A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
הקמת מודל פיזיולוגי למיקרו-גידולים וסקולריים אנושיים לחקר הסרטן
In This Article
Summary
פרוטוקול זה מציג מודל גידול-על-שבב רלוונטי מבחינה פיזיולוגית לביצוע מחקר סרטן אנושי בסיסי ותרגומי בתפוקה גבוהה, קידום בדיקות סקר לתרופות, מידול מחלות וגישות רפואה מותאמת אישית עם תיאור של נהלי העמסה, תחזוקה והערכה.
Abstract
היעדר מודלים מתוקפים של סרטן המשחזרים את המיקרו-סביבה של הגידול של סרטן מוצק במבחנה נותר צוואר בקבוק משמעותי במחקר סרטן פרה-קליני ופיתוח טיפולי. כדי להתגבר על בעיה זו, פיתחנו את המיקרו-גידול הווסקולרי (VMT), או שבב הגידול, מערכת מיקרופיזיולוגית המדמה באופן מציאותי את המיקרו-סביבה האנושית המורכבת. ה-VMT יוצר דה נובו בתוך פלטפורמה מיקרופלואידית על ידי תרבית משותפת של מספר סוגי תאים אנושיים בתנאי זרימה פיזיולוגיים דינמיים. מבנה מיקרו-גידול מהונדס רקמות זה משלב רשת כלי דם מחוררת חיה התומכת במסת הגידול הגדלה, בדיוק כפי שכלי דם חדשים נוצרים עושים in vivo. חשוב לציין, תרופות ותאי מערכת החיסון חייבים לחצות את שכבת האנדותל כדי להגיע לגידול, תוך הדגמת מחסומים פיזיולוגיים in vivo למתן טיפול ויעילות. מכיוון שפלטפורמת VMT שקופה אופטית, ניתן להשיג הדמיה ברזולוציה גבוהה של תהליכים דינמיים כגון אקסטרווזיה של תאי מערכת החיסון וגרורות באמצעות הדמיה ישירה של תאים המסומנים באופן פלואורסצנטי בתוך הרקמה. יתר על כן, VMT שומר על הטרוגניות הגידול in vivo , חתימות ביטוי גנים ותגובות לתרופות. כמעט כל סוג גידול יכול להיות מותאם לפלטפורמה, ותאים ראשוניים מרקמות ניתוח טריות גדלים ומגיבים לטיפול תרופתי ב- VMT, מה שסולל את הדרך לרפואה מותאמת אישית באמת. כאן מתוארות השיטות להקמת ה-VMT ולניצולו למחקר אונקולוגי. גישה חדשנית זו פותחת אפשרויות חדשות לחקר גידולים ותגובות לתרופות, ומספקת לחוקרים כלי רב עוצמה לקידום חקר הסרטן.
Introduction
סרטן נותר דאגה בריאותית מרכזית ברחבי העולם והוא סיבת המוות השנייה בשכיחותה בארצות הברית. עבור שנת 2023 לבדה, המרכז הלאומי לסטטיסטיקה בריאותית צופה יותר מ -1.9 מיליון מקרי סרטן חדשים ומעל 600,000 מקרי מוות מסרטן המתרחשים בארה"ב1, מה שמדגיש את הצורך הדחוף בגישות טיפול יעילות. עם זאת, כיום, רק 5.1% מהתרופות האנטי-סרטניות שנכנסות לניסויים קליניים זוכות בסופו של דבר לאישור ה-FDA. כישלון של מועמדים מבטיחים להתקדם בהצלחה בניסויים קליניים ניתן לייחס באופן חלקי לשימוש במערכות מודל לא פיזיולוגיות, כגון תרביות דו-ממדיות וכדוריות, במהלך פיתוח תרופות פרה-קליניות2. מודלים קלאסיים אלה של סרטן חסרים מרכיבים חיוניים של מיקרו-סביבת הגידול, כגון נישה סטרומלית, תאים חיסוניים הקשורים וכלי דם מחוררים, שהם גורמי מפתח של עמידות טיפולית והתקדמות המחלה. לפיכך, מערכת מודל חדשה המחקה טוב יותר את המיקרו-סביבה האנושית של הגידול in vivo נחוצה כדי לשפר את התרגום הקליני של ממצאים פרה-קליניים.
