
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Иммунофенотипирование и сортировка клеток человеческих МК из первичных источников человека или дифференцированных in vitro от кроветворных прародителей
В этой статье
Резюме
Здесь мы представляем стратегию иммунофенотипирования для характеристики дифференцировки мегакариоцитов и показываем, как эта стратегия позволяет сортировать мегакариоциты на разных стадиях с помощью флуоресцентно-активированного сортировщика клеток. Методика может быть применена к первичным тканям человека, а также к мегакариоцитам, образуемым в культуре in vitro.
Аннотация
Дифференцировка мегакариоцитов (МК) охватывает ряд эндомитотических циклов, которые приводят к высокополиплоидной (достигающей еще >64N) и чрезвычайно большой клетке (40-60 мкм). В отличие от быстро растущих знаний о мегакариопоэсисе на клеточном биологии и молекулярном уровне, характеристика мегакариопоэтиса с помощью проточной цитометрии ограничивается идентификацией зрелых МК с использованием специфических для линии поверхностных маркеров, в то время как более ранние стадии дифференцировки МК остаются неисследованными. Здесь мы представляем стратегию иммунофенотипирования, которая позволяет идентифицировать последовательные стадии дифференцировки МК с возрастающим статусом плоидности в первичных источниках человека или культурах in vitro с панелью, объединяющей специфические и неспецифические поверхностные маркеры МК. Несмотря на свои размеры и хрупкость, МК могут быть иммунофенотипированы с использованием вышеупомянутой панели и обогащены флуоресцентно-активированной сортировкой клеток при определенных условиях давления и диаметра сопла. Этот подход облегчает мультиомические исследования с целью лучшего понимания сложности мегакариопоэтиса и производства тромбоцитов у людей. Лучшая характеристика мегакариопоэтиса может представлять собой фундаментальную в диагностике или прогнозе связанных с линией патологий и злокачественных новообразований.
Введение
Мегакариоциты (МК) развиваются из гемопоэтических стволовых клеток (ГСК) после сложного процесса, называемого мегакариопоэзом, который управляется в основном гормоном тромбопоэтином (ТПО). Классический взгляд на мегакариопоэтизис описывает клеточное путешествие от ГСК через последовательность иерархических стадий преданных предшественников и клеток-предшественников, что в конечном итоге приводит к зрелому МК. Во время созревания МК испытывают множественные раунды эндомитоза, развивают сложную внутриклеточную демаркационную мембранную систему (ДМС), которая обеспечивает достаточную поверхность мембраны для производства тромбоцитов, и эффективно производят и упаковывают множество факторов, которые содержатся в различных гранулах, унаследованных зрелыми тромбоцитами1,2,3. В результате зрелые МК представляют собой крупные клетки (40-60 мкм), характеризующиеся высокополиплоидным ядром (достигающим даже >64N). Недавние исследования предлагают альтернативные пути, по которым ГСК дифференцируются в МК, минуя традиционные контрольные точки приверженности линии в ответ на определенные физио-патологические состояния4,5,6,7,8,9,10,11. Эти результаты подчеркивают, что гемопоэтическая дифференциация в сторону зрелого МК является континуумным и адаптивным процессом, который реагирует на биологические потребности.
С ростом знаний о клеточной биологии и молекулярных аспектах, характеризующих мегакариопоэзис12,большинство исследований, посвященных изучению процесса с помощью проточной цитометрии, ограничены идентификацией зрелых МК с использованием специфических для линии поверхностных маркеров(т. Е. CD42A / B, CD41 / CD61), в то время как более ранние этапы дифференцировки МК остаются неисследованными. Ранее мы задокументировали стратегию постановки мегакариопоэсиса в культурах MK13,14,полученных из костного мозга мышей и костного мозга, которую мы адаптировали и применили к людям15. В настоящей статье показана стратегия иммунофенотипирования, позволяющая характеризовать мегакариопоэтоз, от ГСК до зрелых МК, в первичных источниках человека (костном мозге -BM- и периферической крови -PB-) или культурах in vitro с использованием панели, интегрирующей специфические и неспецифические поверхностные маркеры MK (CD61, CD42B, CD49B, CD31, KIT и CD71, среди прочих). Несмотря на свой большой размер и хрупкость, МК могут быть иммунофенотипированы с использованием вышеупомянутых маркеров клеточной поверхности и обогащены флуоресцентно-активированной сортировкой клеток в определенных условиях давления и диаметра сопла для минимизации разрыва и / или повреждения клеток. Этот метод облегчает мультиомические подходы с целью лучшего понимания сложности мегакариопоэтиса и производства тромбоцитов в здоровье и болезнях человека. Примечательно, что он будет представлять собой полезный инструмент для облегчения диагностики и прогнозирования в клиническом контексте растущего спроса.
В этой рукописи мы документируем стратегию постановки человеческого мегакариопоизиса с помощью панели, объединяющей МК-специфические и неспецифические поверхностные маркеры из первичных источников или сгенерированные in vitro. Кроме того, мы предоставляем протокол для сортировки с помощью сортировщика клеток, активируемого флуоресценцией, предпочтительных фракций и зрелых МК(рисунок 1). Этот шаг не популярен, так как технически сложен из-за больших размеров и хрупкости МК. Тем не менее, он использовался как в образцах костного мозга мышей, так и человека ранее, и благодаря технологическому прогрессу, с лучшим результатом каждый раз16,17,18. Человеческие первичные источники, где могут быть изучены MK или предшественники MK, включают костный мозг, пуповинную кровь и периферическую кровь, среди прочего. Правильная обработка образца для выделения соответствующей клеточной фракции для анализа на каждом образце имеет важное значение. Включены стандартные процедуры, с некоторыми соображениями, которые следует учитывать при изучении мегакариопоизиса.
Access restricted. Please log in or start a trial to view this content.
протокол
Образцы цельной крови и костного мозга были получены и обработаны в соответствии с Хельсинкской декларацией 1964 года. Образцы цельной крови были получены от здоровых доноров после предоставления информированного согласия (ISPA) в рамках исследования, одобренного нашим институциональным медицинским этическим комитетом (Hospital Universitario Central de Asturias -HUCA-). Образцы костного мозга были получены из материала отбрасывания аспирата костного мозга пациентов, управляемых в отделе гематологии больницы Clínico San Carlos (HCSC).

Рисунок 1:Схематическое изображение протокола, задокументированный в этой рукописи. Показаны первичные источники человека или первичные культуры, где дифференциация МК может быть поставлена с помощью иммунофенотипирования. Эта стратегия иммунофенотипирования может быть применена для изучения процесса при различных патологиях, связанных с родословной, или злокачественности в первичных источниках. Кроме того, это делает возможным клеточную сортировку МК и прекурсоров с помощью флуоресцентного активированного клеточного сортировщика, что позволяет проводить дальнейший анализ обогащенных фракций. Используемые изображения являются частью Servier Medical Art (SMART) от Servier и лицензированы в соответствии с CC BY 3.0. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
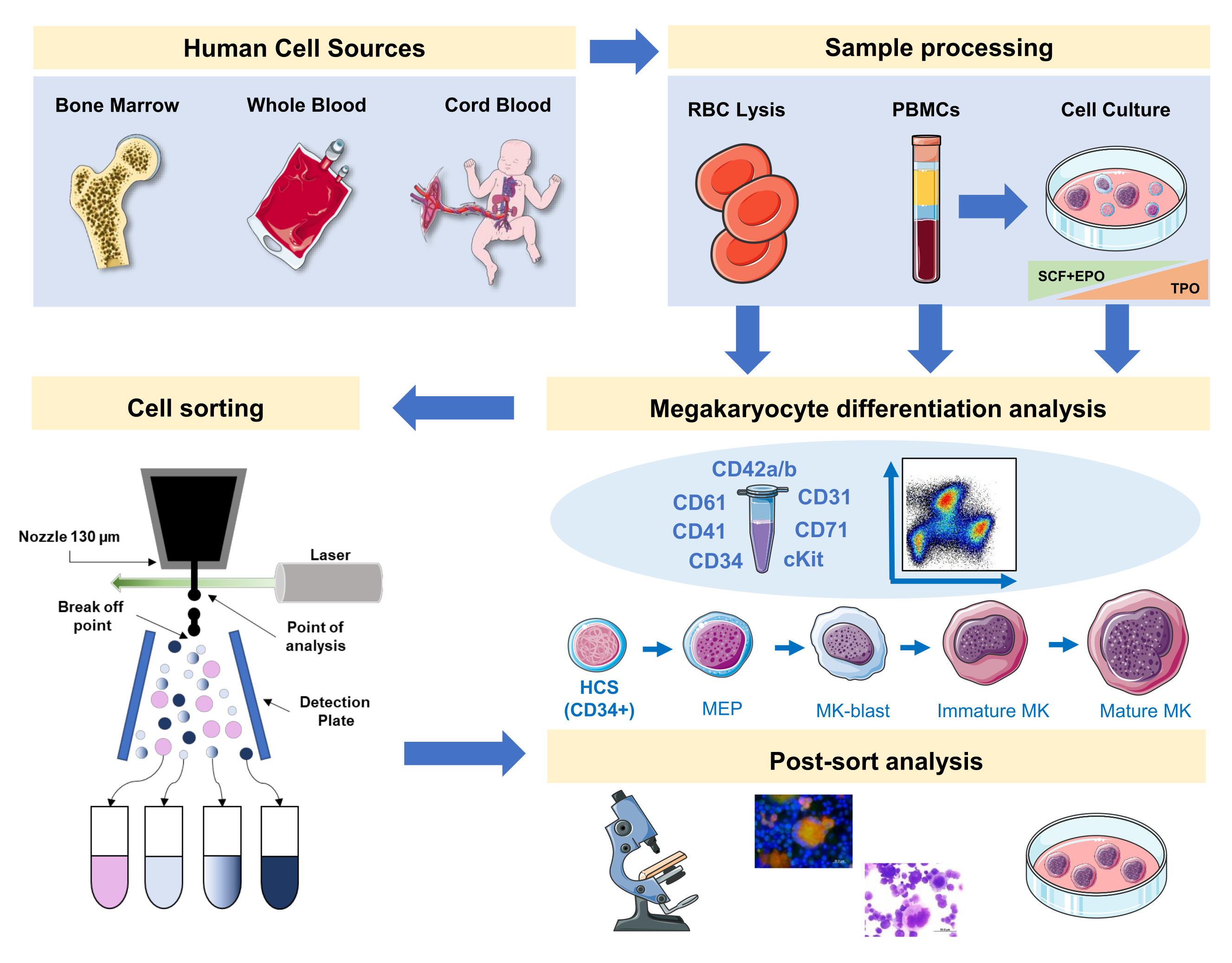{kind=link}
1. Обработка цельной крови и костного мозга перед иммунофенотипированием
- При использовании цельной крови (ВБ) из донорства в качестве первичного источника необязательно выделяют компонент мононуклеарной клетки периферической крови (ПБМК). Это может быть достигнуто путем использования стандартного дифференциального центрифугирования в сочетании с разделением градиентных ячеек плотности, как описаноранее 15.
- Вкратце центрифугируют кровь при 193 х г в течение 15 мин (тормоз 3) при комнатной температуре. Отбросьте верхнюю фракцию плазмы и соберите кольцо баффи. Разбавить 1:1 фосфатным буферным физиологическим раствором (PBS)/Тринатрийцитрат дигидрат (38 г/л, рН 7) буфером и пипеткой объемом 25 мл поверх 15 мл градиентного раствора плотности (1,076 г/мл) в пробирках по 50 мл.
- Центрифуга в течение 20 мин при 1114 х г (акселератор 3, тормоз 3, комнатная температура). Отбросьте плазменную фракцию и соберите пушисткое кольцо, содержащее PBMCs. Промыть, добавив тот же объем PBS, центрифугу при 435 x g в течение 5 мин и повторно использовать в PBS для дальнейшего использования.
- В качестве альтернативы, используйте образец WB (около 100 мкл) для иммунофенотипирования после лизирования эритроцитов (эритроцитов) и тщательной промывки.
