
Method Article
Quantification des modifications post-traductionnelles des histones globales à l’aide de la cytométrie en flux intranucléaire dans la microglie cérébrale isolée de souris
Dans cet article
Résumé
Ce travail décrit un protocole pour la quantification des modifications globales des histones par cytométrie en flux intranucléaire dans des microglies cérébrales isolées. L’ouvrage contient également le protocole d’isolement de la microglie qui a été utilisé pour la collecte de données.
Résumé
Le contrôle de l’expression génique se produit en partie par des modifications de la structure de la chromatine, y compris l’ajout et la suppression de modifications post-traductionnelles des queues d’histones. Les modifications post-traductionnelles des histones (HPTM) peuvent soit faciliter l’expression des gènes, soit leur répression. Par exemple, l’acétylation des résidus de lysine de la queue d’histones neutralise la charge positive et réduit les interactions entre la queue et l’ADN chargé négativement. La diminution des interactions entre les histones et l’ADN de la queue se traduit par une accessibilité accrue de l’ADN sous-jacent, ce qui permet un accès accru aux facteurs de transcription. La marque d’acétylation sert également de site de reconnaissance pour les activateurs transcriptionnels contenant des bromodomaines, ce qui entraîne une amélioration de l’expression des gènes. Les marques d’histones peuvent être régulées dynamiquement au cours de la différenciation cellulaire et en réponse à différents environnements et stimuli cellulaires. Bien que les approches de séquençage de nouvelle génération aient commencé à caractériser les emplacements génomiques des modifications individuelles des histones, une seule modification peut être examinée simultanément. Étant donné qu’il existe des centaines de HPTM différents, nous avons développé une mesure quantitative à haut débit des HPTM globaux qui peut être utilisée pour cribler les modifications des histones avant de mener des approches de séquençage du génome plus étendues. Ce protocole décrit une méthode basée sur la cytométrie en flux pour détecter les HPTM globaux et peut être réalisé à l’aide de cellules en culture ou de cellules isolées de tissus in vivo . Nous présentons des exemples de données provenant de microglies cérébrales de souris isolées pour démontrer la sensibilité du test pour détecter les changements globaux dans les HPTM en réponse à un stimulus immunitaire dérivé de bactéries (lipopolysaccharide). Ce protocole permet l’évaluation rapide et quantitative des HPTM et peut être appliqué à n’importe quel régulateur transcriptionnel ou épigénétique pouvant être détecté par un anticorps.
Introduction
L’épigénétique est l’étude des mécanismes qui régulent l’expression des gènes sans altérer la séquence d’ADN sous-jacente. La régulation épigénétique de l’expression des gènes est dynamique au sein des cellules et peut permettre des réponses rapides et coordonnées à divers stimuli environnementaux. La régulation dynamique se produit en partie en raison de changements dans la structure de la chromatine au niveau du nucléosome, qui est composé de protéines histones (H2A, H2B, H3, H4) assemblées dans un noyau octamère étroitement enroulé par l’ADN1. Les interactions entre les protéines histones et l’ADN peuvent contrôler l’accessibilité de l’ADN à la machinerie de transcription, qui peut finalement contrôler l’expression des gènes et d’autres aspects de la biologie de la chromatine2. Les protéines histones ont des queues non structurées qui comportent des résidus chargés positivement qui forment des interactions électrostatiques avec le squelette de l’ADN chargé négativement. Ces interactions entraînent un compactage serré de l’ADN et une réduction de l’accessibilité de l’ADN. Des modifications covalentes des queues d’histones, appelées modifications post-traductionnelles des histones (HPTM), peuvent réguler ces interactions 3,4. Parmi les HPTM les mieux caractérisés, citons l’acétylation et la méthylation de la queue des histones, qui peuvent modifier l’affinité des interactions électrostatiques entre les queues d’histones et l’ADN, ce qui entraîne une accessibilité différentielle à l’ADN sous-jacent et le recrutement de facteurs de transcription qui reconnaissent ces HPTM sur des sites spécifiques. Les HPTM sont régulés par trois classes importantes d’enzymes appelées lecteurs, qui reconnaissent, écrivains, qui déposent, et effaceurs, qui éliminent les HPTM. Ainsi, le recrutement ou la dissolution d’enzymes lectrices, d’écriture ou d’effaceuses peut finalement changer le paysage des HPTM et régir la structure et la fonction de la chromatine, rendant leur régulation et leur lecture essentielles à la compréhension de la biologie cellulaire et de la fonction 3,4.
Les cellules du système nerveux central (SNC) sont épigénétiquement flexibles car elles modifient leur transcriptome pour s’adapter aux stimuli environnementaux. L’accumulation de preuves suggère que les changements dans l’épigénome, tels que la méthylation de l’ADN, les ARN non codants et les HPTM, jouent un rôle essentiel dans la formation de la mémoire et la fonction synaptique5. Perturber la dynamique HPTM par la manipulation des lecteurs, des écrivains ou des effaceurs pertinents peut bloquer ou améliorer l’apprentissage associatif et la potentialisation à long terme 6,7,8. Les microglies, les cellules immunitaires résidentes du SNC, régulent rapidement leur transcriptome en réponse à une stimulation immunitaire par des changements dynamiques dans leur épigénome 9,10,11. Ce haut niveau d’adaptation à leur environnement cérébral local les rend difficiles à examiner dans un contexte isolé, car des études ont montré que l’épigénome et le transcriptome de la microglie sont altérés après seulement quelques heures dans des milieux de culture après avoir été retirés de l’environnement cérébral11. De plus, comme les microglies ne représentent que 10 % des cellules du cerveau, les mesures examinant les changements au niveau de l’ensemble des tissus manquent de sensibilité et de spécificité12,13. Par conséquent, les microglies doivent être rapidement isolées pour examiner les changements épigénétiques tels que les niveaux de HPTM, ex vivo.
Les méthodes couramment utilisées pour examiner les HPTM comprennent le séquençage par immunoprécipitation de la chromatine (ChIP-seq) et le séquençage par clivage sous cibles et par marquage (CUT&Tag-seq)4. Bien que ces techniques soient très spécifiques à un HPTM individuel et puissent informer sur la présence de HPTM dans un contexte génomique spécifique, elles ne peuvent examiner qu’un seul des nombreux HPTM possibles dans une seule expérience11,14 Par conséquent, avant de procéder à de telles expériences, qui nécessitent un investissement important en temps et en argent, il est très utile de réduire la liste des HPTM potentiellement intéressants pour une étude plus approfondie en examinant d’abord les changements dans les niveaux de HPTM. Les deux principales approches pour examiner les niveaux globaux de HPTM sont l’immunohistochimie et l’analyse par transfert Western, mais les deux approches ne sont que semi-quantitatives, à faible débit et nécessitent un grand nombre de coupes de tissus ou de cellules isolées15,16. Ainsi, nous avons cherché à développer une méthode quantitative très sensible qui pourrait être utilisée pour examiner les niveaux globaux de HPTM rapidement et au niveau de la cellule unique.
Le protocole présenté permet de détecter rapidement les niveaux globaux de HPTM par cytométrie en flux intranucléaire. Des études antérieures sur les cellules cancéreuses ont justifié l’importance d’examiner les niveaux globaux d’un point de vue clinique17,18. Il est également courant que les études utilisent les niveaux globaux comme méthode de dépistage avant d’évaluer la localisation génomique de HPTM spécifiques d’intérêt19,20. Dans le cas de la microglie, il est difficile d’évaluer les niveaux globaux après l’isolement en raison du faible rendement cellulaire. Pan et al. présentent les niveaux globaux de HPTM à partir de microglies isolées, dans lesquelles la microglie de trois animaux a été mise en commun pour permettre la détection des niveaux de protéines par Western blot19. Grâce à notre protocole, nous sommes en mesure de détecter des changements globaux avec des entrées cellulaires beaucoup plus faibles, ce qui permet de dépister plusieurs marques par animal et d’éliminer le besoin de regrouper les échantillons.
Nous décrivons ici un protocole permettant de détecter rapidement les niveaux de HPTM par cytométrie en flux intranucléaire quantitative dans des microglies isolées. Bien que nous nous concentrions spécifiquement sur la quantification HPTM par souci de concision, ce protocole peut être utilisé de la même manière pour quantifier les niveaux globaux d’enzymes de lecture, d’écriture et d’effacement. Le protocole est livré en deux parties : premièrement, la méthode d’isolement de la microglie et, deuxièmement, la méthode basée sur la cytométrie en flux pour déterminer les niveaux de HPTM. La méthode d’isolement permet d’obtenir des cellules qui peuvent être utilisées à la fois pour l’isolement de l’ARN et l’évaluation du niveau HPTM, ce qui permet d’évaluer l’expression génique et les niveaux HPTM à partir du même échantillon. De plus, la méthode d’évaluation HPTM peut être utilisée sur d’autres types de cellules, comme indiqué dans le protocole.
Protocole
Tous les protocoles de soins aux animaux ont été approuvés par le Comité de protection des animaux de l’Université de la Colombie-Britannique, conformément aux lignes directrices du Conseil canadien de protection des animaux.
1. Digestion du cerveau pour l’isolement de la microglie

Figure 1 : Organigramme simple du protocole. Les souris sont d’abord perfusées par voie transcardique avec HBSS, et le cerveau est disséqué. Le cerveau est ensuite dissocié par digestion chimique et perturbation mécanique pour aboutir à un homogénat unicellulaire. La fraction enrichie en système immunitaire est collectée via un gradient de densité discontinu, après quoi les cellules sont colorées pour P2RY12. Les cellules colorées sont soit 1) triées par tri cellulaire activé par fluorescence (FACS) pour conduire à l’analyse de l’ARN ou à l’analyse des protéines en aval et/ou 2) fixées, perméabilisées et colorées pour les protéines intranucléaires. Le niveau de protéine est quantifié par l’intensité médiane de fluorescence dans le canal d’intérêt déterminée par cytométrie en flux. Les cases colorées en bleu font partie de l’étape 1 du protocole) Digestion du cerveau pour l’isolement de la microglie. Les cases colorées en rouge font partie de l’étape 2 du protocole) : Coloration en flux intranucléaire pour l’analyse de l’expression des protéines. Créé avec BioRender.com. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
- Préparation des réactifs
REMARQUE : Si vous planifiez une extraction pour collecter à la fois de l’ARN et des cellules pour l’analyse HPTM, reportez-vous à la section 1.7.1 pour les modifications visant à inclure les inhibiteurs de transcription et de traduction. Cependant, cela n’est pas nécessaire si vous évaluez simplement le signal protéique, car les cellules sont en grande partie au repos lorsqu’elles sont maintenues sur la glace.- Tampon de tri cellulaire activé par fluorescence (FACS) (20 mL par échantillon) : Dissoudre l’albumine sérique bovine (BSA) dans 1 solution saline équilibrée de Hanks (HBSS) pour créer une solution de BSA à 2 %. Dissoudre l’EDTA jusqu’à une concentration finale de 1 mM dans la solution BSA à 2 %. Filtrer : stériliser à l’aide d’un filtre de 0,2 μm et conserver à 4 °C jusqu’à 1 semaine avant utilisation.
- Tampon de digestion (1 mL par échantillon) : Reconstituer un flacon de papaïne dans de l’HBSS jusqu’à une concentration finale de 20 U/mL dans 1 mM de L-cystéine avec 0,5 mM d’EDTA. Activer à 37 °C pendant au moins 10 minutes ou jusqu’à ce que les tissus soient prêts à être digérés. Juste avant l’utilisation, ajouter la DNase I à la solution de papaïne activée jusqu’à une concentration finale de 200 U/mL. Préparez-le le jour de l’expérience et ne le conservez pas.
- Solution de gradient de densité isotonique (5,5 mL par échantillon) : Ajouter 10x HBSS au milieu de gradient de densité à froid jusqu’à une concentration finale de 1x HBSS, ce qui donne une densité finale de 1,117 g/mL. Vortex à mélanger pendant au moins 30 s avant utilisation. Placer sur de la glace jusqu’à utilisation.
- Solution de gradient de densité à 37 % (4 mL par échantillon) : Ajouter le gradient de densité isotonique à 1x HBSS pour obtenir une concentration finale de 37 % avec une densité finale de 1,043 g/mL. Ajouter 20 μL de rouge phénol pour chaque mL de gradient de densité de 37 % pour obtenir une solution rose pour la visualisation pendant la superposition. Vortex pendant au moins 30 s avant utilisation. Placer sur de la glace jusqu’à utilisation.
- Solution de gradient de densité à 70 % (2 mL par échantillon) : Ajouter le gradient de densité isotonique à 1x HBSS pour obtenir une concentration finale de 70 % avec une densité finale de 1,082 g/mL. Ajouter 5 μL de bleu de trypan pour chaque mL de milieu de densité à 70 % pour obtenir une solution bleue pour la visualisation pendant la superposition. Vortex pendant au moins 30 s avant utilisation. Placer sur de la glace jusqu’à utilisation.
- Perfusion et dissection cérébrale
REMARQUE : Le protocole de perfusion est similaire à celui de Posel et al. qui comprend une représentation vidéo de la thoracotomie de souris, de la perfusion transcardique et de l’ablation du cerveau21. Ici, nous utilisons des souris mâles et femelles adultes C57BL/6J (10-15 semaines, 20-30 g), mais ce protocole peut être utilisé pour effectuer une thoracotomie pour n’importe quelle souris. Toutes les procédures sur les animaux doivent être approuvées par le comité d’éthique de l’établissement avant de mener des expériences.- Anesthésie de la souris : Anesthésier les souris avec de l’isoflurane à 4 % dans de l’oxygène à 100 % jusqu’à ce qu’elles aient dépassé le plan d’anesthésie chirurgicale, ce qui peut être confirmé par un pincement de l’orteil ou un manque de réflexe en pinçant fermement le pied de la souris. Placez la souris sur le dos et épinglez fermement ses quatre pattes dans la planche de dissection chirurgicale placée inclinée dans un plateau en plastique, en veillant à ce que le nez soit fixé dans le cône nasal en isofluorane. Après le transfert, assurez-vous que l’animal a toujours dépassé le plan chirurgical de l’anesthésie avant de procéder.
- Thoracomamy de souris : Saisissez et soulevez la peau abdominale à l’aide d’une pince et faites une incision peu profonde à travers la peau et la paroi abdominale pour exposer le xyphoïde sans endommager l’aorte descendante ou les organes sous-jacents.
- Saisissez le xyphoïde avec une pince et faites des incisions latérales sous la cage thoracique pour exposer le diaphragme et le foie. Faites des incisions peu profondes et soigneuses à travers le diaphragme sur toute la longueur de la cage thoracique à l’aide de ciseaux fins et à travers la cage thoracique à l’aide de ciseaux en tissu et épinglez le sternum à la station chirurgicale près de la tête de la souris pour exposer le cœur et les poumons à la perfusion transcardique.
- Perfusion transcardique : Préparez une pompe de perfusion péristaltique et fixez une aiguille de 26,5 G à une extrémité de la tubulure. Amorcez la tubulure pour la procédure en insérant une extrémité de la tubulure dans un flacon de 1x HBSS froid et en allumant la pompe pour remplir complètement la tubulure avec 1x HBSS.
- Tout en maintenant le cœur avec une pince émoussée, insérez la pointe d’une aiguille de 26,5 G avec le tube de perfusion attaché dans le ventricule gauche du cœur et faites une petite incision dans l’oreillette droite. Allumez la pompe à perfusion pour perfuser soigneusement la souris à un débit de ~2-4 mL/min avec au moins 15-20 mL de 1x HBSS froid.
REMARQUE : Une perfusion complète est souvent indiquée lorsque le foie commence à éliminer le sang et devient de la même couleur que le cœur. - Ablation du cerveau : Décapitalisez la souris à l’aide de ciseaux à disséquer les tissus et faites une incision médiane dans le cuir chevelu du cou au nez. Épluchez les lambeaux de peau sur les côtés pour exposer le crâne et retirez l’excès de tissu et d’os à l’extrémité caudale du crâne avec des ciseaux à disséquer.
- Faites glisser avec précaution une lame des ciseaux sous le crâne dans le foramen magnum avec le côté pointu face à l’os et coupez soigneusement la ligne médiane vers le nez. Faites des incisions latérales à la base du crâne et près du nez à l’aide de ciseaux à disséquer. À l’aide d’une pince fine, faites vivre le crâne de la ligne médiane à l’extérieur pour casser les morceaux de crâne et exposer le cerveau. Soulevez doucement le cerveau à l’aide d’une spatule et placez-le sur du papier de dissection.
- Dissection cérébrale : Placez le cerveau sur un morceau de papier buvard de dissection imbibé de 1x HBSS au-dessus d’une boîte de Pétri fermée remplie de glace. Retirez le cervelet et coupez les hémisphères cérébraux en deux à l’aide d’une lame de rasoir propre.
- Retirez le tronc cérébral, le striatum et la substance blanche de chaque hémisphère, tout en gardant l’hippocampe et le cortex superposé intacts. Transférez les hémisphères contenant le cortex et le tissu hippocampique isolés dans un tube de 15 mL avec 5 mL de HBSS 1x froid et conservez sur de la glace.
REMARQUE : Il est important d’effectuer les dissections le plus rapidement possible afin que le tissu reste froid avec pas plus de 2 minutes entre la décapitation et la mise en place finale du tissu disséqué dans 1x HBSS sur glace. Si vous isolez la microglie de plusieurs animaux, les cerveaux peuvent être stockés sur de la glace dans 1x HBSS pendant ~1 h avant de procéder au traitement de l’ensemble de la cohorte d’animaux pour la digestion, etc.
- Digestion et homogénéisation du cerveau
- Dissociation mécanique et chimique : Placez le tissu cérébral de chaque souris et 1 mL de tampon de digestion dans des boîtes de Pétri individuelles sur de la glace. À l’aide d’une lame de scalpel propre, hachez soigneusement la cervelle en petits morceaux (<1 mm).
- Coupez l’embout d’une pipette de transfert en plastique et transférez soigneusement chacun des cerveaux hachés dans des puits séparés à l’intérieur d’une plaque de 24 puits sur de la glace. Couvrir l’assiette d’un film souple transparent et incuber sur glace pendant 30 min.
REMARQUE : Lorsqu’il est haché correctement, le tissu cérébral ressemble à de l’ail bien haché. - Homogénéisation par dounce : Transférer la solution cérébrale digérée de chaque puits dans des homogénéisateurs individuels de 7 ml de verre sur de la glace, chacun rempli de 5 ml de tampon FACS froid. Trempez doucement chaque cervelle avec le pilon lâche (A), environ 30 à 40 fois, jusqu’à l’obtention d’une suspension unicellulaire. Après avoir rebondi avec le pilon A, trempez doucement avec le pilon serré (B) 3 à 4 fois pour assurer une suspension à une seule cellule.
REMARQUE : Ne poussez pas le pilon à plus des 3/4 vers le bas pour éviter d’écraser le tissu au fond de l’homogénéisateur. La solution finale doit être opaque et laiteuse.
REMARQUE : Si vous digérez plusieurs cerveaux dans une seule expérience, chronométrez le transfert de la digestion cérébrale dans la mémoire tampon FACS de manière à ce que chaque échantillon ne soit dans la mémoire tampon de digestion que pendant 30 minutes. La surdigestion peut entraîner un clivage des protéines de surface, ce qui réduit la liaison et le signal des anticorps en aval.
- Obtention d’un fragment enrichi en système immunitaire
- Établissement du gradient de densité : Transférer l’homogénat de chaque cerveau dans des tubes de polypropylène séparés de 15 mL et ajouter 2,125 mL de gradient de densité isotonique et le dessus à 8,5 mL avec tampon FACS pour chacun afin d’obtenir une concentration finale de 25 % de gradient de densité. Retournez délicatement les tubes de 15 ml 20x pour bien mélanger.
- À l’aide d’une pipette de transfert à graduation étroite, appliquer délicatement 4 mL de gradient de densité de 37 % à chaque tube, en prenant soin d’établir des couches propres. Changez de pipette de transfert et sous-posez délicatement 2 mL de gradient de densité de 70 % (Figure 2A). Transférer dans une centrifugeuse refroidie à 4 °C et essorer à 500 x g pendant 20 min avec la rampe de freinage réglée à zéro.
- Prélèvement du fragment enrichi en système immunitaire : À l’aide de pipettes de transfert propres, aspirer doucement la myéline du haut du volume dans le tube de 15 ml à l’aide d’une pipette de transfert propre et jeter. Prélever délicatement le fragment supérieur du gradient de densité dans un tube en polypropylène propre de 15 mL à l’aide d’une pipette de transfert.
- Prélever soigneusement le fragment enrichi en système immunitaire (1,5 mL au-dessus et 1,5 mL au-dessous de l’endroit où les couches de gradient de densité de 70 % et 37 % se rencontrent) dans un nouveau tube de polypropylène de 15 mL (figure 2B). Ajouter 10 mL de tampon FACS à l’échantillon enrichi en système immunitaire pour diluer le milieu de gradient de densité et retourner doucement le tube 20x pour bien mélanger.
REMARQUE : Comme les cellules ont tendance à coller aux côtés du tube, assurez-vous de rassembler toutes les cellules de l’échantillon pendant les étapes de prélèvement en faisant lentement le tour de la pipette le long des côtés du tube tout en recueillant le liquide. - Fragmenter les cellules de l’échantillon enrichi en système immunitaire en centrifugeant les tubes de 15 mL dans une centrifugeuse à 4 °C à 500 x g pendant 10 min avec le frein de rampe de descente réglé à zéro. Immédiatement après la fin de l’essorage, retirez le surnageant avec précaution, en laissant environ 300 μL de liquide dans le tube de 15 mL, en prenant soin de ne pas perturber la pastille (qui peut ne pas être visible).
- Recueillir le surnageant dans un autre tube de 15 mL pour s’assurer que les cellules ont été granulées dans l’essorage (jeter cette fraction une fois la vérification du nombre de cellules de la pastille remise en suspension terminée). Après avoir remis en suspension la pastille cellulaire dans le volume de 300 μL à l’aide d’une pipette P1000, comptez les cellules à l’aide d’un hémacytomètre pour estimer le rendement cellulaire total.

Figure 2 : Obtention du fragment enrichi par gradient de densité discontinu. (A) L’homogénat cérébral est fabriqué à partir d’un milieu de densité de 25 %, avec une sous-couche de 4 mL de densité moyenne de 37 % de couleur rose par l’intermédiaire du rouge phénolique et de 2 mL de couleur moyenne de densité 70 % de couleur bleue par l’intermédiaire du bleu trypan. (B) Après centrifugation, les fractions se sont séparées. La microglie repose à l’interface de fragments de milieux de densité de 37 % et 70 %. Le fragment de myéline se trouve au sommet du tube de 15 ml et sera jeté. Le fragment supérieur est collecté en tant que sauvegarde en cas d’échec du spin, et aucune cellule n’est récupérée. Si cela se produit, le gradient peut être répété à l’aide de cette fraction. La fraction enrichie en système immunitaire est collectée en aval. La fraction inférieure contenant les globules rouges reste dans le tube et est jetée. (C) Exemple de figure représentant des couches complètes. Créé avec BioRender.com. Veuillez cliquer ici pour voir une version agrandie de cette figure.
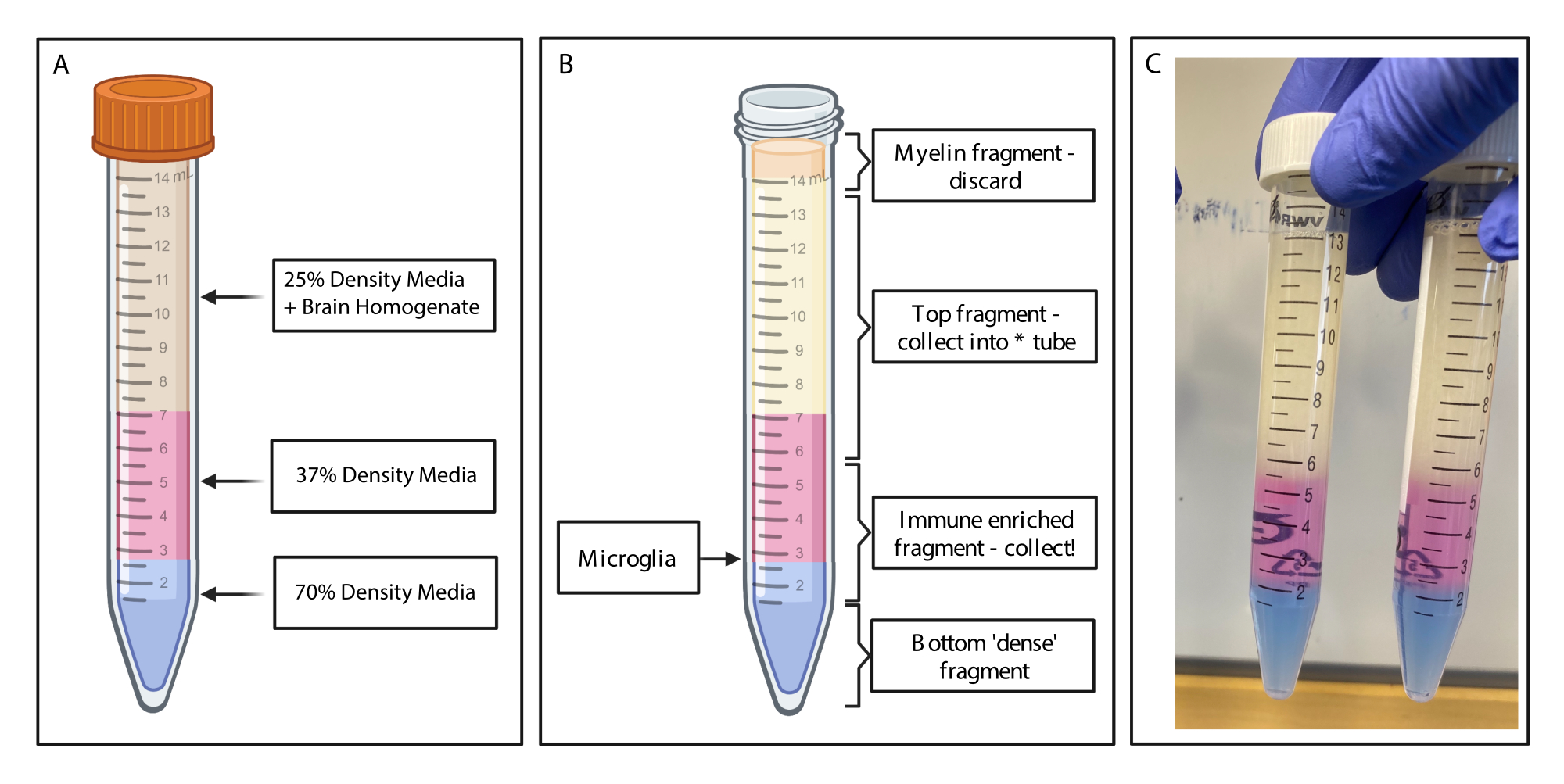{kind=link}
- Coloration des anticorps extracellulaires
- Blocage : Transférer les cellules sur une plaque à fond rond de 96 puits sur de la glace et centrifuger à 500 x g avec frein pour granuler les cellules. Retirez rapidement le surnageant dans l’évier en agitant la plaque pour éliminer le surnageant, en laissant la pastille de cellule intacte au fond du puits.
- Remettre les cellules en suspension dans 50 μL de tampon FACS avec un réactif bloquant le récepteur FC CD16/32 de la souris à l’aide d’une pipette P200 (concentration finale de 10 μg/mL, facteur de dilution de 1 :50) pour empêcher la liaison non spécifique des anticorps aux monocytes ou à d’autres cellules porteuses de FcR. Incuber pendant 10 min sur glace.
- Marquage des anticorps : Préparer le volume approprié d’un mélange maître 2x contenant de la P2RY12-allophycocyanine (APC ; facteur de dilution 1 :50, concentration de 4 μg/mL pour une concentration finale de puits de 1 :100, concentration de 2 μg/mL) et de colorant mort vivant violet 525 (facteur de dilution de 1 :50 pour une concentration finale de puits de 1 :100). Ajouter 50 μL du mélange maître de coloration à la suspension cellulaire (obtenue après blocage à la section 1.5.1) et incuber la plaque pendant 30 min dans l’obscurité sur de la glace.
NOTE : Pour ce protocole, nous présentons la coloration des cellules avec P2RY12. Tout d’abord, P2RY12 est un marqueur homéostatique de la microglie qui peut être régulé à la baisse dans certains contextes pathologiques. Par exemple, les souris modèles 5XFAD atteintes de la maladie d’Alzheimer ont des niveaux de P2RY12 régulés à la baisse, ce qui pourrait les rendre difficiles à identifier22. Les colorants alternatifs qui peuvent être utilisés pour l’isolation comprennent Tmem119, Cd11b et CD4523. Deuxièmement, le fluorochrome conjugué APC peut être ajusté en fonction du panel d’anticorps souhaité. Cependant, le choix d’un fluorochrome brillant, tel que l’APC ou le PE, aidera à s’assurer que les populations positives et négatives sont facilement distinguables24. - Après la coloration, ajoutez 200 μL de tampon FACS directement dans chaque puits pour laver les cellules. Essorer à 500 x g à 4 °C pour éliminer le surnageant par effleurement. Remettre les cellules en suspension dans 200 μL de tampon FACS à l’aide d’une pipette P200, essorer à 500 x g à 4 °C et faire glisser la plaque pour retirer le tampon des puits.
- Préparation des contrôles de débit : Avant de colorer, séparez les volumes nécessaires de cellules de chaque échantillon après avoir bloqué à l’étape 1.5.1 pour les contrôles de débit requis.
REMARQUE : Des contrôles de débit sont nécessaires pour chaque expérience afin d’établir les portes. Les contrôles de débit peuvent être prélevés sur un animal supplémentaire ou sur une fraction de chacun des puits expérimentaux. Lors de la division des cellules, assurez-vous d’attribuer suffisamment de cellules par contrôle, car 10 000 à 30 000 cellules par contrôle sont nécessaires pour établir des portes avec une grande confiance.- Il existe trois contrôles de débit pertinents : pas de tache, mort vivant et contrôle de l’isotype P2RY12. Pour le contrôle sans tache, n’ajoutez pas d’anticorps. Dans le contrôle de l’isotype P2RY12, traiter les cellules avec un colorant de viabilité (1 :100) et un anticorps de contrôle isotype conjugué à l’APC (1 :100).
- Pour préparer le témoin mort-vivant, les cellules aliquotes sont placées dans un puits séparé et la moitié du volume cellulaire est déplacée dans un tube de 500 μL. Placez le tube de 500 μL dans le congélateur à -80 °C pendant 5 minutes, puis placez-le dans un incubateur à 37 °C pendant 5 minutes pour tuer les cellules. Remettez l’aliquote des cellules mortes dans le puits témoin des cellules mortes vivantes et colorez-les avec un colorant de viabilité liant les amines sur le violet 525 (facteur de dilution 1 :100) pour marquer les cellules mortes.
REMARQUE : Le protocole est écrit pour la coloration des plaques avec une méthode de balayage pour l’élimination du surnageant. Cependant, cela nécessite que le surnageant soit retiré immédiatement après la fin de l’essorage et que le mouvement soit effectué avec suffisamment de force pour retirer rapidement le surnageant sans perturber la pastille. Alternativement, des tubes exempts d’ARNase/DNase de 1,5 mL peuvent être utilisés pour la coloration, avec les modifications suivantes : Transférer les cellules dans un tube de microcentrifugation de 1,5 mL et une pastille à 800 x g pendant 5 min à 4 °C. Aspirer le surnageant à l’aide de pipettes. Conseil : Pour plus de rapidité, une pipette de transfert de 5 ml munie d’un embout P200 permet d’aspirer rapidement et avec précision le surnageant. Lors de l’aspiration, vérifiez la présence de granulés. Si la pastille n’est pas visible, laissez 50 μL de surnageant et ajustez les calculs en conséquence. Lors du lavage des anticorps, ajouter des FACS supplémentaires pour augmenter la dilution des anticorps (1000 μL au lieu de 200 μL) pour tenir compte de l’élimination incomplète du surnageant. Selon le cytomètre, utilisez les tubes de 1,5 ml pour le tri, ce qui réduit la quantité de fournitures nécessaires.
- Tri FACS pour la microglie
- Préparation : Remettre chaque puits en suspension dans 200 μL de tampon FACS à l’aide d’une pipette P200 et transférer dans des tubes de tri en flux étiquetés et ajouter du tampon FACS jusqu’à un total de 500 μL pour une concentration d’environ 5 x 105 événements par mL. Conserver sur glace à l’obscurité jusqu’à l’analyse. Préparer les tubes post-tri en ajoutant 100 μL de tampon FACS comme coussin pour les cellules dans des tubes exempts d’ARNase de 1,5 mL.
- Réglages du cytomètre : Triez les cellules sur un trieur de cellules de cytométrie en flux configuré avec la buse de 100 μm. Triez les cellules à l’aide de 18 à 20 psi.
- Gating : Sur le cytomètre, évaluez la taille des cellules à l’aide de la zone de diffusion latérale (SSC) par rapport à la hauteur de diffusion vers l’avant (FSC) à l’aide de la commande sans coloration pour aider à distinguer les débris, placez SSC-A sur un axe logarithmique pour visualiser une population cellulaire et une porte rapprochée pour sélectionner les cellules (porte S1 ; Graphique 3). Pour éliminer les doublets, tracez le FSC-H par rapport au FSC-W et placez-le étroitement autour de la population cellulaire en enlevant tous les débris et les doublets (porte S2). À l’aide de la commande d’isotype P2RY12, examinez les cellules du canal APC et réglez la grille d’autofluorescence pour déterminer les cellules P2RY12+. À l’aide des commandes sans tache et mortes vivantes, les cellules qui ne sont pas fluorescentes sur le violet 525 nm sont considérées comme des cellules vivantes.
- Tri : Tracer le violet 525 nm par rapport à l’APC et déterminer la population qui est P2RY12+ et vivante par FMO (MG). Triez ces cellules dans le tube de post-tri étiqueté (Figure 3). Le pourcentage de tri final est d’environ 50 % du total des événements, la majorité des pertes totales d’événements étant des débris enlevés à la porte S1 (~70 % des événements sont des cellules ; Tableau 1).
- Isolement et analyse de l’ARN
- Inhibiteurs de la transcription et de la traduction : Si vous planifiez l’extraction de l’ARN, afin d’éliminer le risque de signatures transcriptomiques associées à l’isolement, incluez des inhibiteurs de la traduction et de la transcription dans les étapes tampons. Préparez le cocktail d’inhibiteurs tel que décrit par Marsh et al., y compris l’actinomycine D, l’anisomycine et le triptolide25.
- Préparation de l’inhibiteur : Reconstituer les stocks d’inhibiteurs et les conserver comme suit : Reconstituer l’actinomycine D dans le diméthylsulfoxyde (DMSO) à 5 mg/mL et conserver à -20 °C. Reconstituer le triptolide dans du DMSO à 10 mM et le conserver à -20 °C, à l’abri de la lumière. Reconstituer l’anisomycine dans du DMSO à 10 mg/mL et la conserver à 4 °C, à l’abri de la lumière. Conserver tous les stocks d’inhibiteurs pendant 1 mois maximum après leur reconstitution.
- Modifications du tampon : Ajouter des inhibiteurs dans quatre tampons différents dans le protocole comme suit : Lors de la perfusion transcardique, préparer l’HBSS avec de l’actinomycine D (5 μg/mL, 1 :1000 en stock) et du triptolide (10 μM, 1 :1000 en stock). Après la perfusion, transporter les cerveaux au laboratoire dans des HBSS contenant de l’actinomycine D (5 μg/mL, 1 :1000 du stock), du triptolide (10 μM, 1 :1000 du stock) et de l’anisomycine (27,1 μg/mL, 1 :368,5 du stock). Préparer le tampon FACS avec de l’actinomycine D (5 μg/mL, 1 :1000 en stock), du triptolide (10 μM, 1 :1000 en stock) et de l’anisomycine (27,1 μg/mL, 1 :368,5 en stock). Préparer le tampon de digestion avec de l’actinomycine D (5 μg/mL, 1 :1000 en stock), du triptolide (10 μM, 1 :1000 en stock) et de l’anisomycine (27,1 μg/mL, 1 :368,5 en stock). Préparer le tampon de lavage après le tri, avec HBSS contenant de l’actinomycine D (5 μg/mL, 1 :1000 en stock), du triptolide (10 μM, 1 :1000 en stock) et de l’anisomycine (27,1 μg/mL, 1 :368,5 en stock).
REMARQUE : Lors de l’ajout des inhibiteurs, assurez-vous de les ajouter immédiatement avant l’utilisation et de protéger tous les tampons préparés de la lumière pendant l’utilisation. Éviter le gel-dégel des solutions mères.
- Lavages post-tri : Étant donné que les cellules ont été triées dans des tubes exempts de RNase de 1,5 mL dans un tampon FACS, ce qui interférera avec l’isolement de l’ARN, il est nécessaire de laver les cellules. Essorer les cellules à 1000 x g à 4 °C pendant 5 min et retirer le surnageant, en laissant environ 50 μL de liquide.
- Ajouter 200 μL de 1x HBSS contenant de l’actinomycine D (5 μg/mL, 1 :1000 en stock), du triptolide (10 μM, 1 :1000 en stock) et de l’anisomycine (27,1 μg/mL, 1 :368,5 en stock) et bien mélanger. Répétez l’essorage et retirez le surnageant en laissant 50 μL de liquide (lavage 1). Ajouter 200 μL de tampon de lavage après le tri, bien mélanger et répéter l’essorage et retirer le surnageant en laissant 25 μL de liquide (lavage 2).
- Extraction de l’ARN : Pour l’isolement de l’ARN à partir de cellules microgliales, utilisez un kit d’isolement de l’ARN à faible entrée pour obtenir des rendements d’ARN élevés et constants et des scores RIN supérieurs à 9 (voir ci-dessous et le tableau des matériaux pour les recommandations de produits). Ajouter 350 μL du tampon de lyse du kit recommandé + β-mercaptoéthanol (1 :100) et bien mélanger.
REMARQUE : Si nécessaire, le protocole peut être suspendu à ce stade. Les échantillons peuvent être stockés dans le tampon de lyse à -80 °C jusqu’à l’extraction de l’ARN. Si vous extrayez de l’ARN après l’entreposage, décongelez le lysat sur de la glace et suivez les instructions spécifiques à la trousse pour l’isolement. - Transférez le lysat dans un broyeur à cellules à colonne (voir le tableau des matériaux pour les recommandations de produits) et centrifugez à vitesse maximale à 4 °C pendant 2 min. Éluer dans au moins 14 μL d’eau exempte de RNase et déterminer la concentration appropriée. L’ARN peut être utilisé pour n’importe quelle application en aval après ce point.
- Inhibiteurs de la transcription et de la traduction : Si vous planifiez l’extraction de l’ARN, afin d’éliminer le risque de signatures transcriptomiques associées à l’isolement, incluez des inhibiteurs de la traduction et de la transcription dans les étapes tampons. Préparez le cocktail d’inhibiteurs tel que décrit par Marsh et al., y compris l’actinomycine D, l’anisomycine et le triptolide25.

Figure 3 : Stratégie de contrôle pour le tri par flux. Les événements sont contrôlés en fonction de la taille de cellule sur SSC-A par rapport à FSC-H (S1). Ensuite, les cellules sont fermées pour être des singulets sur FSC-H vs FSC-W (S2). Les cellules singulet sont triées comme vivantes si elles sont négatives sur Comp-FL8-A ::405-526-52 (coloration morte vivante violette 525) et comme P2RY12+ si elles sont positives sur Comp-FL32-A ::640-671_30 (P2RY12-APC) à l’aide de la commande d’isotype P2RY12. Les cellules sont étiquetées comme MG et triées si elles sont à la fois vivantes et P2RY12+. Veuillez cliquer ici pour voir une version agrandie de cette figure.
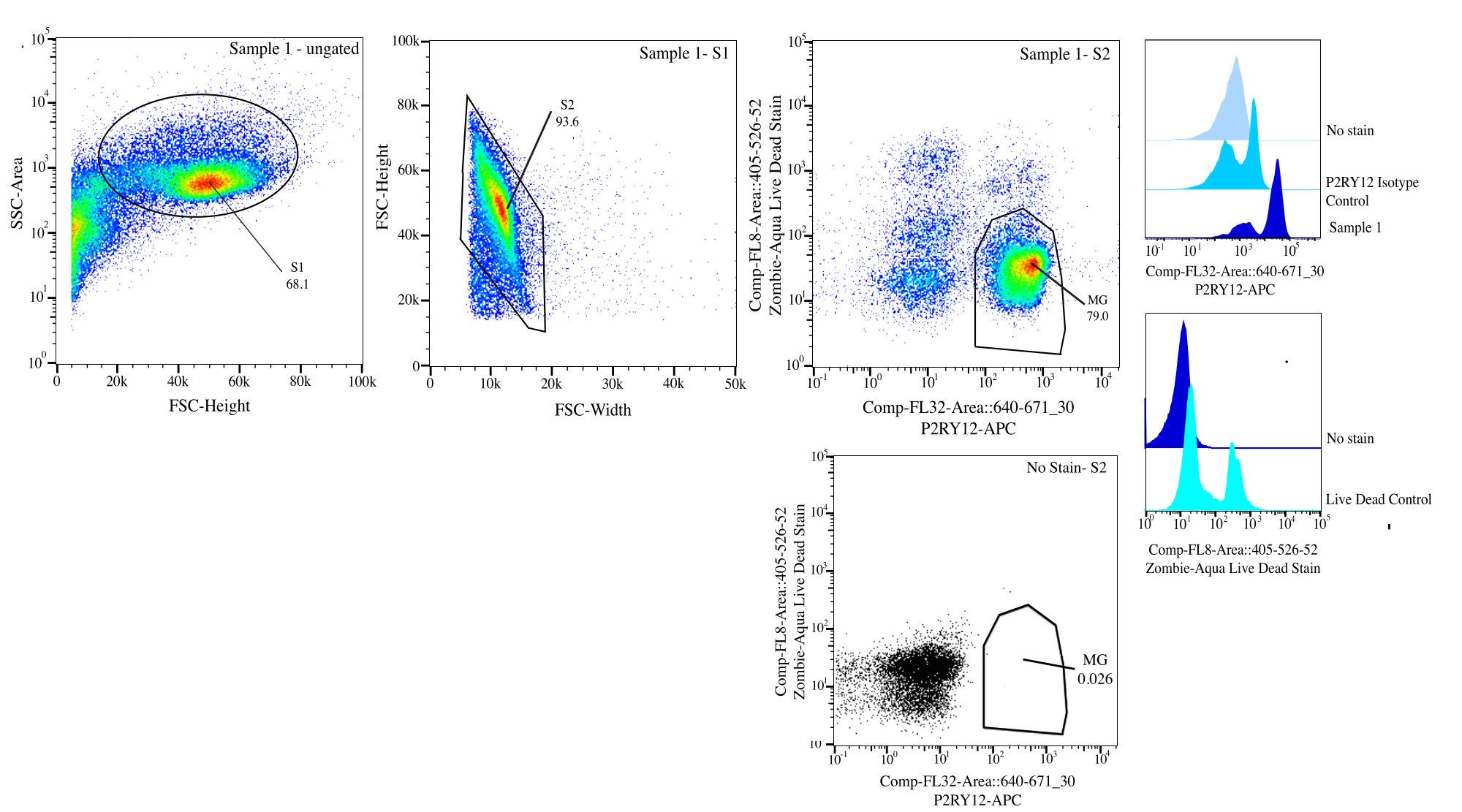{kind=link}
| POPULATION FERMÉE | Fréquence des parents | Fréquence des | Compter |
| S1 (Épisode 1) | 68.10% | 68.10% | 162186 |
| S2>S1 | 93.59% | 63.70% | 151707 |
| P2Ry12+ (670+) > S2 > S1 | 83.05% | 52.90% | 125986 |
| En direct (525-) > S2 > S1 | 92.78% | 59.10% | 140752 |
| MG (P2RY12+ Live) >S2>S1 | 78.96% | 50.30% | 119794 |
Tableau 1 : Exemple de tableau de lignage d’exemple avec les pourcentages de contrôle et les numéros d’événements attendus.
2. Coloration en flux intranucléaire pour l’analyse de l’expression des protéines
REMARQUE : D’autres types de cellules peuvent être démarrés à ce stade, ce protocole est testé avec des cellules en culture, y compris des cellules HEK293, des cellules de type microglie BV2 et des microglies humaines dérivées d’IPSC.
- Fixation et coloration des cellules
REMARQUE : Pour le protocole suivant, utilisez un kit de coloration intracellulaire optimisé pour la coloration nucléaire. Voir Table des matériaux pour obtenir des recommandations de produits.- Cellules aliquotes colorées extracellulairement de la section 1.5.2 dans une plaque à 96 puits (5 x 104- 1 x 106 cellules). Faire tourner les cellules pendant 5 min à 500 x g à 4 °C et effleurer pour retirer le tampon FACS.
NOTA : Pour obtenir des données avec un degré de confiance élevé quant aux niveaux médians, il faut utiliser un minimum de 10 000 cellules par puits. Bien qu’il n’y ait pas de maximum recommandé, il est préférable de maintenir le nombre de cellules constant tout au long de l’expérience pour s’assurer qu’il n’y a pas d’effet significatif des différents coefficients de variation (CV). - Fixation et perméabilisation : Ajouter 200 μL de 1x fix concentré et mélanger délicatement avec la pipette P200 pour remettre les cellules en suspension. Incuber dans l’obscurité pendant 45 à 60 min. Plaque de centrifugation pendant 5 min à 500 x g à température ambiante (RT) et effleurer pour jeter le surnageant.
REMARQUE : Si nécessaire, le protocole peut être suspendu à ce stade. Après avoir éliminé le surnageant, remettre les cellules en suspension dans un tampon de stockage à long terme pour les cellules immunitaires (voir le tableau des matériaux pour les recommandations de produits). Les échantillons peuvent être conservés à 4 °C pendant 12 à 18 h, à l’abri de la lumière et recouverts d’un film transparent pour protéger l’évaporation tampon. - Ajouter 200 μL de tampon de perméabilisation 1x dans chaque puits et pipeter avec un P200 pour mélanger. Plaque de centrifugation pendant 5 min à 500 x g à RT et effleurer pour éliminer le surnageant. Répétez le lavage du tampon de perméabilisation 3 fois au total.
- Préparation des contrôles de débit : Divisez le volume de cellules de chaque échantillon pour les contrôles de débit requis (10 000 à 30 000 cellules par puits de contrôle sont suffisantes).
- Pour préparer le contrôle sans tache, fixez les cellules sans coloration du tri ou les cellules non colorées aliquote dans un puits séparé qui ne recevra aucun anticorps.
- Pour préparer le contrôle de fluorescence moins un (FMO), aliquote des cellules pour chacun des anticorps du panel, à l’exception de celui de ce canal.
- Pour les canaux concernés, inclure l’anticorps de contrôle isotype dans le FMO pour le déclenchement. Par exemple, dans un panneau contenant P2RY12-APC et H3K27Ac-AlexaFluor568, il devrait y avoir deux FMOS : (1) l’APC-FMO qui ne contient que H3K27Ac-AlexaFluor568 et l’anticorps de contrôle de l’isotype P2RY12 et (2) le 568-FMO qui ne contient que P2RY12-APC et le contrôle de l’isotype primaire et le contrôle 568 secondaire.
REMARQUE : Ce protocole est présenté pour tester un seul HPTM, mais des panels peuvent être établis qui contiennent de nombreux HPTM conjugués à différents fluorophores.
- Marquage primaire des anticorps : Ajouter 50 μL de tampon de perméabilisation 1x avec la concentration appropriée d’anticorps primaire dans chaque puits. Incuber pendant 30 min à RT dans l’obscurité. Laver 2x avec 200 μL de 1x tampon de perméabilisation.
REMARQUE : La concentration d’anticorps utilisés pour chaque HPTM est incluse dans le tableau des matériaux. La concentration est déterminée en testant différentes concentrations d’anticorps sur des cellules en culture traitées avec un stimulant qui provoquerait une augmentation spectaculaire, par exemple, un inhibiteur HDAC pour les marques d’acétylation et en s’assurant que les cellules non traitées et traitées étaient bien dans la plage de détection (au-dessus de l’isotype témoin et en dessous de la plage de détection maximale du cytomètre). La concentration optimale d’anticorps pour les HPTM doit avoir une intensité fluorescente médiane moyenne dans le canal fluorophore comprise entre 5 x 104 et 1 x 105. - Coloration secondaire des anticorps : Bloquer avec 200 μL de tampon de perméabilisation 1x avec 2 % de sérum d’ânesse normal (NDS) pendant 10 min à RT. Essorer pendant 5 min à 500 x g à RT et effleurer pour éliminer le surnageant.
- Ajouter 50 μL de tampon de perméabilisation 1x avec 2 % de NDS et la concentration appropriée d’anticorps secondaire et incuber pendant 30 min à RT dans l’obscurité. Ajouter 200 μL de tampon de perméabilisation 1x dans les puits pour diluer, centrifuger la plaque pendant 5 min à 500 x g à RT, et effleurer pour éliminer le surnageant. Laver les cellules 2x avec 200 μL de tampon de perméabilisation 1x.
REMARQUE : Si nécessaire, suspendez le protocole à ce stade. Remettre les cellules en suspension dans 200 μL de tampon de stockage à long terme pour les cellules immunitaires à l’aide d’une pipette P200 (voir le tableau des matériaux pour les recommandations) et les conserver à 4 °C pendant 12 à 24 h à l’abri de la lumière. - Préparation à la cytométrie en flux : Plaque de centrifugation pendant 5 min à 500 x g à RT et effleurement pour éliminer le surnageant. Remettre en suspension des cellules dans 200 μL de tampon FACS à l’aide d’une pipette P200 pour la cytométrie en flux. Sceller avec un film transparent pour le transport vers le cytomètre.
- Cellules aliquotes colorées extracellulairement de la section 1.5.2 dans une plaque à 96 puits (5 x 104- 1 x 106 cellules). Faire tourner les cellules pendant 5 min à 500 x g à 4 °C et effleurer pour retirer le tampon FACS.
- Cytométrie en flux
- Pour analyser le panel d’anticorps proposé, assurez-vous que le cytomètre est équipé d’au moins quatre lasers, dont le violet (405 nm), le bleu (488 nm), le jaune (561 nm) et le rouge (633 nm). Le cytomètre a besoin de filtres pour détecter FITC (bleu-525 nm), KRO (violet-525 nm), PE (jaune-585 nm) et APC (rouge-660 nm). Ajoutez des anticorps supplémentaires en fonction du cytomètre choisi.
- Calibrage et standardisation : Au début de chaque expérience, exécutez des billes fluorescentes arc-en-ciel et ajustez la tension du tube photomultiplicateur (PMT) jusqu’à ce que les pics de perles soient comparables aux valeurs cibles utilisées pour les expériences précédentes. Cette méthode de normalisation permet de s’adapter à la dérive de l’équipement au fil du temps.
- Compensation : Une fois que la tension et le gain PMT ont été réglés pour l’expérience, utilisez des billes de compensation capturées par les anticorps pour établir la matrice de compensation pour le panel d’anticorps. Ce calcul permettra de s’assurer que les fluorophores ne contribuent pas aux changements de signal dans d’autres canaux. Ceci est de plus en plus nécessaire lors du multiplexage de plusieurs anticorps.
- Seuil de taille : Dans un diagramme à points, tracez SSC-A sur log vs FSC-H sur linéaire. Éliminez les débris et sélectionnez la taille de la cellule à l’aide de la porte S1. Sélectionnez les cellules singulet dans un diagramme à points de FSC-W par rapport à FSC-H et portez en tant que S2. (Graphique 4).
- Établissement des grilles de fluorophore : À l’aide de la FMO appropriée pour chaque canal de fluorophore, établissez les portes pour déterminer ce qu’est un signal positif dans chaque canal à l’aide d’histogrammes à paramètre unique (Figure 4).
- Mesure des échantillons : Enregistrez soigneusement les échantillons en utilisant la stratégie de contrôle établie. Identifiez la microglie à l’aide du signal P2RY12+, déterminez l’expression de la protéine dans les canaux respectifs pour la microglie uniquement.
- Analyse des données de cytométrie en flux
- Établissement des portes d’analyse : En utilisant les étapes ci-dessus pour le cytomètre sur l’interface utilisateur du logiciel d’analyse, utilisez les mêmes portes que celles utilisées pour l’enregistrement à des fins d’analyse.
- Obtenir les valeurs de l’IMF à l’aide d’un logiciel d’analyse par cytométrie en flux (voir le tableau des matériaux pour les recommandations) : Récapituler la stratégie de déclenchement du cytomètre pour l’analyse du flux. À l’aide de la fonction d’ajout de statistiques, sélectionnez la médiane de la population d’intérêt (p. ex., 568+) sur la hauteur du canal compensé. À l’aide de l’éditeur de tableaux, exportez les valeurs d’intensité fluorescente médiane (MFI) pour les canaux respectifs dans une feuille de calcul afin de procéder à l’analyse statistique (tableau 2).
NOTE : Le fichier supplémentaire S1 comprend des exemples de données provenant de souris injectées de lipopolysaccharides (LPS) et de solutions salines tamponnées au phosphate (PBS), ainsi qu’un exemple de fichier d’analyse avec la stratégie de déclenchement et les valeurs de l’IMF. - Analyse des valeurs de l’IMF par rapport à la variation du repliement des protéines : Après avoir obtenu les valeurs de l’IMF, calculez le changement de pli de l’IMF par rapport à la population témoin ou non traitée (équation 1). Le changement de pli de l’IMF reflète le changement de pli des niveaux de protéines. À l’aide des valeurs de changement de pli, évaluez le changement d’expression et calculez la signification statistique à l’aide d’un test t ou d’une ANOVA.
 Équation 1
Équation 1

Figure 4 : Stratégie de déclenchement pour l’évaluation de l’IMF des protéines. Les événements sont d’abord contrôlés pour la taille de cellule sur SSC-A par rapport à FSC-H (S1). Les cellules sont ensuite fermées pour les singuliers sur FSC-H vs FSC-W (S2). Les cellules singulet sont ensuite identifiées comme microglies par le signal P2RY12-APC (APC+) avec la grille établie sur la base de la fluorescence dans un témoin APC-FMO qui contient un anticorps de contrôle isotype. Les cellules sont ensuite bloquées pour le signal H3K27Ac-AlexaFluor568 sur Comp-FL5-A ::Y610-mCherry. L’intensité fluorescente des cellules 610+ est déterminée comme une approximation de l’expression des protéines. Veuillez cliquer ici pour voir une version agrandie de cette figure.
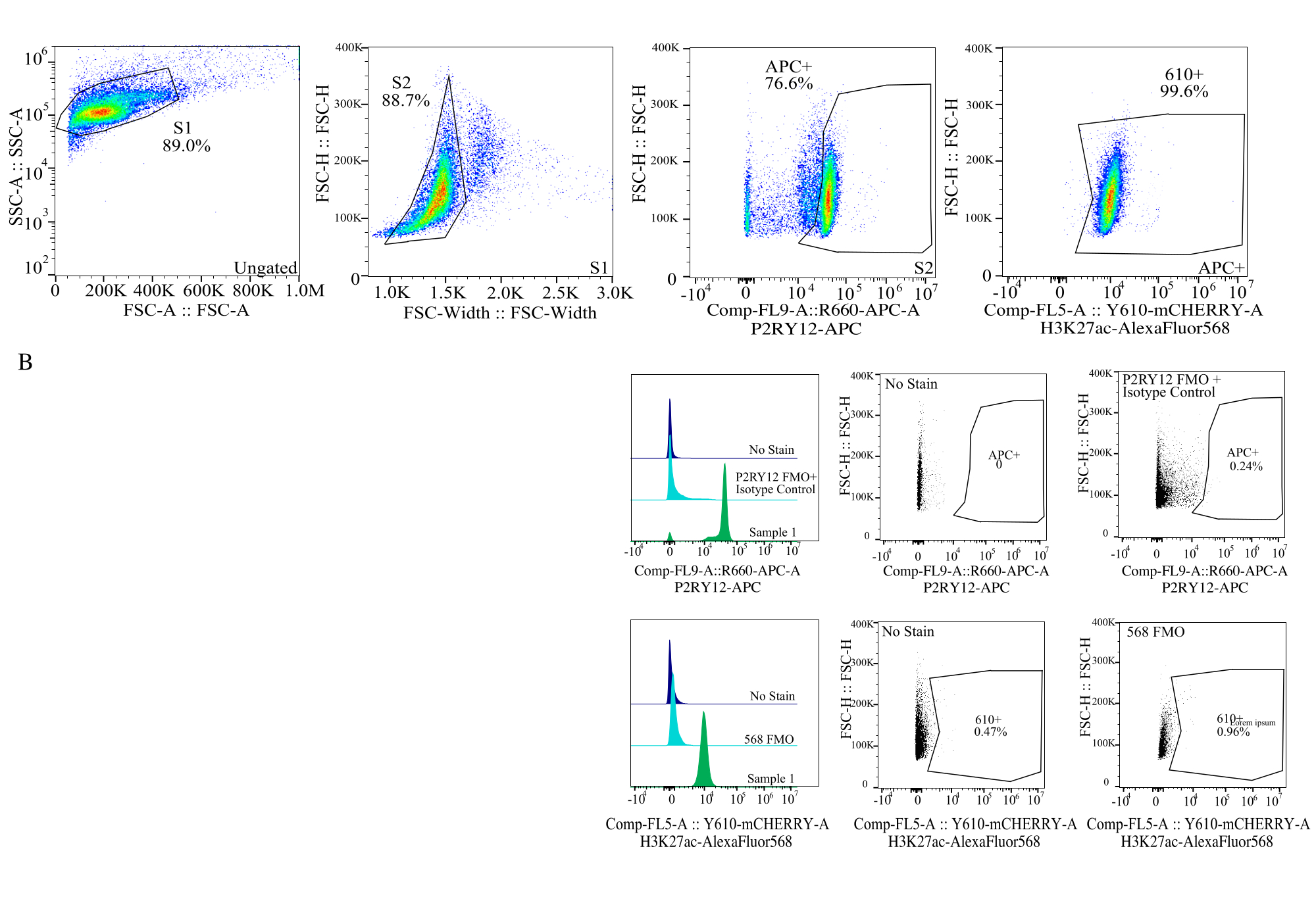{kind=link}
Résultats
Des souris adultes ont été perfusées par voie transcardique et sacrifiées pour l’isolement de la microglie. Les microglies ont été isolées sur de la glace et colorées avec des anticorps vivants P2RY12-APC et violet 525 vivants. Les cellules qui ont été déterminées positives pour P2RY12 et négatives pour la coloration vivante vivante 525 ont été triées comme microglies vivantes. Le rendement moyen de la microglie à partir d’un cerveau de souris disséqué était de 1,28 x 105 ± 0,05 (moyenne ± erreur type de la moyenne (MEB), N = 100). Il n’y a pas de différence dans le rendement de la microglie entre les souris femelles (1,25 x 105 ± 0,09 [moyenne ± MEB, N = 46]) et les souris mâles (1,32 x 105 ± 0,07 [moyenne ± MEB, N = 54]) (t(98)=0,6365, p=0,526). Lors de l’isolement de régions cérébrales spécifiques, le rendement moyen de la microglie à partir du cortex de souris est de 8,3 x 104 ± 0,08 (moyenne ± MEB, N = 15) et de l’hippocampe de souris est de 4,1 x 104 ± 0,02 (moyenne ± MEB, N = 16). Comme on pouvait s’y attendre, il existe une différence significative dans le rendement de la microglie de chaque région du cerveau (F(2, 128)=25,25, P<0,0001). Après l’isolement de la microglie, l’ARN a été extrait des cellules isolées à l’aide d’un kit d’isolement d’ARN à faible entrée. De façon constante, le score d’intégrité de l’ARN (RIN) était supérieur à 9,0 (9,62 ± 0,05) et le rendement moyen en ARN par cellule était de 0,25 ± 0,01 pg (moyenne ± MEB, N = 32 ; Dossier supplémentaire S2).
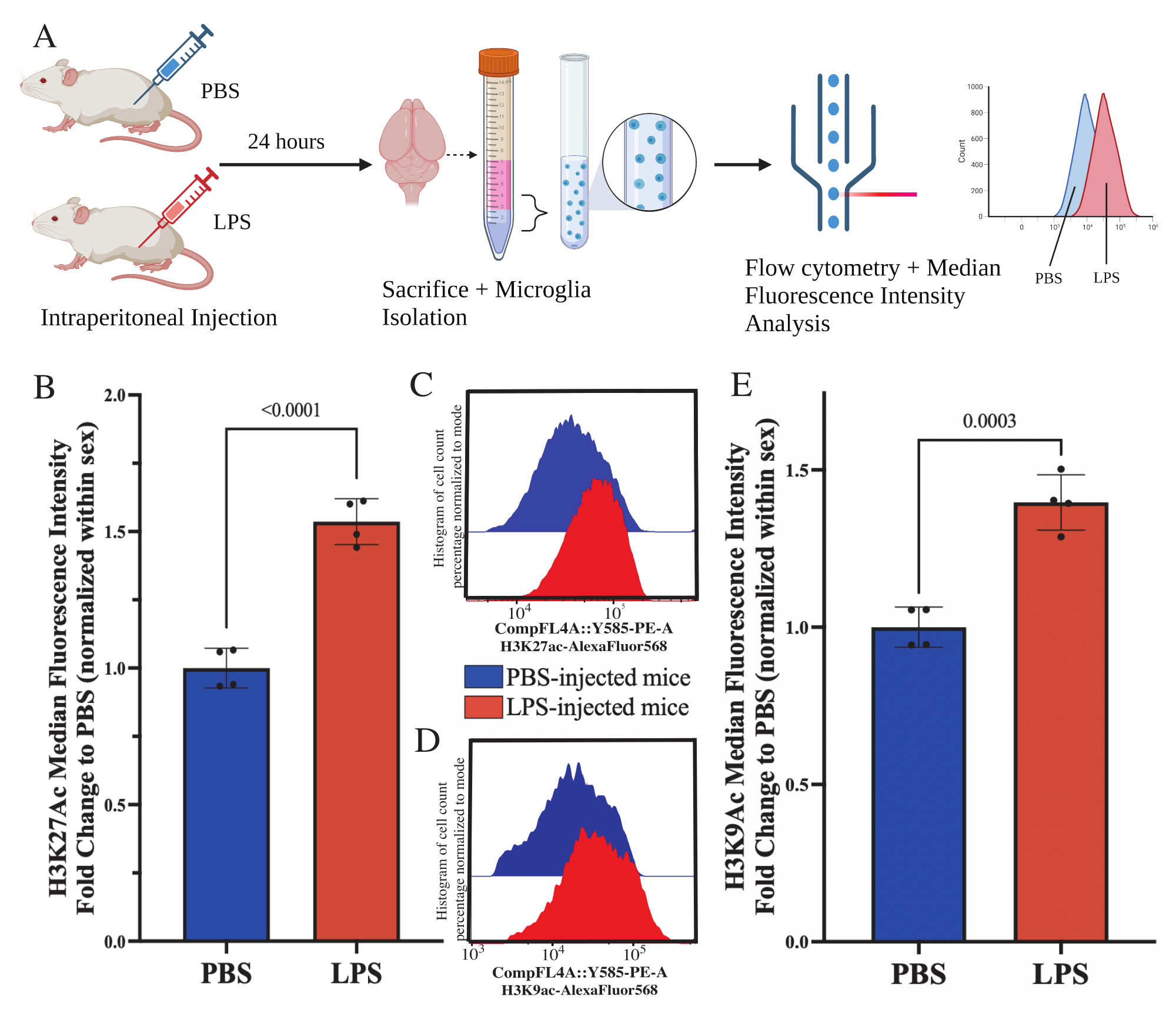
Des souris adultes ont reçu une injection intrapéritonéale de 1 mg/kg de lipopolysaccharide (LPS) 24 h avant le sacrifice. Les souris ont été perfusées par voie transcardique avec HBSS, et la microglie a été isolée de l’ensemble du cerveau selon le protocole décrit (Figure 5A). Pour chaque coloration, 20 000 à 30 000 cellules ont été allouées à chaque panel d’anticorps. Les niveaux globaux d’acétylation de l’histone 3 lysine 27 (H3K27Ac) ont été évalués dans des microglies isolées par cytométrie en flux. Pour les souris mâles et femelles, le traitement LPS a induit une augmentation de H3K27Ac lorsque l’IMF est normalisée dans le sexe (t(6)=9,676, p<0,0001 ; Graphique 5B). Lors de l’examen des histogrammes des cellules colorées, les populations restent normalement distribuées avec des variations similaires ; cependant, les cellules sont passées à une fluorescence accrue, ce qui a entraîné une augmentation de l’IMF (figure 5C). Lors de l’examen de H3K9Ac dans le même traitement, il y a une augmentation similaire de H3K9Ac (t(6)=7,299, p=0,0003 ; Figure 5D,E), cependant, le changement de pli du LPS par rapport au PBS du signal H3K9Ac est inférieur au signal H3K27Ac.

Figure 5 : Changements globaux dans l’acétylation des histones dans la microglie isolée. (A) Les souris sont injectées par voie intrapéritonéale avec une solution saline tamponnée au phosphate (PBS) ou 1 mg/kg de lipopolysaccharide (LPS) 24 h avant le sacrifice. Les microglies sont prélevées à partir de la fraction immuno-enrichie et fixées pour la cytométrie en flux et l’évaluation globale des histones après modification traductionnelle. L’intensité fluorescente médiane est évaluée comme une approximation de l’expression des protéines. Créé avec BioRender.com. (B) Les niveaux globaux de H3K27Ac ont augmenté en réponse au traitement LPS. Changement de pli en PBS normalisé dans l’expérience et le sexe. Test t à deux queues non apparié, t(6)=9,676, p<0,0001. Le graphique à barres représente la moyenne ± MEB. N = 8 animaux ; 2 par condition dans 2 expériences indépendantes. (C) Exemple d’histogrammes illustrant le décalage de l’intensité fluorescente H3K27Ac. La fenêtre modale représente les histogrammes des souris injectées par PBS par rapport aux souris injectées par LPS. (D) Exemple d’histogrammes illustrant le décalage de l’intensité fluorescente H3K9Ac. La fenêtre modale représente les histogrammes des souris injectées par PBS par rapport aux souris injectées par LPS. (E) Les concentrations mondiales de H3K9Ac ont augmenté en réponse au traitement par LPS. Changement de pli en PBS normalisé dans l’expérience et le sexe. Test t à deux queues non apparié, t(6)=7,299, p=0,0003. Le graphique à barres représente la moyenne ± MEB. N = 8 animaux ; 2 par condition dans 2 expériences indépendantes. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Pour confirmer que la méthode décrite était comparable à d’autres méthodes précédemment utilisées pour la quantification globale de la modification des histones, nous avons cherché à utiliser l’immunoblot comme outil comparatif. Cependant, le rendement de la microglie isolée est tout simplement trop faible pour permettre une évaluation raisonnable. Par conséquent, nous avons utilisé des cellules BV2 en culture pour comparer la méthode de cytométrie en flux intracellulaire à un Western blot (WB). Les cellules BV2 ont été cultivées dans des milieux complets (DMEMF12, 10 % de FBS, 1 x pénicilline/streptamycine et 1 x L-glutamine) à 37 °C, 5 % de CO2. Les cellules ont été traitées avec 0,25 % de trypsine-EDTA et plaquées à une densité de 250 000 cellules/puits et traitées dans un milieu sérique réduit (DMEM F12, 2 % FBS, 1x pénicilline/streptamycine et 1x L-glutamine) et laissées récupérer pendant 12 h à 37 °C, 5% CO2. Les cellules ont été traitées avec 25 ng/mL de LPS pendant 24 h avant la fixation comme décrit ci-dessus ou la lyse avec un tampon de lyse WB. Le signal de H3K27Ac a été effectué par les deux méthodes, le GAPDH étant utilisé comme contrôle de charge pour WB. L’analyse de l’intensité fluorescente normalisée par rapport au témoin PBS a été déterminée pour chaque groupe (figure 6A). Lors de l’examen de la variation du signal H3K27Ac normalisé par WB, il y avait une augmentation de 1,527 fois de l’état traité au LPS par rapport au témoin H2O, ce qui a été déterminé comme significatif par le test t non apparié (t = 3,024, df = 5 ; p = 0,0293). Lors de l’examen du changement à l’aide de la cytométrie en flux, il y avait une augmentation de 1,482 fois de l’état traité au LPS, ce qui a été déterminé comme significatif (t = 7,843, df = 10 ; p < 0,0001). À l’aide d’une ANOVA à 2 facteurs pour comparer les méthodes, il a été déterminé qu’il y avait un effet significatif du traitement (F(1,15)=45,21,p<0,0001), mais pas de la méthode (F(1,15)=0,05545, p=0,8697) ou de l’interaction (F(1,15)=0,02785, p=0,8697). De plus, nous vérifions ici qu’il n’y a pas de changement dans les niveaux d’histone H3 à la fois par Western blot et par cytométrie en flux, car l’ANOVA à 2 facteurs n’a révélé aucun effet significatif du traitement LPS (F(1,7)=0,02170,p=0,8870), de la méthode (F(1,7)=0,01191, p=0,9162) ou de l’interaction (F(1,7=0,01191, p=0,9162 ; Graphique 6B). Des exemples de transferts et de décalages d’histogramme pour ces données sont également présentés (Figure 6C,D).

Figure 6 : Comparaison des méthodes de quantification de la modification globale des histones entre la cytométrie en flux et le transfert Western. (A) Les cellules BV2 sont traitées avec 25 ng/mL de lipopolysaccharide (LPS) ou H2O pendant 24 h avant l’analyse. L’intensité fluorescente de H3K27Ac est représentée par un changement de pli du véhicule témoin, la solution saline tamponnée au phosphate (PBS), à la fois pour la cytométrie en flux et le transfert Western. L’ANOVA à 2 facteurs a révélé un effet significatif du traitement LPS (F(1,15)=45,21, p<0,0001), mais pas la méthode (F(1,15)=0,05545, p=0,8697) ou l’interaction (F(1,15)=0,02785, p=0,8697). La correction de Tukey pour les tests d’hypothèses multiples a été appliquée pour les résidus. * présente 0,0332, ** présente 0,0021. (B) L’intensité fluorescente de l’histone H3 est représentée par un changement de pli en PBS pour la cytométrie en flux et le transfert Western. L’ANOVA à 2 facteurs n’a révélé aucun effet significatif du traitement LPS (F(1,7)=0,02170, p=0,8870) ou de la méthode (F(1,7)=0,01191, p=0,9162) ou de l’interaction (F(1,7=0,01191, p=0,9162). (C) Des exemples de transferts et (D) de décalages de cytométrie en flux sont présentés. La taille de l’histogramme est normalisée en pourcentage en fonction du nombre de cellules présentes à l’intensité fluorescente du mode. Le graphique à barres représente le MEB moyen. n = 2 expériences indépendantes, 2 par condition par expérience. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
L’ensemble de ces résultats montre que cette technique peut être utilisée pour évaluer quantitativement les niveaux globaux de HPTM dans la microglie isolée. De plus, la méthode s’est avérée comparable aux techniques précédentes, mais nécessite des entrées de cellules beaucoup plus faibles. De plus, bien qu’elle ne soit pas illustrée, avec une compensation appropriée, la présente technique peut être utilisée avec plusieurs anticorps sur le même panel évaluant différents HPTM.
Fichier supplémentaire S1 : Exemples de fichiers d’analyse. Ce fichier contient un fichier d’analyse wsp et 7 fichiers fcs, y compris le no stain, le P2RY12FMO, le 568FMO, deux animaux traités au PBS et deux animaux traités au LPS colorés au H3K27Ac. Le but de ce fichier est de démontrer l’analyse et le déclenchement d’une expérience qui pourrait décrire à quoi ressemblait une expérience réussie. Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire S2 : Données d’isolement. Le fichier inclus contient les données pertinentes après le tri de la microglie qui contient la microglie et le rendement en ARN du protocole décrit. Veuillez cliquer ici pour télécharger ce fichier.
| POPULATION FERMÉE | Fréquence des parents | Fréquence des | Compter |
| S1 (Épisode 1) | 89.00% | 89.00% | 25672 |
| S2>S1 | 88.73% | 78.97% | 22779 |
| APC+ > S2 > S1 | 76.61% | 60.50% | 17452 |
| 610+ > APC+ > S2 > S1 | 99.56% | 60.24% | 17376 |
Tableau 2 : Un exemple de tableau de lignage d’échantillon illustre le pourcentage et le nombre d’événements requis pour une détection précise des protéines.
Discussion
Le protocole présenté permet une évaluation quantitative des niveaux globaux de HPTM par cytométrie en flux. Bien que ce protocole présente une nouvelle méthode, des études antérieures ont effectué une évaluation quantitative des protéines en utilisant une approche similaire26. Les méthodes précédemment utilisées pour évaluer les niveaux globaux de HPTM comprennent l’immunohistochimie et le western blot 16,17,19,20. La méthode basée sur la cytométrie en flux présentée est une méthode facilement quantifiable, tandis que le western blot et l’immunohistochimie sont semi-quantitatifs et ont un débit plus faible. Le Western blot repose sur la lyse cellulaire et nécessite donc à la fois une normalisation de la protéine et une protéine de contrôle de la charge qui est supposée être inchangée par la condition expérimentale27. L’immunohistochimie est semi-quantitative et à très faible débit car il est difficile d’évaluer quantitativement la quantité de protéines sans examiner au niveau d’une seule cellule16. De même, pour la microglie isolée, il y a un avantage à utiliser la méthode de cytométrie en flux en raison du rendement limité, car le transfert Western nécessite un apport protéique beaucoup plus important19. Les exigences en matière de faible nombre de cellules permettent d’exécuter plusieurs panneaux de coloration à partir du même animal.
Cependant, comme pour toute autre méthode, cette technique présente des limites, notamment le coût et la disponibilité des anticorps, car tous les anticorps ne fonctionnent pas bien dans un contexte de cytométrie en flux. De plus, par rapport à l’immunoblot, la concentration d’anticorps requise est beaucoup plus élevée. Bien que le multiplexage permette d’utiliser plusieurs anticorps sur le même panel de cellules, les cellules ne peuvent pas être débarrassées de l’anticorps après analyse, limitant ainsi l’utilisation des cellules à une par espèce d’anticorps. Ceci est différent de l’immunoblot dans lequel le même blot peut être utilisé à plusieurs reprises. Cependant, en fonction de la disponibilité des anticorps et du nombre de canaux de détection sur un cytomètre, il serait possible d’examiner jusqu’à une douzaine de marques simultanément.
La méthode actuelle ne saisit que les niveaux globaux d’expression des protéines et non l’emplacement génomique spécifique, et les changements dans les niveaux globaux peuvent ne pas refléter les changements au niveau des loci génomiques individuels. De même, l’absence de changement dans les niveaux globaux peut ne pas signifier qu’il n’y a pas de changements génomiques, mais simplement que la somme des changements globaux ne correspond à aucune différence entre les traitements. En tant que telle, cette technique est destinée à être utilisée comme un crible pour identifier les HPTM d’intérêt pour l’analyse génomique. De plus, cette méthode ne permet pas de comparer différentes marques de protéines, sauf lorsqu’elles sont évaluées comme un changement de pli pour le contrôle. Par conséquent, cela est limité par rapport à une méthode standard basée sur des courbes telle que ELISA pour la détermination des protéines.
Le protocole présenté propose une stratégie d’isolement de la microglie cérébrale vivante. Ce protocole s’appuie sur l’expression de la protéine P2RY12 pour l’isolement de la microglie. Cependant, P2RY12 est un marqueur homéostatique de la microglie et peut être régulé à la baisse dans des modèles de maladie, tels que 5XFAD22. Par conséquent, lors de l’utilisation d’un animal modèle de maladie, assurez-vous de choisir d’autres protéines marqueurs telles que TMEM119, CD11b ou CD45 pour aider à isoler la microglie23. De même, nous présentons ce protocole comme un isolement de l’hippocampe et/ou du cortex. Ce protocole permettrait d’isoler la microglie d’autres régions du cerveau, y compris les régions de la substance blanche, mais plusieurs animaux peuvent être nécessaires pour obtenir suffisamment de microglie en fonction de la taille des régions d’intérêt.
Le protocole présenté permet d’isoler de manière robuste des microglies cérébrales vivantes, mais il existe plusieurs étapes, décrites ci-dessous, dans la phase d’isolement qui peuvent diminuer le rendement cellulaire si elles ne sont pas effectuées correctement.
Les perfusions pour ce protocole se traduisent par un pourcentage plus élevé de microglie dans le fragment enrichi par le système immunitaire, ce qui réduira le temps passé au trieur. Cependant, la perfusion n’est pas requise et d’autres méthodes d’euthanasie peuvent être utilisées si nécessaire.
Lors de l’isolement de la microglie, la myéline doit être complètement éliminée. Les cytomètres en flux reposent sur la capacité des cellules à se déplacer à travers des tubes étroits à un rythme rapide. En raison de sa viscosité et de sa tendance à s’agglutiner, la myéline cause des problèmes avec les cytomètres, provoquant souvent des obstructions qui peuvent à la fois endommager l’équipement et détruire l’échantillon, réduisant considérablement le rendement. Veillez à retirer toute la myéline lors de la collecte de fragments enrichis par le système immunitaire pour éviter d’avoir des problèmes en aval.
Coloration sur plaque ou coloration sur tube : Dans ce protocole, nous avons décrit deux options pour colorer les cellules dans des tubes de 1,5 ml ou une plaque à 96 puits. Le cas d’utilisation de chacun dépend de l’expérience ; Cependant, en général, la coloration en tube présente un risque plus faible d’impact sur le rendement que la coloration sur plaque, car la coloration risque de perdre des cellules si elle n’est pas effectuée correctement. La coloration des plaques est beaucoup plus rapide car l’aspiration du surnageant pour chaque tube prend du temps. Avant la fixation (pour le tri, etc.), utilisez la coloration des tubes pour maximiser le rendement et réduire le risque de perte. Cependant, pour l’analyse HPTM, une fois que les cellules sont fixées pour la coloration intranucléaire, la pastille est plus stable et le risque de perte est réduit avec le scintillement.
Etablir le gradient de densité discontinu : Lors de l’établissement de la stratification, il est essentiel de mettre en place correctement les couches pour obtenir la fraction enrichie en système immunitaire. Si les couches sont perturbées ou mélangées et apparaissent troubles, les cellules ne seront pas triées à l’emplacement souhaité et il sera difficile d’obtenir la fraction cellulaire immuno-enrichie. Si cela se produit, essorez avec le milieu de densité pour éliminer la myéline, puis collectez toutes les fractions restantes, diluez avec 3 mL de tampon FACS à 1 mL de milieu de densité et mélangez bien (cela nécessitera plusieurs tubes). Tourner à 500 x g pendant 10 min avec le frein sur 0. Jeter le surnageant en ne laissant que ~300 μL de solution. Prélevez l’ensemble de l’échantillon et de la tache. Cela donnera des pourcentages de tri réduits et un temps passé plus longtemps au cytomètre, mais le rendement peut toujours être comparable.
Lors de l’utilisation de la méthode d’isolement, il est avantageux de pouvoir collecter des cellules pour l’ARN et l’évaluation HPTM du même cerveau de souris. Dans cette situation, après avoir trié la microglie vivante, les cellules peuvent être divisées pour allouer une partie à l’évaluation de l’ARN (le nombre minimum de cellules d’entrée pour obtenir un rendement en ARN décent est de 75 000 cellules) et une partie pour une analyse plus approfondie de la cytométrie en flux (minimum 10 000 cellules par puits pour une bonne détermination de l’IMF). Dans ce cas, un tri par cytomètre en flux est nécessaire. Cependant, lorsque l’on prévoit uniquement d’utiliser les cellules pour l’analyse HPTM, le tri n’est pas nécessaire et la fraction immunitaire peut être colorée avec l’anticorps P2RY12 et l’anticorps HPTM. Le déclenchement du cytomètre peut alors être réglé pour la microglie P2RY12+, comme cela serait fait pour le tri en flux, afin d’analyser uniquement le signal HPTM dans la microglie. L’élimination du tri permet au protocole d’être plus rapide et plus rentable. De plus, si l’on évalue les HPTM à partir de cellules en culture, il suffit de commencer par le protocole de coloration et aucun anticorps marqueur cellulaire n’est nécessaire, comme le montre la figure 6. Le protocole d’évaluation HPTM peut être utilisé pour de nombreux types de cellules, y compris les cellules en culture, les cellules primaires et les cellules dérivées de l’IPSC.
Enfin, si nous n’avons présenté que deux utilisations potentielles de la microglie en aval de l’isolement, il en existe de nombreuses autres dont des techniques épigénétiques telles que ChIP, CUT&Tag et CUT&RUN. Dans le cas des techniques épigénétiques génomiques, où la caractérisation des changements à des loci spécifiques est intéressante, choisir des inhibiteurs spécifiques pour les auteurs et les effaceurs de marques de chromatine11 adaptés aux expériences afin de s’assurer que les modifications épigénétiques microgliales profilées ne sont pas des artefacts techniques provenant de n’importe quelle étape de la procédure d’isolement telle que la digestion enzymatique. Lors de l’évaluation des changements dans les niveaux globaux de marques épigénétiques, par exemple en utilisant la cytométrie en flux quantitative, les changements induits par la procédure ne sont pas censés être si importants qu’ils soient détectés au niveau mondial.
Dans l’ensemble, les méthodes discutées fournissent une nouvelle méthode unicellulaire pour quantifier les niveaux globaux de modifications des histones et d’autres changements épigénétiques par cytométrie en flux. Nous avons démontré que cette méthode est suffisamment sensible pour détecter des changements globaux dans le marqueur enhancer H3K27ac dans la microglie en réponse au LPS in vivo. Ceci est cohérent avec le séquençage ChIP précédent de H3K27ac après stimulation LPS montrant un remodelage spectaculaire des amplificateurs sensibles à LPS28. Les applications de cette méthode permettront d’examiner les changements épigénétiques globaux dans différents types de cellules cérébrales au cours du développement et de la maladie.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Merci à Yanyang Bai pour son aide avec l’immunoblot de la figure 5. Ces travaux ont été financés par les Instituts de recherche en santé du Canada [CRC-RS 950-232402 à AC] ; Conseil de recherches en sciences naturelles et en génie du Canada [RGPIN-2019-04450, DGECR-2019-00069 à AC] ; la Scottish Rite Charitable Foundation [21103 à AC] et la Fondation Brain Canada [AWD-023132 à AC] ; Bourse d’études supérieures pour les Autochtones de l’Université de la Colombie-Britannique (6481 à MT) ; Bourse d’études supérieures de la Colombie-Britannique (6768 à MT) ; Bourse d’études de la Plateforme canadienne ouverte de neurosciences (10901 à JK) ; Bourse de doctorat de quatre ans de l’Université de la Colombie-Britannique (6569 à JK). Les bailleurs de fonds n’ont joué aucun rôle dans la conception de l’étude, la collecte et l’analyse des données, la décision de publier ou la préparation du manuscrit.
matériels
| Name | Company | Catalog Number | Comments |
| 0.5M EDTA | Invitrogen | AM9260G | |
| 15 mL Falcon Centrifuge Tubes, Polypropylene, Sterile | Falcon | 352196 | |
| 24-well Clear Not Treated Plates | Costar | 3738 | |
| 2-Mercaptoethanol | Gibco | 21985023 | |
| 96 Well Clear Polystyrene Microplate, clear round bottom, non treated surface | Corning | 3788 | |
| Acetyl Histone 3 K9 (C5B11) | Cell Signalling Technology | 9649S | Dilution: 1:100 |
| Acetyl Histone H4 K8 (2594) | Cell Signalling Technology | 2594S | Dilution: 1:100 |
| Acetyl-Histone H3 K27 (D5E4) | Cell Signalling Technology | 8173S | Dilution: 1:100 |
| Acetyl-Histone H3 Lys27 (MA523516) | Invitrogen | MA523516 | Dilution: 1:100 |
| Actinomycin D | New England Biolabs | 15021S | |
| Anisomycin | New England Biolabs | 2222S | |
| Anti-Histone H3 (tri methyl K4) | Abcam | ab213224 | Dilution: 1:100 |
| Anti-Lactyl-Histone H4 (Lys 12) Rabbit mAb | PTM Biolabs | PTM-1411RM | Dilution: 1:250 |
| Anti-L-Lactyllysine Rabbit pAb | PRM Biolabs | PTM-1401RM | Dilution: 1:250 |
| Apc anti-P2RY12 Antibody, Clone: S16007D | BioLegend | 848006 | |
| BSA | Tocris | 5217 | |
| Cyto-Last Buffer | BioLegend | 422501 | |
| dimethylsulfoxide, sterile | Cell Signalling Technology | 12611S | |
| DNAse I | STEMCELL Technologies | 07900 | |
| Donkey Anti Mouse AlexaFluor488 | Jackson ImmunoResearch | 715-546-150 | Dilution: 1:500 |
| Donkey Anti Rabbit AlexaFluor488 | ABclonal | AS035 | Dilution: 1:500 |
| Donkey Anti Rabbit AlexaFluor568 | Invitrogen | A10042 | Dilution: 1:500 |
| Donkey Anti Rabbit Brilliant Violet 421 | BioLegend | 406410 | Dilution: 1:500 |
| Fisherbrand Disposable Graduated Transfer Pipettes | Fisherbrand | 13-711-9AM | |
| Fisherbrand Disposable PES Filter Unit, 250mL | Fisherbrand | FB12566502 | |
| H3K18ac Polyclonal Antibody | Invitrogen | 720095 | Dilution: 1:100 |
| HBSS (10X), no calcium, no magnesium, no phenol red | Gibco | 14185052 | |
| HBSS, no calcium, no magnesium, no phenol red | Gibco | 14175103 | |
| Histone 3 Trimethyl K27 (ab6002) | Abcam | ab6002 | Dilution: 1:100 |
| KONTES Dounce Tissue Grinders 125mm 7mL | VWR | 885300-0007 | |
| Lactyl-Histone H3 (Lys 18) Rabbit mAb | PTM BIolabs | PTM-1406RM | Dilution: 1:250 |
| Lipopolysacharide | Sigma-Aldrich | L5418 | |
| Normal Donkey Serum | Jackson ImmunoResearch | 017-000-121 | |
| OneComp eBeads Compensation Beads | Invitrogen | 01-1111-41 | |
| PDS Kit, Papain Vial - Worthington Biochemical | Cedarlane | LK003178 | |
| Percoll | Sigma-Aldrich | GE17-0891-02 | |
| Phenol Red | VWR | RC57004 | |
| QIAshredder | Qiagen | 79656 | |
| Rainbow Fluorescent Particles, 1 peak (3.0-3.4 uM - Mid Range Intensity | BioLegend | 422905 | |
| RNase-free Microfuge Tubes, 1.5 mL | Invitrogen | AM12400 | |
| Rneasy Plus Micro Kit | Qiagen | 74034 | |
| Round Bottom Polypropylene Tubes with Caps, 5 mL | Corning | 352063 | |
| Triptolide | New England Biolabs | 97539 | |
| True Nuclear Transcription Factor Buffer Set | BioLegend | 424401 | |
| TruStain FcX PLUS (anti-mouse CD16/32) Antibody | BioLegend | 156604 | |
| Trypan Blue | VWR | 97063-702 | |
| Zombie Aqua Fixable Viability Kit | BioLegend | 423102 |
Références
- Miller, J. L., Grant, P. A. The Role of DNA Methylation and Histone Modifications in Transcriptional Regulation in Humans. Epigenetics: Development and Disease. 61, 289-317 (2013).
- Kouzarides, T. Chromatin Modifications and Their Function. Cell. 128 (4), 693-705 (2007).
- Bannister, A. J., Kouzarides, T. Regulation of chromatin by histone modifications. Cell Research. 21 (3), 381-395 (2011).
- Barski, A., et al. High-resolution profiling of histone methylations in the human genome. Cell. 129 (4), 823-837 (2007).
- Vogel Ciernia, A., LaSalle, J. The landscape of DNA methylation amid a perfect storm of autism aetiologies. Nature Reviews. Neuroscience. 17 (7), 411-423 (2016).
- Keiser, A. A., et al. Systemic HDAC3 inhibition ameliorates impairments in synaptic plasticity caused by simulated galactic cosmic radiation exposure in male mice. Neurobiology of Learning and Memory. 178, 107367 (2021).
- McQuown, S. C., et al. HDAC3 is a critical negative regulator of long-term memory formation. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 31 (2), 764-774 (2011).
- Barrett, R. M., et al. Hippocampal Focal Knockout of CBP Affects Specific Histone Modifications, Long-Term Potentiation, and Long-Term Memory. Neuropsychopharmacology. 36 (8), 1545-1556 (2011).
- Datta, M., et al. Histone Deacetylases 1 and 2 Regulate Microglia Function during Development, Homeostasis, and Neurodegeneration in a Context-Dependent Manner. Immunity. 48 (3), 514.e6-529.e6 (2018).
- Belhocine, S., et al. Context-dependent transcriptional regulation of microglial proliferation. Glia. 70 (3), 572-589 (2022).
- Gosselin, D., et al. An environment-dependent transcriptional network specifies human microglia identity. Science (New York, N.Y.). 356 (6344), eaal3222 (2017).
- Kettenmann, H., Hanisch, U. -. K., Noda, M., Verkhratsky, A. Physiology of Microglia. Physiological Reviews. 91 (2), 461-553 (2011).
- Sullivan, O., Ciernia, A. V. Work hard, play hard: how sexually differentiated microglia work to shape social play and reproductive behavior. Frontiers in Behavioral Neuroscience. 16, 989011 (2022).
- Das, P. M., Ramachandran, K., vanWert, J., Singal, R. Chromatin immunoprecipitation assay. BioTechniques. 37 (6), 961-969 (2004).
- Mahmood, T., Yang, P. C. Western blot: technique, theory, and trouble shooting. North American Journal of Medical Sciences. 4 (9), 429-434 (2012).
- Crowe, A., Yue, W. Semi-quantitative Determination of Protein Expression Using Immunohistochemistry Staining and Analysis: An Integrated Protocol. BIO-PROTOCOL. 9 (24), (2019).
- Seligson, D. B., et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 435 (7046), 1262-1266 (2005).
- Liu, B., et al. Global Histone Modification Patterns as Prognostic Markers to Classify Glioma Patients. Cancer Epidemiology, Biomarkers & Prevention. 19 (11), 2888-2896 (2010).
- Pan, R. Y., et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer's disease. Cell Metabolism. 34 (4), 634.e6-648.e6 (2022).
- Zhang, D., et al. Metabolic regulation of gene expression by histone lactylation. Nature. 574 (7779), 575-580 (2019).
- Pösel, C., Möller, K., Boltze, J., Wagner, D. C., Weise, G. Isolation and Flow Cytometric Analysis of Immune Cells from the Ischemic Mouse Brain. Journal of Visualized Experiments. (108), 53658 (2016).
- Oblak, A. L., et al. Comprehensive Evaluation of the 5XFAD Mouse Model for Preclinical Testing Applications: A MODEL-AD Study. Frontiers in Aging Neuroscience. 13, 713726 (2021).
- Bohlen, C. J., Bennett, F. C., Bennett, M. L. Isolation and Culture of Microglia. Current Protocols in Immunology. 125 (1), e70 (2019).
- McKinnon, K. M. Multiparameter Conventional Flow Cytometry. Flow Cytometry Protocols. 1678, 139-150 (2018).
- Marsh, S. E., et al. Dissection of artifactual and confounding glial signatures by single-cell sequencing of mouse and human brain. Nature Neuroscience. 25 (3), 306-316 (2022).
- Wang, L., Gaigalas, A. K., Marti, G., Abbasi, F., Hoffman, R. A. Toward quantitative fluorescence measurements with multicolor flow cytometry. Cytometry Part A. 73A (4), 279-288 (2008).
- Rumbaugh, G., Miller, C. A. Epigenetic changes in the brain: measuring global histone modifications. Methods in Molecular Biology (Clifton, N.J). 670, 263-274 (2011).
- Xavier, A. M., et al. Systematic delineation of signaling and epigenomic mechanisms underlying microglia inflammatory activity in acute and chronic brain pathologies. BioRvix. , (2022).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.