
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Immunofenotipizzazione e smistamento cellulare di MK umani da fonti primarie umane o differenziati in vitro da progenitori ematopoietici
In questo articolo
Riepilogo
Qui presentiamo una strategia di immunofenotipizzazione per la caratterizzazione della differenziazione dei megacariociti, e mostriamo come tale strategia consente lo smistamento dei megacariociti in diverse fasi con uno smistatore di cellule attivato dalla fluorescenza. La metodologia può essere applicata ai tessuti primari umani, ma anche ai megacariociti generati in coltura in vitro.
Abstract
La differenziazione dei megacariociti (MK) comprende un certo numero di cicli endomitotici che si traducono in una cellula altamente poliploide (raggiungendo anche >64N) ed estremamente grande (40-60 μm). A differenza della conoscenza in rapida crescita della megacariopoiesi a livello cellulare e molecolare, la caratterizzazione della megacariopoiesi per citometria a flusso è limitata all'identificazione di MK maturi usando marcatori di superficie specifici del lignaggio, mentre i precedenti stadi di differenziazione MK rimangono inesplorate. Qui presentiamo una strategia di immunofenotipizzazione che consente l'identificazione di fasi successive di differenziazione MK, con crescente stato di ploidia, in fonti primarie umane o colture in vitro con un pannello che integra marcatori superficiali specifici e non specifici di MK. Nonostante le sue dimensioni e fragilità, gli MK possono essere immunofenotipati utilizzando il pannello sopra menzionato e arricchiti da smistamento cellulare attivato dalla fluorescenza in specifiche condizioni di pressione e diametro dell'ugello. Questo approccio facilita gli studi multi-omici, con l'obiettivo di comprendere meglio la complessità della megacariopoiesi e della produzione piastrinica nell'uomo. Una migliore caratterizzazione della megacariopoiesi può essere fondamentale nella diagnosi o nella prognosi di patologie legate al lignaggio e malignità.
Introduzione
I megacariociti (MK) si sviluppano da cellule staminali ematopoietiche (HSC) a seguito di un processo complesso chiamato megacariopoiesi, che è orchestrato principalmente dall'ormone trombopoietina (TPO). La visione classica della megacariopoiesi descrive il viaggio cellulare dagli HSC attraverso una successione di fasi gerarchiche di progenitori impegnati e cellule precursori, portando infine a un MK maturo. Durante la maturazione, gli MK sperimentano più cicli di endomitosi, sviluppano un intricato sistema di membrana di demarcazione intracellulare (DMS), che fornisce una superficie di membrana sufficiente per la produzione di piastrine, e producono e confezionano in modo efficiente la pletora di fattori contenuti nei diversi granuli ereditati dalle piastrine mature1,2,3. Di conseguenza, gli MK maturi sono celle di grandi dimensioni (40-60 μm) caratterizzate da un nucleo altamente poliploide (che raggiunge anche >64N). Studi recenti suggeriscono percorsi alternativi attraverso i quali gli HSC si differenziano in MK bypassando i tradizionali checkpoint di impegno del lignaggio in risposta a determinate condizioni fisiopatologica4,5,6,7,8,9,10,11. Questi risultati evidenziano che la differenziazione ematopoietica verso l'MK maturo è un processo continuo e adattivo che risponde alle esigenze biologiche.
Con la crescente conoscenza della biologia cellulare e degli aspetti molecolari che caratterizzano la megacariopoiesi12, la maggior parte della ricerca dedicata allo studio del processo per citometria a flusso si limitaall'identificazione di MK maturi utilizzando marcatori di superficie specifici del lignaggio (cioè CD42A/B, CD41/CD61), mentre le precedenti fasi di differenziazione MK rimangono inesplorate. In precedenza abbiamo documentato una strategia per stadioe la megacariopoiesi nelle colture MK derivate dal midollo osseo e osseo13,14, che abbiamo adattato e applicato all'uomo15. Nel presente articolo mostriamo una strategia di immunofenotipizzazione che consente la caratterizzazione della megacariopoiesi, dagli HSC agli MK maturi, nelle fonti primarie umane (midollo osseo -BM- e sangue periferico -PB-) o nelle colture in vitro utilizzando un pannello che integra marcatori di superficie specifici e non specifici di MK (CD61, CD42B, CD49B, CD31, KIT e CD71, tra gli altri). Nonostante le sue grandi dimensioni e fragilità, gli MK possono essere immunofenotipati utilizzando i marcatori di superficie cellulare sopra menzionati e arricchiti dallo smistamento cellulare attivato dalla fluorescenza in specifiche condizioni di pressione e diametro dell'ugello per ridurre al minimo la rottura cellulare e / o i danni. Questa tecnica facilita gli approcci multi-omici, con l'obiettivo di comprendere meglio la complessità della megacariopoiesi e della produzione piastrinica nella salute e nelle malattie umane. Degno di nota, si porrà come uno strumento utile per aiutare la diagnosi e la prognosi in un contesto clinico di crescente domanda.
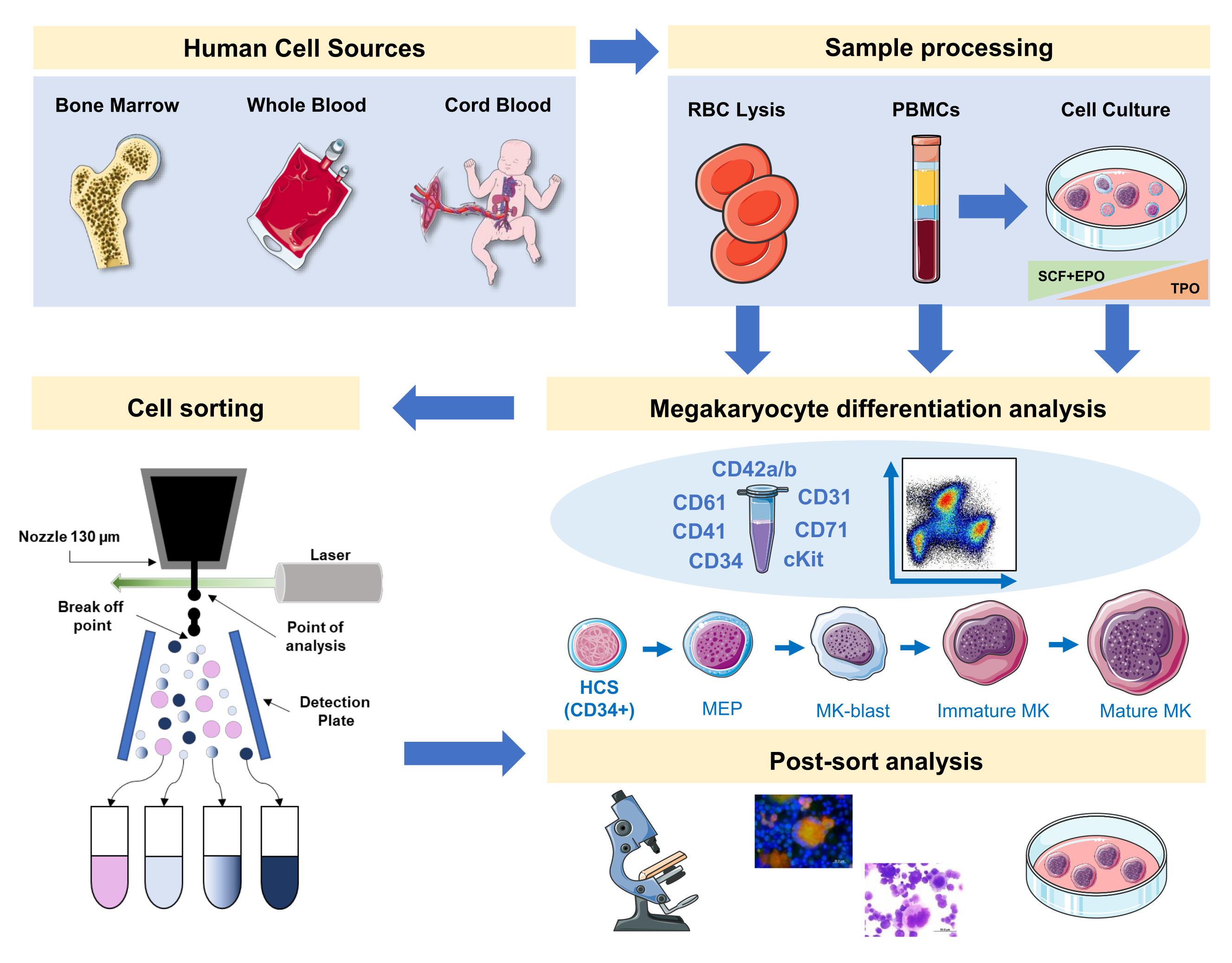
In questo manoscritto documentiamo una strategia per in scena la megacariopoiesi umana con un pannello che integra marcatori di superficie specifici e non specifici di MK da fonti primarie o generati in vitro. Inoltre, forniamo un protocollo per ordinare, con uno smistatore di celle attivato dalla fluorescenza, le frazioni preferite e gli MK maturi (Figura 1). Questo passaggio non è popolare, in quanto è tecnicamente difficile a causa delle grandi dimensioni e fragilità degli MK. Tuttavia, è stato impiegato sia in campioni di midollo osseo del topo che umano in precedenza, e a causa del progresso tecnologico, con unrisultato migliore ogni volta 16,17,18. Le fonti primarie umane in cui gli MK o i precursori MK possono essere studiati includono, tra gli altri, midollo osseo, sangue del cordone ombelicale e sangue periferico. È importante l'elaborazione corretta del campione per isolare la frazione cellulare pertinente per l'analisi su ciascun campione. Sono incorporate procedure standard, con alcune considerazioni da tenere in considerazione quando si mira allo studio della megacariopoiesi.
Protocollo
Campioni di sangue intero e di midollo osseo sono stati ottenuti e trattati conformemente alla dichiarazione di Helsinki del 1964. Campioni di sangue intero sono stati ottenuti da donatori sani dopo aver dato il consenso informato (ISPA), nell'ambito di uno studio approvato dal nostro comitato medico etico istituzionale (Hospital Universitario Central de Asturias -HUCA-). Campioni di midollo osseo sono stati ottenuti da materiale di scarto aspirato al midollo osseo di pazienti gestiti presso il Dipartimento di Ematologia dell'Ospedale Clínico San Carlos (HCSC).

Figura 1: Rappresentazione schematica del protocollo documentato in questo manoscritto. Sono indicate le fonti umane primarie o le colture primarie in cui la differenziazione MK può essere messa in scena usando l'immunofenotipizzazione. Questa strategia di immunofenotipizzazione può essere applicata allo studio del processo in diverse patologie correlate al lignaggio o malignità nelle fonti primarie. Inoltre, rende possibile lo smistamento cellulare di MK e precursori con uno smistatore di cellule attivato dalla fluorescenza, che consente un'ulteriore analisi delle frazioni arricchite. Le immagini utilizzate fanno parte di Servier Medical Art (SMART) di Servier e sono concesse in licenza ai sensi di CC BY 3.0. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
1. Trattamento del sangue intero e del midollo osseo prima dell'immunofenotipizzazione
- Quando si utilizza sangue intero (WB) dalle donazioni come fonte primaria, facoltativamente isolare il componente della cellula mononucleare del sangue periferico (PBMC). Questo può essere ottenuto utilizzando la centrifugazione differenziale standard combinata con la separazione cellulare gradiente di densità, come descritto inprecedenza 15.
- In breve, centrifugare il sangue a 193 x g per 15 min (freno 3) a temperatura ambiente. Scartare la frazione plasma superiore e raccogliere l'anello buffy. Diluire 1:1 con tampone fosfato saline (PBS)/Trisodium Citrato Diidrato (38 g/L, pH 7) tampone e pipetta con cura un volume di 25 mL sopra 15 mL di una soluzione gradiente di densità (1,076 g/mL) in tubi da 50 mL.
- Centrifuga per 20 min a 1114 x g (acceleratore 3, freno 3, temperatura ambiente). Scartare la frazione plasmatica e raccogliere l'anello di buffy contenente PBFC. Lavare aggiungendo lo stesso volume di PBS, centrifugare a 435 x g per 5 minuti e rimorsi in PBS per un ulteriore utilizzo.
- In alternativa, utilizzare un campione WB (circa 100 μL) per l'immunofenotipizzazione dopo aver lisciviato i globuli rossi (RBC) e un lavaggio accurato.
- In breve, diluire 1:1 nel tampone di llysing RBC ghiacciato (4,15 g di NH4Cl, 0,5 g di KHCO3 e 18,5 mg di EDTA (triplex III) a 500 mL di H2O, pH 7,1-7,4). Attendere che la sospensione cellulare diventi rosso traslucido (3-5 min).
- Centrifugare a 435 x g per 5 min, a 4 °C, e rimosoppo le cellule in PBS. Ripetere la procedura tutte le volte che è necessario per ottenere un pellet di cellule bianche.
- Analogamente, elaborare direttamente i campioni ottenuti dal midollo osseo (aspirazione) con tampone di llysing RBC (cfr. punto 1.2) e lavaggio accurato, come iniziare con una sospensione a cella singola chiara(figura 1).
- Evitare l'uso di vortici per mescolare i campioni durante la lavorazione, in quanto potrebbe danneggiare i fragili MK. Mescolare facendo scorrere o invertendo il tubo.
NOTA: Il gradiente di densità per ottenere PBBC può comportare una frazione cellulare più ricca e pulita rispetto al WB con litoro RBC. Tuttavia, dovremmo tenere presente che gli MK maturi ad alta densità potrebbero andare persi nella frazione "neutrofilo". Se ne discuterà nei risultati rappresentativi.
- Evitare l'uso di vortici per mescolare i campioni durante la lavorazione, in quanto potrebbe danneggiare i fragili MK. Mescolare facendo scorrere o invertendo il tubo.
2. Differenziazione in vitro di MK dai PBMC
NOTA: Gli MK possono essere differenziati in vitro dai precedenti precursori, come le cellule CD34+, presenti in diverse fonti primarie(ad esempio, WB/PBBC, sangue del cordone ombelicale, midollo osseo) e da iPSC. A tal fine sono stati applicati diversi protocolli. Qui utilizziamo un metodo di coltura da noi sviluppato che consente la differenziazione MK dai PBMC, senza la necessità di arricchire per CD34+ precursori15,19,20,21,22.
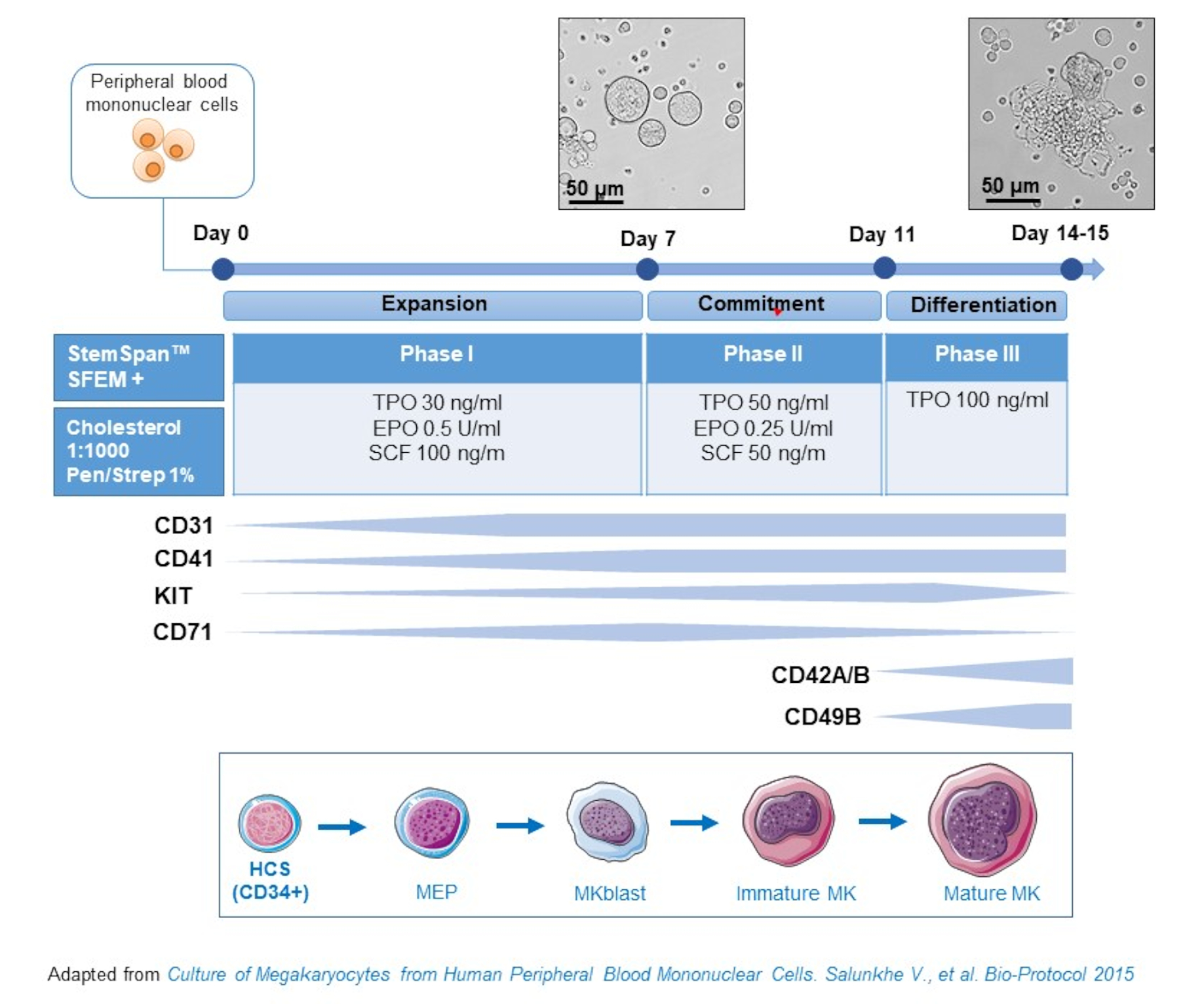
- Questo protocollo si compone di tre fasi di coltura, in cui la concentrazione di trombopoietina (TPO) aumenta gradualmente a scapito dei fattori di crescitache favoriscono la proliferazione di precursoriprecedenti ( ad esempio, SCF, EPO), che diminuiscono gradualmente(Figura 2)15.
- Per il mezzo di base, utilizzare StemSpan SFEM integrato con lo 0,4% di miscela lipidica ricca di colesterolo e l'1% di penicillina / streptomicina.
- Per il mezzo di fase I, utilizzare il mezzo di base integrato con SCF (100 ng/mL), eritropoietina (EPO, 0,5 U/mL) e trombopoietina (TPO, 30 ng/mL). Il mezzo di fase II è il mezzo di base integrato con SCF (50 ng/mL), EPO (0,25 U/mL) e TPO (50 ng/mL). Per il mezzo di fase III, utilizzare il mezzo di base integrato con TPO (100 ng/mL).
- Coltura PBPC nel mezzo di fase I. Al giorno 6-8, posizionare i PBBC nel mezzo di fase II e al giorno 9-12 posizionare i PBBC nel mezzo di fase III.
- Sostituire il mezzo centrifugando le cellule a 435 x g per 5 minuti a temperatura ambiente dalla fase I alla fase II, e a 95 x g per 5 minuti a temperatura ambiente dalla fase II alla fase III e rimotivandole in mezzo fresco.
- Cellule di coltura in un incubatore a 37 °C, 5% CO2. In queste culture primarie, la differenziazione MK dura 10-14 giorni e i campioni possono essere disegnati in diversi punti di tempo durante il periodo della coltura, in modo da seguire la differenziazione MK.
- Per il mezzo di base, utilizzare StemSpan SFEM integrato con lo 0,4% di miscela lipidica ricca di colesterolo e l'1% di penicillina / streptomicina.

Figura 2: Rappresentazione schematica del metodo delle impostazioni cultura MK derivato da PBMC. I PBMC di donatori sani sono stati coltivati secondo il protocollo in tre fasi da noi sviluppato per generare MK in vitro (schema adattato da Salunkhe et al. Vengono mostrate 15 foto scattate al giorno 10 e al giorno 13 della cultura. Le foto vengono scattate con un obiettivo 20X. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Raccolta del campione: Lavare le cellule applicando centrifugazione a bassa velocità per 5 min (95 x g) e rimornerne in PBS o PBS contenenti l'1% di albumina di siero bovino (BSA). Per l'immunofenotipizzazione, la densità ideale è di 105-106 cellule/100 μL (cfr. punto 3.1). A seconda dello stato dellecolture (cioè, presenza di cellule morte, detriti, ecc.), possono essere necessari 1 o 2 lavaggi. Raccogli le tue cellule nei punti di interesse durante la coltura.
3. Immunofenotipizzazione della differenziazione MK - incubazione con un pannello di anticorpi taggati
- Incubare i campioni cellulari con un pannello di anticorpi taggati seguendo procedure standard, prestando attenzione alla centrifuga a bassa velocità (95 x g) durante la lavorazione dei megacariociti. Normalmente incubamo i campioni con 1% di BSA in PBS in volumi di 100 μL a 4 °C, 20 min, con un intervallo di 105-106 cellule.
- Scalare quando necessario.
- Dopo l'incubazione, aggiungere 5 mL di 1% di BSA in PBS, centrifugare a bassa velocità (95 x g), aspirare il supernatante e rimescolare il campione in 2% BSA in PBS per preservare la vitalità MK (2 mL). Aggiungere 1-5 mM EDTA per interrompere gli aggregati cellulare-cella (che sono naturalmente visti nelle colture MK).
- Trasferire campioni in un tubo o in una piastra inferiore rotonda da 12 x 75 mm (tubo FACS), mantenendoli al buio fino all'analisi della citometria del flusso o allo smistamento delle celle.
- Preparazione dei singoli anticorpi e miscele di pannelli anticorpali; impostazione del citometro di flusso:
- Titolare gli anticorpi prima del loro uso per determinare la concentrazione ottimale nei pannelli anticorpali. La concentrazione ottimale di anticorpi è la concentrazione più bassa che separa chiaramente positivo dalle cellule negative (e consente la distinzione dei livelli intermedi di espressione). Ad esempio, la maggior parte degli anticorpi sono utilizzati in una diluizione 1:200 (stock 100 μg/mL), salvo diversa titolazione o indicazione da parte del produttore.
- Una volta determinata la titolazione dell'anticorpo, preparare una diluizione 10x di ogni anticorpo. Queste diluizioni vengono utilizzate per i controlli monocolore e per preparare le miscele di pannelli. Le diluizioni e le miscele di pannelli vanno bene anche un mese dopo la preparazione (conservate a 4 °C, a meno che le indicazioni del produttore non precludano queste condizioni di conservazione). Ciò consente la colorazione dei campioni con lo stesso pannello per un periodo di tempo.
- Utilizzare 10 μL per 100 μL della diluizione 10x sia per i controlli monocolore che per la miscela di pannelli.
- Per i controlli monocolore, utilizzare perline di affinità anticorpale, che possono essere misurate direttamente dopo aver aggiunto l'anticorpo. I controlli monocolore devono essere misurati ad ogni esperimento, per consentire una corretta regolazione della compensazione (e la messa a punto della post-misurazione con il software di analisi).
- In alternativa, eseguire i controlli a colori singolo con campioni di cella. Tuttavia, le perline consentono la misurazione rapida di un dato numero di eventi che, a seconda dell'anticorpo / marcatore di superficie, potrebbero non essere possibili ottenere su fonti cellulari primarie o coltivate complesse. Si consiglia inoltre di eseguire campioni di cellule macchiati con miscele di pannelli "Fluorescence Minus One" (FMO) per impostare le impostazioni di compensazione appropriate (prima di eseguire esperimenti). Ciò è rilevante per identificare attentamente i problemi di compensazione e, in particolare, negli MK coltivati, per identificare le interferenze di autofluorescenza (che saranno presenti se si utilizza un mezzo di coltura contenente rosso fenolo).
- Preparare un volume sufficiente della miscela di pannelli, a seconda del numero di campioni, contenente gli anticorpi del pannello progettato. La maggior parte dei nostri pannelli include sei anticorpi (pannelli a 6 colori, vedi tabelle 1-2).
- Per questi pannelli, utilizzare laser a 488 nm e 633 nm del citometro a flusso, tuttavia, i pannelli possono essere adattati ad altri scenari tecnici. Inoltre, le considerazioni di compensazione possono essere evitate quando si utilizza citometria di flusso basata sulla spettrometria di massa o citometri con tecnologia di messa a fuoco acustica.
- I coloranti per misurare la vitalità possono fornire informazioni false sugli MK, specialmente quando maturano. Gli MK sono cellule di ucchi molto attive e la positività con Hoechst, 7-AAD o PE, potrebbe non sempre riflettere la morte cellulare effettiva. Un'alternativa (se è richiesta la misurazione della morte cellulare) potrebbe essere l'uso di macchie di mitocondri (CMX Ros) o coloranti reattivi all'ammina (coloranti Zombie o Ghost).
Tabella 1: Note sui marcatori di superficie cellulare del lignaggio megacariocitico Fare clic qui per scaricare questa tabella.
Tabella 2: Pannelli anticorpali Clicca qui per scaricare questa tabella.
4. Analisi di Ploidy combinata con pannelli a 6 colori
- Per l'analisi della ploidia, in combinazione con un pannello anticorpale a 6 colori, procedere con la fissazione e la permeabilizzazione delle cellule dopo l'incubazione con il pannello anticorpale. Questa strategia consentirà la conservazione della colorazione del marcatore superficiale, consentendo al contempo la colorazione del DNA delle cellule. Usiamo Hoechst 33342 per macchiare il DNA, in quanto può essere visualizzato con il laser viola 405 nm disponibile.
- Per 105-106 cellule, dopo l'incubazione con il pannello anticorpale, le cellule centrifughe (95 x g per 5 minuti), risosopende in 200 μL di tampone di fissazione e incubano 10 minuti a temperatura ambiente (RT).
- Cellule di centrifuga come indicato sopra, risortizzate per la seconda volta in tampone di fissazione di 200 μL e incubare altri 10 minuti a RT.
- Preparare tampone di permeabilizzazione, contenente 0,1% Tritone X-100, 200 mg/mL RNasi e 20 mg/mL Hoechst 33342 (permeabilizzazione Hoechst MIX).
- Centrifugare le cellule come sopra, sosostierle in 300 μL della permeabilizzazione Hoechst MIX e incubare 30 min a 37 °C. Questo passaggio è molto importante, poiché, per ottenere una misurazione pulita della ploidia, l'RNA deve essere degradato.
- Dopo il tempo di incubazione, misurare i campioni direttamente con un citometro a flusso. Altrimenti, tenere i campioni a 4 °C, al buio. Misurali prontamente. Tuttavia, poiché questi campioni sono fissi, la misurazione può essere ritardata anche di 24-48 ore. Assicurarsi che il campione venga sfogliato accuratamente prima della misurazione o passato attraverso un filtro cellulare, per garantire una sospensione a singola cella.
- I parametri morfometrici come Avanti e Dispersione laterale non vengono mantenuti dopo la fissazione delle celle. Il plottaggio Dispersione avanti/laterale mostrerà un restringimento della distribuzione della cella dopo la fissazione. Tuttavia, la colorazione del marcatore di superficie è per lo più preservata, e la strategia di gating è appena alterata, consentendo l'analisi dello stato di ploidia nelle diverse fasi di differenziazione definite dalle combinazioni di marcatori di superficie.
5. Analisi della differenziazione MK
NOTA: Abbiamo visto che la combinazione di CD31/CD71 permette di impostare un certo numero di porte che corrispondono a diversi stadi di differenziazione MK. Un ulteriore back-gating con marcatori specifici di MK consente la separazione di MK maturi e immaturi. Inoltre, nei campioni freschi, il back-gating per verificare la presenza di altri marcatori utilizzati, o per posizionare le popolazioni negli assi Disperso Avanti/Laterale, affina la valutazione delle fasi di differenziazione MK e consente di scartare altri tipi di cellule che potrebbero essere presenti sulle stesse popolazioni.
- Utilizzare un pannello di anticorpi che includa marcatori precursori precoci (KIT, CD34), marcatori precursori comuni (CD31, CD71) e marcatori di lignaggio, alcuni dei quali specifici (CD42A/CD42B, CD49B, CD41/CD61, CLEC2, GPVI, ecc.) (cfr. tabelle 1-2). L'uso del cocktail Lineage (Lin) (CD3, CD14, CD16, CD19, CD20 e CD56), consente anche di "filtrare" le cellule ematopoietiche mature che potrebbero aggiungere rumore all'analisi (quando si seleziona la Lin- popolazione). Ad esempio, passeremo attraverso l'analisi degli MK nei PBMC, nel midollo osseo e attraverso colture cellulari derivate da PBMC nei risultati rappresentativi.
6. Smistamento delle cellule precursori MK e MK
NOTA: Le celle macchiate sono state analizzate e ordinate su uno smistatore di celle attivato a fluorescenza FACS Aria IIu dotato di laser standard a stato solido da 488 nm e 633 nm utilizzando il software FACSDiva; i dati sono stati inoltre analizzati e presentati utilizzando il software FlowJo e Cytobank (analisi viSNE). La purezza delle frazioni ordinate è stata confermata dall'analisi citometrica del flusso di ciascuna delle frazioni ordinate (purezza superiore all'85%).
- Eseguire lo smistamento cellulare il prima possibile o entro 1 ora dall'incubazione degli anticorpi al fine di evitare il deterioramento delle cellule.
- Filtrare il campione con un filtro cellulare da 100 μm per garantire la sospensione a cella singola e l'integrità di MK di grandi dimensioni.