- Вкратце, разбавляют 1:1 в ледяном буфере лизирования эритроцитов (4,15 г NH4Cl, 0,5 г KHCO3 и 18,5 мг ЭДТА (триплекс III) до 500 млH2O, рН 7,1-7,4). Подождите, пока клеточная суспензия станет полупрозрачная красного цвета (3-5 мин).
- Центрифуга при 435 х г в течение 5 мин, при 4 °C, и повторное суспендировать ячейки в PBS. Повторите процедуру столько раз, сколько необходимо, чтобы получить гранулу белых клеток.
- Аналогично непосредственно обрабатывать образцы, полученные из костного мозга (аспирация) с помощью буфера лизирования эрицитов (см. пункт 1.2) и тщательного промывания, как для начала с прозрачной одноклеточной суспензии(рисунок 1).
- Избегайте использования вихрей для смешивания образцов во время обработки, так как это может повредить хрупкие МК. Смешайте путем щелчка или инвертирования трубки.
ПРИМЕЧАНИЕ: Градиент плотности для получения PBMCs может привести к более богатой и чистой клеточной фракции по сравнению с RBC-лизированной WB. Однако мы должны иметь в виду, что зрелые МК высокой плотности могут быть потеряны во фракции «нейтрофилов». Это будет обсуждаться в репрезентативных результатах.
- Избегайте использования вихрей для смешивания образцов во время обработки, так как это может повредить хрупкие МК. Смешайте путем щелчка или инвертирования трубки.
2. Дифференциация IN vitro МК от НБМК
ПРИМЕЧАНИЕ: МК могут быть дифференцированы in vitro от более ранних предшественников, таких как CD34+ клетки, присутствующие в различных первичных источниках(т.е. WB / PBMCs, пуповинная кровь, костный мозг) и из IPSCs. Существуют различные протоколы, которые были применены с этой целью. Здесь мы используем разработанный нами метод культивирования, позволяющий дифференцировать МК от НБМК, без необходимости обогащения на CD34+ прекурсоры15,19,20,21,22.
- Данный протокол состоит из трех фаз культивирования, где концентрация тромбопоэтина (ТПО) постепенно увеличивается за счет факторов роста, благоприятствующих распространению более ранних предшественников(т.е.SCF, EPO), которые постепенно снижаются(Рис.2)15.
- Для базовой среды используйте StemSpan SFEM, дополненный 0,4% богатой холестерином липидной смесью и 1% пенициллина / стрептомицина.
- Для среды фазы I используйте базовую среду, дополненную SCF (100 нг/мл), эритропоэтином (EPO, 0,5 ЕД/мл) и тромбопоэтином (TPO, 30 нг/мл). Среда фазы II является базовой средой, дополненной SCF (50 нг/мл), EPO (0,25 U/mL) и TPO (50 нг/мл). Для среды фазы III используйте базовую среду, дополненную ТПО (100 нг/мл).
- Культивные НБМК в среде фазы I. На 6-8-й день поместите МПК в среду фазы II, а на 9-12 день поместите НБМК в среду фазы III.
- Заменить среду центрифугированием клеток при 435 х г в течение 5 мин при комнатной температуре от фазы I до фазы II и при 95 х г в течение 5 мин при комнатной температуре от фазы II до фазы III и повторно использовать их в свежей среде.
- Культививная культура клеток в инкубаторе при 37 °C, 5% CO2. В этих первичных культурах дифференциация МК длится 10-14 дней, и образцы могут быть взяты в разные моменты времени в течение всего периода культуры, чтобы следовать дифференциации МК.
- Для базовой среды используйте StemSpan SFEM, дополненный 0,4% богатой холестерином липидной смесью и 1% пенициллина / стрептомицина.

Рисунок 2:Схематическое представление метода культуры MK, полученного из PBMC. НБМК от здоровых доноров культивировали в соответствии с трехфазным протоколом, разработанным нами для генерации МК in vitro (схема, адаптированная из Salunkhe et al). Показаны 15 фотографий, сделанных на 10-й и 13-й день культуры. Снимки делаются с объективом 20X. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
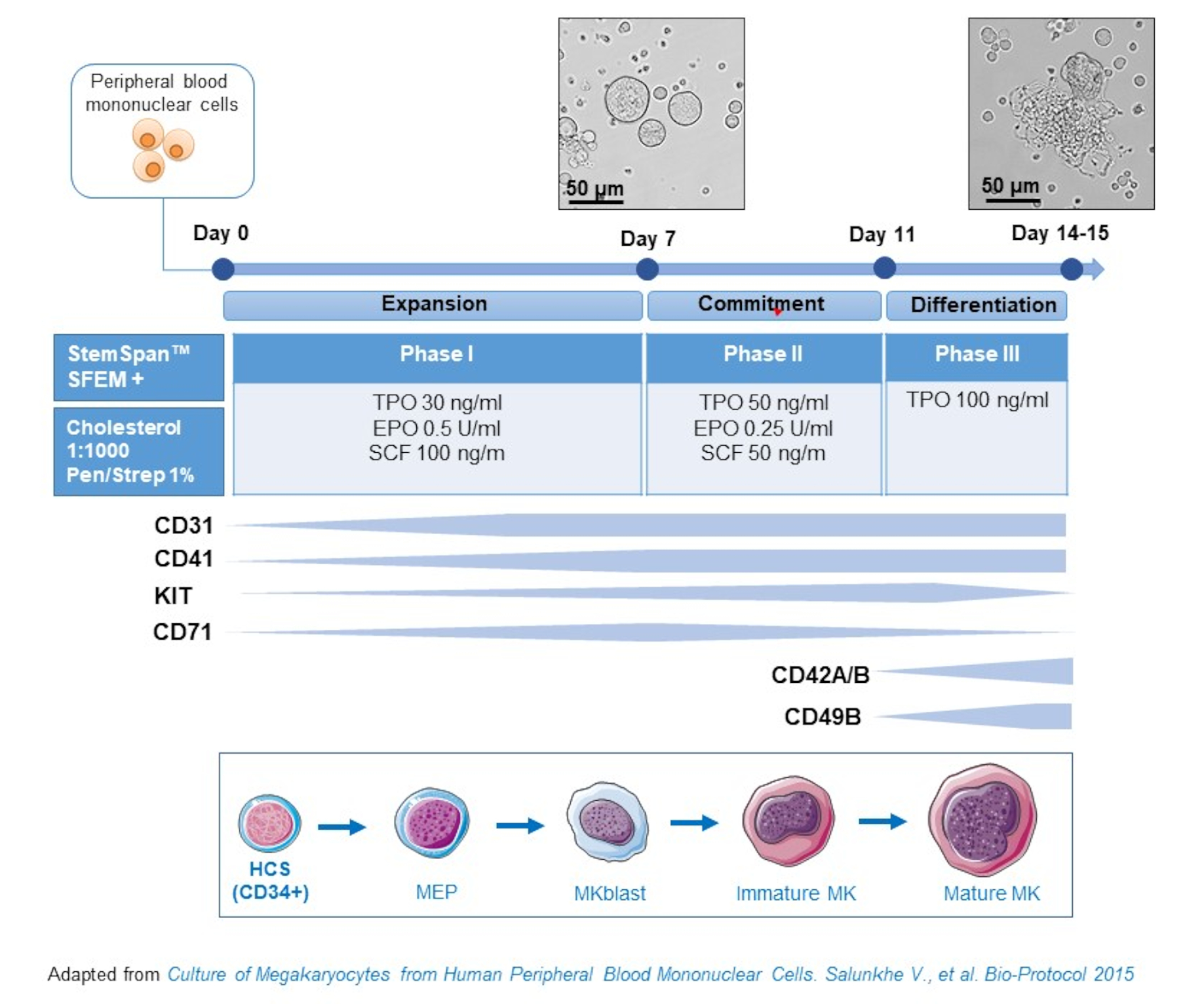{kind=link}
- Отбор проб: Промывайте клетки путем применения низкоскоростного центрифугирования в течение 5 мин (95 х г) и повторно суспендировать их в PBS или PBS, содержащем 1% бытового сывороточного альбумина (BSA). Для иммунофенотипирования идеальная плотность составляет 105-106 клеток/100 мкл (см. пункт 3.1). В зависимости от состояния культур(т.е. наличия мертвых клеток, мусора и т.д.), может потребоваться 1 или 2 промывки. Собирайте свои клетки в моменты интереса во время культивирования.
3. Иммунофенотипирование дифференцировки МК - инкубация с панелью метки-антител
- Инкубируют образцы клеток с помощью панели помеченных антител, следуя стандартным процедурам, обращая внимание на центрифугу на низкой скорости (95 х г) при обработке мегакариоцитов. Обычно мы инкубировали образцы с 1% BSA в PBS в объемах 100 мкл при 4 °C, 20 мин, с диапазоном 105-106 клеток.
- При необходимости масштабируйте масштаб.
- После инкубации добавляют 5 мл 1% BSA в PBS, центрифугу на низкой скорости (95 x g), аспирируют супернатант и повторно суспендируют образец в 2% BSA в PBS для сохранения жизнеспособности MK (2 мл). Добавьте 1-5 мМ ЭДТА для разрушения клеточно-клеточных агрегатов (которые естественным образом наблюдаются в культурах МК).
- Передавайте образцы в трубку с круглым дном размером 12 x 75 мм (трубку FACS) или пластину, сохраняя их в темноте до анализа проточной цитометрии или сортировки клеток.
- Получение одиночных антител и панели антител смесей; настройка проточного цитометра:
- Титруют антитела перед их применением для определения оптимальной концентрации в панелях антител. Оптимальная концентрация антител — это самая низкая концентрация, которая отделяет явно положительные клетки от отрицательных (и позволяет различать промежуточные уровни экспрессии). Например, большинство антител используются в разведении 1:200 (запас 100 мкг/мл), если иное не титруется или не указывается производителем.
- Как только титрование антител определено, приготовьте 10-кратное разведение каждого антитела. Эти разбавления используются для одноцветных элементов управления и для приготовления панельных смесей. Разбавления и панельные смеси подходят для использования даже через месяц после приготовления (хранятся при температуре 4 °C, если только показания производителя не исключают эти условия хранения). Это позволяет окрашивать образцы одной и той же панелью в течение определенного периода времени.
- Используйте 10 мкл на 100 мкл 10-кратного разбавления как для одноцветных элементов управления, так и для панельной смеси.
- Для одноцветных контрольных элементов используют шарики сродства антител, которые можно непосредственно измерить после добавления антитела. Одноцветные элементы управления должны измеряться с каждым экспериментом, чтобы обеспечить правильную регулировку компенсации (и тонкую настройку после измерения с помощью программного обеспечения для анализа).
- Кроме того, можно выполнить одноцветные элементы управления с образцами ячеек. Однако шарики позволяют быстро измерять заданное количество событий, которые, в зависимости от маркера антитела/поверхности, могут быть невозможны для получения на сложных первичных или культивированных клеточных источниках. Мы также советуем запускать образцы клеток, окрашенные панельными смесями «Флуоресценция минус один» (FMO), чтобы настроить соответствующие настройки компенсации (перед запуском экспериментов). Это актуально как для тщательного выявления проблем компенсации, и особенно, в культивированных МК, для выявления аутофлуоресцентной интерференции (которая будет присутствовать при использовании культурально-питательной среды, содержащей фенол красный).
- Подготовьте достаточный объем панельной смеси в зависимости от количества образцов, содержащих антитела проектной панели. Большинство наших панелей включают шесть антител (6-цветные панели, см. Таблицы 1-2).
- Для этих панелей используют 488-нм и 633-нм лазеры проточных цитометров, однако панели могут быть адаптированы к другим техническим сценариям. Кроме того, компенсационные соображения могут быть устранены при использовании проточной цитометрии на основе масс-спектрометрии или цитометров с технологией акустической фокусировки.
- Красители для измерения жизнеспособности могут давать ложную информацию о МК, особенно когда они созревают. МК очень активны, поднявая клетки, и позитивность с Hoechst, 7-AAD или PE, не всегда может отражать фактическую гибель клеток. Альтернативой (если требуется измерение гибели клеток) может быть использование пятен митохондрий (CMX Ros) или аминных реактивных красителей (красители Zombie или Ghost).
Таблица 1: Примечания к маркерам клеточной поверхности линии мегакариоцитов Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
Таблица 2: Панели антител Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
4. Анализ плоидности в сочетании с 6-цветными панелями
- Для анализа плоидности в сочетании с 6-цветной панелью антител приступают к фиксации и пермеабилизации клеток после инкубации с панелью антител. Эта стратегия позволит сохранить поверхностное маркерное окрашивание, при этом позволяя окрашивать ДНК клеток. Мы используем Hoechst 33342 для окрашивания ДНК, так как его можно визуализировать с помощью доступного фиолетового 405-нм лазера.
- На 105-106 клеток, после инкубации с антителом, центрифужные клетки (95 х г в течение 5 мин) повторно суспендируют в 200 мкл буфера фиксации и инкубируют 10 мин при комнатной температуре (РТ).
- Центрифужные клетки, как указано выше, повторно суспендируют во второй раз в буфере фиксации 200 мкл и инкубируют еще 10 мин при РТ.
- Готовят буфер пермеабилизации, содержащий 0,1% тритона X-100, 200 мг/мл РНКазы и 20 мг/мл Hoechst 33342 (пермеабилизация Hoechst MIX).
- Центрифужные клетки, как указано выше, повторно суспендируют их в 300 мкл пермеабилизации Hoechst MIX и инкубируют 30 мин при 37 °C. Этот шаг очень важен, так как, чтобы получить чистое измерение плоидности, РНК должна быть деградирована.
- По истечении инкубационного времени измеряйте образцы непосредственно с помощью проточная цитометра. В противном случае держите образцы при 4 °C, в темноте. Своевременно измеряйте их. Однако, поскольку эти образцы фиксированы, измерение может затянуться даже на 24-48 часов. Убедитесь, что образец тщательно щелкает перед измерением или пропускается через клеточный ситечко, чтобы обеспечить одноклеточную суспензию.
- Морфометрические параметры, такие как прямое и боковое рассеяние, не поддерживаются после фиксации клеток. Диаграмма рассеяния вперед/сбоку покажет сокращение распределения ячеек после фиксации. Тем не менее, окрашивание поверхностных маркеров в основном сохраняется, а стратегия решетки практически не изменяется, что позволяет анализировать статус плоидности на разных этапах дифференциации, определяемых комбинациями поверхностных маркеров.
5. Анализ дифференциации МК
ПРИМЕЧАНИЕ: Мы видели, что комбинация CD31/CD71 позволяет установить ряд затворов, которые соответствуют различным стадиям дифференциации МК. Дальнейшая обратное заделывание маркерами, специфичными для МК, позволяет разделять зрелых и незрелых МК. Кроме того, в свежих образцах обратный контроль для проверки наличия других используемых маркеров или для размещения популяций в осях прямого/бокового рассеяния уточняет оценку стадий дифференцировки МК и позволяет отбрасывать другие типы клеток, которые могут присутствовать в тех же популяциях.
- Используйте панель антител, которая включает ранние маркеры-предшественники (KIT, CD34), общие маркеры-предшественники (CD31, CD71) и маркеры родословной, некоторые из которых специфичны (CD42A / CD42B, CD49B, CD41 / CD61, CLEC2, GPVI и т. Д.) (см. Таблицы 1-2). Использование коктейля Lineage (Lin) (CD3, CD14, CD16, CD19, CD20 и CD56), также позволяет «отфильтровать» зрелые кроветворные клетки, которые могут добавить шум в анализ (при выборе Lin- популяции). В качестве примера мы рассмотрим анализ МК в НБМК, костном мозге и через клеточные культуры, полученные из ПБМК, в репрезентативных результатах.
6. Сортировка клеток прекурсоров МК и МК
ПРИМЕЧАНИЕ: Окрашенные ячейки были проанализированы и отсортированы на флуоресцентно-активированном сортировщике клеток FACS Aria IIu, оснащенном стандартными твердотельными лазерами 488 и 633 нм с использованием программного обеспечения FACSDiva; данные были дополнительно проанализированы и представлены с использованием программного обеспечения FlowJo и Cytobank (анализ viSNE). Чистота отсортированных фракций подтверждена проточным цитометрическим анализом каждой из отсортированных фракций (чистота выше 85%).
- Выполните сортировку клеток как можно скорее или в течение 1 часа после инкубации антител, чтобы избежать ухудшения состояния клеток.
- Фильтруйте образец с помощью клеточного ситечка 100 мкм, чтобы обеспечить одноэлектную суспензию и целостность большого МК.
- Используйте керамическое сопло 130 мкм, давление в оболочке, установленное на 11 фунтов на квадратный дюйм (PSI), и частоту капельного привода, установленную на 12 кГц, чтобы разбить поток на капли.
- Перед сортировкой стерилизуйте сопло, оболочку и линии для отбора проб, выполнив 30-минутное приобретение пенициллином / стрептомицином, разбавленным 1: 5 в стерильной воде, с последующим 10-минутным приобретением стерильной водой для удаления оставшегося дезактиванта.
- Как только поток стабилизируется, отрегулируйте задержку падения с рекомендуемыми шариками, чтобы отсортировать в режиме тонкой настройки более 97,5% отраженных капель со скоростью потока 400-1200 событий в секунду.
- Подготовьте сборные пробирки FACS с 500 мкл 2% BSA в PBS. Процент BSA может быть увеличен до 5-10%.
- Сгенерируйте шаблон эксперимента с соответствующими параметрами матрицы компенсации.
- Загрузите трубку FACS в цитометр.
- Выполните измерение образца, чтобы установить желаемые ворота и чистоту целевых клеточных популяций. Поддерживайте активированную запись, чтобы отображать до 200 000 событий в выбранных шлюзах популяции во время сортировки ячеек.
- FACS Aria IIu позволяет разделять до 4 различных клеточных популяций одновременно. Создайте новую схему сортировки и выберите устройство сбора (4 трубки) и соответствующий режим точности (рекомендуется промежуточная маска чистоты и восстановления). Наконец, добавьте интересующих население в каждое поле расположения сортировки(рисунок 3).

Рисунок 3:Схематическое представление принципа флуоресцентно-активированной сортировки клеток (FACS). Частицы проходят через 130 мкм-сопло и вынуждены распадаться на поток правильных капель из-за приложения вибрации к соплу. Затем капли опрашиваются лазером (точкой анализа), и сигналы обрабатываются, чтобы дать «решение о сортировке», применяя заряд к этим каплям. Когда капля заряда проходит через высоковольтное электростатическое поле (детектионную пластину), она отклоняется и собирается в соответствующую коллекторную трубку. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
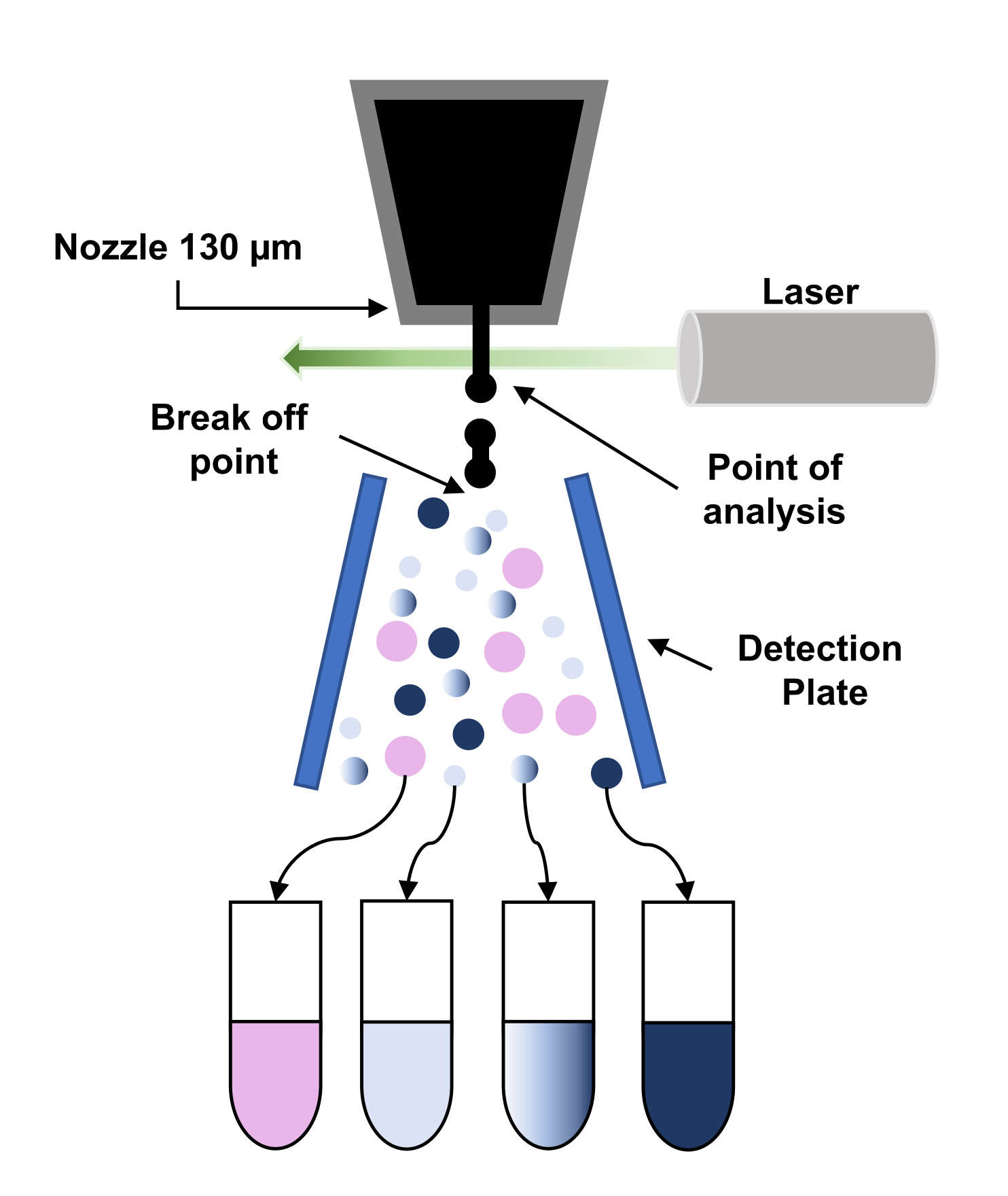{kind=link}
- Загрузите коллекционные трубки и начните сортировать целевые популяции.
- Центрифугируют сборные трубки в течение 5 минут при 95 х г и повторно суспендируют ячейку гранулы в соответствующем объеме 2% BSA в PBS.
- Измерьте еще раз долю каждого отсортированного образца, чтобы рассчитать чистоту.
- Храните ячейки соответствующим образом для дальнейшего использования. Отсортированные клетки могут быть использованы для цитологического и молекулярного анализа или могут быть повторно культивированы с целью изучения процесса дифференцировки выбранной клеточной популяции.
7. Послесортировочная пробоподготовка
- Подготовка цитоспинов к цитологическом анализу с помощью цитоцентрифуги
- Доведите отсортированные ячейки до удобного в обращении рабочего объема 100-200 мкл. Примите во внимание, что плотность клеток будет зависеть от отсортированного урожая популяции в каждом конкретном случае.
- Поместите чистый слайд на металлический держатель и поместите верхнюю часть фильтра. Не забудьте пометить слайд и фильтр, чтобы избежать смешивания образцов.
- Добавьте 100 мкл PBS на отверстие фильтра против затвора, чтобы фильтр увлажнял на ободке отверстия.
- Поместите воронку, закройте металлический держатель и поместите его на свое место в центрифуге.
- Добавьте образец (100-200 мкл) внутрь воронки.
- Центрифуга при 36 х г в течение 5 минут. Цитоспиновые слайды могут высохнуть на воздухе при RT (должным образом покрыты для предотвращения пыли) и могут храниться в RT в течение 1 недели до иммуноокрашивания или гистохимии.
- Для иммуноокрашивания
- Зафиксируйте слайды в 2% параформальдегиде (PFA), разведенном в PBS, и инкубируйте в течение 5 мин.
- Может использоваться диапазон 0,5-4% PFA в PBS. В наших руках мы используем 4% для получения правильной фиксации тканей или некоторых типов клеток, а 0,5% PFA в PBS достаточно для тромбоцитов. При настройке этого метода правильный процент PFA требует оптимизации для каждого типа ячейки / источника.
- Инкубировать 5 мин в PBS.
- Инкубировать 5 мин в 50% этаноле (EtOH).
- Инкубировать 5 мин в 70% EtOH.
- Хранить в 70% EtOH при -20ºC.
- При выполнении иммуноокрашивания регидратируют и соблюдают стандартные процедуры (пермеабилизация, промывка, блокирование, первичная и вторичная инкубация антител, консервирование и т.д.).
- Зафиксируйте слайды в 2% параформальдегиде (PFA), разведенном в PBS, и инкубируйте в течение 5 мин.
- Для цитохимии:
ПРИМЕЧАНИЕ: Слайды могут быть окрашены окрашиванием May-Grünwald Giemsa или удобным окрашиванием для каждой цели. - Для немедленного исследования морфологии
- Добавьте одну каплю монтажной среды на клеточно-содержащее место цитоспина и поместите крышку.
- Держите слайды при 4 °C не дольше недели, если они не герметизированы, что позволяет длительно хранить даже при RT. Монтажная фиксация среды позволяет длительно хранить в RT.
Access restricted. Please log in or start a trial to view this content.
Результаты
Костный мозг и плоидия
Нарисунке 4показан репрезентативный иммунофенотипический анализ мегакариопоэтиса в БМ-образцах (аспирация) пациентов. При построении клеточной фракции против CD71 и CD31 мы определили шесть основных популяций: CD31- CD71- ...
Access restricted. Please log in or start a trial to view this content.
Обсуждение
Большинство исследований, сосредоточенных на изучении мегакариопоэзиса с помощью проточной цитометрии, на сегодняшний день ограничены идентификацией подмножеств МК с использованием только специфических для линии поверхностных маркеров(т.е.CD42A / CD42B, CD41 / CD61), в то время как более р...
Access restricted. Please log in or start a trial to view this content.
Раскрытие информации
Производство аудиовизуальных материалов поддерживалось BD Biosciences.
Благодарности
Мы благодарим Маркоса Переса Бастерречеа, Лорену Родригес Лоренцо и Бегонью Гарсию Мендес (HUCA) и Палому Сересо, Альмудену Пайеро и Марию де ла Поведа-Коломо (HCSC) за техническую поддержку. Эта работа была частично поддержана медицинскими грантами (Roche SP200221001) для A.B., стипендии RYC (RYC-2013-12587; Ministerio de Economía y Competitividad, Испания) и грант I+D 2017 (SAF2017-85489-P; Ministerio de Ciencia, Innovación y Universidades, Испания и Fondos FEDER) Л.Г., грант Северо Очоа (PA-20-PF-BP19-014; Consejería de Ciencia, Innovación y Universidades del Principado de Asturias, Spain) до P.M.-B. и очный постдокторский грант 2018 года (Fundación para la Investigación y la Innovación Biosanitaria de Asturias - FINBA, Oviedo, Spain) для A.A.-H. Мы благодарим Рейнира ван дер Линдена за то, что он поделился своими знаниями (и временем), особенно за его мудрые советы по многоцветной смеси антител с меткими антителами и подготовке к контролю над одноцветными шариками.
Access restricted. Please log in or start a trial to view this content.
Материалы
| Name | Company | Catalog Number | Comments |
| 130 micron Nozzle | BD | 643943 | required for MK sorting |
| 5810R Centrifuge | Eppendorf | Cell isolation and washes | |
| A-4-62 Swing Bucket Rotor | Eppendorf | Cell isolation and washes | |
| Aerospray Pro Hematology Slide Stainer / Cytocentrifuge | ELITech Group | Automatized cytology devise, where slides are stained with Mat-Grünwald Giemsa | |
| CO2 Incubator Galaxy 170 S | Eppendorf | Cell Incubation | |
| Cytospin 4 Cytocentrifuge | Thermo Scientific | To prepare cytospins | |
| FACSAria IIu sorter | BD | Lasers 488-nm and 633-nm | |
| FACSCanto II flow cytometer | BD | Lasers 488-nm , 633-nm and 405-nm | |
| Olympus Microscope BX 41 | Olympus | Microphotographs | |
| Olympus Microscope BX 61 | Olympus | Microphotographs | |
| Zoe Fluorescent Cell Imager | BioRad | Microphotographs | |
| To obtain PBMCs | |||
| Lipids Cholesterol Rich from adult bovine serum | Sigma-Aldrich | L4646 | or similar |
| Lymphoprep | Stem Cell Technologies | #07801 | or similar |
| Penicillin-Streptomycin | Sigma-Aldrich | P4333 | or similar |
| Recombinant human Erythropoietin (EPO) | R&D Systems | 287-TC-500 | or similar |
| Recombinant human stem cell factor (SCF) | Thermo Fisher Scientific, Gibco™ | PHC2115 | or similar |
| Recombinant human thrombopoietin (TPO) | Thermo Fisher Scientific, Gibco™ | PHC9514 | or TPO receptor agonists |
| StemSpan SFEM | Stem Cell Technologies | #09650 | |
| Flow Cytometry Analyses | |||
| Bovine Serum Albumin | Merck | A7906-100G | or similar |
| BD CompBead Anti-Mouse Ig, κ/Negative Control Compensation Particles Set | BD | 552843 | Antibodies for human cells are generally from mouse. |
| BD Cytofix/Cytoperm | BD | 554714 | or similar |
| BD FACS Accudrop Beads | BD | 345249 | |
| CD31 AF-647 | BD | 561654 | Mouse anti-human |
| CD31 FITC | Immunostep | 31F-100T | |
| CD34 FITC | BD | 555821 | Mouse anti-human |
| CD41 PE | BD | 555467 | Mouse anti-human |
| CD41 PerCP-Cy5.5 | BD | 333148 | Mouse anti-human |
| CD42A APC | Immunostep | 42AA-100T | We observed unspecific binding... that needs to be assessed |
| CD42A PE | BD | 558819 | Mouse anti-human |
| CD42B PerCP | Biolegend | 303910 | Mouse anti-human |
| CD49B PE | BD | 555669 | Mouse anti-human |
| CD61 FITC | BD | 555753 | Mouse anti-human |
| CD71 APC-Cy7 | Biolegend | 334109 | Mouse anti-human |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | |
| Human BD Fc Block | BD | 564219 | Fc blocking - control |
| KIT PE-Cy7 | Biolegend | 313212 | Mouse anti-human |
| Lineage Cocktail 2 FITC | BD | 643397 | Mouse anti-human |
| RNAse | Merck | R6513 | or similar |
| Triton X-500 | Merck | 93443-500ML | or similar |
| Cell strainers for sorting | |||
| CellTrics Filters 100 micrometers | Sysmex | 04-004-2328 | Cell strainers |
| Note: we do not specify general reagents/chemicals (PBS, EDTA, etc) or disposables (tubes, etc), or reagents specified in previous published and standard protocols - unless otherwise specified. |
Ссылки
- Italiano, J. E. Unraveling Mechanisms That Control Platelet Production. Semin Thrombosis And Haemostasis. 39 (1), 15-24 (2013).
- Machlus, K. R., Italiano, J. E. The Incredible Journey: From Megakaryocyte Development To Platelet Formation. Journal Of Cell Biology. 201 (6), 785-796 (2013).
- Eckly, A., et al. Biogenesis Of The Demarcation Membrane System (DMS) In Megakaryocytes. Blood. 123 (6), 921-930 (2014).
- Couldwell, G., Machlus, K. R. Modulation Of Megakaryopoiesis And Platelet Production During Inflammation. Thrombosis Research. 179, 114-120 (2019).
- Kosaki, G. In Vivo Platelet Production From Mature Megakaryocytes: Does Platelet Release Occur Via Proplatelets. International Journal Of Hematology. 81 (3), 208-219 (2005).
- Lefrancais, E., Looney, M. R. Platelet Biogenesis In The Lung Circulation. Physiology (Bethesda). 34 (6), 392-401 (2019).
- Nieswandt, B., Stritt, S. Megakaryocyte Rupture For Acute Platelet Needs. Journal Of Cell Biology. 209 (3), 327-328 (2015).
- Nishimura, S., et al. IL-1alpha Induces Thrombopoiesis Through Megakaryocyte Rupture In Response To Acute Platelet Needs. Journal Of Cell Biology. 209 (3), 453-466 (2015).
- Sanjuan-Pla, A., et al. Platelet-Biased Stem Cells Reside At The Apex Of The Haematopoietic Stem-Cell Hierarchy. Nature. 502 (7470), 232-236 (2013).
- Notta, F., et al. Distinct Routes Of Lineage Development Reshape The Human Blood Hierarchy Across Ontogeny. Science. 351 (6269), 2116(2016).
- Yamamoto, R., et al. Clonal Analysis Unveils Self-Renewing Lineage-Restricted Progenitors Generated Directly From Hematopoietic Stem Cells. Cell. 154 (5), 1112-1126 (2013).
- Wang, H., et al. Decoding Human Megakaryocyte Development. Cell Stem Cell. , (2020).
- Meinders, M., et al. Repercussion Of Megakaryocyte-Specific Gata1 Loss On Megakaryopoiesis And The Hematopoietic Precursor Compartment. Plos One. 11 (5), 0154342(2016).
- Meinders, M., et al. Sp1/Sp3 Transcription Factors Regulate Hallmarks Of Megakaryocyte Maturation And Platelet Formation And Function. Blood. 125 (12), 1957-1967 (2015).
- Salunkhe, V. P., Gutiérrez, L. Culture Of Megakaryocytes From Human Peripheral Blood Mononuclear Cells. Bio-Protocol. 5 (21), 1639(2015).
- Choudry, F. A., et al. Transcriptional Characterization Of Human Megakaryocyte Polyploidization And Lineage Commitment. Journal Of Thrombosis And Haemostasis. , 15271(2021).
- Heazlewood, S. Y., Williams, B., Storan, M. J., Nilsson, S. K. The Prospective Isolation Of Viable, High Ploidy Megakaryocytes From Adult Murine Bone Marrow By Fluorescence Activated Cell Sorting. Methods In Molecular Biology. 1035, 121-133 (2013).
- Tomer, A., Harker, L. A., Burstein, S. A. Purification Of Human Megakaryocytes By Fluorescence-Activated Cell Sorting. Blood. 70 (6), 1735-1742 (1987).
- Martinez-Botia, P., Acebes-Huerta, A., Seghatchian, J., Gutierrez, L. On The Quest For In Vitro Platelet Production By Re-Tailoring The Concepts Of Megakaryocyte Differentiation. Medicina. 56 (12), Kaunas. (2020).
- Martinez-Botia, P., Acebes-Huerta, A., Seghatchian, J., Gutierrez, L. In Vitro Platelet Production For Transfusion Purposes: Where Are We Now. Transfusion And Apheresis Science. 59 (4), 102864(2020).
- Butov, K. R., et al. In Vitro Megakaryocyte Culture From Human Bone Marrow Aspirates As A Research And Diagnostic Tool. Platelets. , 1-8 (2020).
- Di Buduo, C. A., et al. A Gold Standard Protocol For Human Megakaryocyte Culture Based On The Analysis Of 1,500 Umbilical Cord Blood Samples. Thrombosis And Haemostasis. , (2020).
Access restricted. Please log in or start a trial to view this content.
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены