
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Immunophénotypage et tri cellulaire des MKs humains provenant de sources primaires humaines ou différenciés in vitro à partir de progéniteurs hématopoïétiques
Dans cet article
Résumé
Ici, nous présentons une stratégie d’immunophenotyping pour la caractérisation de la différenciation de megakaryocyte, et montrons comment cette stratégie permet le tri des megakaryocytes à différentes étapes avec un trieur de cellules fluorescence-activé. La méthodologie peut être appliquée aux tissus primaires humains, mais aussi aux mégacaryocytes générés en culture in vitro.
Résumé
La différenciation des mégacaryocytes (MK) englobe un certain nombre de cycles endomitotiques qui se traduisent par une cellule très polyploïde (atteignant même >64N) et extrêmement grande (40-60 μm). Contrairement à l’augmentation rapide des connaissances sur la mégakaryopoïèse au niveau de la biologie cellulaire et moléculaire, la caractérisation de la mégakaryopoïèse par cytométrie en flux se limite à l’identification des MKs matures à l’aide de marqueurs de surface spécifiques à la lignée, tandis que les stades antérieurs de différenciation mk restent inexplorés. Ici, nous présentons une stratégie d’immunophenotyping qui permet l’identification des étapes successives de différenciation de MK, avec le statut mentoïdique croissant, dans les sources primaires humaines ou les cultures in vitro avec un panneau intégrant des marqueurs de surface spécifiques et non spécifiques de MK. Malgré sa taille et sa fragilité, les MKs peuvent être immunophénotypés à l’aide du panneau mentionné ci-dessus et enrichis par tri cellulaire activé par fluorescence dans des conditions spécifiques de pression et de diamètre de buse. Cette approche facilite les études multi-omiques, dans le but de mieux comprendre la complexité de la mégacaryopoïèse et de la production de plaquettes chez l’homme. Une meilleure caractérisation de megakaryopoiesis peut poser fondamental dans le diagnostic ou le pronostic des pathologies et de la malignité Lignage-connexes.
Introduction
Les mégacaryocytes (MKs) se développent à partir de cellules souches hématopoïétiques (CSH) à la suite d’un processus complexe appelé mégacaryopoïèse, qui est orchestré principalement par l’hormone thrombopoïétine (TPO). La vision classique de la mégacaryopoïèse décrit le voyage cellulaire des CSH à travers une succession d’étapes hiérarchiques de progéniteurs engagés et de cellules précurseurs, conduisant finalement à un MK mature. Au cours de la maturation, les MKs subissent plusieurs cycles d’endomitosis, développent un système complexe de membrane de démarcation intracellulaire (DMS), qui fournit suffisamment de surface membranaire pour la production de plaquettes, et produisent et emballent efficacement la pléthore de facteurs qui sont contenus dans les différents granules hérités par les plaquettes matures1,2,3. En conséquence, les MKs matures sont de grandes cellules (40-60 μm) caractérisées par un noyau hautement polyploïde (atteignant même >64N). Des études récentes suggèrent des voies alternatives par lesquelles les CSH se différencient en MKs en contournant les points de contrôle traditionnels d’engagement de lignée en réponse à certaines conditions physiopathologiques4,5,6,7,8,9,10,11. Ces résultats soulignent que la différenciation hématopoïétique vers le MC mature est un continuum et un processus adaptatif qui répond aux besoins biologiques.
Avec les connaissances croissantes sur la biologie cellulaire et les aspects moléculaires caractérisant la mégakaryopoïèse12,la plupart des recherches consacrées à l’étude du processus par cytométrie en flux se limitent à l’identification des MKs matures à l’aide de marqueurs de surface spécifiques à la lignée(c’est-à-dire CD42A / B, CD41 / CD61), tandis que les stades de différenciation mk antérieurs restent inexplorés. Nous avons précédemment documenté une stratégie pour mettre en scène la mégakaryopoïèse dans la moelle osseuse de souris et les cultures mk dérivées de la moelle osseuse13,14, que nous avons adaptée et appliquée à l’homme15. Dans le présent article, nous montrons une stratégie d’immunophénotypage qui permet la caractérisation de la mégakaryopoïèse, des CSH aux MKs matures, dans des sources primaires humaines (moelle osseuse -BM- et sang périphérique -PB-) ou des cultures in vitro à l’aide d’un panel intégrant des marqueurs de surface spécifiques et non spécifiques à MK (CD61, CD42B, CD49B, CD31, KIT et CD71, entre autres). Malgré sa grande taille et sa fragilité, les MKs peuvent être immunophénotypés à l’aide des marqueurs de surface cellulaire mentionnés ci-dessus et enrichis par le tri cellulaire activé par fluorescence dans des conditions spécifiques de pression et de diamètre de buse pour minimiser la rupture et / ou les dommages cellulaires. Cette technique facilite les approches multi-omiques, dans le but de mieux comprendre la complexité de la mégakaryopoïèse et de la production plaquettaire dans la santé humaine et la maladie. Il convient de noter qu’il constituera un outil utile pour faciliter le diagnostic et le pronostic dans un contexte clinique de demande croissante.
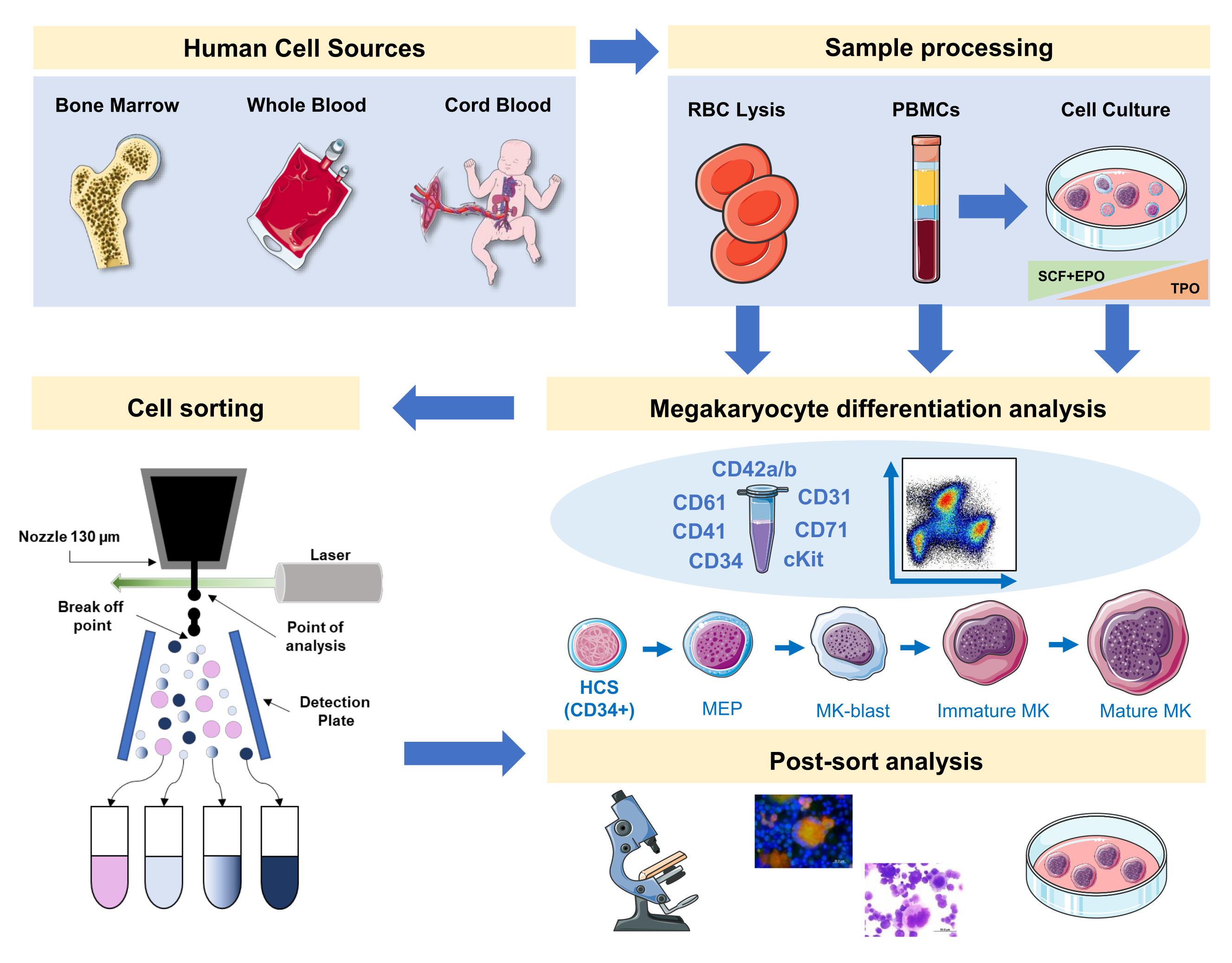
Dans ce manuscrit, nous documentons une stratégie pour mettre en scène la mégacaryopoïèse humaine avec un panneau intégrant des marqueurs de surface spécifiques et non spécifiques au MC provenant de sources primaires ou générés in vitro. De plus, nous fournissons un protocole pour trier, avec un trieur de cellules activé par fluorescence, les fractions préférées et les MKs matures (Figure 1). Cette étape n’est pas populaire, car elle est techniquement difficile en raison de la grande taille et de la fragilité des Clés. Cependant, il a été utilisé à la fois dans des échantillons de moelle osseuse de souris et d’homme précédemment, et en raison des progrès technologiques, avec un meilleur résultat à chaque fois16,17,18. Les sources primaires humaines où les précurseurs de MKs ou de MK peuvent être étudiés incluent la moelle osseuse, le sang de cordon et le sang périphérique, entre autres. Le traitement approprié de l’échantillon pour isoler la fraction cellulaire pertinente pour l’analyse sur chaque échantillon est important. Des procédures standard sont incorporées, avec certaines considérations à prendre en compte lors de l’étude de la mégakaryopoïèse.
Protocole
Des échantillons de sang total et de moelle osseuse ont été obtenus et traités conformément à la Déclaration d’Helsinki de 1964. Des échantillons de sang total ont été obtenus auprès de donneurs sains après avoir donné leur consentement éclairé (ISPA), dans le cadre d’une étude approuvée par notre comité d’éthique médicale institutionnel (Hospital Universitario Central de Asturias -HUCA-). Des échantillons de moelle osseuse ont été obtenus à partir de matériel de rejet aspiré de moelle osseuse de patients pris en charge au département d’hématologie de l’hôpital Clínico San Carlos (HCSC).

Figure 1: Représentation schématique du protocole documenté dans ce manuscrit. Les sources humaines primaires ou les cultures primaires où la différenciation du MC peut être mise en scène à l’aide de l’immunophénotypage sont indiquées. Cette stratégie d’immunophénotypage peut être appliquée à l’étude du processus dans différentes pathologies liées à la lignée ou malignité dans les sources primaires. En outre, il permet le tri cellulaire des MKs et des précurseurs avec un trieur cellulaire activé par fluorescence, ce qui permet une analyse plus approfondie des fractions enrichies. Les images utilisées font partie de Servier Medical Art (SMART) de Servier et sont sous licence CC BY 3.0. Veuillez cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
1. Traitement du sang total et de la moelle osseuse avant l’immunophénotypage
- Lorsque vous utilisez du sang total (WB) provenant de dons comme source primaire, isolez éventuellement le composant de cellules mononucléaires de sang périphérique (PBMC). Ceci peut être réalisé en utilisant la centrifugation différentielle standard combinée à la séparation des cellules de gradient de densité, comme décrit précédemment15.
- En bref, centrifuger le sang à 193 x g pendant 15 min (frein 3) à température ambiante. Jetez la fraction plasmatique supérieure et collectez l’anneau buffy. Diluer 1:1 avec un tampon phosphate salin (PBS)/Citrate trisodique dihydraté (38 g/L, pH 7) tampon et pipetter soigneusement un volume de 25 mL au-dessus de 15 mL d’une solution à gradient de densité (1,076 g/mL) dans des tubes de 50 mL.
- Centrifuger pendant 20 min à 1114 x g (accélérateur 3, frein 3, température ambiante). Jeter la fraction plasmatique et recueillir l’anneau chamois contenant des PBMC. Laver en ajoutant le même volume de PBS, centrifuger à 435 x g pendant 5 min et ressusciter dans du PBS pour une utilisation ultérieure.
- Vous pouvez également utiliser un échantillon de WB (environ 100 μL) pour l’immunophénotypage après avoir lysé les globules rouges (GR) et un lavage complet.
- En bref, diluer 1:1 dans un tampon de lysement RBC glacé (4,15 g deNH4Cl, 0,5 g de KHCO3 et 18,5 mg d’EDTA (triplex III) à 500 mL deH2O,pH 7,1-7,4). Attendez que la suspension cellulaire devienne rouge translucide (3-5 min).
- Centrifuger à 435 x g pendant 5 min, à 4 °C, et ressusciter les cellules dans du PBS. Répétez la procédure autant de fois que nécessaire pour obtenir une pastille de globules blancs.
- De même, traiter directement les échantillons obtenus à partir de moelle osseuse (aspiration) avec un tampon de lysement RBC (voir point 1.2) et un lavage approfondi, comme pour commencer par une suspension unicellulaire claire(figure 1).
- Évitez l’utilisation du vortexing pour mélanger les échantillons pendant le traitement, car il peut endommager les MKs fragiles. Mélanger en feuilletant ou en inversant le tube.
REMARQUE: Le gradient de densité pour obtenir des PBMC peut entraîner une fraction cellulaire plus riche et plus propre par rapport à la WB lysée par RBC. Cependant, nous devons garder à l’esprit que les MKs matures à haute densité pourraient être perdus dans la fraction « neutrophile ». Cela sera discuté dans les résultats représentatifs.
- Évitez l’utilisation du vortexing pour mélanger les échantillons pendant le traitement, car il peut endommager les MKs fragiles. Mélanger en feuilletant ou en inversant le tube.
2. Différenciation in vitro du MC par rapport aux PBMC
NOTA : Les MKs peuvent être différenciés in vitro des précurseurs antérieurs, tels que les cellules CD34+, présents dans différentes sources primaires(c.-à-d. WB/PBMCCs, sang de cordon ombilical, moelle osseuse) et des iPSCs. Différents protocoles ont été appliqués à cette fin. Ici, nous utilisons une méthode de culture développée par nos services qui permet la différenciation mk des PBMC, sans avoir besoin d’enrichir pour les précurseurs CD34+ 15,19,20,21,22.
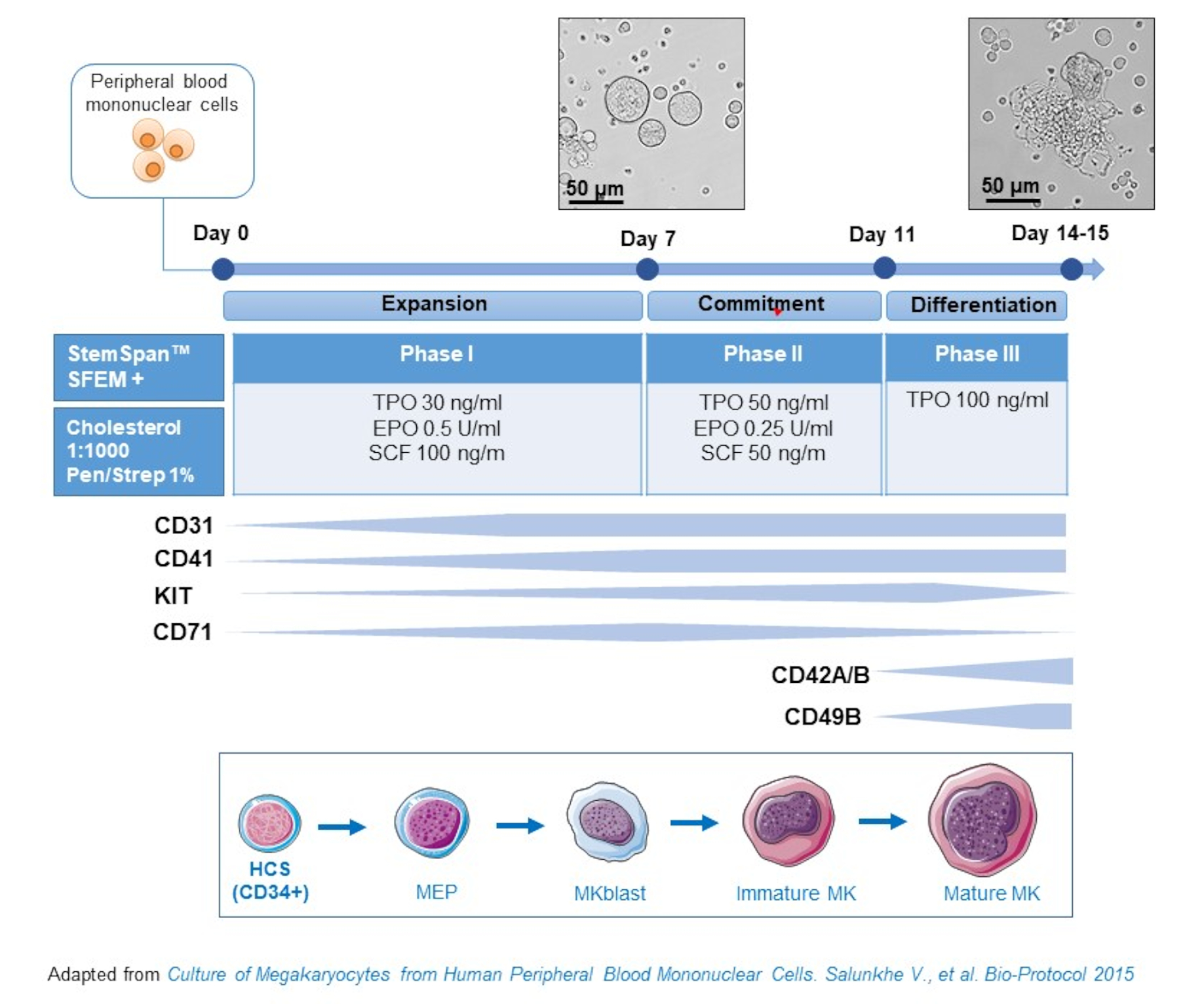
- Ce protocole se compose de trois phases de culture, où la concentration de thrombopoïétine (TPO) augmente progressivement au détriment des facteurs de croissance favorisant la prolifération de précurseurs antérieurs(c’est-à-dire,SCF, EPO), qui diminuent progressivement(figure 2)15.
- Pour le milieu de base, utilisez StemSpan SFEM complété par 0,4% de mélange lipidique riche en cholestérol et 1% de pénicilline / streptomycine.
- Pour le milieu de phase I, utiliser le milieu de base complété par du SCF (100 ng/mL), de l’érythropoïétine (EPO, 0,5 U/mL) et de la thrombopoïétine (TPO, 30 ng/mL). Le milieu de phase II est le milieu de base complété par le SCF (50 ng/mL), l’EPO (0,25 U/mL) et le TPO (50 ng/mL). Pour le milieu de phase III, utiliser le milieu de base complété par du TPO (100 ng/mL).
- Culture pbmccs dans le milieu de phase I. Aux jours 6-8, placez les PBMC dans le milieu de la phase II et, au jour 9-12, placez les PBMC dans le milieu de la phase III.
- Remplacer les cellules de centrifugation par 435 x g pendant 5 min à température ambiante de la phase I à la phase II, et à 95 x g pendant 5 min à température ambiante de la phase II à la phase III, et les ressusciter en milieu frais.
- Cellules de culture dans un incubateur à 37 °C, 5 % deCO2. Dans ces cultures primaires, la différenciation du MC dure de 10 à 14 jours, et des échantillons peuvent être prélevés à différents moments tout au long de la période de culture, comme pour suivre la différenciation du MC.
- Pour le milieu de base, utilisez StemSpan SFEM complété par 0,4% de mélange lipidique riche en cholestérol et 1% de pénicilline / streptomycine.

Figure 2: Représentation schématique de la méthode de culture du MC dérivée du PBMC. Les PBMC provenant de donneurs sains ont été cultivés selon le protocole triphasé que nous avons développé pour générer du MC in vitro (schéma adapté de Salunkhe et al). 15 photos prises au jour 10 et au jour 13 de la culture sont montrées. Les photos sont prises avec un objectif 20X. Veuillez cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
- Prélèvement d’échantillons : Laver les cellules en appliquant une centrifugation à basse vitesse pendant 5 min (95 x g) et les réinsérer dans du PBS ou du PBS contenant 1 % d’albumine sérique bovine (BSA). Pour l’immunophénotypage, la densité idéale est de10 5à10 6 cellules/100 μL (voir point 3.1). Selon l’état des cultures(c.-à-d. présence de cellules mortes, de débris, etc.), 1 ou 2 lavages peuvent être nécessaires. Collectez vos cellules aux points d’intérêt temporels pendant la culture.
3. Immunophénotypage de la différenciation du MC - incubation avec un panel d’anticorps marqués
- Incuber les échantillons cellulaires avec un panel d’anticorps marqués en suivant les procédures standard, en faisant attention à centrifuger à basse vitesse (95 x g) lors du traitement des mégacaryocytes. Nous incubons normalement les échantillons avec 1% BSA dans PBS en volumes de 100 μL à 4 °C, 20 min, avec une plage de 105-106 cellules.
- Augmentez la taille si nécessaire.
- Après incubation, ajouter 5 mL de BSA à 1 % dans du PBS, centrifuger à basse vitesse (95 x g), aspirer le surnageant et ressusciter l’échantillon dans 2 % de BSA dans du PBS pour préserver la viabilité du MC (2 mL). Ajouter de l’EDTA de 1 à 5 mM pour perturber les agrégats cellule-cellule (qui sont naturellement observés dans les cultures de MC).
- Transférer les échantillons dans un tube ou une plaque de fond rond de 12 x 75 mm (tube FACS), en les gardant dans l’obscurité jusqu’à l’analyse cytométrique en flux ou le tri cellulaire.
- Préparation des mélanges d’anticorps simples et de panneaux d’anticorps; mise en place du cytomètre de flux:
- Titrer les anticorps avant leur utilisation pour déterminer la concentration optimale dans les panneaux d’anticorps. La concentration optimale d’anticorps est la concentration la plus faible qui sépare les cellules clairement positives des cellules négatives (et permet de distinguer les niveaux intermédiaires d’expression). À titre d’exemple, la plupart des anticorps sont utilisés dans une dilution de 1:200 (stock 100 μg/mL), sauf indication contraire du fabricant.
- Une fois le titrage des anticorps déterminé, préparez une dilution 10x de chaque anticorps. Ces dilutions sont utilisées pour les contrôles unicolores et pour la préparation des mélanges de panneaux. Les dilutions et les mélanges de panneaux peuvent être utilisés même un mois après la préparation (conservés à 4 °C, à moins que les indications du fabricant n’excluent ces conditions d’entreposage). Cela permet la coloration d’échantillons avec le même panneau sur une période de temps.
- Utilisez 10 μL par 100 μL de dilution 10x à la fois pour les contrôles unicolores et pour le mélange de panneaux.
- Pour les contrôles unicolores, utilisez des billes d’affinité d’anticorps, qui peuvent être mesurées directement après l’ajout de l’anticorps. Les contrôles unicolores doivent être mesurés à chaque expérience, pour permettre un ajustement approprié de la compensation (et un réglage fin après la mesure avec le logiciel d’analyse).
- Vous pouvez également effectuer les contrôles unicolores avec des échantillons de cellules. Cependant, les billes permettent la mesure rapide d’un nombre donné d’événements, qui, selon l’anticorps /marqueur de surface, pourraient ne pas être possibles à obtenir sur des sources cellulaires primaires ou cultivées complexes. Nous vous conseillons également d’exécuter des échantillons de cellules colorés avec des mélanges de panneaux « Fluorescence Minus One » (FMO) pour configurer les paramètres de compensation appropriés (avant d’exécuter des expériences). Ceci est pertinent pour identifier soigneusement les problèmes de compensation, et en particulier, dans les MKs cultivés, pour identifier les interférences d’autofluorescence (qui seront présentes si vous utilisez un milieu de culture contenant du rouge de phénol).
- Préparer un volume suffisant du mélange de panneaux, en fonction du nombre d’échantillons, contenant les anticorps du panneau conçu. La plupart de nos panneaux comprennent six anticorps (panneaux de 6 couleurs, voir tableaux 1-2).
- Pour ces panneaux, utilisez des lasers de 488 nm et 633 nm du cytomètre de flux, cependant, les panneaux peuvent être adaptés à d’autres scénarios techniques. En outre, les considérations de compensation peuvent être évités lors de l’utilisation de la cytométrie de flux basée sur la spectrométrie de masse ou des cytomètres avec la technologie de mise au point acoustique.
- Les colorants pour mesurer la viabilité peuvent donner de fausses informations concernant les MKs, en particulier lorsqu’ils arrivent à maturité. Les MKs sont des cellules de prise de contrôle très actives, et la positivité avec Hoechst, 7-AAD ou PE, pourrait ne pas toujours refléter la mort cellulaire réelle. Une alternative (si la mesure de la mort cellulaire est requise) pourrait être l’utilisation de taches de mitochondries (CMX Ros) ou de colorants réactifs aux amines (colorants Zombie ou Ghost).
Tableau 1 : Notes sur les marqueurs de surface cellulaire de la lignée mégacaryocytaire Veuillez cliquer ici pour télécharger ce tableau.
Tableau 2 : Panneaux sur les anticorps Veuillez cliquer ici pour télécharger ce tableau.
4. Analyse mentoïdale combinée avec des panneaux à 6 couleurs
- Pour l’analyse rachitaire, en combinaison avec un panneau d’anticorps de 6 couleurs, procéder à la fixation et à la perméabilisation des cellules après incubation avec le panneau d’anticorps. Cette stratégie permettra la préservation de la coloration du marqueur de surface, tout en permettant la coloration de l’ADN des cellules. Nous utilisons Hoechst 33342 pour colorer l’ADN, car il peut être visualisé avec le laser violet 405 nm disponible.
- Pour 105-106 cellules, après incubation avec le panneau d’anticorps, centrifuger les cellules (95 x g pendant 5 min), ressusciter dans 200 μL de tampon de fixation et incuber 10 min à température ambiante (RT).
- Cellules centrifuges comme indiqué ci-dessus, remises en suspension une deuxième fois dans un tampon de fixation de 200 μL et incuber encore 10 min à TA.
- Préparer un tampon de perméabilisation contenant 0,1 % de Triton X-100, 200 mg/mL de RNase et 20 mg/mL de Hoechst 33342 (perméabilisation Hoechst MIX).
- Cellules centrifugeuses comme ci-dessus, les ressusciter dans 300 μL de la perméabilisation Hoechst MIX et incuber 30 min à 37 °C. Cette étape est très importante, car, pour obtenir une mesure de ploidy propre, l’ARN doit être dégradé.
- Après le temps d’incubation, mesurer les échantillons directement avec un cytomètre de flux. Sinon, gardez les échantillons à 4 °C, dans l’obscurité. Mesurez-les rapidement. Cependant, comme ces échantillons sont fixes, la mesure peut être retardée même de 24 à 48 heures. Assurez-vous que l’échantillon est bien feuilleté avant de le mesurer, ou passé à travers une passoire cellulaire, pour assurer une suspension à cellule unique.
- Les paramètres morphométriques tels que la diffusion vers l’avant et latérale ne sont pas maintenus après la fixation cellulaire. Le nuage de points avant/côté montrera un rétrécissement de la distribution cellulaire après la fixation. Cependant, la coloration du marqueur de surface est principalement préservée, et la stratégie de gating est à peine modifiée, permettant l’analyse du statut prpoidy aux différentes étapes de différenciation définies par des combinaisons de marqueurs de surface.
5. Analyse de différenciation mk
NOTE: Nous avons vu que la combinaison de CD31/CD71 permet de définir un certain nombre de portes qui correspondent à différentes étapes de différenciation MK. D’autres rétro-gating avec des marqueurs spécifiques au MK permettent la séparation des MKs matures et immatures. De plus, dans les échantillons frais, le rétro-contrôle pour vérifier la présence d’autres marqueurs utilisés, ou pour placer les populations dans les axes de diffusion avant/côté, affine l’évaluation des stades de différenciation du MC et permet de rejeter d’autres types de cellules qui pourraient être présents sur les mêmes populations.
- Utilisez un panel d’anticorps qui comprend des marqueurs précurseurs précoces (KIT, CD34), des marqueurs précurseurs communs (CD31, CD71) et des marqueurs de lignée, dont certains sont spécifiques (CD42A/CD42B, CD49B, CD41/CD61, CLEC2, GPVI, etc.) (voir les tableaux 1-2). L’utilisation du cocktail Lineage (Lin) (CD3, CD14, CD16, CD19, CD20 et CD56) permet également de « filtrer » les cellules hématopoïétiques matures qui pourraient ajouter du bruit à l’analyse (lors de la sélection de la population Lin). À titre d’exemple, nous allons passer en revue l’analyse des MKs dans les PBMC, la moelle osseuse et les cultures cellulaires dérivées du PBMC dans les résultats représentatifs.
6. Tri des cellules précurseurs MK et MK
REMARQUE: Les cellules colorées ont été analysées et triées sur un trieur de cellules activé par fluorescence FACS Aria IIu équipé de lasers à semi-conducteurs standard de 488 nm et 633 nm à l’aide du logiciel FACSDiva; les données ont en outre été analysées et présentées à l’aide du logiciel FlowJo et de la cytobanque (analyse viSNE). La pureté des fractions triées a été confirmée par l’analyse cytométrique de flux de chacune des fractions triées (pureté supérieure à 85%).
- Effectuer le tri cellulaire dès que possible ou dans l’heure 1 après l’incubation des anticorps afin d’éviter la détérioration cellulaire.
- Filtrer l’échantillon avec une passoire cellulaire de 100 μm pour assurer la suspension à cellule unique et l’intégrité du grand MK.
- Utilisez une buse en céramique de 130 μm, une pression de gaine réglée à 11 livres par pouce carré (PSI) et la fréquence d’entraînement de chute réglée sur 12 kHz pour diviser le flux en gouttes.
- Avant le tri, stérilisez la buse, la gaine et les lignes d’échantillonnage en effectuant une acquisition de 30 minutes avec de la pénicilline/streptomycine diluée 1:5 dans de l’eau stérile, suivie d’une acquisition de 10 minutes avec de l’eau stérile pour éliminer le décontaminant restant.
- Une fois que le flux s’est stabilisé, ajustez le drop-delay avec les perles recommandées afin de trier en mode de réglage fin plus de 97,5% des gouttes réfléchies à un débit de 400-1200 événements par seconde.
- Préparer les tubes FACS de collecte avec 500 μL de BSA à 2 % dans du PBS. Le pourcentage de BSA peut être augmenté jusqu’à 5-10%.
- Générez le modèle d’expérience avec les paramètres de matrice de compensation appropriés.
- Charger le tube FACS dans le cytomètre.
- Effectuer une mesure de l’échantillon pour définir les portes souhaitées et la pureté des populations de cellules cibles. Maintenez l’enregistrement activé pour afficher jusqu’à 200 000 événements dans les portes de population sélectionnées pendant le tri des cellules.
- FACS Aria IIu permet la séparation de jusqu’à 4 populations cellulaires différentes en même temps. Créez une nouvelle disposition de tri et sélectionnez le dispositif de collecte (4 tubes) et le mode de précision approprié (masque intermédiaire de pureté et de récupération est recommandé). Enfin, ajoutez la ou les populations d’intérêt à chaque champ d’emplacement de tri(Figure 3).

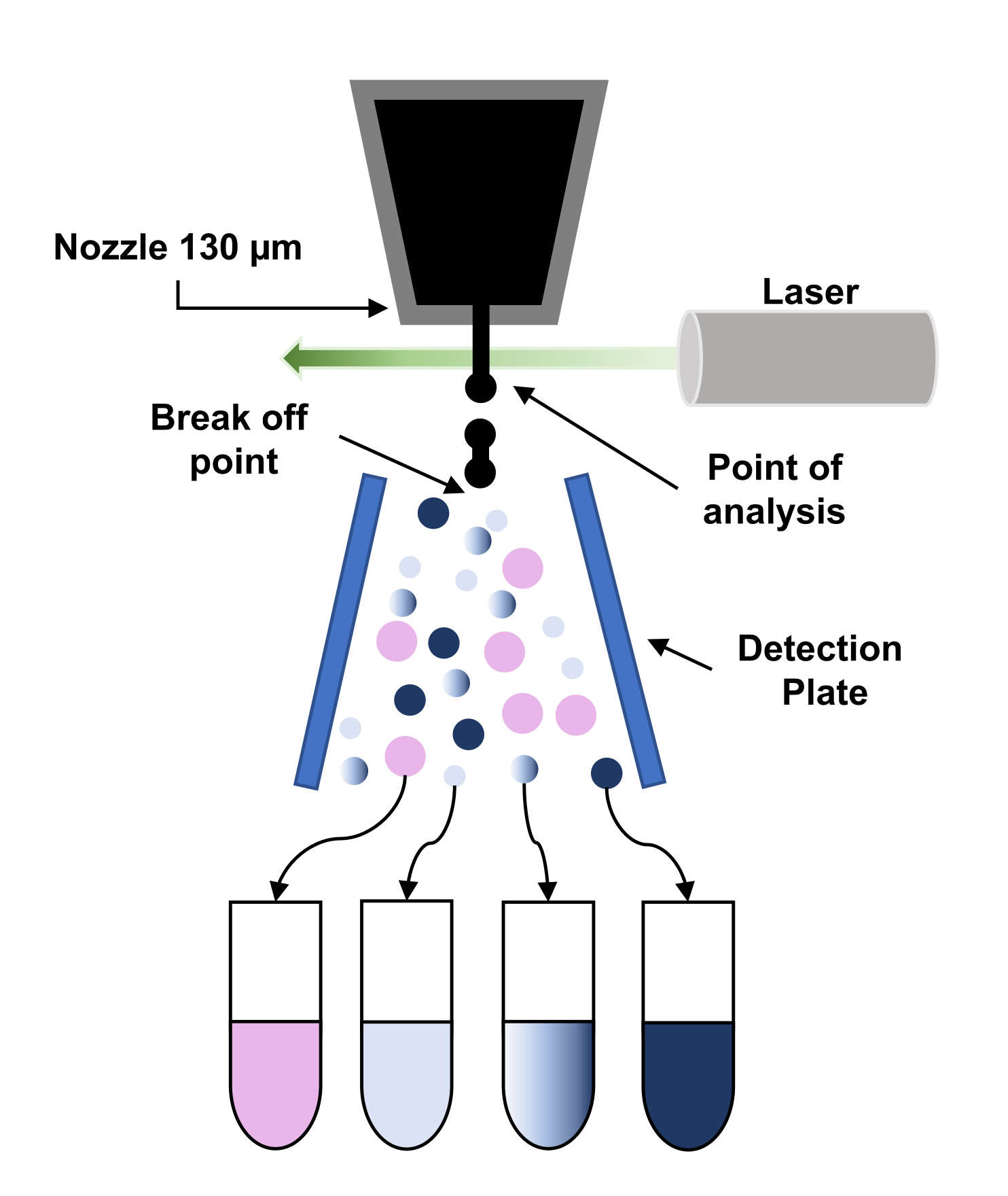
Figure 3: Représentation schématique du principe du tri cellulaire activé par fluorescence (FACS). Les particules traversent la buse de 130 μm et sont forcées de se décomposer en un flux de gouttelettes régulières en raison de l’application de vibrations sur la buse. Ensuite, les gouttelettes sont interrogées par le laser (point d’analyse) et les signaux sont traités pour donner la « décision de tri » en appliquant une charge à ces gouttelettes. Lorsqu’une gouttelette de charge traverse un champ électrostatique haute tension (plaque de détection), elle est déviée et recueillie dans le tube de collecte correspondant. Veuillez cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
- Chargez les tubes de collecte et commencez à trier les populations cibles.
- Centrifuger les tubes de collecte pendant 5 minutes à 95 x g et ressusciter la pastille cellulaire dans le volume approprié de BSA à 2 % dans du PBS.
- Mesurez à nouveau une fraction de chaque échantillon trié, pour calculer la pureté.
- Stockez les cellules de manière appropriée pour une utilisation ultérieure. Les cellules triées peuvent être utilisées pour des analyses cytologiques et moléculaires, ou peuvent être re-cultivées dans le but d’étudier le processus de différenciation d’une population cellulaire sélectionnée.
7. Préparation de l’échantillon après le tri
- Préparation de cytospins pour l’analyse cytologique avec une cytocentrifugation
- Amenez les cellules triées à un volume de travail facile à manipuler de 100 à 200 μL. Prenez en considération que la densité cellulaire dépendra du rendement de la population triée dans chaque cas.
- Placez une lame propre sur le support métallique et placez un dessus de filtre. N’oubliez pas d’étiqueter la lame et le filtre pour éviter de mélanger les échantillons.
- Ajoutez 100 μL de PBS sur le trou du filtre contre la glissière, de sorte que le filtre soit humidifié sur le bord du trou.
- Placez l’entonnoir, fermez le support métallique et placez-le à son endroit dans la centrifugeuse.
- Ajouter l’échantillon (100-200 μL) à l’intérieur de l’entonnoir.
- Centrifuger à 36 x g pendant 5 minutes. Les lames de cytospin peuvent être laissées sécher à l’air à TA (correctement couvertes pour prévenir la poussière) et peuvent être conservées à TA pendant 1 semaine avant l’immunomarquage ou l’histochimie.
- Pour l’immunomarquage
- Fixer les lames dans du paraformaldéhyde à 2% (PFA) dilué dans du PBS et incuber pendant 5min.
- Une fourchette de 0,5 à 4 % de PFA dans le PBS peut être utilisée. Dans nos mains, nous utilisons 4% pour obtenir la fixation appropriée des tissus ou de certains types de cellules, et 0,5% PFA dans PBS est suffisant pour les plaquettes. Lors de la configuration de cette technique, le bon pourcentage de PFA nécessite une optimisation par type/source de cellule.
- Incuber 5 min dans pbs.
- Incuber 5 min dans de l’éthanol à 50% (EtOH).
- Incuber 5 min dans 70% EtOH.
- Conserver dans 70% EtOH à -20ºC.
- Lors de l’exécution de l’immunomarquage, réhydrater et suivre les procédures standard (perméabilisation, lavage, blocage, incubations d’anticorps primaires et secondaires, conservation, etc.).
- Fixer les lames dans du paraformaldéhyde à 2% (PFA) dilué dans du PBS et incuber pendant 5min.
- Pour la cytochimie :
REMARQUE: Les lames peuvent être colorées avec la coloration May-Grünwald Giemsa ou la coloration pratique pour chaque but. - Pour un examen morphologique immédiat
- Ajoutez une goutte de support de montage sur la tache contenant des cellules du cytospin et placez une lamelle.
- Maintenir les lames à 4 °C au plus tard une semaine, à moins qu’elles ne soient scellées, ce qui permet un stockage à long terme même à TA. La fixation moyenne de montage permet un stockage à long terme à TA.
Résultats
Moelle osseuse et onoïdie
Sur la figure 4, nous montrons une analyse immunophénotypage représentative de la mégakaryopoïèse dans des échantillons de BM (aspiration) provenant de patients. En traçant la fraction cellulaire par rapport à CD71 et CD31, nous avons rated six populations principales: CD31- CD71- (rouge), CD31- CD71+ (bleu), CD31+ CD71- (orange), CD31+ CD71
Discussion
La plupart des recherches portant sur l’étude de la mégakaryopoïèse par cytométrie en flux se limitent à ce jour à l’identification de sous-ensembles de MC en utilisant uniquement des marqueurs de surface spécifiques à la lignée(c’est-à-direCD42A/CD42B, CD41/CD61), tandis que les stades antérieurs de différenciation du MC ont été mal examinés. Dans le présent article nous montrons une stratégie immunophenotyping pour adresser une caractérisation cytometry complète d’écoulement de meg...
Déclarations de divulgation
La production de matériel audiovisuel a été soutenue par BD Biosciences.
Remerciements
Nous remercions Marcos Pérez Basterrechea, Lorena Rodríguez Lorenzo et Begoña García Méndez (HUCA) et Paloma Cerezo, Almudena Payero et María de la Poveda-Colomo (HCSC) pour leur soutien technique. Ce travail a été partiellement soutenu par des subventions médicales (Roche SP200221001) à A.B., une bourse RYC (RYC-2013-12587; Ministerio de Economía y Competitividad, Espagne) et une subvention I+D 2017 (SAF2017-85489-P; Ministerio de Ciencia, Innovación y Universidades, Espagne et Fondos FEDER) à L.G., une subvention Severo Ochoa (PA-20-PF-BP19-014; Consejería de Ciencia, Innovación y Universidades del Principado de Asturias, Espagne) à P.M.-B. et une bourse postdoctorale intra-muros 2018 (Fundación para la Investigación y la Innovación Biosanitaria de Asturias - FINBA, Oviedo, Espagne) à A.A.-H. Nous remercions Reinier van der Linden d’avoir partagé ses connaissances (et son temps), en particulier ses sages conseils sur le mélange de panneaux d’anticorps marqués multicolores et la préparation du contrôle des perles unicolores.
matériels
| Name | Company | Catalog Number | Comments |
| 130 micron Nozzle | BD | 643943 | required for MK sorting |
| 5810R Centrifuge | Eppendorf | Cell isolation and washes | |
| A-4-62 Swing Bucket Rotor | Eppendorf | Cell isolation and washes | |
| Aerospray Pro Hematology Slide Stainer / Cytocentrifuge | ELITech Group | Automatized cytology devise, where slides are stained with Mat-Grünwald Giemsa | |
| CO2 Incubator Galaxy 170 S | Eppendorf | Cell Incubation | |
| Cytospin 4 Cytocentrifuge | Thermo Scientific | To prepare cytospins | |
| FACSAria IIu sorter | BD | Lasers 488-nm and 633-nm | |
| FACSCanto II flow cytometer | BD | Lasers 488-nm , 633-nm and 405-nm | |
| Olympus Microscope BX 41 | Olympus | Microphotographs | |
| Olympus Microscope BX 61 | Olympus | Microphotographs | |
| Zoe Fluorescent Cell Imager | BioRad | Microphotographs | |
| To obtain PBMCs | |||
| Lipids Cholesterol Rich from adult bovine serum | Sigma-Aldrich | L4646 | or similar |
| Lymphoprep | Stem Cell Technologies | #07801 | or similar |
| Penicillin-Streptomycin | Sigma-Aldrich | P4333 | or similar |
| Recombinant human Erythropoietin (EPO) | R&D Systems | 287-TC-500 | or similar |
| Recombinant human stem cell factor (SCF) | Thermo Fisher Scientific, Gibco™ | PHC2115 | or similar |
| Recombinant human thrombopoietin (TPO) | Thermo Fisher Scientific, Gibco™ | PHC9514 | or TPO receptor agonists |
| StemSpan SFEM | Stem Cell Technologies | #09650 | |
| Flow Cytometry Analyses | |||
| Bovine Serum Albumin | Merck | A7906-100G | or similar |
| BD CompBead Anti-Mouse Ig, κ/Negative Control Compensation Particles Set | BD | 552843 | Antibodies for human cells are generally from mouse. |
| BD Cytofix/Cytoperm | BD | 554714 | or similar |
| BD FACS Accudrop Beads | BD | 345249 | |
| CD31 AF-647 | BD | 561654 | Mouse anti-human |
| CD31 FITC | Immunostep | 31F-100T | |
| CD34 FITC | BD | 555821 | Mouse anti-human |
| CD41 PE | BD | 555467 | Mouse anti-human |
| CD41 PerCP-Cy5.5 | BD | 333148 | Mouse anti-human |
| CD42A APC | Immunostep | 42AA-100T | We observed unspecific binding... that needs to be assessed |
| CD42A PE | BD | 558819 | Mouse anti-human |
| CD42B PerCP | Biolegend | 303910 | Mouse anti-human |
| CD49B PE | BD | 555669 | Mouse anti-human |
| CD61 FITC | BD | 555753 | Mouse anti-human |
| CD71 APC-Cy7 | Biolegend | 334109 | Mouse anti-human |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | |
| Human BD Fc Block | BD | 564219 | Fc blocking - control |
| KIT PE-Cy7 | Biolegend | 313212 | Mouse anti-human |
| Lineage Cocktail 2 FITC | BD | 643397 | Mouse anti-human |
| RNAse | Merck | R6513 | or similar |
| Triton X-500 | Merck | 93443-500ML | or similar |
| Cell strainers for sorting | |||
| CellTrics Filters 100 micrometers | Sysmex | 04-004-2328 | Cell strainers |
| Note: we do not specify general reagents/chemicals (PBS, EDTA, etc) or disposables (tubes, etc), or reagents specified in previous published and standard protocols - unless otherwise specified. |
Références
- Italiano, J. E. Unraveling Mechanisms That Control Platelet Production. Semin Thrombosis And Haemostasis. 39 (1), 15-24 (2013).
- Machlus, K. R., Italiano, J. E. The Incredible Journey: From Megakaryocyte Development To Platelet Formation. Journal Of Cell Biology. 201 (6), 785-796 (2013).
- Eckly, A., et al. Biogenesis Of The Demarcation Membrane System (DMS) In Megakaryocytes. Blood. 123 (6), 921-930 (2014).
- Couldwell, G., Machlus, K. R. Modulation Of Megakaryopoiesis And Platelet Production During Inflammation. Thrombosis Research. 179, 114-120 (2019).
- Kosaki, G. In Vivo Platelet Production From Mature Megakaryocytes: Does Platelet Release Occur Via Proplatelets. International Journal Of Hematology. 81 (3), 208-219 (2005).
- Lefrancais, E., Looney, M. R. Platelet Biogenesis In The Lung Circulation. Physiology (Bethesda). 34 (6), 392-401 (2019).
- Nieswandt, B., Stritt, S. Megakaryocyte Rupture For Acute Platelet Needs. Journal Of Cell Biology. 209 (3), 327-328 (2015).
- Nishimura, S., et al. IL-1alpha Induces Thrombopoiesis Through Megakaryocyte Rupture In Response To Acute Platelet Needs. Journal Of Cell Biology. 209 (3), 453-466 (2015).
- Sanjuan-Pla, A., et al. Platelet-Biased Stem Cells Reside At The Apex Of The Haematopoietic Stem-Cell Hierarchy. Nature. 502 (7470), 232-236 (2013).
- Notta, F., et al. Distinct Routes Of Lineage Development Reshape The Human Blood Hierarchy Across Ontogeny. Science. 351 (6269), 2116 (2016).
- Yamamoto, R., et al. Clonal Analysis Unveils Self-Renewing Lineage-Restricted Progenitors Generated Directly From Hematopoietic Stem Cells. Cell. 154 (5), 1112-1126 (2013).
- Wang, H., et al. Decoding Human Megakaryocyte Development. Cell Stem Cell. , (2020).
- Meinders, M., et al. Repercussion Of Megakaryocyte-Specific Gata1 Loss On Megakaryopoiesis And The Hematopoietic Precursor Compartment. Plos One. 11 (5), 0154342 (2016).
- Meinders, M., et al. Sp1/Sp3 Transcription Factors Regulate Hallmarks Of Megakaryocyte Maturation And Platelet Formation And Function. Blood. 125 (12), 1957-1967 (2015).
- Salunkhe, V. P., Gutiérrez, L. Culture Of Megakaryocytes From Human Peripheral Blood Mononuclear Cells. Bio-Protocol. 5 (21), 1639 (2015).
- Choudry, F. A., et al. Transcriptional Characterization Of Human Megakaryocyte Polyploidization And Lineage Commitment. Journal Of Thrombosis And Haemostasis. , 15271 (2021).
- Heazlewood, S. Y., Williams, B., Storan, M. J., Nilsson, S. K. The Prospective Isolation Of Viable, High Ploidy Megakaryocytes From Adult Murine Bone Marrow By Fluorescence Activated Cell Sorting. Methods In Molecular Biology. 1035, 121-133 (2013).
- Tomer, A., Harker, L. A., Burstein, S. A. Purification Of Human Megakaryocytes By Fluorescence-Activated Cell Sorting. Blood. 70 (6), 1735-1742 (1987).
- Martinez-Botia, P., Acebes-Huerta, A., Seghatchian, J., Gutierrez, L. On The Quest For In Vitro Platelet Production By Re-Tailoring The Concepts Of Megakaryocyte Differentiation. Medicina. 56 (12), (2020).
- Martinez-Botia, P., Acebes-Huerta, A., Seghatchian, J., Gutierrez, L. In Vitro Platelet Production For Transfusion Purposes: Where Are We Now. Transfusion And Apheresis Science. 59 (4), 102864 (2020).
- Butov, K. R., et al. In Vitro Megakaryocyte Culture From Human Bone Marrow Aspirates As A Research And Diagnostic Tool. Platelets. , 1-8 (2020).
- Di Buduo, C. A., et al. A Gold Standard Protocol For Human Megakaryocyte Culture Based On The Analysis Of 1,500 Umbilical Cord Blood Samples. Thrombosis And Haemostasis. , (2020).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.