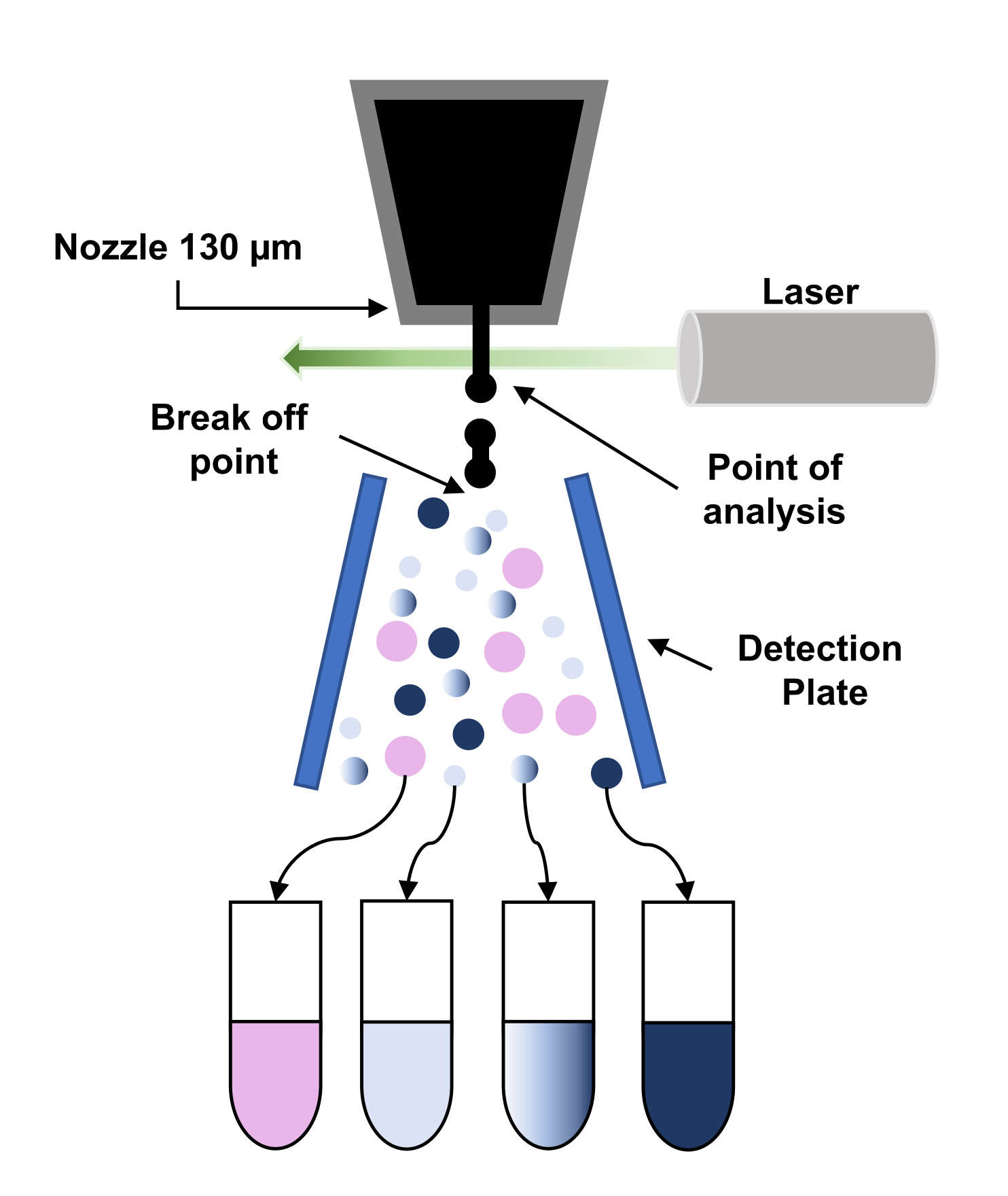
- Utilizzare un ugello in ceramica da 130 μm, una pressione di suona impostata su 11 libbre per pollice quadrato (PSI) e la frequenza di azionamento a discesa impostata su 12 kHz per rompere il flusso in gocce.
- Prima dello smistamento, sterilizzare l'ugello, la fodera e le linee del campione eseguendo un'acquisizione di 30 minuti con Penicillina / Streptomicina diluita 1:5 in acqua sterile, seguita da un'acquisizione di 10 minuti con acqua sterile per rimuovere il decontaminante rimanente.
- Una volta stabilizzato il flusso, regolare il ritardo di caduta con perline consigliate per ordinare in modalità di messa a punto più del 97,5% delle gocce riflesse a una portata di 400-1200 eventi al secondo.
- Preparare i tubi FACS di raccolta con 500 μL di 2% di BSA in PBS. La percentuale di BSA può essere aumentata fino al 5-10%.
- Generare il modello di esperimento con i parametri della matrice di compensazione appropriati.
- Caricare il tubo FACS nel citometro.
- Eseguire una misurazione del campione per impostare i gate e la purezza desiderati delle popolazioni di celle di destinazione. Mantenere attivato il record per visualizzare fino a 200.000 eventi nei gate di popolazione selezionati durante l'ordinamento delle celle.
- FACS Aria IIu consente la separazione di un massimo di 4 diverse popolazioni cellulari contemporaneamente. Creare un nuovo layout di ordinamento e selezionare il dispositivo di raccolta (4 tubi) e la modalità di precisione appropriata (si consiglia la maschera intermedia di purezza e recupero). Aggiungere infine le popolazioni di interesse a ciascun campo di ubicazione di ordinamento (Figura 3).

Figura 3: Rappresentazione schematica del principio dello smistamento cellulare attivato dalla fluorescenza (FACS). Le particelle attraversano l'ugello da 130 μm e sono costrette a rompersi in un flusso di goccioline regolari a causa dell'applicazione di vibrazioni all'ugello. Successivamente, le goccioline vengono interrogate dal laser (punto di analisi) e i segnali vengono elaborati per dare la "decisione di ordinamento" applicando una carica a quelle goccioline. Quando una goccia di carica passa attraverso un campo elettrostatico ad alta tensione (piastra di rilevamento), viene deviata e raccolta nel corrispondente tubo di raccolta. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Caricare i tubi di raccolta e iniziare a ordinare le popolazioni target.
- Centrifugare i tubi di raccolta per 5 minuti a 95 x g e rimosogliere il pellet di cella nel volume appropriato del 2% di BSA in PBS.
- Misurare di nuovo una frazione di ogni campione ordinato, per calcolare la purezza.
- Conservare le celle in modo appropriato per un ulteriore utilizzo. Le cellule ordinate possono essere utilizzate per analisi citologiche e molecolari, o possono essere ri-coltivate con l'obiettivo di studiare il processo di differenziazione di una popolazione cellulare selezionata.
7. Preparazione del campione post-ordinamento
- Preparazione dei citospini per l'analisi citologica con un citocentrifugo
- Portare le celle ordinate a un volume di lavoro facile da maneggiare di 100-200 μL. Prendi in considerazione che la densità cellulare dipenderà dalla resa della popolazione ordinata in ogni caso.
- Posizionare uno scivolo pulito sul supporto metallico e posizionare un piano del filtro. Ricordarsi di etichettare la diapositiva e il filtro per evitare di mescolare i campioni.
- Aggiungere 100 μL PBS sul foro del filtro contro lo scivolo, in modo che il filtro venga umidificato sul bordo del foro.
- Posizionare l'imbuto, chiudere il supporto metallico e posizionarlo sul suo punto nella centrifuga.
- Aggiungere il campione (100-200 μL) all'interno dell'imbuto.
- Centrifuga a 36 x g per 5 minuti. I vetrini di citospino possono essere autorizzati ad asciugarsi all'aria a RT (adeguatamente coperti per prevenire la polvere) e possono essere conservati a RT per 1 settimana prima dell'immunosottenizione o dell'istochimica.
- Per l'immunosocienza
- Fissare i vetrini in paraformaldeide al 2% (PFA) diluita in PBS e incubare durante 5 minuti.
- Può essere utilizzato un intervallo di PFA dello 0,5-4% in PBS. Nelle nostre mani, usiamo il 4% per ottenere una corretta fissazione dei tessuti o di alcuni tipi di cellule e lo 0,5% di PFA in PBS è sufficiente per le piastrine. Quando si imposta questa tecnica, la giusta percentuale di PFA richiede l'ottimizzazione per tipo di cella/origine.
- Incubare 5 min in PBS.
- Incubare 5 min in 50% di etanolo (EtOH).
- Incubare 5 min in 70% EtOH.
- Conservare in 70% EtOH a -20ºC.
- Quando si esegue l'immunostaining, reidratare e seguire le procedure standard (permeabilizzazione, lavaggio, blocco, incubazioni di anticorpi primari e secondari, conservazione, ecc.).
- Fissare i vetrini in paraformaldeide al 2% (PFA) diluita in PBS e incubare durante 5 minuti.
- Per la citochimica:
NOTA: Le diapositive possono essere macchiate con la colorazione May-Grünwald Giemsa o la comoda colorazione per ogni scopo. - Per un esame morfologico immediato
- Aggiungere una goccia di supporto di montaggio sul punto contenente celle del citospino e posizionare un copripasci.
- Mantenere le diapositive a 4 °C non più di una settimana, a meno che non siano sigillate, il che consente la conservazione a lungo termine anche a RT. Il montaggio della fissazione media consente lo stoccaggio a lungo termine a RT.
Risultati
Midollo osseo e Ploidia
Nella figura 4, mostriamo un'analisi immunofenotipica rappresentativa della megacariopoiesi nei campioni di BM (aspirazione) dei pazienti. Quando tracciamo la frazione cellulare rispetto a CD71 e CD31, abbiamo recintato sei popolazioni principali: CD31- CD71- (rosso), CD31- CD71+ (blu), CD31+ CD71- (arancione), CD31+ CD71mid (verde chiaro), CD31...
Discussione
La maggior parte della ricerca incentrata sullo studio della megacariopoiesi per citometria a flusso è finora limitataall'identificazione disottoinsiemi MK utilizzando solo marcatori di superficie specifici del lignaggio (cioè CD42A/CD42B, CD41/CD61), mentre le precedenti fasi di differenziazione MK sono state esaminate male. Nel presente articolo mostriamo una strategia di immunofenotipizzazione per affrontare una caratterizzazione citometrica a flusso completo della megacariopoiesi umana. Nel complesso, vorr...
Divulgazioni
La produzione di materiale audiovisivo è stata sostenuta da BD Biosciences.
Riconoscimenti
Ringraziamo Marcos Pérez Basterrechea, Lorena Rodríguez Lorenzo e Begoña García Méndez (HUCA) e Paloma Cerezo, Almudena Payero e María de la Poveda-Colomo (HCSC) per il supporto tecnico. Questo lavoro è stato parzialmente supportato da Medical Grants (Roche SP200221001) ad A.B., una borsa di studio RYC (RYC-2013-12587; Ministerio de Economía y Competitividad, Spagna) e una sovvenzione I+D 2017 (SAF2017-85489-P; Ministerio de Ciencia, Innovación y Universidades, Spagna e Fondos FEDER) a L.G., una Borsa di Studio Severo Ochoa (PA-20-PF-BP19-014; Da Consejería de Ciencia, Innovación y Universidades del Principado de Asturias, Spagna) a P.M.-B. e una sovvenzione post-dottorato intramurale 2018 (Fundación para la Investigación y la Innovación Biosanitaria de Asturias - FINBA, Oviedo, Spagna) ad A.A.-H. Ringraziamo Reinier van der Linden per aver condiviso le sue conoscenze (e il suo tempo), in particolare i suoi saggi consigli sul mix di pannelli con anticorpi taggati multicolore e sulla preparazione del controllo delle perline monocolore.
Materiali
| Name | Company | Catalog Number | Comments |
| 130 micron Nozzle | BD | 643943 | required for MK sorting |
| 5810R Centrifuge | Eppendorf | Cell isolation and washes | |
| A-4-62 Swing Bucket Rotor | Eppendorf | Cell isolation and washes | |
| Aerospray Pro Hematology Slide Stainer / Cytocentrifuge | ELITech Group | Automatized cytology devise, where slides are stained with Mat-Grünwald Giemsa | |
| CO2 Incubator Galaxy 170 S | Eppendorf | Cell Incubation | |
| Cytospin 4 Cytocentrifuge | Thermo Scientific | To prepare cytospins | |
| FACSAria IIu sorter | BD | Lasers 488-nm and 633-nm | |
| FACSCanto II flow cytometer | BD | Lasers 488-nm , 633-nm and 405-nm | |
| Olympus Microscope BX 41 | Olympus | Microphotographs | |
| Olympus Microscope BX 61 | Olympus | Microphotographs | |
| Zoe Fluorescent Cell Imager | BioRad | Microphotographs | |
| To obtain PBMCs | |||
| Lipids Cholesterol Rich from adult bovine serum | Sigma-Aldrich | L4646 | or similar |
| Lymphoprep | Stem Cell Technologies | #07801 | or similar |
| Penicillin-Streptomycin | Sigma-Aldrich | P4333 | or similar |
| Recombinant human Erythropoietin (EPO) | R&D Systems | 287-TC-500 | or similar |
| Recombinant human stem cell factor (SCF) | Thermo Fisher Scientific, Gibco™ | PHC2115 | or similar |
| Recombinant human thrombopoietin (TPO) | Thermo Fisher Scientific, Gibco™ | PHC9514 | or TPO receptor agonists |
| StemSpan SFEM | Stem Cell Technologies | #09650 | |
| Flow Cytometry Analyses | |||
| Bovine Serum Albumin | Merck | A7906-100G | or similar |
| BD CompBead Anti-Mouse Ig, κ/Negative Control Compensation Particles Set | BD | 552843 | Antibodies for human cells are generally from mouse. |
| BD Cytofix/Cytoperm | BD | 554714 | or similar |
| BD FACS Accudrop Beads | BD | 345249 | |
| CD31 AF-647 | BD | 561654 | Mouse anti-human |
| CD31 FITC | Immunostep | 31F-100T | |
| CD34 FITC | BD | 555821 | Mouse anti-human |
| CD41 PE | BD | 555467 | Mouse anti-human |
| CD41 PerCP-Cy5.5 | BD | 333148 | Mouse anti-human |
| CD42A APC | Immunostep | 42AA-100T | We observed unspecific binding... that needs to be assessed |
| CD42A PE | BD | 558819 | Mouse anti-human |
| CD42B PerCP | Biolegend | 303910 | Mouse anti-human |
| CD49B PE | BD | 555669 | Mouse anti-human |
| CD61 FITC | BD | 555753 | Mouse anti-human |
| CD71 APC-Cy7 | Biolegend | 334109 | Mouse anti-human |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | |
| Human BD Fc Block | BD | 564219 | Fc blocking - control |
| KIT PE-Cy7 | Biolegend | 313212 | Mouse anti-human |
| Lineage Cocktail 2 FITC | BD | 643397 | Mouse anti-human |
| RNAse | Merck | R6513 | or similar |
| Triton X-500 | Merck | 93443-500ML | or similar |
| Cell strainers for sorting | |||
| CellTrics Filters 100 micrometers | Sysmex | 04-004-2328 | Cell strainers |
| Note: we do not specify general reagents/chemicals (PBS, EDTA, etc) or disposables (tubes, etc), or reagents specified in previous published and standard protocols - unless otherwise specified. |
Riferimenti
- Italiano, J. E. Unraveling Mechanisms That Control Platelet Production. Semin Thrombosis And Haemostasis. 39 (1), 15-24 (2013).
- Machlus, K. R., Italiano, J. E. The Incredible Journey: From Megakaryocyte Development To Platelet Formation. Journal Of Cell Biology. 201 (6), 785-796 (2013).
- Eckly, A., et al. Biogenesis Of The Demarcation Membrane System (DMS) In Megakaryocytes. Blood. 123 (6), 921-930 (2014).
- Couldwell, G., Machlus, K. R. Modulation Of Megakaryopoiesis And Platelet Production During Inflammation. Thrombosis Research. 179, 114-120 (2019).
- Kosaki, G. In Vivo Platelet Production From Mature Megakaryocytes: Does Platelet Release Occur Via Proplatelets. International Journal Of Hematology. 81 (3), 208-219 (2005).
- Lefrancais, E., Looney, M. R. Platelet Biogenesis In The Lung Circulation. Physiology (Bethesda). 34 (6), 392-401 (2019).
- Nieswandt, B., Stritt, S. Megakaryocyte Rupture For Acute Platelet Needs. Journal Of Cell Biology. 209 (3), 327-328 (2015).
- Nishimura, S., et al. IL-1alpha Induces Thrombopoiesis Through Megakaryocyte Rupture In Response To Acute Platelet Needs. Journal Of Cell Biology. 209 (3), 453-466 (2015).
- Sanjuan-Pla, A., et al. Platelet-Biased Stem Cells Reside At The Apex Of The Haematopoietic Stem-Cell Hierarchy. Nature. 502 (7470), 232-236 (2013).
- Notta, F., et al. Distinct Routes Of Lineage Development Reshape The Human Blood Hierarchy Across Ontogeny. Science. 351 (6269), 2116 (2016).
- Yamamoto, R., et al. Clonal Analysis Unveils Self-Renewing Lineage-Restricted Progenitors Generated Directly From Hematopoietic Stem Cells. Cell. 154 (5), 1112-1126 (2013).
- Wang, H., et al. Decoding Human Megakaryocyte Development. Cell Stem Cell. , (2020).
- Meinders, M., et al. Repercussion Of Megakaryocyte-Specific Gata1 Loss On Megakaryopoiesis And The Hematopoietic Precursor Compartment. Plos One. 11 (5), 0154342 (2016).
- Meinders, M., et al. Sp1/Sp3 Transcription Factors Regulate Hallmarks Of Megakaryocyte Maturation And Platelet Formation And Function. Blood. 125 (12), 1957-1967 (2015).
- Salunkhe, V. P., Gutiérrez, L. Culture Of Megakaryocytes From Human Peripheral Blood Mononuclear Cells. Bio-Protocol. 5 (21), 1639 (2015).
- Choudry, F. A., et al. Transcriptional Characterization Of Human Megakaryocyte Polyploidization And Lineage Commitment. Journal Of Thrombosis And Haemostasis. , 15271 (2021).
- Heazlewood, S. Y., Williams, B., Storan, M. J., Nilsson, S. K. The Prospective Isolation Of Viable, High Ploidy Megakaryocytes From Adult Murine Bone Marrow By Fluorescence Activated Cell Sorting. Methods In Molecular Biology. 1035, 121-133 (2013).
- Tomer, A., Harker, L. A., Burstein, S. A. Purification Of Human Megakaryocytes By Fluorescence-Activated Cell Sorting. Blood. 70 (6), 1735-1742 (1987).
- Martinez-Botia, P., Acebes-Huerta, A., Seghatchian, J., Gutierrez, L. On The Quest For In Vitro Platelet Production By Re-Tailoring The Concepts Of Megakaryocyte Differentiation. Medicina. 56 (12), (2020).
- Martinez-Botia, P., Acebes-Huerta, A., Seghatchian, J., Gutierrez, L. In Vitro Platelet Production For Transfusion Purposes: Where Are We Now. Transfusion And Apheresis Science. 59 (4), 102864 (2020).
- Butov, K. R., et al. In Vitro Megakaryocyte Culture From Human Bone Marrow Aspirates As A Research And Diagnostic Tool. Platelets. , 1-8 (2020).
- Di Buduo, C. A., et al. A Gold Standard Protocol For Human Megakaryocyte Culture Based On The Analysis Of 1,500 Umbilical Cord Blood Samples. Thrombosis And Haemostasis. , (2020).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati