps primaires dirigés contre la cible (p. ex., anti-BrdU chez la souris et anti-GFAP chez le lapin) dans PBS+. Utilisez des hôtes différents pour chaque anticorps primaire utilisé. Incuber pendant une nuit à 4 °C. Passez aux mêmes étapes pour chaque protocole.
- Préparer un cocktail avec les anticorps secondaires correspondants pour chaque anticorps primaire utilisé (p. ex. FITC anti-souris caprine, TRITC anti-lapin caprin) dans la même solution de diluant pour chaque protocole. Passez aux mêmes étapes pour chaque protocole. Idéalement, utilisez des anticorps secondaires provenant des mêmes hôtes pour éviter les réactions croisées.
- Immunocolorations multiples séquentielles.
- Suivez le protocole pour la première cible d’anticorps (par exemple, souris anti-BrdU) et arrêtez-vous avant le montage des tranches. Incuber pendant 2 h à température ambiante avec PBS++ (solution bloquante).
- Incuber le deuxième anticorps primaire (par exemple, anti-GFAP chez le lapin) dans PBS+ pendant une nuit à 4 °C. Suivez les étapes suivantes pour chaque protocole, y compris l’incubation du deuxième anticorps secondaire (p. ex., TRITC anti-lapin de chèvre). Passez aux étapes suivantes pour chaque protocole jusqu’à la fin.
7. Contre-coloration (facultatif)
- Pour les protocoles utilisant la réaction de peroxydase, préchauffer la solution de violet crésyl à 60 °C. Hydrater les lames avec ddH2O pendant 1 min. Incuber les lames dans la solution chaude de violet crésyle pendant 5 à min.
- Rincer les lames avec ddH2O pendant 1 min. Rincer les lames avec de l’alcool éthylique à 70 %, 80 %, 90 % et 100 % pendant 1 à 123 min chacune. Rincer les lames avec du xylène pendant 1 à 1 min.
- Ajouter un support de montage hydrophobe permanent et placer des lamelles de couverture.
REMARQUE : Conserver à 4 °C jusqu’à 6 mois. Un support de montage fabriqué par vous-même contenant du PVA (alcool polyvinylique)-DABCO peut être utilisé.
- Pour les protocoles utilisant l’immunofluorescence, ajouter un petit volume (25\u201250 μL) de support de montage hydrophile avec DAPI, iodure de propidium ou similaire. Scellez autour du périmètre avec du vernis à ongles ou un scellant en plastique. Conserver à 4 °C jusqu’à 6 mois.
8. Imagerie et analyse
REMARQUE : Voir le tableau 2 pour les spécifications de configuration du microscope. Habituellement, le comptage des nouvelles cellules colorées est effectué à l’aide des tranches colorées par réaction de peroxydase (méthode moins chère), mais il peut également être effectué par immunofluorescence.
- Pour quantifier les cellules, il faut d’abord identifier correctement le gyrus denté avec la lentille de grossissement 4x (pour plus d’instructions sur les détails anatomiques DG, voir Amaral et al.16).
- Recherchez dans la couche cellulaire granulaire du gyrus denté des noyaux marqués avec BrdU (à l’aide de la lentille de grossissement 40x). Effectuez une recherche de cellules exhaustive le long de l’axe z, car les nouvelles cellules peuvent être distribuées en différentes couches (voir la vidéo 1).
- Sélectionnez une section d’intervalle pour la recherche cellulaire sur tout le gyrus denté (par exemple, chaque 6ème section de tissu, équivalent à tous les 240 μm).
- Comptez toutes les cellules BrdU positives. La morphologie du noyau marqué peut changer en fonction de la quantité de BrdU incorporée par la cellule (voir la figure 3 à titre indicatif). Déplacez-vous lentement sur l’axe z pour quantifier les différents noyaux qui intègrent un amas (voir la vidéo 2).
- Multipliez le nombre total de cellules comptées par la section d’intervalle sélectionnée (par exemple, 6) pour estimer le nombre total de nouvelles cellules marquées BrdU.
- Idéalement, dans une expérience régulière, comptez au moins dix sections par animal et au moins cinq animaux par groupe.
- Déconvolution d’image (facultatif)
REMARQUE : reportez-vous à la section d’introduction pour obtenir des informations importantes sur cette étape. Cette procédure nécessite des images monochromatiques (niveaux de gris). Transformez les images couleur en niveaux de gris. Si les images sont un composite RVB, divisez d’abord les couches et fusionnez-les en une seule image (non composite), puis transformez-les en niveaux de gris 8 bits.- Créez un fichier z-stack à partir de micrographies.
- Créez un fichier de fonction d’étalement de points (PSF) en ouvrant le plug-in Diffraction PSF 3D (https://imagej.net/Diffraction_PSF_3D) à partir du menu d’options Plugins . Remplissez toutes les données requises (voir le tableau 2). Appuyez sur OK et enregistrez le fichier.
- Ouvrez le plugin DeconvolutionLab29 à partir du menu Plugins (http://bigwww.epfl.ch/deconvolution/deconvolutionlab2/). Faites glisser l’image z-stack et le fichier PDF correspondants vers l’emplacement de fenêtre correspondant.
- Sélectionnez l’algorithme de déconvolution (par exemple, Richardson-Lucy) et le nombre d’itérations (par exemple, 20). Appuyez sur RUN.
- Combinez les images déconvolutées en une seule image z-stack en sélectionnant Piles dans le menu Image en haut. Cliquez ensuite sur Projet Z. Sélectionnez Intensité maximale dans le menu déroulant Type de projection , appuyez sur OK et enregistrez le fichier.
- Créez une image RVB à l’aide du fichier image z-stack unique créé à l’étape ci-dessus avec la pseudocouleur souhaitée en sélectionnant Couleur dans le menu Image en haut. Cliquez ensuite sur Fusionner les chaînes. Définissez l’image correspondante sur le canal de couleur souhaité dans le menu déroulant. Décochez la case Créer un composite, appuyez sur OK et enregistrez le fichier (voir la figure 4).
- S’il existe plusieurs images de canal, répétez les étapes 8.2.1\u20128.2.5. Créez un fichier image RVB à l’étape 8.2.6, en ouvrant au moins deux fichiers image et en sélectionnant différents canaux de couleur pour chaque fichier image (voir la figure 4).
Résultats
Les méthodes décrites ci-dessus ont été appliquées pour quantifier les cellules nouveau-nées dans l’hippocampe de rat adulte après une activité physique volontaire, contrairement à un groupe témoin sans activité physique supplémentaire. Nous avons utilisé l’hippocampe postnatal du rat comme témoin positif. Les rats mâles âgés de 3 mois ont été soumis à un protocole d’activité physique volontaire (roue sans fin) pendant sept jours. Le jour 6, des rats ont reçu une injection de BrdU (section 2), puis toutes les 12 heures jusqu’à trois injections complètes. Pour compléter trois divisions du cycle cellulaire, les animaux ont été perfusés par voie transcardielle (section 3) 8 heures après la dernière injection de BrdU. La même procédure a été utilisée sur des rats âgés de trois mois qui n’ont pas subi d’activité physique pour être utilisés comme témoin comparatif. En tant que contrôle positif, des ratons d’un jour (jour 1 postnatal) ont reçu une injection de BrdU une fois, comme décrit dans la section 2 ci-dessus. Un jour après l’injection (jour 2 postnatal), les petits ont été euthanasiés et leur tête a été immergée dans une solution de PFA, comme décrit à l’étape 3.4. Les rats adultes ont été anesthésiés profondément (étape 3.1), perfusés par voie transcardiale, comme décrit à l’étape 3.2. Les cerveaux ont été disséqués et post-fixés (étape 3.4). Les cerveaux ont été coupés en sections coronales de 40 μm (étape 3.5). Des coupes ont été traitées pour l’immunohistochimie BrdU, comme décrit à l’étape 4.
Nous avons utilisé la réaction de la peroxydase de raifort avec DAB IHC pour la coloration (étape 4.1) et le comptage des cellules BrdU positives dans DG. La figure 5 montre une section DG avec des cellules marquées BrdU. La figure 5C,D montre une partie représentative de la section DG à un grossissement plus élevé. Les cellules marquées présentaient une coloration sombre intense, marquée par des flèches. L’encart montre le nombre moyen de cellules marquées dans les groupes expérimental et témoin (cellules positives comptées multipliées par six comme décrit à l’étape 8.1). Le test t d’un Student a révélé des différences significatives entre le nombre de cellules BrdU positives (t(10) = 2,704, p = 0,0222). Le groupe témoin qui n’a pas subi d’activité physique a montré 2 040 ± 314 cellules (n = 6 rats). En comparaison, le groupe d’activité physique a montré, en moyenne, 3 606 ± 486 (n = 6 rats) cellules BrdU positives. Comme on l’a observé, l’exposition à l’activité physique augmente les cellules BrdU positives. Par conséquent, ces résultats sont cohérents avec d’autres résultats rapportés qui montrent que l’activité physique a augmenté la prolifération cellulaire dans le gyrus dentéadulte 17.

Figure 3 : Exemples de morphologie différente du noyau cellulaire marqué BrdU. BrdU est un marqueur de synthèse de l’ADN qui marque le noyau. Dans la région de l’hippocampe, les noyaux BrdU positifs avaient une forme semi-ovale située dans la zone sous-granulaire du gyrus denté. Puisque BrdU est incorporé par compétition, la quantité incorporée pour chaque cellule aura une variation qui se reflétera plus tard dans la façon dont le noyau sera visualisé. (A) Image d’immunofluorescence. (B) Une image utilisant la réaction de peroxydase sans méthode d’amplification supplémentaire est présentée. Les flèches jaunes montrent des artefacts et des signaux non spécifiques. Les flèches noires ou blanches indiquent les cellules BrdU+. 1 – Noyau entièrement limposé, noyaux semi-ovales très colorés. 2 – Noyaux à points, le bord des noyaux est marqué et a à l’intérieur plusieurs points. 3 – Les noyaux avec peu de points, la bordure des noyaux sont marqués et ont un petit nombre de points à l’intérieur. 4 – Le petit noyau est une cellule possible à un stade de différenciation différent mais faisant toujours partie de la niche. 5 – Les grappes sont des cellules précurseurs en cours de division, donc plusieurs cellules ensemble en groupes condensés peuvent être observées. Au sein de ces groupes, le comptage doit être fait particulièrement soigneusement pour éviter d’étiqueter mal les cellules positives. Les flèches rouges montrent le noyau en division qui peut être confondu avec une seule cellule. Chaque cellule est enfermée dans une boîte et peut être distinguée dans un plan de l’axe Z en temps réel. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Image RVB représentative pour les canaux simples et fusionnés. L’image du haut montre l’image z-stack d’origine, et l’image inférieure montre l’image z-stack 3D déconvolutée. (A) Faible grossissement de la DG. (B) image RVB pour chaque canal, et (C) image fusionnée RVB. C’était un cerveau du groupe témoin. L’immunofluorescence a été utilisée sans méthode d’amplification supplémentaire. BrdU (rouge), DAPI comme contre-coloration (bleu) et GFAP (protéine acide fibrillaire gliale) comme marqueur astroglial (vert). ML = couche moléculaire; GCL = couche cellulaire granulaire; SGZ = zone sous-granulaire. Veuillez cliquer ici pour voir une version agrandie de cette figure.
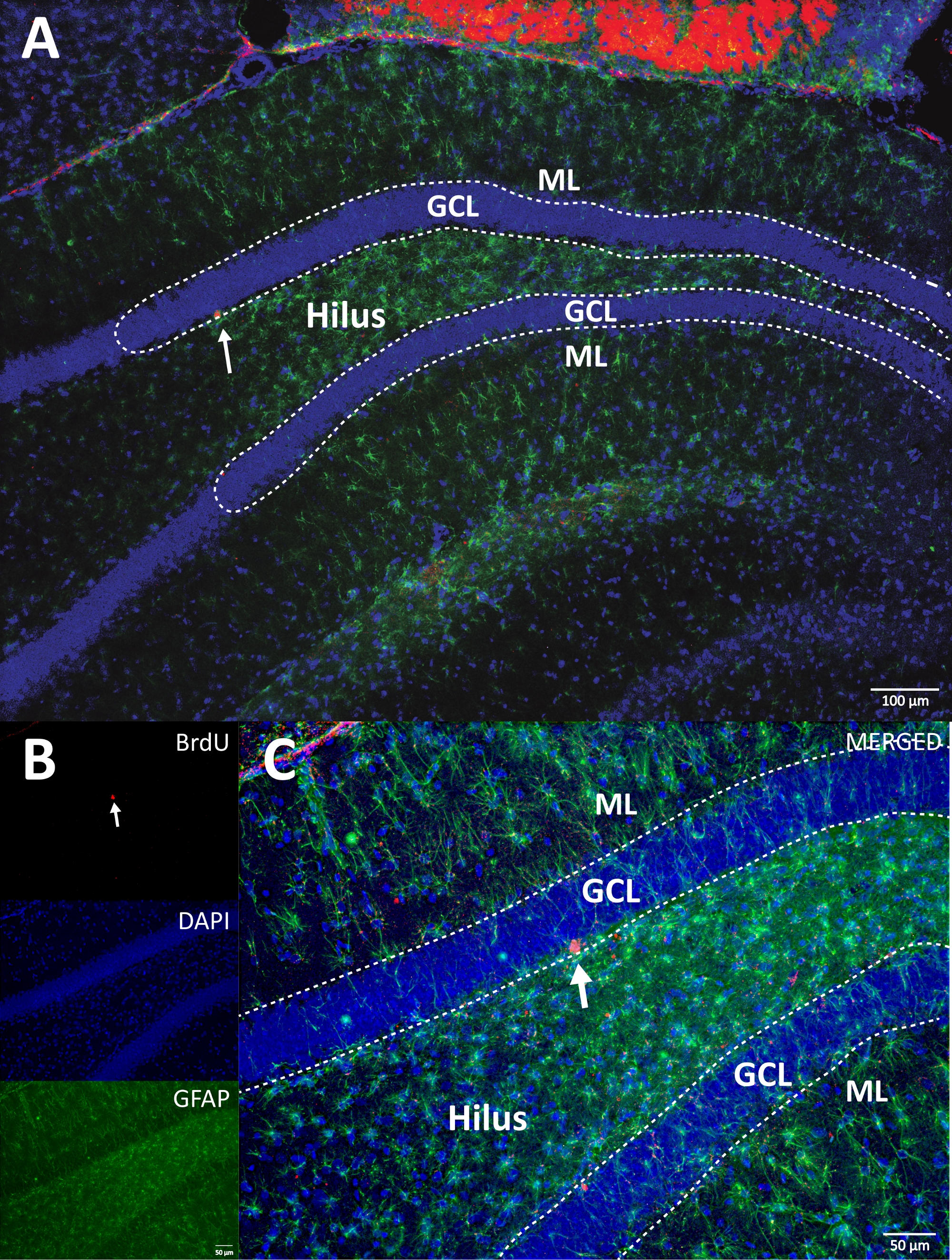{kind=link}

Figure 5 : Section représentative du DG avec des cellules marquées BrdU (foncé intense) pour chaque groupe expérimental. La réaction de peroxydase a été utilisée avec la méthode d’amplification du complexe avidine-biotine-peroxydase. (A, B) Afficher un faible grossissement du DG, et (C, D) montrer la zone de la boîte à un grossissement plus élevé. Les panels A et C sont des tissus du groupe d’activité physique, les panels B et D sont du groupe témoin. L’encart montre le nombre moyen de cellules marquées dans les groupes d’activité physique et de contrôle (cellules positives comptées multipliées par six comme décrit à l’étape 8.1). ML = couche moléculaire; GCL = couche cellulaire granulaire; SGZ = zone sous-granulaire; les flèches indiquent les cellules BrdU+. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Vidéo 1: Vidéo montrant un foyer différent de cellules positives le long de l’axe z réparties en différentes couches. Veuillez cliquer ici pour télécharger cette vidéo.
Vidéo 2 : Vidéo montrant un foyer différent d’amas de cellules positives le long de l’axe z répartis en différentes couches. Déplacez-vous lentement sur l’axe z pour quantifier les différents noyaux qui intègrent un amas. Veuillez cliquer ici pour télécharger cette vidéo.
| Type de microscope: | Microscope à épifluorescence Olympus BX53 | ||||
| Source lumineuse: | Lampe à arc au mercure haute pression de 130 W (U-HGLGPS) | ||||
| Logiciel d’acquisition : | CellSens Standard | ||||
| Jeux de filtres : | Numéro de catalogue | Plage d’excitation | Miroir dichromatique | Plage de suppression | |
| U-FUW | 340 - 490 nm | 410 nm | 420 nm | ||
| U-FBW | 460 - 495 nm | 505 nm | 510 nm | ||
| U-FGW | 530 - 550 nm | 570 nm | 575 nm | ||
| Caméra: | Modèle: | Caméra CCD UC50 | |||
| Gamme spectrale: | 290 – 1000 nm | ||||
| Taille de puce CCD : | 2/3 po, 2588 (largeur *7) x 1960 (hauteur *8) pixels | ||||
| Taille de pixel: | 3,4 X 3,4 μm | ||||
| Fluorochrome: | Nom | Longueur d’onde d’excitation (nm) | Émission *3 Longueur d’onde (nm) | Couleur d’émission | |
| 4, 6-diamidino-2-phényl-indole HCI (DAPI) | 345 | 455 | Bleu | ||
| Tétraméthylrhodamine-isothyocyanate (TRITC) | 541 | 572 | Rouge | ||
| Isothiocyanate de fluorescéine (FITC) | 494 | 519 | Vert | ||
| Cy3 | 552 | 565 | Rouge | ||
| Support de montage et huile d’immersion: | Nom | Indice de réfraction du milieu *1 | |||
| Air (rien entre la diapositive et l’objectif) | 1.00029 | ||||
| Support de montage antifade avec DAPI | 1.45 | ||||
| Support de montage par montage | 1.519 | ||||
| Huile d’immersion à faible autofluorescence (MOIL-30 Type F) | 1.518 | ||||
| Lentille grossissante (Plan Fluorite) | Grossissement | Ouverture numérique (NA) *2 | Résolution (μm) | Espacement des pixels de l’image (nm) *5 | Espacement des tranches axe Z (nm) *6 |
| 4X | 0.13 | 2.12 | 850 | 3000 | |
| 10X | 0.3 | 0.92 | 340 | 3000 | |
| 20X | 0.5 | 0.55 | 27S | 2000 | |
| 40X | 0.75 | 0.37 | 85 | 1000 | |
| 100X | 1.3 | 0.21 | 34 | 1000 | |
Tableau 2 : spécifications de configuration du microscope et exigences de création de fichiers PSF (Point Spread Function). Il y a 11 emplacements dans la fenêtre du plugin diffraction PSF 3D pour créer le fichier PSF. Chaque emplacement est décrit comme suit : *1 - Indice de réfraction du milieu : indice de réfraction pour le milieu entre la lame et la lentille (par exemple, air = 1,00029). *2 - Ouverture numérique : NA de l’objectif utilisé (il doit être corrigé lorsqu’un média d’immersion différent est utilisé et que l’objectif a été assigné). *3 - Longueur d’onde: Longueur d’onde d’émission maximale fluorochrome (nm). *4 - Aberration sphérique longitudinale : 0,00. *5 - Espacement des pixels de l’image : taille de pixel CCD (nm)/grossissement (par exemple, objectif 3,4 μm et 100X, 3400/100 = 34 nm). *6 - Distance entre les images axe Z. *7 - Largeur : Entrez la largeur de l’image à décomposer en pixels. *8 - Hauteur : Entrez la hauteur de l’image à décomposer en pixels. *9 - Profondeur, tranches : le nombre d’images dans la z-stack. *10 - Normalisation : Somme des valeurs de pixels = 1. *11 - Titre : Nom souhaité pour le fichier PSF. Le fichier doit correspondre à l’image z-stack unique donnée.
Discussion
La neurogenèse adulte est un processus qui se produit le plus souvent dans des niches de cellules précurseurs neuronales adultes qui ont le potentiel de générer de nouveaux neurones tout au long de leur vie. Le marquage de la bromodésoxyuridine (BrdU) est largement utilisé en immunologie pour caractériser le nombre de cellules nouvellement générées dans un cerveau adulte. BrdU sera principalement incorporé dans les cellules de régions cérébrales discrètes (zones neurogène). Ces cellules sont situées dans la zone sous-ventriculaire (SVZ), le gyrus denté de l’hippocampe, entre le hile et les cellules granulaires connues sous le nom de zone sous-granulaire (SGZ)1,2,18. De plus, il existe différentes régions du cerveau caractérisées par une capacité proliférative plus faible à l’âge adulte, notamment l’hypothalamus, le striatum, le néocortex et l’amygdale19. Comme mentionné précédemment, la coloration BrdU est la méthode couramment utilisée pour la recherche sur la neurogenèse chez l’adulte afin de détecter la prolifération cellulaire. Cependant, l’utilisation de BrdU comme marqueur a des limites et des pièges. La première est que BrdU est un marqueur du cycle cellulaire. Par conséquent, une double ou triple coloration doit être effectuée pour identifier le devenir cellulaire et inclure des marqueurs cellulaires pour détecter le stade de développement spécifique des cellules marquées. Une autre préoccupation au sujet de BrdU est qu’il s’agit d’une solution toxique et mutagène qui modifie la stabilité de l’ADN et peut altérer la fonction cellulaire et les cycles cellulaires. Il faut tenir compte des informations précédentes lors de la décision de suivre un protocole d’administration et des doses d’administration (50\u2012600 mg/kg). Une autre caractéristique cruciale est que BrdU est un marqueur de synthèse de l’ADN, pas un marqueur de prolifération cellulaire14. Par conséquent, il est pertinent de distinguer la prolifération cellulaire d’autres événements tels qu’une réparation de l’ADN, une rentrée avortée du cycle cellulaire et une duplication de gènes. Les chercheurs doivent suivre les contrôles appropriés pour assurer l’utilisation appropriée de BrdU. Pour une discussion plus détaillée sur ces problèmes et limites, nous vous recommandons de revoir le travail de Taupin14. Le processus de normalisation d’un protocole d’immunohistochimie pourrait être lent et difficile. Dans ce travail, nous avons présenté toutes les étapes générales pour gérer un protocole IHC réussi. Cependant, nous recommandons que chaque groupe de recherche teste et évalue les tissus, les anticorps et les conditions à l’avance. Les tests et les évaluations doivent être effectués avec au moins trois niveaux différents d’incubation, d’étapes de lavage et de concentration pour chaque anticorps et tissu testé. Nous recommandons également aux chercheurs d’examiner les protocoles supplémentaires afin de pouvoir choisir le meilleur qui répond aux besoins et exigences spécifiques 20,21,22,23,24,25.
Comme mentionné précédemment, la procédure implique plusieurs étapes et considérations méthodologiques qui sont couramment utilisées et mentionnées dans les articles scientifiques, qui seront discutées plus tard. Nous recommandons aux chercheurs de choisir les anticorps avec soin et correctement en termes de technique, de budget, d’équipement, de configuration et d’objectif principal de recherche. Les anticorps doivent être testés avec le même type de tissu qui sera testé plus tard dans l’expérience. Nous recommandons également l’utilisation d’un anticorps qui a été testé dans le même but (IHC) (c.-à-d. pas seulement dans les techniques de Western blot ou de cytométrie en flux) pour tester sa compatibilité avec la technique de fixation. Différentes voies pourraient être utilisées pour administrer la coloration BrdU, comme l’injection intrapéritonéale, la perfusion intrapéritonéale, l’ingestion orale ou la perfusion intraventriculaire (pour une description plus détaillée de chaque technique, voir la référence26). Si l’injection intrapéritonéale est sélectionnée, assurez-vous que BrdU est administré dans la cavité péritonéale en évitant la région de l’intestin. Puisque l’intestin a plusieurs cellules en duplication qui peuvent épuiser le BrdU avant qu’il n’atteigne le cerveau, ce qui affectera le nombre de cellules marquées. Il est crucial d’obtenir des sections minces car elles permettent une meilleure pénétration des solutions. Des tranches coronales de 40 μm d’épaisseur ont été coupées rostro-caudale et transférées dans une plaque de culture cellulaire de 24 puits, suivant la procédure stéréologique proposée par Kempermann et al.27. L’immunohistochimie peut être réalisée avec des tissus montés sur des lames ou sous forme de sections flottantes. Puisque BrdU est situé profondément dans les noyaux cellulaires, il permet la pénétration de solutions dans des sections flottantes libres, ce qui donne de meilleurs résultats et un meilleur accès à la zone d’intérêt. Il est important d’ouvrir les liaisons ADN (dénaturation de l’ADN) pour permettre l’accès à l’anticorps anti-BrdU primaire. Dans ce travail, nous avons effectué ces procédures spécifiques en utilisant l’incubation HCI. D’autre part, le processus de blocage d’épitopes non spécifiques a permis une identification plus précise du signal cellulaire.
La bonne perméabilisation membranaire permet aux anticorps de pénétrer correctement dans la zone d’intérêt. L’ajout d’un perméabilisant tel que Triton X-100 aux solutions PBS++ et PBS+ améliore la perméabilisation de la membrane. Les réactifs PBS et Tris-buffered saline (TBS) peuvent être utilisés dans ce protocole. En termes de budget, le SCT pourrait être relativement moins cher que le PBS. Cependant, le PBS pourrait interférer avec les anticorps anti-phosphate et inhiber les anticorps conjugués à la phosphatase alcaline, alors évitez l’utilisation de PBS si la cible est modifiée post-traductionnellement par phosphorylation (c.-à-d. phosphorylée). Nous avons utilisé le PBS pour ce travail, et nous avons découvert que les étapes de lavage des tissus donnaient un signal plus spécifique. Nous recommandons également aux chercheurs d’effectuer au moins trois cycles de lavage en utilisant TBS ou PBS. Les solutions doivent être fraîchement préparées. Le prélèvement d’antigènes (RA) est une méthode destinée à réduire la perte d’antigénicité causée par la fixation qui modifie la structure des antigènes tertiaires et quaternaires. Cette réduction rend les antigènes indétectables par les anticorps28,29. Le prélèvement d’épitopes induit par la chaleur (HIER) utilisé dans ce protocole a tenté d’inverser les réactions chimiques entre le formaldéhyde et les protéines par hydrolyse alcaline à haute température ou forte (avec d’autres solutions tampons comme l’EDTA pH 8,5 ou Tris pH 9,5). Il est essentiel de tester de nouveaux anticorps avec différents protocoles AR pour comparer les résultats et choisir le meilleur pour le protocole. Cette dernière étape peut être facultative dans un protocole régulier; Cependant, nous avons traité les tissus avec un protocole de récupération d’antigènes pour fournir de meilleurs résultats pour ce protocole.
Il est crucial de sélectionner la couleur contrastante finale correcte et la technique de contre-coloration en tenant compte de la couleur de coloration primaire et de la méthode utilisée pour rendre visible une structure non tachante et éviter de masquer la couleur de coloration primaire de la réaction immunitaire. Pour la microscopie à fluorescence, le DAPI (4', 6-diamidino-2-phénylindole) est une contre-coloration nucléaire et chromosomique très populaire qui émet une fluorescence bleue (absorption: 360 nm, émission: 460 nm) lors de la liaison aux régions AT de l’ADN. Un support de montage contenant du DAPI est disponible et facile à utiliser; Cela fournit une excellente rétention du signal pour l’acquisition d’images. Pour la réaction au peroxyde, l’IHC était disponible dans différentes options telles que la coloration au violet de crésyle, à l’hématoxyline, au rouge neutre ou au vert de méthyle. Pour les techniques d’immunomarquage multiples, il est crucial de choisir un anticorps compatible avec la technique de fixation utilisée pour éviter la réactivité croisée30. Lorsque les problèmes et les complications liés à la coloration unique sont résolus, administrez une autre coloration de couleur si nécessaire. Il est crucial de contrôler la liaison non spécifique entre les anticorps secondaires. Cela pourrait être fait en saturant les anticorps primaires avant d’utiliser un anticorps secondaire produit dans la même espèce hôte des anticorps primaires. Par exemple, lors de l’utilisation d’anticorps anti-souris produits chez le lapin et d’anticorps anti-lapin produits chez les anticorps secondaires de chèvre, l’anti-lapin produit dans l’anticorps de chèvre doit être utilisé avant l’anticorps anti-souris produit dans l’anticorps de lapin. Lorsque la méthode séquentielle est complètement dominée, le processus d’immunomarquage simultané peut être lancé. Dans cette méthode, il est essentiel de choisir les anticorps secondaires de manière appropriée. Idéalement, tous ces anticorps doivent provenir du même animal hôte pour éviter la réactivité croisée. Nous recommandons d’effectuer un contrôle positif pour confirmer que la méthode de coloration fonctionne avec précision dans le tissu postnatal de l’hippocampe (neurogenèse abondante autour de cet âge). Si le tissu témoin positif présente des problèmes de coloration, revoir et revoir la procédure, apporter des corrections et des ajustements, et répéter jusqu’à ce qu’une bonne coloration soit produite. Ensuite, exécutez un contrôle négatif pour vérifier que l’anticorps fonctionne correctement en omettant ou en remplaçant un anticorps primaire particulier par du sérum normal (même espèce que l’anticorps primaire). Comme mentionné dans l’introduction, la déconvolution d’image est un outil puissant et offre une alternative lorsqu’un microscope confocal n’est pas disponible. Il peut appliquer la déconvolution d’image à toutes les images obtenues à l’aide de la microscopie à fond clair transmise, de fluorescence à grand champ et de fluorescence confocale. Le but ultime de la déconvolution d’image est de reconstruire le signal original que le système d’acquisition se détériore10.
En résumé, l’identification des cellules nouvellement générées visualisées par l’immunodétection de l’analogue de thymidine BrdU est une technique compliquée mais puissante. Ce travail est une tentative d’aider les scientifiques, en particulier dans le domaine de la neurogenèse hippocampique adulte, à quantifier les nouvelles cellules avec plus de précision. Nous espérons que cet effort a été utile à la communauté scientifique et a facilité l’étude de la prolifération cellulaire par la technique d’immunohistochimie.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous tenons à remercier M. Miguel Burgos et Gustavo Lago pour leur assistance technique. Nous tenons également à remercier le Dr Clorinda Arias, le Dr Karla Hernandez et le Dr Oscar Galicia pour leur aimable soutien dans la fourniture de réactifs et de matériel. Nous remercions également la División de Investigación y Posgrado de l’Universidad Iberoamericana Ciudad de México d’avoir financé l’exécution de ce travail et d’avoir couvert les dépenses de production vidéo.
matériels
| Name | Company | Catalog Number | Comments |
| REAGENT PREPARATION AND SETUP | |||
| Donor Horse Serum | BioWest | S0900 | Blocking and incubation solutions in PBS |
| Paraformaldehyde reagent grade, crystalline (PFA) | Sigma-Aldrich | P6148 | toxic, flammable |
| Potassium Chloride | Sigma-Aldrich | 746436-500G | |
| Potassium Phosphate, monobasic | J.T Bker | 3246-01 | |
| Sacarose | J.T Baker | 4072-01 | |
| Sodium Chloride | Meyer | 2365-500G | |
| Sodium Hydroxide | Sigma-Aldrich | S5881-500G | Corrisive, to calibrate pH |
| Sodium Phophate Dibasic | Sigma-Aldrich | S9763-5KG | |
| Triton-x 100 | Sigma-Aldrich | T8787 | |
| THYMIDINE ANALOGUE BRDU ADMINISTRATION | |||
| 5-Bromo-2′-deoxyuridine, BrdU | Sigma-Aldrich | B9285 | toxic (mutagenic, teratogenic) |
| 23–27G hypodermic needle | BD PrecisionGlide | ||
| Saline solution | PiSA | 30130032 | |
| Syringes 1 mL | NIPRO | ||
| TISSUE PREPARATION | |||
| 15-ml polypropylene conical tube | Thermo Scientific | 339650 | |
| 50-ml polypropylene conical tube | Thermo Scientific | 339652 | |
| Dissecting tools | |||
| Guillotine | Stoelting | ||
| Microtome Cryostat | MICROM | HM525 | |
| Netwell 15 mm polyester mesh membrane inserts | Corning | 3477 | pre-loaded in 12-well culture plates |
| Netwell plastic 12-well carrier kit | Corning | 3520 | for 15 mm polyester mesh membrane inserts |
| Netwell plastic 6-well carrier kit | Corning | 3521 | |
| Perfusion pump | Cole-Parmer | 7553-70 | |
| Shaker | IKA | ROKCER 3D Digital | |
| IMMUNOSTAINING | |||
| Cresyl violet | Sigma-Aldrich | C5042 | 1%, Light sensitive |
| DAB Peroxidase (HRP) Substrate Kit (with Nickel), 3,3’-diaminobenzidine | Vector Laboratories | SK-4100 | carcinogenic, light sensitive |
| Hydochloric Acid | J.T.Baker | 9535-05 | Corrosive, to calibrate pH |
| Hydrogen Peroxide, 50% | Meyer | 5375-1L | Toxic, oxidative |
| Permount Mounting Medium | Fisher Chemical | SP15-500 | |
| VECTASHIELD Antifade Mounting Medium with DAPI | Vector Laboratories | H-1200 | Light sensitive |
| VECTASTAIN® Elite® ABC Kit Peroxidase (HRP) | Vector Laboratories | PK-6100 | enzymatic, avidin/biotin based amplification system |
| Primary antibodies | |||
| Anti-GFAP antibody produced in rabbit | Sigma-Aldrich | HPA056030 | 1:500 |
| Monoclonal Anti-BrdU antibody produced in Mouse | Sigma-Aldrich | B2531 | 1:250 |
| Secondary antibodies | |||
| Biotin-SP (long spacer) AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch | 715-065-151 | 1:250 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, HRP | Invitrogen | G-21040 | 1:1000 |
| Goat Anti-Mouse IgG (whole molecule), TRITC | Sigma-Aldrich | T5393 | 1:250 |
| Goat Anti-Rabbit IgG (H+L) Cross-Adsorbed, FITC | Invitrogen | F-2765 | 1:250 |
| Streptavidin, Cy3 | Vector Laboratories | SA-1300 | 1:250 |
| IMAGING AND ANALYSIS | |||
| Computer | Dell Computer Company | T8P8T-7G8MR-4YPQV-96C2F-7THHB | For controlling and monitoring protocols’ processes |
| CCD-camera | Olympus | UC50 | |
| CellSens Standard software | Olympus | cellSens Standard Edition | Acquisition Software |
| Epifluorescent microscope | Olympus | BX53 | |
| High-pressure 130 W mercury arc lamp | Olympus | U-HGLGPS | |
| low autofluorescence immersion oil | Olympus | MOIL-30 Type F | |
| Micro cover-glasses (VWR, cat no 48404 454; 24 60 mm) | |||
| Microscope slides (VWR, cat no 48323-185; 76 26 mm) | |||
| U-FBW filter cube | Olympus | U-FBW | excitation 460 - 495 nm, dichroic mirror 505 nm, suppression 510 nm |
| U-FGW filter cube | Olympus | U-FGW | excitation 530 - 550 nm, dichroic mirror 570 nm, suppression 575 nm |
| U-FUW filter cube | Olympus | U-FUW | excitation 340 - 490 nm, dichroic mirror 410 nm, suppression 420 nm |
Références
- Altman, J. Are new neurons formed in the brains of adult mammals. Science. 135 (3509), 1127-1128 (1962).
- Altman, J., Das, G. D. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. The Journal of Comparative Neurology. 124 (3), 319-335 (1965).
- Miller, M. W., Nowakowski, R. S. Use of bromodeoxyuridine-immunohistochemistry to examine the proliferation, migration and time of origin of cells in the central nervous system. Brain Research. 457 (1), 44-52 (1988).
- Eriksson, P. S., et al. Neurogenesis in the adult human hippocampus. Nature Medicine. 4 (11), 1313-1317 (1998).
- Filippov, V., et al. Subpopulation of nestin-expressing progenitor cells in the adult murine hippocampus shows electrophysiological and morphological characteristics of astrocytes. Molecular and Cellular Neuroscience. 23 (3), 373-382 (2003).
- Kempermann, G., Jessberger, S., Steiner, B., Kronenberg, G. Milestones of neuronal development in the adult hippocampus. Trends in Neurosciences. 27 (8), 447-452 (2004).
- Matiašová, A., et al. Flow cytometric determination of 5-bromo-2'-deoxyuridine pharmacokinetics in blood serum after intraperitoneal administration to rats and mice. Histochemistry and Cell Biology. 142 (6), 703-712 (2014).
- Von Bohlen Und Halbach, O. Immunohistological markers for proliferative events, gliogenesis, and neurogenesis within the adult hippocampus. Cell and Tissue Research. 345 (1), 1-19 (2011).
- Sage, D., et al. DeconvolutionLab2: An open-source software for deconvolution microscopy. Methods. 115, 28-41 (2017).
- Manz, W., Arp, G., Schumann-Kindel, G., Szewzyk, U., Reitner, J. Widefield deconvolution epifluorescence microscopy combined with fluorescence in situ hybridization reveals the spatial arrangement of bacteria in sponge tissue. Journal of Microbiological Methods. 40 (2), 125-134 (2000).
- Schindelin, J., et al. Fiji: An open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Abràmoff, M. D., Magalhães, P. J., Ram, S. J. Image processing with imageJ. Biophotonics International. 11 (7), 36-41 (2004).
- . 2D and 3D Fluorescence Deconvolution Manual Available from: https://pages.stolaf.edu/wp-content/uploads/sites/803/2016/12/Giannini_Giannini_Deconvolution_Manual_20161215.p (2016)
- Taupin, P. BrdU immunohistochemistry for studying adult neurogenesis: Paradigms, pitfalls, limitations, and validation. Brain Research Reviews. 53 (1), 198-214 (2007).
- Revilla, V., Jones, A. Cryostat sectioning of brains. International Review of Neurobiology. 47, 61-70 (2002).
- Amaral, D. G., Scharfman, H. E., Lavenex, P. The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies). Progress in brain research. 163, 3-22 (2007).
- Eadie, B. D., Redila, V. A., Christie, B. R. Voluntary exercise alters the cytoarchitecture of the adult dentate gyrus by increasing cellular proliferation, dendritic complexity, and spine density. Journal of Comparative Neurology. 486 (1), 39-47 (2005).
- Leal-Galicia, P., Romo-Parra, H., Rodríguez-Serrano, L. M., Buenrostro-Jáuregui, M. Regulation of adult hippocampal neurogenesis exerted by sexual, cognitive and physical activity: An update. Journal of Chemical Neuroanatomy. 101, 101667 (2019).
- Gould, E. How widespread is adult neurogenesis in mammals. Nature Reviews Neuroscience. 8 (6), 481-488 (2007).
- Ngwenya, L. B., Peters, A., Rosene, D. L. Light and electron microscopic immunohistochemical detection of bromodeoxyuridine-labeled cells in the brain: Different fixation and processing protocols. Journal of Histochemistry and Cytochemistry. 53 (7), 821-832 (2005).
- Ansorg, A., Bornkessel, K., Witte, O. W., Urbach, A. Immunohistochemistry and multiple labeling with antibodies from the same host species to study adult hippocampal neurogenesis. Journal of Visualized Experiments. (98), e52551 (2015).
- Tuttle, A. H., et al. Immunofluorescent detection of two thymidine analogues (CldU and IdU) in primary tissue. Journal of Visualized Experiments. (46), e2166 (2010).
- Tischler, A. S. Triple immunohistochemical staining for bromodeoxyuridine and catecholamine biosynthetic enzymes using microwave antigen retrieval. Journal of Histochemistry and Cytochemistry. 43 (1), 1-4 (1995).
- Taupin, P. Protocols for studying adult neurogenesis: Insights and recent developments. Regenerative Medicine. 2 (1), 51-62 (2007).
- Leuner, B., Glasper, E. R., Gould, E. Thymidine analog methods for studies of adult neurogenesis are not equally sensitive. Biosystems. 517 (2), 123-133 (2010).
- Magavi, S. S., MacKlis, J. D. Identification of newborn cells by BrdU labeling and immunocytochemistry in vivo. Methods in Molecular Biology. 438, 335-343 (2008).
- Kempermann, G., Gast, D., Kronenberg, G., Yamaguchi, M., Gage, F. H. Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development. , 391-399 (2003).
- Ramos-Vara, J. A., Beissenherz, M. E. Optimization of immunohistochemical methods using two different antigen retrieval methods on formalin-fixed, paraffin-embedded tissues: Experience with 63 markers. Journal of Veterinary Diagnostic Investigation. 12 (4), 307-311 (2000).
- Ramos-Vara, J. A. Principles and methods of immunohistochemistry. Methods in Molecular Biology. 1641, 115-128 (2017).
- Wojtowicz, J. M., Kee, N. BrdU assay for neurogenesis in rodents. Nature Protocols. 1 (3), 1399-1405 (2006).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.