
Method Article
Préparation, administration et évaluation de l’absorption cellulaire in vivo de liposomes marqués par un colorant fluorescent
Dans cet article
Résumé
L’objectif de ce protocole est de synthétiser des liposomes marqués par fluorescence et d’utiliser la cytométrie en flux pour identifier la localisation in vivo des liposomes au niveau cellulaire.
Résumé
Il existe un intérêt croissant pour l’utilisation des liposomes pour délivrer des composés in vivo, en particulier pour les approches de traitement ciblées. Selon la formulation des liposomes, les liposomes peuvent être absorbés préférentiellement par différents types de cellules dans le corps. Cela peut influencer l’efficacité de la particule thérapeutique car la progression de différentes maladies est spécifique au tissu et au type cellulaire. Dans ce protocole, nous présentons une méthode de synthèse et de marquage fluorescent des liposomes à l’aide de DSPC, de cholestérol et de PEG-2000 DSPE et du colorant lipidique DiD en tant que marqueur fluorescent. Ce protocole présente également une approche pour administrer des liposomes in vivo et évaluer l’absorption spécifique des liposomes par cytométrie en flux. Cette approche peut être utilisée pour déterminer les types de cellules qui absorbent les liposomes et quantifier la distribution et la proportion de l’absorption des liposomes entre les types de cellules et les tissus. Bien que cela ne soit pas mentionné dans ce protocole, des tests supplémentaires tels que l’immunofluorescence et l’imagerie par fluorescence unicellulaire sur un cytomètre renforceront les résultats ou les conclusions formulés car ils permettent d’évaluer la coloration intracellulaire. Les protocoles peuvent également devoir être adaptés en fonction du ou des tissus d’intérêt.
Introduction
À mesure que l’intérêt pour le développement de thérapies utilisant l’administration de médicaments par nanoparticules augmente, les méthodes de préparation et d’évaluation de la distribution et de l’absorption des particules doivent continuer à progresser, à se développer et à être accessibles au milieu de la recherche 1,2. Ce protocole a été développé pour évaluer les types de cellules exactes qui ont absorbé les liposomes in vivo après un traitement avec des liposomes marqués DiD chargés de tésaglitazar, unagoniste 3,4 du récepteur activé par les proliférateurs de peroxysomes (PPAR)-α/γ. Dans ces études, nous avons pu évaluer quels types de cellules étaient directement touchés par le traitement par tésaglitazar liposomal, l’efficacité du ciblage des fractions et générer des hypothèses pour expliquer les résultats du traitement que nous avons observés. De plus, les fonctions biologiques établies dans une variété de types cellulaires suggèrent que les cellules phagocytaires telles que les macrophages, les cellules dendritiques et les cellules de Kupffer spécifiques au foie absorbent la plupart des liposomes 5,6,7. En utilisant ce protocole, nous avons démontré que les phagocytes non classiques pouvaient également absorber les liposomes 3,4.
Ce protocole présente une méthode optimisée pour solubiliser le tesaglitazar, préparer les liposomes par évaporation en phase inverse et utiliser l’acétate de calcium comme attractif pour la charge de médicaments à distance. Les méthodes présentées sont accessibles à de nombreux laboratoires et manquent de matériaux difficiles à acquérir et d’étapes nécessitant des températures élevées. Le protocole produit des liposomes de tailles optimales pour une circulation accrue in vivo8. En outre, comme l’ont résumé Su et coll., à ce jour, des méthodes d’évaluation de la distribution in vivo des liposomes et de l’absorption tissulaire ont été étudiées et testées en profondeur9. Les méthodes de tomographie par émission de positons (TEP), d’imagerie par résonance magnétique (IRM) et de tomographie moléculaire par fluorescence (FMT) sont appliquées pour quantifier la biodistribution et l’absorption spécifiques aux tissus 9,10,11. Bien que ces méthodes aient été optimisées pour maximiser la détection in vivo, elles n’ont toujours pas la capacité de quantifier l’absorption des liposomes in vivo à la résolution cellulaire. Le protocole présenté ici vise à répondre à ce besoin grâce à l’utilisation de la cytométrie en flux. Enfin, pour ce protocole, l’absorption cellulaire a été réduite à quelques tissus, y compris le tissu adipeux. Il existe de plus en plus de littérature sur le potentiel d’utilisation des nanoparticules pour administrer des thérapies dans le contexte de l’obésité, du dysmétabolisme et de l’inflammation 12,13,14,15,16,17. À ce titre, nous avons jugé important de partager un protocole avec des méthodes efficaces de traitement et d’analyse du tissu adipeux, l’un des tissus qui joue un rôle important dans ces pathologies.
Protocole
Toutes les étapes de ce protocole sont approuvées par le Comité de soin et d’utilisation des animaux de l’Université de Virginie et suivent les directives de celui-ci.
REMARQUE : Il y a quelques contrôles importants à considérer pour les étapes d’analyse ultérieures, qui sont résumés dans le tableau 1 et devraient être pris en compte avant l’administration de liposomes.
1. Préparation de liposomes marqués par fluorescence, chargés d’acétate de calcium et de tésaglitazar
- Combinez DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine), cholestérol, PEG-2000-DSPE et DiD. Pour cela, combinez le DSPC, le cholestérol et le PEG-2000 DSPE à un rapport de masse de 2: 1: 1. Ajouter un colorant lipidique DiD à une concentration de 1 mg de DiD par 1 mL de liposomes (rapport molaire de 46:1 de DSPC:DiD).
REMARQUE : DiD est une abréviation acceptée pour le colorant 1,1'-dioctadécyl-3,3,3',3'tétraméthylindocarbocyanine. Comme il a deux octadécyles « queues grasses » de longueur égale au DSPC utilisé dans cette formulation, il devrait principalement être incorporé dans la membrane lipidique. Les colorants lipidiques comme DiO, DiD et DiI sont couramment utilisés pour la recherche sur les liposomes8 et ils sont considérés comme non échangeables18. - Utilisez un flacon de scintillation de 20 ml pour l’émulsion en phase inversée et la préparation des liposomes. Dans ce flacon, mélanger une solution éther-chloroforme 2:1 de lipides avec de l’acétate de calcium aqueux (acétate de Ca, 1 M, pH 7,4). Le rapport entre la phase organique et la phase aqueuse devrait être de 4:1, p. ex. 4 mL de phase organique et 1 mL de phase aqueuse.
- Émulsionner la solution éther-chloroforme de lipides par sonication pendant 30 s à température ambiante. Faites fonctionner le sonicateur à 20 KHz et 50% de puissance et utilisez un 1/2 po. sonde.
REMARQUE: Gardez l’extrémité de la sonde sonicator plus près du fond du flacon pour éviter la formation de mousse. Ne touchez pas le verre avec la pointe de la sonde pendant la sonication, il pourrait se briser. De plus, le chloroforme doit être ajouté à l’éther en tant que co-solvant: en présence de cholestérol, une émulsion d’éther seul se sépare rapidement, ce qui rend cette étape de la procédure impossible. - Placez immédiatement le flacon avec émulsion eau dans huile homogénéisée sur un évaporateur rotatif doté d’un adaptateur spécial, d’une jauge de manomètre et d’une vanne de régulateur de pression. L’évaporateur doit être connecté à une conduite de vide pour éliminer les solvants organiques. Réglez la vitesse de rotation à 100 tr/min et le vide à 0,5 atm, et relâchez si la mousse de l’émulsion semble excessive. Après la formation et la disparition d’un gel, augmentez le vide à 0,9 atm.
REMARQUE: Lors de l’élimination de la phase organique volatile, le niveau de vide doit être ajusté progressivement, pour éviter un moussage rapide, car cela peut entraîner une perte de contenu du flacon dans le corps de l’évaporateur rotatif. Finalement, lorsque l’éther et le chloroforme s’évaporent partiellement et que le rapport volumique entre la phase aqueuse et la phase de solvant organique est proche de 1:1, un gel se forme. L’évaporation doit se poursuivre jusqu’à ce que le gel disparaisse et que le milieu aqueux restant soit à nouveau complètement liquide. Un mélange supplémentaire peut aider à accélérer l’élimination des solvants organiques. Ceci peut être réalisé en plaçant une barre d’agitation en polytétrafluoroéthylène dans le ballon d’évaporation, afin d’améliorer la convection du gel visqueux pendant l’évaporation rotative. - Filtrer les liposomes résultants à l’aide de membranes en polycarbonate gravées pour obtenir une distribution granulométrique homogène.
- Effectuer la filtration en faisant passer la dispersion aqueuse des liposomes plusieurs fois à travers un filtre en polycarbonate à pores de 200 nm dans une extrudeuse de liposomes équipée de deux seringues étanches aux gaz.
REMARQUE : Les seringues plus petites sont préférables (p. ex. 0,5 mL), car elles assurent une pression suffisante pour la filtration. Avec une teneur élevée en cholestérol dans la membrane liposomique, une température élevée n’est pas nécessaire et la procédure peut être effectuée à température ambiante. Un nombre impair de filtrations (par exemple, 21) est effectué, de sorte que le matériau résultant se retrouve du côté opposé du filtre dès le début et s’il est pré-stérilisé, l’échantillon stérile de liposomes filtrés ajustés peut être collecté. La taille des liposomes résultants est généralement proche de la taille des pores filtrants sélectionnés. Deux filtres peuvent être empilés (au lieu d’un) pour effectuer un réglage précis à la taille inférieure des particules. - Vérifier la distribution de taille à l’aide de la diffusion dynamique de la lumière laser (DLS)3,4.
- Ajouter 1 à 3 mL de solution saline dans une cuvette de 1 cm à quatre côtés transparents. À cela, ajoutez 10 μL de liposomes et mélangez soigneusement. Placez l’échantillon dans l’appareil et sélectionnez les paramètres suivants à mesurer: viscosité du solvant, indice de réfraction, indice de réfraction des lipides. Cliquez sur le bouton Démarrer . Les mesures dureront plusieurs minutes et consisteront en 100 passages ou plus.
- Effectuer la filtration en faisant passer la dispersion aqueuse des liposomes plusieurs fois à travers un filtre en polycarbonate à pores de 200 nm dans une extrudeuse de liposomes équipée de deux seringues étanches aux gaz.
- Retirez l’acétate de Ca externe à l’aide d’une colonne de dessalage de la colonne. Ajouter à la moitié du lot, ajouter le tésaglitazar aqueux dans un tampon HEPES 10 mM (pH 7,4) et incuber en mélangeant à 37 °C pendant 1 h. Utilisez la seconde moitié du lot comme formulation de liposomes de contrôle sans médicament.
REMARQUE : Prééquilibrer la colonne de spin dessalante de 2 mL avec un tampon HEPES de 10 mM, pH 7,4, avant utilisation. Pour ce faire, placez 1 mL de tampon HEPES dans la colonne et faites tourner dans une centrifugeuse à 1000 x g pendant 2 min. Retirez la mémoire tampon pass-through et répétez cette opération quatre fois. - Retirer le tesaglitazar non piégé des liposomes à l’aide d’une colonne de spin de 2 mL et déterminer la concentration du médicament piégé par spectrophotométrique.
- N’ajoutez pas plus de 0,5 mL d’échantillon de liposomes dans le lit de gel de la colonne sèche et attendez que tout l’échantillon pénètre dans le gel. Centrifuger exactement dans les mêmes conditions que précédemment (1000 x g, 2 min) et recueillir l’échantillon de liposomes dans le pass-through purifié à partir de composés de petite masse moléculaire.
- Quantifier les caractéristiques finales des particules: taille et concentration des particules en utilisant le potentiel DLS et zêta avec un système combiné DLS-électrophorectic light scattering (ELS)3,4 dans 10 mM de tampon HEPES pH 7,4 et à 25 °C.
- Comme à l’étape 1.5.2, diluer la dispersion des liposomes dans le tampon de mesure (p. ex. 10 μL de liposomes par 1 mL de solution tampon) dans une cuvette en forme de U à l’aide d’une seringue Luer jetable ou d’une pipette munie d’un embout coupé. Assurez-vous qu’il n’y a pas de bulles dans le « U » afin qu’il y ait une solution ininterrompue pour le flux de courant électrique.
- Placez la cuvette dans l’appareil (veuillez faire attention à l’avant et à l’arrière de la cuvette, afin que les électrodes soient correctement connectées à l’unité). Fermez la porte de l’instrument; Après cela, la mesure a lieu (avec plusieurs répétitions), sous le contrôle du logiciel de guidage.
2. Préparer les liposomes pour l’administration in vivo
- Dans une enceinte de biosécurité, diluer les liposomes dans une solution saline stérile à la concentration appropriée dans un volume final de 50 μL pour administration in vivo.
REMARQUE: Dans des études antérieures, notre préparation de liposomes contenait 2 mg / mL de tesaglitazar, ce qui équivaut à environ 4,89 μmol de tesaglitazar / mL, et nous avons administré des liposomes à une dose de 1 μmol de médicament / kg. Pour une souris de 40 g, on apporterait 8,2 μL de liposomes jusqu’à un volume final de 50 μL dans une solution saline. À l’aide du DLS/ELS, le nombre de liposomes par unité de volume doit également être quantifié pour les préparations de liposomes chargés de médicaments et de véhicules afin de garantir qu’un nombre égal de liposomes de véhicule est administré par gramme de poids de souris par rapport aux liposomes chargés de médicaments. - Chargez la solution liposomique dans une aiguille de 27 G dans l’enceinte de biosécurité. Conservez-le à température ambiante pour éviter d’injecter une solution froide dans la souris.
3. Administrer des liposomes par injection intraveineuse rétro-orbitaire
REMARQUE: Il est également approprié d’effectuer l’injection intraveineuse par d’autres méthodes, telles que les injections de veines de queue si elle est préférée. Bien que non couverts par ce protocole, des protocoles publiés expliquant cette méthode19 sont disponibles.
- Configurez l’espace de travail pour l’administration des liposomes.
- Nettoyez l’établi avec de l’éthanol à 70 %. Assurez-vous de choisir un espace qui permet l’utilisation d’un système d’anesthésie à l’isoflurane.
- Allumez un coussin chauffant et placez un tampon ou une serviette propre dessus pour garder la souris sur une surface propre. Laissez suffisamment de temps au tampon pour se réchauffer avant de commencer à travailler avec des souris.
- Configurez le système d’anesthésie de sorte que la chambre soit à proximité et que le cône nasal soit sur le coussin chauffant.
- Assurez-vous que tous les autres aspects du système sont prêts (par exemple, le niveau d’isoflurane est suffisamment élevé dans le vaporisateur, le filtre à charbon a été pesé, le tube est correctement connecté).
- Rassemblez les autres matériaux nécessaires à cette section du protocole : gel lubrifiant ophtalmique, anesthésique local pour le traitement post-administration, compresses de gaze stériles.
- Sédatif la souris à l’aide d’isoflurane dans la chambre d’induction. Une fois qu’il ne répond pas à un léger tapotement du pied, transférez rapidement la souris dans l’espace de travail tout en maintenant la sédation par un cône nasal.
REMARQUE : L’animal doit être maintenu à 1,5 % à 2,5 % d’isoflurane et évalué pour une profondeur d’anesthésie appropriée (par l’absence de réponse au pincement des orteils) avant de procéder à l’intervention. - Déplacez la souris d’un côté pour l’administration de liposomes. Parce que la souris ne cligne pas des yeux pendant l’anesthésie, appliquez une petite quantité de lubrifiant ophtalmique sur les deux yeux pour les garder hydratés pendant le reste de la procédure.
- Appuyez doucement sur la peau au-dessus et au-dessous de l’œil exposé. L’œil doit se soulever au-dessus du plan du visage.
- Insérez soigneusement la pointe de l’aiguille au niveau du canthus médial, en vous assurant que l’aiguille est sous l’œil et en ne la touchant pas. Une fois l’aiguille insérée sous l’œil, injectez lentement les liposomes dans l’espace rétro-orbitaire. Lors du retrait de l’aiguille, il peut être nécessaire de fermer les paupières pendant quelques secondes pour obtenir une hémostase.
- Si l’aiguille n’est pas insérée assez loin, la solution peut émerger autour de l’œil. Arrêtez immédiatement l’injection si cela est vu et repositionnez l’aiguille.
- Appliquez un anesthésique local, comme la proparacaïne, sur l’œil pour prévenir la douleur et l’inconfort post-intervention.
- Gardez la souris sur un coussin chauffant et surveillez jusqu’à ce qu’elle se réveille pour vous assurer qu’elle va bien et maintient une température corporelle appropriée.
- Remettez la souris dans sa cage et son environnement de logement normal jusqu’à ce que le point d’intérêt temporel arrive.
REMARQUE: Cela doit être fait conformément aux directives locales de l’IACUC.
4. Préparer le matériel pour le prélèvement des tissus, le traitement des tissus et la coloration par cytométrie en flux
- Préparer des solutions pour la récolte, le traitement et la coloration (sections 5 à 127) : solution saline tamponnée au phosphate (PBS)-Héparine, tampon HEPES, collagénase de type I à 2 mg/mL, tampon de lyse AKC, tampon FACS, PBS, tampon de fixation (tableau 2). Conserver toutes les solutions sauf le tampon de fixation à 4 °C ou sur de la glace pendant l’intervention.
- Préparer des tubes avec des tampons et d’autres matériaux pour la récolte et le traitement des tissus.
- Pour le sang de chaque souris, ajouter 10 μL d’EDTA 0,5 M dans un tube microcentrifuge de 1,5 ou 1,7 mL pour recueillir le sang. L’EDTA empêchera le sang de coaguler. Une seringue de 1 mL avec une aiguille de 25 G et un tube conique de 15 mL sont également nécessaires.
- Pour la rate, prélever un tube microcentrifuge de 1,5 ou 1,7 mL avec 1 mL de tampon HEPES, une seringue de 1 mL, deux tubes coniques de 50 mL et deux filtres de 70 μm par rate.
- Pour chaque dépôt de tissu adipeux, prélever un flacon de polyéthylène de 20 mL avec 1,5 mL de tampon HEPES pour hacher le tissu, un tube conique de 50 mL et un filtre de 70 μm par type de tissu adipeux par souris.
- Préparez l’espace de travail pour la récolte.
- Nettoyez l’espace de la paillasse avec de l’éthanol à 70%. Préparez un plateau en caoutchouc pour épingler la souris pendant la récolte en la nettoyant avec de l’éthanol à 70% et en la recouvrant d’un tampon absorbant ou de serviettes en papier. Assurez-vous qu’au moins 5 broches sont disponibles pour travailler.
- Remplissez une seringue de 10 ml avec de l’héparine PBS et fixez une aiguille de 25 g pour la perfusion.
- Rassemblez les outils et le matériel à utiliser pendant la récolte. Des pinces (deux paires), des ciseaux, des essuie-tout, des lingettes non pelucheuses, le(s) tube(s) microcentrifuge(s) avec EDTA, le(s) tube(s) microcentrifuge(s) avec tampon HEPES et le(s) flacon(s) en polyéthylène avec tampon HEPES sont nécessaires.
5. Récolter les tissus
- Euthanasier la souris par asphyxie au CO2 . Ne pas effectuer une luxation cervicale car cela pourrait empêcher une collecte de sang efficace et une perfusion tissulaire à des étapes ultérieures.
- Dans une zone de paillasse nettoyée avec suffisamment d’espace de travail et d’éclairage pour bien voir la souris, installez un plateau de dissection en caoutchouc, un seau de glace pour stocker les échantillons et un flacon pulvérisateur contenant 70% d’éthanol. Vaporisez la souris avec de l’éthanol à 70% pour réduire la contamination et contrôler la propagation des poils. Placez la souris sur son dos sur le plateau en caoutchouc et épinglez ses pattes écartées de son corps.
- Pour vous préparer à la collecte de sang, faites soigneusement une incision dans la peau au bord de l’extrémité caudale de la cage thoracique de la souris. Coupez une petite ligne droite vers la tête de la souris (environ 1 cm) jusqu’à ce que les muscles pectoraux soient exposés.
- Au site d’incision initial, faites deux petites coupures perpendiculaires à la ligne vers la tête. Ensuite, coupez soigneusement le muscle pectoral d’un côté de la cage thoracique dans la zone exposée. Cela permet un meilleur accès et une meilleure visualisation de l’endroit où l’aiguille doit être insérée.
- Pour recueillir du sang, insérez l’aiguille entre les troisième et quatrième côtes du côté où le muscle a été enlevé. Puisque le cœur de la souris se trouve au centre de la cavité thoracique, gardez l’aiguille aussi près que possible de la ligne centrale de la cage thoracique. Une fois inséré, tirez doucement sur la seringue pour commencer à prélever du sang.
- Une fois recueilli, transférer le sang dans le tube microcentrifuge préparé avec de l’EDTA et le conserver sur de la glace.
REMARQUE: Si environ 100 μL de volume est tiré vers le haut et qu’aucun sang ne pénètre dans la seringue, essayez de faire pivoter la seringue vers la droite ou la gauche au cas où l’ouverture de l’aiguille serait pressée contre la paroi du cœur. Si cela ne vous aide pas, déplacez lentement l’aiguille plus loin dans la cavité thoracique ou commencez à la retirer. Si le sang commence à s’accumuler dans la seringue à ce stade, continuez à retirer lentement sur la seringue. Envisagez de faire pivoter la seringue et l’aiguille pour une extraction réussie. Enfin, si aucun sang n’est prélevé, retirez l’aiguille car elle a peut-être manqué le cœur. Essayez de réinsérer l’aiguille et de répéter le processus susmentionné.
- Ensuite, pour percer la souris, ouvrez la cavité thoracique pour accéder au cœur.
- Pour ce faire, coupez la peau le long de l’extrémité de la cage thoracique jusqu’au côté de la souris de chaque côté. Ensuite, utilisez la pince pour maintenir le sternum loin de la surface de travail. Faites une petite incision peu profonde juste en dessous de l’extrémité du sternum pour couper à travers la cavité péritonéale. Couper le long de la membrane péritonéale le long de l’extrémité de la cage thoracique sur chacun des côtés de la souris. Cela devrait exposer le foie et la vésicule biliaire. Veillez à ne pas couper dans l’un ou l’autre de ces tissus.
- Ensuite, faites une petite coupe peu profonde dans le diaphragme, crânien au foie. Ensuite, coupez le diaphragme le long du bord de la cage thoracique pour ouvrir la cavité thoracique. Assurez-vous d’éviter de couper l’un des organes dans la cavité thoracique.
- Faites deux coupes le long de la cage thoracique vers la tête à environ 2-3 mm de la ligne médiane de la souris et environ 0,75 cm de long.
REMARQUE: Si elles sont coupées trop haut, les artères situées au sommet de la cage thoracique seront coupées. Cela interférera avec l’efficacité de la perfusion. - Soulevez la pièce centrale de la cage thoracique pour exposer la cavité thoracique. Déplacez toute graisse ou tissu pour accéder au cœur.
- Faites une petite incision dans l’oreillette droite du cœur de la souris pour créer une ouverture à travers laquelle pousser le sang.
- À l’aide d’une seringue de 10 ml d’héparine PBS, insérez l’aiguille dans le ventricule gauche du cœur de la souris.
- Commencez doucement à pousser le PBS dans le cœur aussi lentement que possible.
REMARQUE: Le sang doit être observé émergeant des oreillettes droites et remplissant la cavité thoracique. Assurez-vous de garder le cœur dans son emplacement physiologique pour éviter d’inhiber le flux d’héparine PBS du cœur à travers l’aorte. - Une fois que les 10 ml d’héparine PBS ont été perfusés à travers la souris, jetez la seringue et l’aiguille et retirez l’excès de sang et d’héparine PBS de la cavité thoracique à l’aide de serviettes en papier ou de lingettes non pelucheuses.
- Ensuite, pour commencer à extraire les tissus, coupez la peau et la membrane péritonéale vers la queue de la souris pour ouvrir la cavité péritonéale.
- Tout d’abord, extrayez le tampon de tissu adipeux inguinal de chaque côté de la souris.
REMARQUE: Lisez attentivement ce processus: assurez-vous d’extraire le ganglion lymphatique inguinal de chaque dépôt pour éviter de fausser la composition cellulaire du tissu adipeux dans les résultats.- À l’aide d’un deuxième jeu de pinces, tenez la membrane péritonéale avec un jeu de pinces et le bord de la peau superposé au-dessus de la membrane de ce côté avec l’autre forceps. Retirez doucement la peau de la membrane péritonéale pour séparer ces couches les unes des autres. Recherchez le dépôt de tissu adipeux inguinal le long de la peau. Épinglez le bord extérieur de la peau pour mieux accéder au dépôt adipeux.
- Avant l’extraction, localisez le ganglion lymphatique inguinal au centre du dépôt adipeux et retirez-le à l’aide d’une pince et de ciseaux au besoin.
REMARQUE: Si possible, localisez les trois plus grandes artères qui partent des bords extérieurs du dépôt vers le centre. Le ganglion lymphatique est situé autour de l’endroit où ces artères se rencontrent. - Une fois le ganglion lymphatique enlevé, tenez soigneusement l’extrémité du dépôt adipeux le plus proche du point épinglé avec la pince et commencez à faire de petites incisions à la membrane conjonctive entre le tissu adipeux et la peau. Soulevez le tissu adipeux loin de la peau tout en faisant des coupures pour mieux accéder à la membrane et s’assurer que tout le dépôt est extrait.
- Placez le dépôt adipeux dans un flacon de polyéthylène préparé avec tampon HEPES sur de la glace pour garder le tissu viable pendant le reste de la récolte.
- Répétez ce processus de l’autre côté de la souris pour extraire les deux dépôts. Les dépôts peuvent être digérés et traités ensemble ou séparément. Si chaque dépôt doit être traité séparément, d’autres tubes doivent être préparés.
- Ensuite, extrayez les dépôts adipeux épididymaires de l’extrémité caudale de la cavité péritonéale. À l’aide d’une pince, tirez doucement le premier dépôt adipeux épididymaire de l’extrémité dorsale de la souris et localisez l’épididyme et le canal déférent attachés à ce dépôt.
NOTE: Il y a deux dépôts adipeux épididymaires: un attaché à chaque épididyme et canal déférent.- Couper soigneusement entre le dépôt adipeux et l’épididyme et le canal déférent pour séparer le tissu adipeux de ces autres tissus. Placez le dépôt adipeux dans un flacon de polyéthylène avec tampon HEPES sur glace pour garder le tissu viable pendant le reste de la récolte.
- Enfin, extrayez la rate, qui se trouve à gauche de l’estomac près du diaphragme. À l’aide d’une pince, tirez doucement l’estomac vers le centre de la cavité péritonéale pour exposer la rate.
- Tenez doucement une extrémité de la rate et éloignez-la légèrement de l’estomac. Couper la membrane entre la rate et son tissu adjacent jusqu’à ce que l’organe soit détaché. Placer la rate dans le tube microcentrifuge préparé avec un tampon HEPES et conserver sur de la glace.
- Avant de traiter les mouchoirs ou de prélever les tissus de la souris suivante, jetez la carcasse et les serviettes en papier ou serviettes souillées. Essuyez également les outils.
REMARQUE: S’il y a plusieurs souris, répétez ces étapes de récolte pour chaque souris avant de passer à l’étape de traitement suivante. Si une ou plusieurs souris témoins sont incluses, envisagez de les prélever avant de les prélever sur les souris traitées aux liposomes afin d’éviter toute contamination.
6. Traiter les tissus
REMARQUE: Étant donné que le tissu adipeux a une longue incubation de digestion, il est recommandé de commencer par ce processus et de travailler sur le traitement du sang et de la rate pendant la période de digestion.
- Tout d’abord, hachez et digérez les tissus adipeux. À l’aide d’une ou deux paires de ciseaux, hacher le tissu adipeux dans chaque flacon de polyéthylène jusqu’à ce que le tissu soit en petits morceaux de moins de 0,5 mm. Cela permet une digestion plus efficace.
- Une fois les mouchoirs de tous les flacons hachés, ajouter 1,5 mL de tampon collagénase à 2 mg/mL dans chaque flacon. Placez les flacons dans un incubateur à secousses réglé à 37 °C et 150 tr/min. Incuber pendant 30 à 45 min.
REMARQUE : Si les tissus adipeux sont particulièrement gros, envisagez d’ajouter un autre tampon de 0,5 mL à 1,5 mL de tampon HEPES et un volume égal de tampon collagénase dans le(s) flacon(s) pour s’assurer que les tissus sont complètement immergés et qu’il y a suffisamment d’enzyme. La concentration finale de collagénase de type I à la digestion doit être de 1 mg/mL, quel que soit le volume final de la solution. De plus, si un incubateur à agitation n’est pas disponible, les échantillons peuvent être placés dans un bain-marie chauffé à 37 °C. Secouez doucement les échantillons toutes les 5 minutes pour mélanger et remettre en suspension la digestion. - Vérifiez les échantillons à 30 min. Utilisez une pipette de 1 mL pour pipeter l’échantillon de haut en bas. Si les morceaux de tissu sont encore trop gros pour faciliter le pipetage, remettez les échantillons dans l’incubateur pendant 15 minutes supplémentaires.
- Une fois que les échantillons sont complètement digérés, continuez à monter et descendre l’échantillon 10 fois de plus pour vous assurer qu’une suspension unicellulaire a été créée.
NOTE: (Facultatif) Vérifiez les échantillons à 30 min. Utilisez une pipette de 1 mL pour pipeter l’échantillon de haut en bas. Si les morceaux de tissu sont encore trop gros pour faciliter le pipetage, remettez les échantillons dans l’incubateur pendant 15 minutes supplémentaires. - Pipeter la suspension cellulaire à travers un filtre de 70 μm dans un tube conique de 50 mL. Ajouter 5 ml de tampon FACS dans le flacon de digestion vide pour laver le flacon. Transférer ce tampon de lavage à travers le filtre pour l’ajouter à la suspension cellulaire.
- Conservez les échantillons sur de la glace pendant que d’autres sont en cours de traitement. Une fois tous les échantillons filtrés, faites-les tourner vers le bas à 400 x g, 4 °C pendant 5 min.
- Retirer le surnageant adipocytaire par aspiration, puis retirer délicatement l’infranageant entre le surnageant adipocytaire et la pastille par aspiration pour laisser la pastille de fraction stroco-vasculaire (SVF).
- Remettez cette pastille en suspension dans 1 mL de tampon FACS et transférez-la dans un tube microcentrifuge propre de 1,5 ou 1,7 mL. Cellules aliquotes maintenant si désiré ou nécessaire. Gardez sur la glace jusqu’à ce que tous les échantillons soient prêts pour la coloration par cytométrie en flux.
REMARQUE: Si les dépôts adipeux digérés étaient importants, envisager d’utiliser seulement 50% ou 25% de l’échantillon pour la coloration et l’analyse cytométriques en flux. De plus, si des témoins de fluorescence moins un (FMO) ou des contrôles supplémentaires pour l’analyse de cytométrie en flux (tableau 1) sont nécessaires, assurez-vous de mettre l’échantillon supplémentaire dans un tube séparé pour le traitement. Les FMO sont utilisés pour distinguer le signal négatif du signal positif pour un anticorps conjugué fluorophore individuel dans le panel autrement complet utilisé dans l’expérience.
- Une fois les mouchoirs de tous les flacons hachés, ajouter 1,5 mL de tampon collagénase à 2 mg/mL dans chaque flacon. Placez les flacons dans un incubateur à secousses réglé à 37 °C et 150 tr/min. Incuber pendant 30 à 45 min.
- Deuxièmement, traitez le sang.
- Transférer 50 μL de sang dans un tube conique de 15 mL.
- Ajouter 1 mL de tampon de lyse AKC à chaque tube et pipeter de haut en bas pour obtenir une suspension unicellulaire. Ajouter 4 mL supplémentaires de tampon de lyse AKC à chaque tube et incuber pendant 5 à min. Si un agitateur ou un rotateur est disponible, scellez hermétiquement les bouchons du tube et placez les tubes sur l’un d’eux pour améliorer le mélange.
- Ajouter 5 mL de tampon FACS pour éteindre le processus de lyse et faire tourner les échantillons à 400 x g, 4 °C pendant 5 min. Retirez le surnageant et vérifiez la pastille. S’il est encore assez rouge, répétez le processus de lyse. Sinon, remettre les pastilles en suspension dans 1 mL de tampon FACS et transférer dans un tube microcentrifuge propre de 1,5 ou 1,7 mL. Gardez sur la glace jusqu’à ce que tous les échantillons soient prêts pour la coloration par cytométrie en flux.
- Enfin, traitez la rate. Transférer la rate sur un filtre de 70 μm sur un tube conique de 50 mL. Laver le mouchoir avec 1 mL de tampon FACS, puis écraser la rate à travers le filtre à l’aide de l’extrémité piston d’une seringue de 1 mL. Tout au long du processus de brassage, laver les cellules dans le tube conique de 50 ml en utilisant plus de tampon FACS. Le volume final dans le tube conique doit être de 10 mL.
- Faire tourner les cellules à 300 x g à 4 °C pendant 5 min. Retirer le surnageant et remettre en suspension dans 1 mL de tampon de lyse AKC. Ajouter 4 mL supplémentaires de tampon de lyse AKC et incuber pendant 5 min. Ajouter 5 mL de tampon FACS pour éteindre le processus de lyse et faire tourner les échantillons à 300 x g à 4 °C pendant 5 min.
- Retirer le surnageant et remettre en suspension la pastille dans 1 mL de tampon FACS. Transférer la suspension à travers un deuxième filtre propre de 70 μm dans un tube conique de 50 mL. Ajouter 4 mL de tampon FACS pour laver le tube d’origine et transférer le tampon à travers le filtre pour un volume final de 5 mL.
- Transférer 50 μL de la suspension cellulaire dans un tube microcentrifuge propre de 1,5 ou 1,7 mL et garder sur la glace jusqu’à ce que tous les échantillons soient prêts pour la coloration par cytométrie en flux. Des aliquotes supplémentaires peuvent être transférées dans des tubes si d’autres sont souhaitées ou requises.
REMARQUE: Les splénocytes sont d’excellentes cellules à utiliser pour une coloration unique vivante / morte. Envisager de transférer une partie aliquote supplémentaire pour ce contrôle.
7. Coloration des cellules des tissus pour la cytométrie en flux
- Faire tourner les échantillons aliquotes à 400 x g, 4 °C pendant 5 min.
- Retirer le surnageant et remettre les échantillons en suspension dans 50 μL de bloc Fc (dilué) (tableau 2). Incuber sur la glace pendant 5 min.
- Ajouter 50 μL de 2x mélange d’anticorps (tableau 3) à chaque échantillon. Incuber sur la glace dans l’obscurité pendant 20 min.
REMARQUE: Toute tache unique ne doit PAS être colorée avec ce mélange d’anticorps. De plus, si des FMO doivent être utilisés, les mélanges d’anticorps FMO doivent être préparés séparément. - Laver les échantillons avec 1 mL de PBS et essorer à 400 x g, 4 °C pendant 5 min. Retirer le surnageant et remettre les échantillons en suspension dans 200 μL de coloration de viabilité (tableau 3). Incuber sur la glace dans l’obscurité pendant 20 min.
REMARQUE: N’oubliez pas de colorer les cellules qui ont été mises de côté pour une tache unique Live/Dead au cours de cette étape. - Laver les échantillons avec 1 mL de tampon FACS et essorer à 400 x g, 4 °C pendant 5 min. Retirer le surnageant et remettre les échantillons en suspension (à l’exception de la coloration simple vivante/morte) dans 50 μL de milieu de fixation (réactif A) pour fixer les échantillons. Incuber à température ambiante dans l’obscurité pendant 15 min.
- Remettez en suspension la coloration simple vivante/morte dans 100 μL de PFA à 2 %. Incuber à température ambiante dans l’obscurité pendant 5 min.
- Laver l’échantillon avec 1 mL de tampon FACS et essorer à 800 x g, 4 °C pendant 5 min. Retirer le surnageant et remettre les échantillons en suspension dans 250 à 500 μL de tampon FACS. Conserver à 4 °C jusqu’à ce que les échantillons puissent être analysés sur le cytomètre en flux.
- Laver les échantillons avec 1 mL de tampon FACS et essorer à 800 x g, 4 °C pendant 5 min. Retirer le surnageant et remettre les échantillons en suspension dans 50 μL de milieu de perméabilisation (réactif B) plus des anticorps dirigés contre les protéines intracellulaires. Incuber à température ambiante dans l’obscurité pendant 20 min.
- Laver les échantillons avec 1 mL de tampon FACS et les faire tourner à 800 x g à 4 °C pendant 5 min. Retirer le surnageant et remettre les échantillons en suspension dans 100 μL de paraformaldéhyde (PFA) à 2 %. Incuber à température ambiante dans l’obscurité pendant 5 min.
- Laver les échantillons avec 1 mL de tampon FACS et essorer à 800 x g, 4 °C pendant 5 min. Retirer le surnageant et remettre les échantillons en suspension dans 250 à 500 μL de tampon FACS. Conserver à 4 °C jusqu’à ce que les échantillons puissent être analysés sur le cytomètre en flux.
Résultats
Production de liposomes
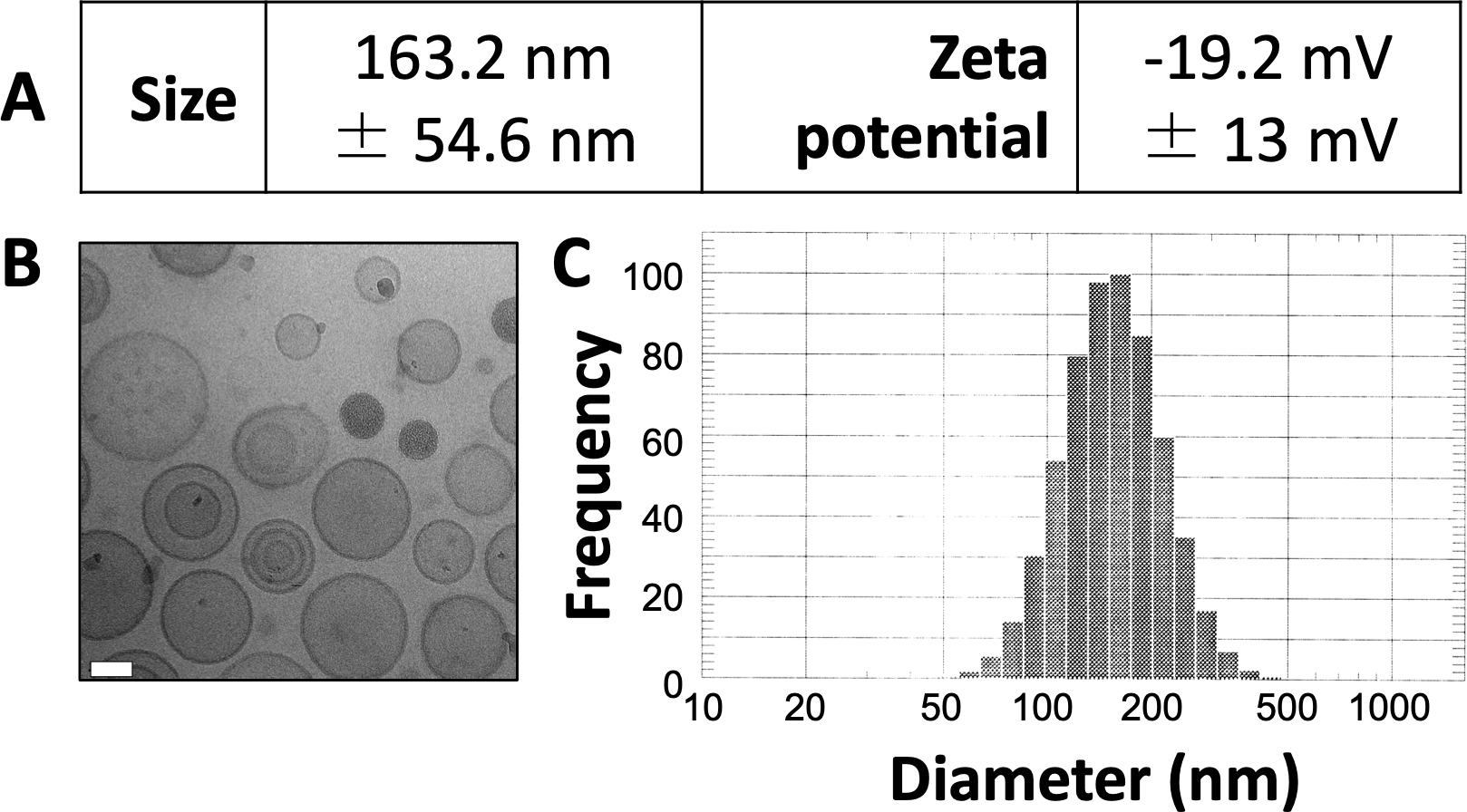
Les résultats publiés ici sont similaires à ceux de nos travaux précédemment publiés 3,4,20. En utilisant le protocole présenté ici, nous prévoyons de produire des liposomes d’une taille d’environ 150 à 150 nm. Le DLS révèle un diamètre moyen de liposomes de 163,2 nm et un potentiel zêta de -19,2 mV (Figure 1A). L’imagerie par microscopie électronique cryogénique (cryo-EM) révèle des liposomes circulaires (Figure 1B) et le diagramme DLS révèle un écart type relativement faible par rapport au diamètre moyen (Figure 1C).
La liaison positive aux liposomes nécessite un contrôle traité par PBS
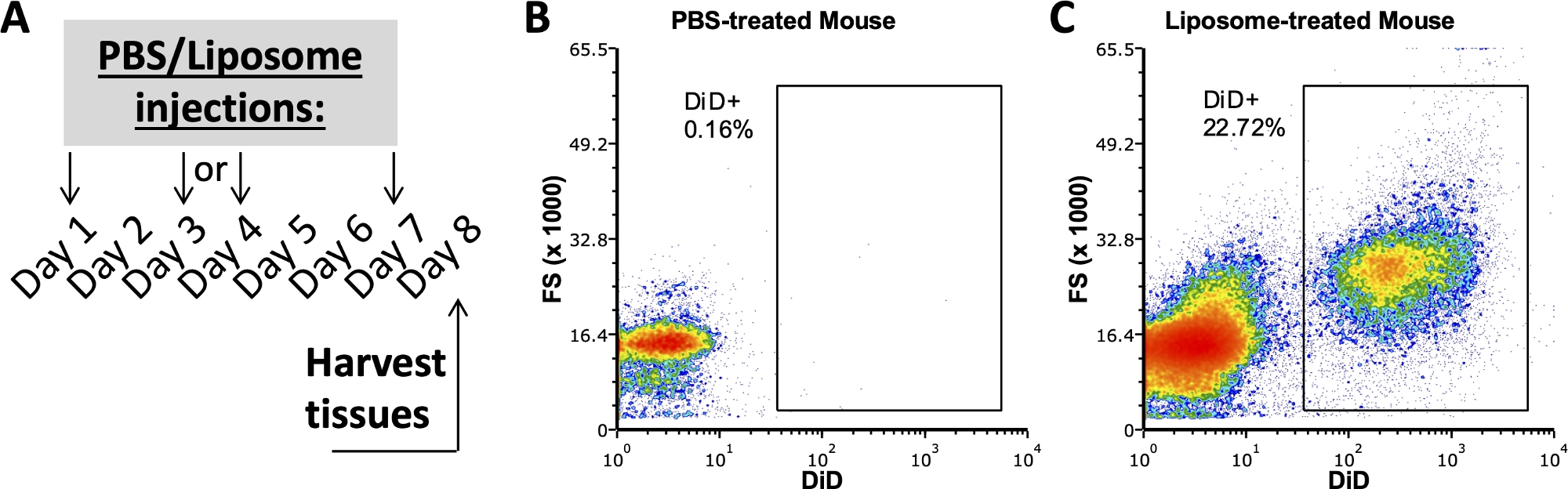
Des études antérieures de notre groupe utilisant ce protocole ont examiné quels sous-ensembles cellulaires dans la SVF adipeuse, la rate et le sang liés aux liposomes après une semaine d’administration in vivo 3,4. À l’aide d’une souris traitée au PBS, les cellules de la cavité péritonéale et de la rate ont été colorées avec le même panel d’anticorps utilisé sur des échantillons de souris traitées par liposomes. Les tissus ont été prélevés après une semaine de traitement (figure 2A). Les échantillons de la souris traitée au PBS ont servi de FMO DiD avec lequel créer des portes DiD positives (Figure 2B,C). Une porte positive peut être créée à l’aide d’un signal DiD-positif, mais les échantillons dépourvus de signal DiD doivent également être utilisés pour vérifier que la porte positive ne comprend pas d’échantillons DiD négatifs.
Des titrages sont nécessaires pour optimiser les signaux de fluorescence
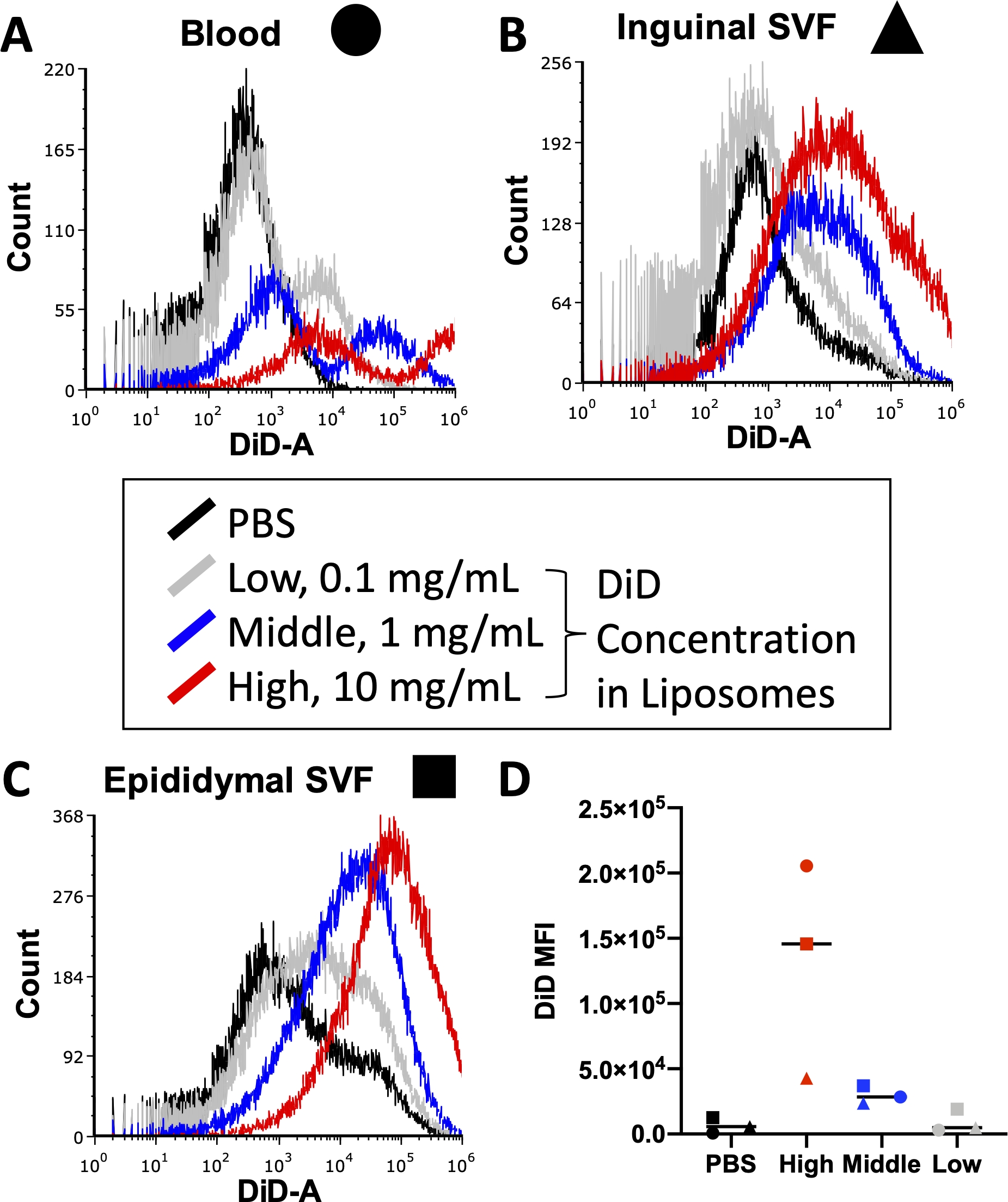
Avant d’exécuter une expérience complète, diverses conditions, y compris la concentration d’anticorps conjugués par fluorescence utilisés lors de la coloration cellulaire et de colorant lipidique utilisé pendant la préparation des liposomes, doivent être optimisées. Les cytomètres de flux ont une limite supérieure de détection pour l’intensité de fluorescence, de sorte que trop de colorant incorporé dans les liposomes conduira à des niveaux non quantifiables de signal DiD dans les échantillons traversant le cytomètre. De plus, trop de DiD dans les liposomes peut entraîner des niveaux élevés de transfert de colorant non spécifique, ce qui pourrait fausser les résultats de l’absorption cellulaire. La figure 3 présente les résultats d’une expérience dans laquelle les concentrations de colorant lipidique ont été titrées pour identifier la concentration qui produirait un signal optimal dans la plage de détection du cytomètre en flux utilisé. Cela a été réalisé sur les tissus d’intérêt pour l’expérience finale: sang (Figure 3A), SVF adipeux inguinal (Figure 3B) et SVF adipeux épididymaire (Figure 3C). Les concentrations sélectionnées pour les tests étaient de 10 mg de DiD (élevé, rouge), 1 mg de DiD (moyen, bleu) ou 0,1 mg de DiD (faible, gris) par 1 mL de liposomes. La concentration la plus élevée utilisée dans les liposomes était trop élevée et dépassait la plage quantifiable du cytomètre dans les trois tissus (Figure 3A\u2012C, rouge). La concentration la plus faible de DiD a montré un certain signal (Figure 3 A\u2012C, gris), mais une population claire au-delà des cellules traitées par PBS (Figure 3 A\u2012C, noir) n’a pas été observée. Lorsqu’elle a été quantifiée, la moyenne arithmétique de l’IFM DiD pour chaque tissu et concentration a démontré une distinction claire entre les témoins PBS et la concentration moyenne de DiD (Figure 3D). Ainsi, comme indiqué dans le protocole, nous avons sélectionné la concentration moyenne (Figure 3, bleu) à utiliser dans notre préparation de liposomes.
L’utilisation d’un panel multi-anticorps permet d’identifier l’absorption des liposomes par différents sous-ensembles cellulaires
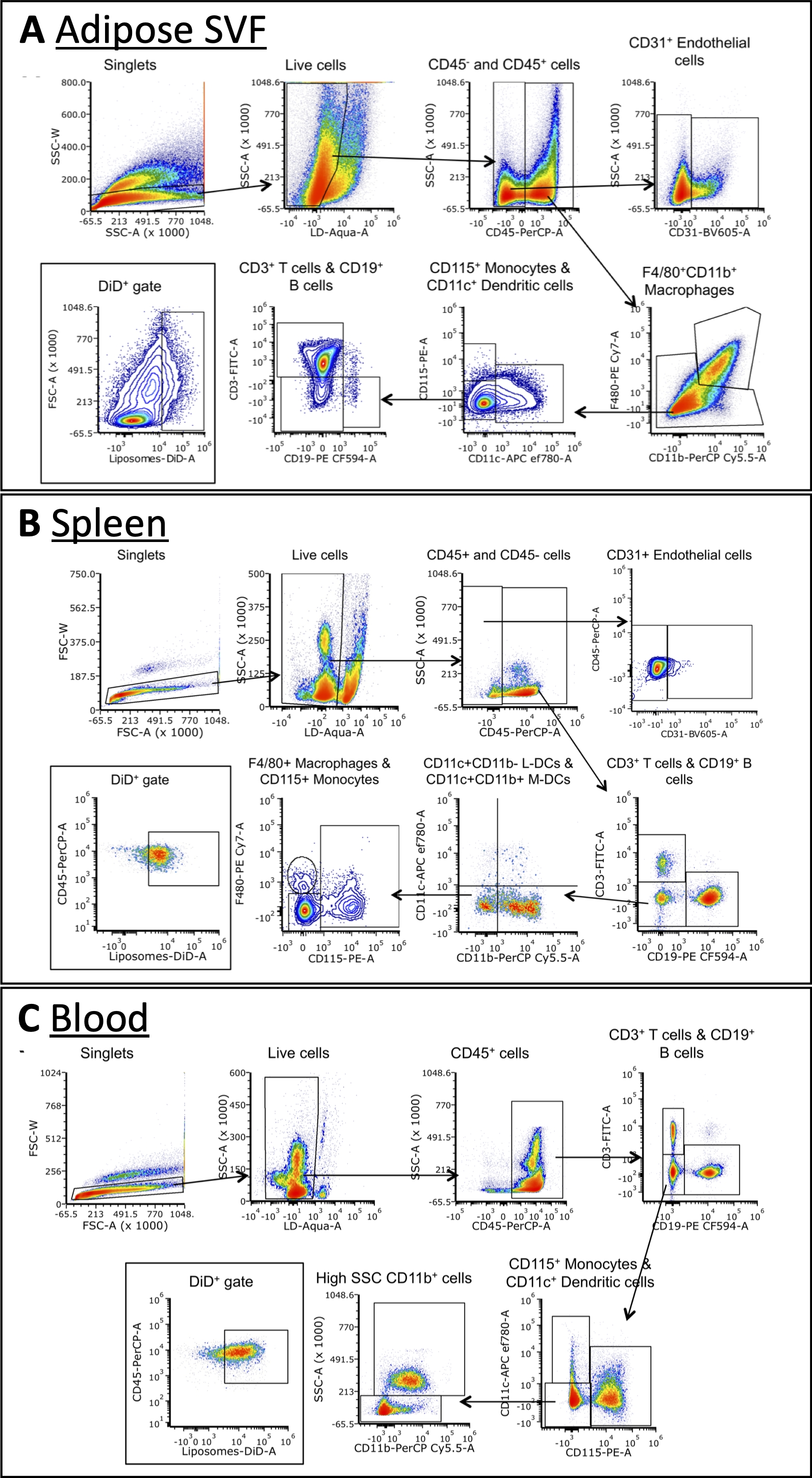
À l’aide du panel décrit dans le tableau 3, les cellules ont été colorées avec des anticorps contre les marqueurs des macrophages A, des cellules B, des cellules T, des cellules dendritiques, des monocytes et des cellules endothéliales (Figure 4). Des stratégies de déclenchement légèrement différentes sont nécessaires pour chaque type de tissu, mais la plupart des mêmes types de cellules peuvent être identifiés dans chacun. Certaines exceptions comprennent les cellules endothéliales, qui ne se trouvent normalement pas dans le sang, et les monocytes, qui sont généralement à une fréquence plus élevée dans le sang que les autres tissus. Une fois les populations identifiées, la taille totale de chaque population cellulaire et la fréquence à laquelle elles sont DiD+ peuvent être quantifiées. D’autres calculs peuvent être effectués pour caractériser la population DiD+ : quel pourcentage de cellules DiD+ sont des macrophages, des cellules endothéliales, etc. Veuillez noter qu’il s’agit d’exemples de stratégies de contrôle, mais ce n’est pas la seule façon d’analyser les échantillons. L’analyse sera dictée par le panel sélectionné et le(s) cytomètre(s) de flux disponible(s).

Figure 1 : Exemples de caractéristiques des liposomes préparés.
(A) La taille et le potentiel zêta ont été mesurés comme décrit ci-dessus et ont été présentés sous forme de tableau. Chaque paramètre est présenté comme la moyenne ± l’écart-type. (B) Cryo-EM a été utilisé pour imager les liposomes préparés. La barre d’échelle blanche mesure 50 nm de longueur. (C) DLS a été utilisé pour générer un histogramme du diamètre des liposomes dans cette préparation. Cette figure est adaptée d’Osinski et al.3. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Coloration DiD représentative de souris traitées au PBS ou aux liposomes.
(A) Schéma expérimental pour les traitements PBS et liposomes. PBS ou liposomes ont été injectés trois fois au cours d’une semaine. Les tissus ont été prélevés le jour 8 du traitement. (B, C) Des diagrammes de flux représentatifs révèlent une coloration positive au DiD chez les souris traitées par liposomes (C), mais pas par PBS (B). FSC, diffusion vers l’avant. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Titrage de DiD dans les liposomes.
Les liposomes ont été préparés avec trois concentrations différentes de DiD et injectés à des souris. Le gris indique la faible concentration à 0,1 mg DiD par 1 mL de liposomes, le bleu indique la concentration moyenne à 1 mg DiD/mL de liposomes et le rouge indique la concentration élevée à 10 mg DiD/mL de liposomes. Une souris traitée par PBS a été utilisée comme témoin négatif (noire). Le sang (A, cercle), l’adipeux inguinal (B, triange) et l’adipeux épididymaire (C, carré) ont été prélevés 24 heures après l’injection et traités pour isoler une suspension unicellulaire. Ces échantillons ont été exécutés sur un cytomètre en flux au niveau de DiD détectable. Des histogrammes spécifiques aux tissus avec des superpositions de chaque groupe de traitement sont présentés pour démontrer l’intensité de fluorescence par concentration (A\u2012C). La moyenne arithmétique de DiD a également été quantifiée pour chaque tissu et concentration et tracée (D). SSC = dispersion latérale. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Analyse par cytométrie en flux représentative de sous-ensembles cellulaires dans la SVF adipeuse, le sang et la rate.
(A\u2012C) Schéma représentatif de la stratégie de déclenchement pour identifier les sous-ensembles cellulaires et les cellules DiD+ dans le SVF adipeux (A), la rate (B) et le sang (C). Abréviations : FSC = diffusion vers l’avant; LD = vivant/mort; L-DCs = cellules dendritiques lymphoïdes; M-DCs = cellules dendritiques myéloïdes; SSC = dispersion latérale. Cette figure est adaptée d’Osinski et al.3. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
| Contrôle | But |
| Souris traitée avec PBS ou solution saline | Utilisez les cellules de cette souris pour les contrôles de cytométrie en flux suivants : |
| 1. Cellules non colorées | |
| 2. Tache unique vivante / morte | |
| 3. Cellules colorées avec le panneau complet, mais dépourvues de fluorescence liposomique pour déterminer le signal liposomique positif pendant l’analyse | |
| Cette / ces souris / souris seront également utilisées pour déterminer si les liposomes ont des effets in vivo, car vous aurez un contrôle non liposomique dans votre expérience. | |
| Liposomes déchargés | Si vous chargez un composé dans vos liposomes, une partie de votre lot de liposomes doit être synthétisée sans le composé. Cela explique tous les effets in vivo des liposomes seuls. |
| DiD seul | Étant donné que le DiD peut également être absorbé par les membranes cellulaires, l’allocation de certaines souris pour recevoir un colorant libre à une quantité égale à celle trouvée dans les liposomes aidera à tenir compte de toute coloration de la membrane de fond. |
| Contrôles de fluorescence moins un (FMO) | Ce sont des cellules colorées avec tous les anticorps de votre panel sauf un. Comme #3 dans l’encadré ci-dessus, cela aide à déterminer le véritable signal positif pour cet anticorps pendant l’analyse |
Tableau 1 : Contrôles à utiliser dans ce protocole.
| Solution | Composants | Volume approximatif nécessaire par lot/souris |
| Préparation des liposomes | ||
| Acétate de calcium | 1 M acétate de calcium dansH2O | 50 mL |
| Tampon HEPES | 10 mM HEPES enH2O, pH 7,4 | 50 mL |
| Tesaglitazar dans HEPES | en 10 mM HEPES | 10 mL |
| Récolte, traitement et coloration des tissus | ||
| Solution tamponnée au phosphate (PBS) | 137 mM NaCl, 2,7 mM KCl, 10 mMNa2HPO4, 1,8 mM KH 2PO4 dansH2Odistillé | 2 mL |
| PBS-Héparine | 0,1 mM d’héparine dans PBS | 10 mL |
| Tampon HEPES | 20 mM HEPES dans PBS | 5 mL |
| Tampon de digestion | 2 mg/mL de collagénase de type I dans le tampon HEPES | 5 mL |
| Tampon de lyse AKC | 0,158 MNH3Cl, 10 mM KHCO3, 0,1 mMNa2 EDTA dansddH2O, pH 7,2 | 15 mL |
| Tampon FACS | 1% BSA, 0,05% NaN3 dans PBS | 15 mL |
| Bloc Fc (dilué) | Bloc 1:50 Fc dans le tampon FACS | 250 μL |
| Tampon de fixation | 2% de paraformaldéhyde dans le PBS | 200 μL |
Tableau 2 : Solutions à préparer.
| Un | B | C | D |
| Coloration extracellulaire (2x mélange d’anticorps) | |||
| Antigène | Fluorophore | Volume Ab par test de 100 μL | Volume total nécessaire : |
| CD45 | PerCP | 0,5 μL | Colonne C x 1,2 x # total d’échantillons |
| CD11b | PerCP Cy5.5 | 0,25 μL | (0,5 μL/essai) x (1,2) x (# échantillons) |
| F4/80 | PE Cy7 | 0,25 μL | (0,25 μL/essai) x (1,2) x (# échantillons) |
| CD19 | PE-CF594 | 1 μL | (0,25 μL/essai) x (1,2) x (# échantillons) |
| CD3 | FITC | 1 μL | (1,0 μL/essai) x (1,2) x (# échantillons) |
| DC31 | BV605 | 0,25 μL | etc... |
| CD11c | APC ef780 | 1 μL | |
| DC115 | PE | 1,5 μL | |
| Pour créer votre mélange d’anticorps, combinez les anticorps calculés dans la colonne D avec le tampon FACS ou le tampon de coloration violet brillant* jusqu’à un volume final de (50 μL x 1,2 x # échantillons totaux) | |||
| Taches vivantes/mortes (1x) | |||
| Vivant/Mort | Fluorophore | Volume L/D par essai de 200 uL | Volume total nécessaire : |
| Vivant/Mort | Aqua | 0,67 μL | Colonne C x 1,2 x # total d’échantillons |
| Coloration intracellulaire (1x) | |||
| Antigène | Fluorophore | Volume Ab par test de 50 μL | Volume total nécessaire : |
| αSMA | FITC | 0.125 | Colonne C x 1,2 x # total d’échantillons |
| *Un tampon de coloration violette brillante doit être utilisé si plus d’un anticorps conjugué à un fluorophore violet brillant est utilisé dans votre panel. | |||
Tableau 3 : Exemple de panel d’anticorps et calculs des mélanges de coloration à utiliser pour la coloration en flux.
Discussion
Nous décrivons ici un protocole en trois parties pour (i) préparer des liposomes marqués avec un colorant lipidique fluorescent et chargés d’un composé antidiabétique, le tesaglitazar, (ii) administrer des liposomes à une souris par injection rétro-orbitale, et (iii) récolter, traiter et colorer les tissus pour détecter l’absorption des liposomes au niveau cellulaire par cytométrie en flux. Ce protocole examine la préparation de liposomes d’environ 150 μm et l’évaluation de l’absorption dans le tissu adipeux, le sang et la rate. La préparation des liposomes est évolutive, réalisée principalement à température ambiante et utilise l’évaporation en phase inverse pour maximiser la charge médicamenteuse et l’élimination des solvants organiques. En utilisant ce protocole, une concentration de tésaglitazar allant jusqu’à 2 mg/mL peut être atteinte dans l’échantillon de liposomes purifiés. Les liposomes préparés peuvent être conservés dans un tampon HEPES à 4 °C pendant plus d’un an. D’après notre expérience, ils ont démontré une variation minimale de la taille moyenne des particules. Moins de 10% de la perte de contenu du médicament a été démontrée par spectrophotométrique, après séparation par ultrafiltration des liposomes du médicament externe avec un filtre centrifuge de 10 kDa.
Lors de la préparation des liposomes, il y a quelques étapes et facteurs critiques à considérer. Tout d’abord, l’ordre des étapes du protocole est important et doit être respecté. Deuxièmement, le pH de la solution utilisée lors du chargement du tésaglitazar doit être maintenu à 7,4 afin de maximiser la solubilité et la charge efficace. Troisièmement, l’assemblage approprié de l’équipement et des filtres garantit que le rendement de chaque étape est de la taille et de la pureté appropriées. Par exemple, si les filtres de 100 et 200 nm ne sont pas assemblés correctement, un lot de liposomes plus hétérogène et mal dimensionné peut en résulter. Quatrièmement, l’élimination complète de l’acétate de Ca-acétate avant la charge médicamenteuse est nécessaire pour maximiser le transfert du tésaglitazar dans les liposomes. Pour tester l’élimination complète de l’acétate de Ca, utilisez la sédimentation à grande vitesse pour éliminer les liposomes, puis mesurez les niveaux d’acétate de Ca-dans la solution non liposomale. Cinquièmement, il est important de peser et d’enregistrer la masse de tous les matériaux ajoutés à la préparation des liposomes à chaque étape. Cela garantit que les concentrations appropriées peuvent être calculées et que les ratios de matériaux nécessaires sont maintenus. Enfin, si la technique n’est pas correctement exécutée, il peut y avoir un niveau indésirable d’hétérogénéité. Il est important de vérifier soigneusement ce paramètre à l’aide de DLS et d’autres approches telles que la microscopie électronique. Pour améliorer l’homogénéité, envisagez d’ajuster la taille du filtre sélectionné ou d’empiler deux filtres.
De plus, il est essentiel que les témoins et un panel d’anticorps pour la cytométrie en flux soient planifiés et optimisés avant d’exécuter ce protocole dans son intégralité (Tableau 1, Tableau 3). Les anticorps doivent être testés pour s’assurer que les concentrations appropriées sont utilisées pour la coloration et que le chevauchement entre les fluorophores est minime. L’excitation et l’émission du colorant utilisé lors de la préparation des liposomes doivent également être prises en compte dans la planification du panel. Dans nos résultats, nous avons utilisé DiD, qui a une excitation et une émission similaires aux fluorophores tels que l’allophycocyanine (APC) et l’AlexaFluor 647. Ainsi, nous n’avons pas sélectionné d’anticorps conjugués à ces fluorophores dans notre panel d’anticorps. De plus, les contrôles d’isotypes ne sont pas inclus dans ce protocole. En effet, les anticorps sélectionnés pour ce protocole sont des anticorps bien validés et disponibles dans le commerce. Cependant, si vous souhaitez utiliser un anticorps qui n’a pas été optimisé auparavant, veuillez envisager de tester l’anticorps contre un témoin d’isotype sur les tissus d’intérêt avant de mener l’expérience complète.
Bien que ce protocole démontre comment extraire et traiter le sang, la rate, le tissu adipeux inguinal et les tissus adipeux épididymaires de la souris après le traitement, cette approche générale peut être appliquée à d’autres tissus. Selon le tissu d’intérêt, les protocoles de traitement et de digestion peuvent devoir être modifiés comme cela est publié pour les tissus suivants: poumon21, foie 22, cavité péritonéale 3, moelle osseuse3,23, cerveau 24.
Une limite importante de cette méthode à considérer est que l’absorption ne peut être évaluée qu’à un seul moment par animal. Ainsi, il peut être avantageux de coupler ce protocole avec d’autres techniques d’imagerie non invasives ou de planifier en conséquence pour assurer des ressources suffisantes pour effectuer l’évaluation. Le moment de l’absorption cellulaire et le renouvellement cellulaire sont des facteurs importants à prendre en compte : les liposomes circuleront dans tout le corps au cours des premières 24 heures et, en fonction de la durée de vie des cellules qui absorbent les liposomes ou de la façon dont elles réagissent à l’absorption, la mort cellulaire ou une phagocytose supplémentaire peut survenir. Notre étude précédente a démontré des changements dans les caractéristiques de la population des populations DiD+ à différents moments3. Pour cette raison, il est important d’évaluer l’adoption à des moments antérieurs ou aux points temporels les plus pertinents pour la biologie du mécanisme d’intérêt. De plus, alors que la quantification de l’absorption cellulaire dans l’ensemble du tissu peut être effectuée avec ce protocole, la cytométrie en flux ne peut pas révéler la localisation tissulaire. Le couplage de cette approche avec des méthodes histologiques peut aider à remédier à cette limitation.
En général, ce protocole complète les méthodologies existantes telles que l’histologie et l’imagerie par fluorescence du corps entier. Avec les progrès continus des outils et des méthodes de cytométrie en flux, le développement de panels plus grands pour des populations cellulaires de plus en plus spécifiques deviendra possible. Nous suggérons que ce protocole soit utilisé en plus des méthodes susmentionnées, car cela améliorera l’évaluation de l’absorption cellulaire et fournira également la possibilité de valider les résultats observés par cytométrie en flux. Par exemple, faut-il constater que la majorité des particules dans le tissu adipeux ont été absorbées par les macrophages par cytométrie en flux. L’immunofluorescence d’une partie aliquote supplémentaire du même tissu adipeux pourrait être sauvegardée, fixée, sectionnée et colorée pour les marqueurs de macrophages afin de vérifier que le type de cellule absorbe bien les liposomes. Cette approche devrait ajouter de la rigueur aux essais de biodistribution des nanoparticules menés : validation du ciblage spécifique des cellules, quantification de l’absorption cellulaire, identification de l’absorption hors cible et, espérons-le, fourniture d’informations pour générer des hypothèses mécanistes pour les résultats thérapeutiques observés. Ce protocole peut également être adapté pour de futures études utilisant différents liposomes, étudiant l’absorption dans d’autres tissus et testant de nouveaux composés dans le contexte de l’obésité et du dysmétabolisme ou de toute autre maladie dans laquelle l’administration de nanoparticules est une option thérapeutique réalisable.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Les auteurs tiennent à remercier Michael Solga et le reste du personnel de base de cytométrie en flux pour avoir fourni une formation et des services en cytométrie en flux. Les auteurs aimeraient également remercier Shiva Sai Krishna Dasa, Dustin K. Bauknight, Melissa A. Marshall, James C. Garmey, Chantel McSkimming, Aditi Upadhye et Prasad Srikakulapu pour leur aide à la préparation des liposomes (SSKD, DKB), aux prélèvements de tissus (MAM, JCG), à la coloration par cytométrie en flux et à l’acquisition d’échantillons (AU, PS, CM). Ce travail a été soutenu par des subventions AstraZeneca, R01HL 136098, R01HL 141123 et R01HL 148109, AHA 16PRE30770007 et T32 HL007284.
matériels
| Name | Company | Catalog Number | Comments |
| 1-mL syringe | BD | 309659 | |
| 10-mL syringe | BD | 302995 | |
| 25-gauge needle, sterile for retro-orbital injection | BD | 305122 | |
| 27-gauge needle, sterile for retro-orbital injection | BD | 305620 | |
| Anti-mouse B220 BV421 | Biolegend | 103251 | Clone RA3-6B2 |
| Anti-mouse CD115 PE | eBioscience | 12-1152-82 | Clone AFS98 |
| Anti-mouse CD11b PerCP Cy5.5 antibody | BD Biosciences | 550993 | Clone M1/70 |
| Anti-mouse CD11c APC ef780 antibody | eBioscience | 47-0114-82 | Clone N418 |
| Anti-mouse CD19 PE CF594 | BD Biosciences | 562291 | Clone 1D3 |
| Anti-mouse CD3 FITC antibody | BD Biosciences | 553061 | Clone 145-2C11 |
| Anti-mouse CD31 BV605 | Biolegend | 102427 | Clone 390 |
| Anti-mouse CD45 PerCP | BD Biosciences | 557235 | Clone 30-F11 |
| Anti-mouse F4/80 PE Cy7 | Biolegend | 123114 | Clone BM8 |
| Bovine serum albumin | Gemini Bio-products | 700-107P | |
| Desalting spin-column | ThermoFisher | 89889, 89890 | Zeba spin column |
| DPBS | Gibco | 14190-144 | |
| Dynamic Light Scattering, Nicomp 370 | Particle Sizing System, Inc | ||
| FIX & PERM Cell Permeabilization Kit | ThermoFisher Scientific | GAS004 | |
| Gauze sponges | Dermacea | 441211 | |
| Heparin | Sigma | 3393-1MU | |
| Liposome extruder | Millipore Sigma | Z373400 | LiposoFast |
| Live/Dead Aqua | ThermoFisher Scientific | L34957 | |
| Nanosight | Malvern Instruments Ltd | NS300 | |
| Ophthalmic lubricant | Optixcare | 20g/70 oz Sterile | |
| Paraformaldehyde, 16% w/v aq. soln., methanol free | Alfa Aesar | 433689L | |
| Polyethylene vial for mincing | Wheaton | 986701 | |
| Rotary evaporator | Buchi | Re111 | |
| Sonicator | Misonix | XL2020 | |
| T/Pump Heat therapy pump and pad | Gaymer Industries | TP-500 | |
| Tesaglitazar | Tocris | 3965 | |
| Track-etched polycarbonate membranes | Thomas Scientific | 1141Z** | Whatman, Nuclepore Polycarbonate hydrophilic membranes |
| ZetaSizer/DLS-ELS system | Malvern Instruments Ltd |
Références
- Allen, T. M., Cullis, P. R. Liposomal drug delivery systems: from concept to clinical applications. Advanced Drug Delivery Reviews. 65 (1), 36-48 (2013).
- Sercombe, L., et al. Advances and Challenges of Liposome Assisted Drug Delivery. Frontiers in Pharmacology. 6, 286 (2015).
- Osinski, V., et al. In vivo liposomal delivery of PPARα/γ dual agonist tesaglitazar in a model of obesity enriches macrophage targeting and limits liver and kidney drug effects. Theranostics. 10 (2), (2020).
- Bauknight, D. K., et al. Importance of thorough tissue and cellular level characterization of targeted drugs in the evaluation of pharmacodynamic effects. PLoS ONE. 14 (11), (2019).
- Rosales, C., Uribe-Querol, E. Phagocytosis: A Fundamental Process in Immunity. BioMed Research International. , (2017).
- He, H., Ghosh, S., Yang, H. Nanomedicines for dysfunctional macrophage-associated diseases. Journal of Controlled Release. 247, 106-126 (2017).
- Song, G., Petschauer, J. S., Madden, A. J., Zamboni, W. C. Nanoparticles and the mononuclear phagocyte system: pharmacokinetics and applications for inflammatory diseases. Current Rheumatology Reviews. 10 (1), 22-34 (2014).
- Litzinger, D. C., Buiting, A. M. J., van Rooijen, N., Huang, L. Effect of liposome size on the circulation time and intraorgan distribution of amphipathic poly(ethylene glycol)-containing liposomes. BBA - Biomembranes. , (1994).
- Su, C., Liu, Y., He, Y., Gu, J. Analytical methods for investigating in vivo fate of nanoliposomes: A review. Journal of Pharmaceutical Analysis. , (2018).
- Vasquez, K. O., Casavant, C., Peterson, J. D. Quantitative whole body biodistribution of fluorescent-labeled agents by non-invasive tomographic imaging. PLoS One. 6 (6), 20594 (2011).
- Larmann, J., et al. In vivo fluorescence-mediated tomography for quantification of urokinase receptor-dependent leukocyte trafficking in inflammation. Anesthesiology. 113 (3), 610-618 (2010).
- Feng, B., et al. Clodronate liposomes improve metabolic profile and reduce visceral adipose macrophage content in diet-induced obese mice. PLoS One. 6 (9), 1-11 (2011).
- Bu, L., Gao, M., Qu, S., Liu, D. Intraperitoneal injection of clodronate liposomes eliminates visceral adipose macrophages and blocks high-fat diet-induced weight gain and development of insulin resistance. The AAPS Journal. 15 (4), 1001-1011 (2013).
- Toita, R., Kawano, T., Murata, M., Kang, J. H. Anti-obesity and anti-inflammatory effects of macrophage-targeted interleukin-10-conjugated liposomes in obese mice. Biomaterials. 110, 81-88 (2016).
- Sakurai, Y., Kajimoto, K., Hatakeyama, H., Harashima, H. Advances in an active and passive targeting to tumor and adipose tissues. Expert Opin Drug Deliv. 12 (1), 41-52 (2015).
- Nakhlband, A., et al. Combating atherosclerosis with targeted nanomedicines: Recent advances and future prospective. BioImpacts. , (2018).
- Sibuyi, N. R. S., Meyer, M., Onani, M. O., Skepu, A., Madiehe, A. M. Vascular targeted nanotherapeutic approach for obesity treatment. International Journal of Nanomedicine. , (2018).
- Honig, M. G., Hume, R. I. Fluorescent carbocyanine dyes allow living neurons of identified origin to be studied in long-term cultures. Journal of Cell Biology. , (1986).
- Warren, J. S. A., Feustel, P. J., Lamar, J. M. Combined Use of Tail Vein Metastasis Assays and Real-Time In Vivo Imaging to Quantify Breast Cancer Metastatic Colonization and Burden in the Lungs. Journal of visualized experiments : JoVE. , (2019).
- Dasa, S. S. K., et al. Plectin-targeted liposomes enhance the therapeutic efficacy of a PARP inhibitor in the treatment of ovarian cancer. Theranostics. , (2018).
- Morrison, B. E., Park, S. J., Mooney, J. M., Mehrad, B. Chemokine-mediated recruitment of NK cells is a critical host defense mechanism in invasive aspergillosis. Journal of Clinical Investigation. , (2003).
- Finlon, J. M., Burchill, M. A., Jirón Tamburini, B. A. Digestion of the murine liver for a flow cytometric analysis of lymphatic endothelial cells. Journal of Visualized Experiments. , e58621 (2019).
- Upadhye, A., et al. Diversification and CXCR4-Dependent Establishment of the Bone Marrow B-1a Cell Pool Governs Atheroprotective IgM Production Linked to Human Coronary Atherosclerosis. Circulation research. , (2019).
- O'Brien, C. A., Harris, T. H. ICOS-deficient and ICOS YF mutant mice fail to control Toxoplasma gondii infection of the brain. PLoS ONE. , (2020).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.