תחום הנדסת הרקמות מתקדם במהירות, ומספק שיטות משופרות לחקר מחלות אנושיות בתנאי מעבדה. התפתחות משמעותית אחת היא הופעתן של מערכות מיקרופיזיולוגיות (MPS), הידועות גם בשם שבבי איברים או שבבי רקמות, שהם איברים אנושיים פונקציונליים וממוזערים המסוגלים לשכפל מצבים בריאים או חולים 3,4,5. בהקשר זה, שבבי גידול, שהם מודלים תלת מימדיים מבוססי מיקרופלואידים במבחנה של גידולים אנושיים, פותחו עבור מחקר אונקולוגי 2,3,4,5,6,7,8,9,10,11,12,13 . מודלים מתקדמים אלה משלבים רמזים ביוכימיים וביופיזיים בתוך מיקרו-סביבה דינמית של גידול, ומאפשרים לחוקרים לחקור את התנהגות הגידול ואת תגובותיו לטיפולים בהקשר פיזיולוגי רלוונטי יותר. עם זאת, למרות התקדמות זו, קבוצות מעטות שילבו בהצלחה כלי דם חי ומתפקד, במיוחד כזה שדפוסים עצמיים בתגובה לזרימה פיזיולוגית 3,4,5,6. הכללתה של רשת כלי דם תפקודית היא חיונית מכיוון שהיא מאפשרת מידול מחסומים פיזיים המשפיעים על העברת תרופות או תאים, ביות תאים למיקרו-סביבות נפרדות ונדידה טרנסאנדותלית של גידולים, סטרומה ותאי מערכת החיסון. על ידי הכללת תכונה זו, שבב הגידול יכול לייצג טוב יותר את המורכבויות שנצפו במיקרו-סביבה של הגידול in vivo.
כדי לענות על צורך זה שלא נענה, פיתחנו פלטפורמה חדשנית לסינון תרופות המאפשרת לרשתות כלי מיקרו להיווצר בתוך מכשיר מיקרופלואידי 8,9,10,11,12,13,14,15,16. פלטפורמת שבבי איברי בסיס זו, המכונה מיקרו-איבר וסקולרי (VMO), ניתנת להתאמה כמעט לכל מערכת איברים כדי לשכפל את הפיזיולוגיה המקורית של הרקמה לצורך מידול מחלות, בדיקות סקר לתרופות ויישומי רפואה מותאמת אישית. VMOs נקבעים על ידי תרבית משותפת של תאי אנדותל יוצרי תאים שמקורם בתאים (ECFC-EC), HUVEC או iPSC-EC (להלן EC), ותאי סטרומה מרובים בחדר, כולל פיברובלסטים ריאתיים אנושיים רגילים (NHLF), אשר מעצבים מחדש את המטריצה, ופריציטים העוטפים ומייצבים את כלי הדם. ה-VMO יכול גם להתבסס כמערכת מודל לסרטן על ידי תרבית משותפת של תאי גידול עם הסטרומה הקשורה ליצירת מיקרו-גידול וסקולרי (VMT)8,9,10,11,12,13, או שבב גידול, מודל. באמצעות תרבית משותפת של סוגי תאים מרובים בסביבת זרימה דינמית, רשתות מיקרו-וסקולריות מחוררות יוצרות דה נובו בחדרי הרקמה של המכשיר, שם כלי הדם מווסתים באופן הדוק על ידי קצבי זרימה אינטרסטיציאליים14,15. המדיום מונע דרך התעלות המיקרופלואידיות של המכשיר על ידי ראש לחץ הידרוסטטי המספק לתאים הסובבים את תא הרקמה חומרי מזון אך ורק דרך כלי הדם הזעירים, עם מקדם חדירות של 1.2 x 10-7 ס"מ לשנייה, בדומה למה שנראה עבור נימים in vivo8.
השילוב של מיקרו-כלי דם בעלי ארגון עצמי במודל VMT מהווה פריצת דרך משמעותית מכיוון שהוא: 1) מחקה את המבנה והתפקוד של מסות גידול וסקולריות in vivo; 2) יכול למדל שלבים מרכזיים של גרורות, כולל אינטראקציות בין תאי גידול-אנדותל וסטרומה; 3) קובע מחסומים סלקטיביים פיזיולוגית לאספקת חומרים מזינים ותרופות, שיפור סינון תרופות; ו-4) מאפשר הערכה ישירה של תרופות בעלות יכולות אנטי-אנגיוגניות ואנטי-גרורתיות. על ידי שכפול אספקת in vivo של חומרים מזינים, תרופות ותאי חיסון במיקרו-סביבה תלת-ממדית מורכבת, פלטפורמת VMO/VMT היא מודל רלוונטי מבחינה פיזיולוגית שניתן להשתמש בו לביצוע בדיקות סקר של תרופות ולחקר סרטן, ביולוגיה של כלי דם או איברים ספציפיים. חשוב לציין, VMT תומך בצמיחה של סוגים שונים של גידולים, כולל סרטן המעי הגס, מלנומה, סרטן השד, גליובלסטומה, סרטן ריאות, קרצינומטוזיס הצפק, סרטן השחלות, וסרטן הלבלב 8,9,10,11,12,13. בנוסף להיותה זולה, מבוססת בקלות וערוכה לניסויים בתפוקה גבוהה, הפלטפורמה המיקרופלואידית תואמת אופטית לחלוטין לניתוח תמונה בזמן אמת של אינטראקציות גידול-סטרומה ותגובה לגירויים או טיפולים. כל סוג תא במערכת מסומן בסמן פלואורסצנטי שונה כדי לאפשר הדמיה ישירה ומעקב אחר התנהגות התא לאורך כל הניסוי, וליצור חלון למיקרו-סביבה הדינמית של הגידול. הראינו בעבר שה-VMT מודל בצורה נאמנה יותר גידול גידול in vivo, ארכיטקטורה, הטרוגניות, חתימות ביטוי גנים ותגובות לתרופות מאשר שיטות תרבית סטנדרטיות10. חשוב לציין, ה-VMT תומך בצמיחה ובמחקר של תאים שמקורם בחולה, כולל תאים סרטניים, אשר מדגים טוב יותר את הפתולוגיה של גידולי האב מאשר תרביות ספרואידים סטנדרטיות ומקדם עוד יותר את מאמצי הרפואה המותאמת אישית11. כתב יד זה מתאר את השיטות להקמת VMT, ומציג את התועלת שלו לחקר סרטן אנושי.
Protocol
1. עיצוב וייצור
- עיצוב המכשיר
- לייצור התקנים מיקרופלואידים, צור תבנית SU-8 באמצעות שכבה של 200 מיקרומטר של SU-8 מצופה ספין על פרוסת Si (RCA-1 מנוקה ו-2% מימן פלואוריד (HF) מטופל), ולאחר מכן שלב פוטוליתוגרפיה של מסכה אחת כפי שתוארקודם 8,9.
- יצקו העתק פולידימתילסילוקסאן בעובי 4 מ"מ (PDMS) מתבנית SU-8 כדי ליצור תבנית פוליאוריתן עמידה לשלבי ייצור במורד הזרם. ניתן להשתמש באיטרציות עיצוב שונות 8,9,10,11,12,13,14,15.
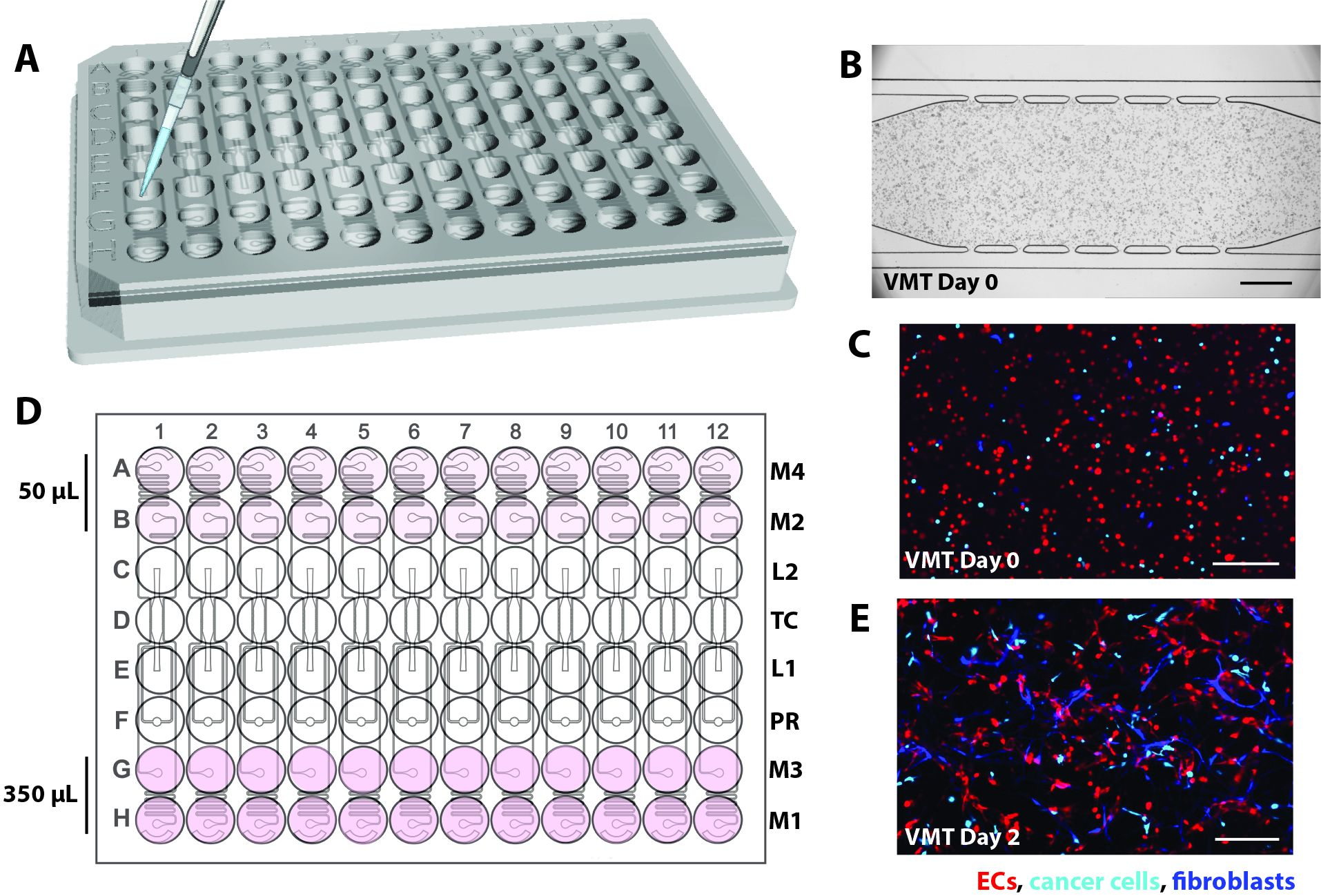
- באיטרציה הנוכחית, תכננו את המכשיר המיקרופלואידי כך שיותקן בהתאמה אישית לפורמט לוח סטנדרטי של 96 בארות ויכלול שכבת PDMS בעובי 2 מ"מ עם 12 יחידות התקן מיקרופלואידיות המוקפות בשכבת קרום פולימרי שקופה דקה (1/16 אינץ') בתחתית (איור 1A).
- ודא שיחידות רקמה נפרדות מורכבות מתא רקמה שמשני צדדיו פתח טעינת ג'ל (L1) ושקע (L2), וסת לחץ (PR)16, ותעלות מיקרופלואידיות מנותקות המחוברות לשני פתחי מדיה ושקעים בכל צד (M1-M2, M3-M4; איור 1B).
- מקם כל כניסה ומוצא בתוך באר אחת המשמשת כמאגר בינוני ליצירת לחץ הידרוסטטי (10 מ"מ H2O) על פני התעלה המיקרופלואידית. כדי לאפשר אנסטומוזות של רשת כלי הדם עם התעלות החיצוניות, חבר תעלות מיקרופלואידיות לתא הרקמה באמצעות נקבוביות תקשורת ברוחב 50 מיקרומטר (6 למעלה, 6 למטה).
הערה: נגדים מיקרופלואידים יוצרים שיפוע לחץ אינטרסטיציאלי של 5 מ"מ H2O על פני תא הרקמה שהופך תוך לומינלי ברגע שרשת כלי הדם נוצרת במלואה 8,10. ההליכים הבאים מתחילים עם צלחת תפוקה גבוהה שהורכבה במלואה.

איור 1. תכנון פלטפורמה מיקרופלואידית. (A) הסכימה של מכלול הפלטפורמה מראה את שכבת תכונת PDMS עם 12 יחידות התקן המחוברות לצלחת ללא תחתית של 96 בארות ואטומה בקרום פולימרי שקוף דק. כל יחידת מכשיר תופסת טור של בארות על הצלחת. יחידת ההתקן היחידה המסומנת באדום מוצגת עם הפרטים ב- (B). (B) סכמה של יחידת התקן אחת מראה תא רקמה יחיד הממוקם בתוך באר אחת של לוח 96 בארות ושתי יציאות טעינה עם חור כניסה ויציאה (L1-L2) מנוקב כדי לאפשר הכנסת תערובת מטריצת תאים. פתחים ושקעים בינוניים (M1-M2, M3-M4) מנוקבים חורים וממוקמים בתוך בארות המשמשות כמאגרי מדיה. נפחים שונים של מדיה יוצרים שיפוע לחץ הידרוסטטי על פני תא הרקמה באמצעות תעלות מיקרופלואידיות מנותקות. יחידת וסת הלחץ (PR) משמשת כשסתום פיצוץ ג'ל כדי להגביר את קלות ההעמסה. שים לב שעומק המכשיר הוא 200 מיקרומטר, ותא הרקמה הוא 2 מ"מ x 6 מ"מ. אנא לחץ כאן כדי להציג גרסה גדולה יותר של איור זה.
{kind=link}
2. הכנות לפני העמסה
- תרבית תאים
- לשמור על תאים בהתאם להמלצות היצרן באינקובטור לח של 37 מעלות צלזיוס ו-5% CO2 .
- צלחת T75 צלוחיות של EC, NHLF או תא פיברובלסט/סטרומה אחר ותאי סרטן רצויים 3-4 ימים לפני הטעינה בצפיפות המודעת על ידי פרוטוקולי היצרן והמשתמש. עבור פרוטוקול זה, צלחת 1 x 106 תאים לכל בקבוק עבור כל סוג תא. תרבית EC במדיה צמיחה אנדותל 2 (EGM2) מדיה מלאה, NHLF במדיום הנשר המתוקן של דולבקו (DMEM) עם 10% FBS, ותאים סרטניים במדיה המתאימה בהתאם לסוג התא.
- שמור על תאים על ידי הזנה עם המדיה המתאימה כל 2-3 ימים ולאשר מחדש את יעילות ההתמרה או התיוג על ידי הדמיה של תאים תחת מיקרוסקופ פלואורסצנטי. ביום הטעינה, ודא שה- EC הוא 80%-100% ב- confluent, בעוד NHLF הוא תת-שוטף ב- 70%-80%.
- הכנת פיברינוגן
- הכן את תמיסת הפיברינוגן לריכוז הרצוי (בדרך כלל, 5-8 מ"ג / מ"ל תומך בהיווצרות רשת כלי דם חזקה), תוך התחשבות באחוז קרישת הפיברינוגן. חשב את כמות הפיברינוגן הדרושה באמצעות המשוואה הבאה:
פיברינוגן (מ"ג) = (נפח (מ"ל)) x (ריכוז (מ"ג/מ"ל)/(קרישה%) - יש להמיס את הפיברינוגן בנפח מתאים של מדיה בסיסית אנדותל 2 (EBM2), המחוממת ל-37°C, על ידי תנועה עדינה של הצינור (אין מערבולת). לדגור פיברינוגן באמבט מים של 37 מעלות צלזיוס כדי לאפשר לו להיכנס לחלוטין לתמיסה. חשוב לציין, אין להשתמש במדיום שלם.
- תמיסת פיברינוגן סטרילית עם מסנן 0.22 מיקרומטר ואליציטוט לנפח הרצוי, בדרך כלל 400 μL לכל צינור מיקרוצנטריפוגה.
הערה: חלבוני מטריצה אחרים (למשל, קולגן, פיברונקטין או למינין) יכולים להיות מתובלים לתוך תערובת פיברינוגן.
- הכן את תמיסת הפיברינוגן לריכוז הרצוי (בדרך כלל, 5-8 מ"ג / מ"ל תומך בהיווצרות רשת כלי דם חזקה), תוך התחשבות באחוז קרישת הפיברינוגן. חשב את כמות הפיברינוגן הדרושה באמצעות המשוואה הבאה:
3. טעינת דגימות
הערה: הטעינה היא תלוית זמן ויש להשלים אותה מתחילתה (הרמת תאים) ועד סופה (הוספת מדיה למכשירים) תוך כ-1.5-1.75 שעות כדי להבטיח תוצאות מיטביות. כל שלב מצוין עם טיימר מוצע כדי לעזור לשמור על המשתמש במסלול.
- הכנת חומרים (יום העמסה)
- הכניסו את הפריטים הבאים לאמבט מים בטמפרטורה של 37°C למשך 10-15 דקות: תמיסת מלח מאוזנת של Hank (HBSS) או מלוחים חוצצי פוספט (PBS) לשטיפת תאים, מגיב דיסוציאציה של תאים, מדיה (למשל, EGM2, DMEM)
- יש לשמור את הריאגנטים הבאים במקרר בטמפרטורה של 4°C עד שהם מוכנים לשימוש: טרומבין, למינין (מופשר למשך הלילה ב-4°C).
- להפשיר פיברינוגן aliquot בטמפרטורת החדר. הכינו אליציטוטים של 1.5 מיקרו-ליטר של תרומבין לתוך צינורות מיקרוצנטריפוגות של 500 מיקרו-ליטר, עם צינור אחד לכל יחידת מכשיר. ודא כי aliquot תרומבין הוא בתחתית של כל צינור כדי להקל על העמסה.
- הניחו לוחות בעלי תפוקה גבוהה מעוקרים UV לתוך מייבש למשך 30 דקות לפחות לפני ההעמסה כדי להסיר אוויר הכלוא במיקרופלואידיקה.
- הכנת תאים (טיימר התחלה = להתחיל ב 0 דקות)
- בדוק תאים מתחת למיקרוסקופ בהגדלה של פי 4 כדי לאשר את המפגש ואת יעילות הטרנסדוקציה.
- לשטוף כל בקבוק T75 של תאים 2x עם 5 מ"ל של HBSS ולשאוף לחלוטין. יש להוסיף 1 מ"ל של מגיב דיסוציאציה לכל בקבוק ולדגור ב-37°C, 5% CO2 למשך 1-2 דקות.
- טפחו בעדינות על הצלחת באמצעות כף היד ובדקו שכל התאים הורמו.
- לשטוף את התאים מן הבקבוק עם 9 מ"ל של מדיה מתאימה ולאסוף אותם לתוך חרוט 15 מ"ל. הסר מיד aliquot קטן לספירת תאים.
- צנטריפוגה את התאים ב 300 x גרם במשך 3-5 דקות ב 4 ° C. תוך כדי צנטריפוגה של תאים, ספרו את התאים. בקבוק T75 של EC או NHLF אמור להניב לפחות 2 x 106 תאים.
- לאחר הצנטריפוגה, יש לשאוף מדיה ולהשהות מחדש את הגלולה במדיה מתאימה בריכוז של 1 x 106 תאים/מ"ל. שמור תאים על קרח.
- הכנת תערובת תאים ופיברינוגנים (טיימר = להתחיל ב 20 דקות)
- קבע כמה מכשירים ייטענו, הוסף 1-2 כדי להסביר אובדן צנרת, והכפל בנפח תערובת התא/פיברינוגן הדרושה לכל מכשיר. הדבר תלוי בתצורת המכשיר, אך עבור עיצוב המכשיר המוצג במאמר זה, נדרש 6 μL לכל מכשיר.
- הריכוז של כל סוג תא צריך להיקבע באופן ניסיוני. עבור נקודת התחלה, לטעון EC בריכוז של כ 7 x 106 תאים / מ"ל ו NHLF בריכוז של 3.5 x 106 תאים / מ"ל. ריכוז התאים הסרטניים יכול להשתנות במידה ניכרת בהתבסס על קצב הצמיחה שלהם, אך בדרך כלל נמצא בטווח של 0.5-2 x 106 תאים / מ"ל. השתמש במשוואה זו כדי לחשב את מספר התאים הדרוש:
מספר התאים הדרושים = (נפח פיברין (μL))/1000 μL x (ריכוז תאים) - השהה מחדש את התאים בריכוז של 1 x 106 תאים/מ"ל והשתמש במשוואה הבאה כדי לקבוע את נפח התאים הדרוש:
נפח התאים הדרוש (μL) = (מספר התאים הדרושים)/1000 - ערבב נפחים מתאימים של EC, NHLF ותאים סרטניים (עבור VMT בלבד) בצינור חרוטי ובצנטריפוגה במהירות של 300 x גרם למשך 3-5 דקות ב- 4 ° C.
- לאחר הסיבוב, יש לשאוף בזהירות את המדיה ולסלק כל שארית מדיה ליד הכדור. בעדינות אך ביסודיות להשעות את הגלולה לתוך נפח מחושב של פיברינוגן, תוך הקפדה יתרה לא להציג בועות אוויר. שמור על קרח.
- הביאו צלחות מעוקרות ואליציטוטים של תרופת טרומבין לתוך מכסה המנוע של תרבית הרקמות.
- טעינת מכשירים (טיימר = להתחיל ב 30-35 דקות)
- באמצעות פיפטה P20, פיפט 6 μL של נפח מתערובת התא / פיברין. הקפד לזלף את התערובת למעלה ולמטה לפחות 5x כדי להבטיח תרחיף תאים אחיד. שמרו את התערובת על קרח כדי להאט את הקרישה.
- ערבבו בעדינות את התא/פיברין לתוך צינור אחד של תרומבין על ידי הכנסת קצה הפיפט ישירות לתוך אליקוט התרומבין בתחתית הצינור. מיד צינור למעלה ולמטה לפחות 2x, נזהר לא להציג בועות אוויר. הפיברין יתחיל להיקרש פעם מעורבב עם טרומבין, כל כך מהר אבל בכוונה להשלים את שלבים 3.4.3. ו-3.4.4. לפני ג'ל הפיברין בקצה פיפטה (~ 3 שניות).
- הרם את לוח התפוקה הגבוהה בזווית והכנס במהירות את קצה הצינור לאחת מיציאות הטעינה של ההתקן (L1 או L2). ראו איור 2A לסכמה.
- דחפו את בוכנת הפיפט מטה לתחנה הראשונה בתנועה חלקה וזורמת כדי להזריק את תערובת התא/פיברין לתא הרקמה. שימו לב שהג'ל יעבור לחלוטין דרך החדר.
הערה: הפעלת לחץ רב מדי במהלך שלב זה עלולה להוביל להתפוצצות הג'ל לתעלות המיקרופלואידיות בחלק העליון ו / או התחתון של תאי הרקמה. - הניחו בעדינות את הצלחת חזרה שטוחה במכסה המנוע של תרבית הרקמה מבלי להסיר את קצה הפיפטה, לשחרר את בוכנה הפיפטה, או להפריע לפיפטה. השתמש בידך כדי לסובב ולהסיר את קצה פיפטה מה-P20 והשאר אותו בחור פתח הטעינה. אל תשתמש בלחצן השליפה כדי להסיר את הקצה מכיוון שהדבר יגרום ללחץ רב מדי.
- המשך עם שלבים 3.4.1-3.4.5 עבור המכשירים הנותרים.
- בסיום ההעמסה, הניחו לצלחת לשבת במשך 2 דקות ללא הפרעה במכסה המנוע של תרבית הרקמה.
- הסר את קצות פיפטה על ידי סיבוב עדין ומשיכה שלהם מיציאות הטעינה. החלף את המכסה בצלחת.
- דוגרים על הצלחת כולה למשך 15-20 דקות באינקובטור של 37 מעלות צלזיוס כדי לאפשר לג'ל להתפלמר במלואו.
- לאחר הדגירה, בדוק כל יחידת מכשיר מתחת למיקרוסקופ. בדקו שהתאים מפוזרים באופן שווה בכל החדר ללא בועות אוויר, ושקיים ממשק ג'ל שנראה בבירור בין תא הרקמה לבין התעלות המיקרופלואידיות, כמו באיור 2B-C.
- ציפוי תעלה עם למינין (טיימר = להתחיל ב 45-50 דקות)
- לאחר שהג'לים מוצקים לחלוטין, הכניסו למינין לתעלות המיקרופלואידיות כדי לקדם אנסטומוזה וסקולרית.
- באמצעות P20, הכנס 4 μL של למינין לכל תעלה מיקרופלואידית (עליונה ותחתונה) של המכשיר. הכנס את קצה הצינור ל- M1 או M3 והרחק את הלמינין באיטיות, תוך התבוננות כדי להבטיח שהלמינין מצפה את כל הערוץ העליון, ולאחר מכן חזור על הפעולה עבור M2 או M4 כדי לצפות את כל הערוץ התחתון.
- קבע את הכיוון על ידי pipetting laminin מהצד הנגדי לווסת הלחץ כדי לאפשר לחץ מספיק כדי לדחוף אותו. עם זאת, אם הלמינין אינו נע בקלות מצד אחד, הסר את הקצה מצד אחד ודחוף את הלמינין מצד שני. הליכה לתחנה השנייה של פיפטה (כלומר, לדחוף את הבוכנה כל הדרך למטה) עשוי להיות נחוץ כדי ליצור לחץ מספיק כדי לדחוף את הלמינין דרך התעלה כולה.
- הסר את הקצה בעדינות מפתח הכניסה/שקע המדיה. אל תשתמש בלחצן השליפה ב-P20.
- חזור על שלבים 3.5.1-3.5.4 עבור כל מכשיר ודגור על הצלחת בטמפרטורה של 37°C, 5% CO2 למשך 10 דקות.
- תוספת מדיה (טיימר = להתחיל בערך שעה ו-10 דקות)
- הוסף 275 μL של מדיה מלאה EGM2 לתוך מאגרי מדיה לא מצומדים של בארות בשורות A ו- B או G ו- H. זה יהיה הצד הגבוה, ואת הכיוון צריך להיקבע על ידי הפיכת הבארות בצד שממול לווסת הלחץ נפח גבוה להתחיל. התקשורת תידחף מהצד הגבוה על ידי כוח הכבידה.
- באמצעות פיפטה P200, להכניס 75 μL של מדיה לתוך כניסות בינוניות / שקעים של בארות המכילים 275 μL של EGM2. הכניסו את הקצה לחור כניסת המדיה וגרשו לאט לאט את המדיום, תוך צפייה בכך שהמדיה עוברת דרך הערוץ ומבעבעת בצד השני.
- הסר את קצה הצינור ודחוף את המדיה הנותרת מהקצה לתוך מאגר המדיה כך שהנפח הכולל בצד הגבוה הוא 350 μL.
- חזור על שלבים 3.6.1-3.6.3 עבור כל יחידת מכשיר, ערוצים עליונים ותחתונים.
- הוסף 50 μL של מדיה כדי לכסות לחלוטין את הצד הנמוך, בארות בשורות A ו- B או G ו- H, בהתאם לכיוון המתואר לעיל. ודא שיש שכבה אחידה של מדיה המכסה את תחתית הבאר. עיינו באיור 2D לקבלת סכמה המציגה נפחי מדיה במאגרים.
- הסרת בועות אוויר (לאחר העמסה)
הערה: הסרת בועות היא שלב קריטי כדי להבטיח זרימה תקינה בכל מכשיר. ביום השני, תאי אנדותל ופיברובלסטים יתחילו להימתח בתגובה לזרימה (איור 2E).- לאחר הוספת כל המדיה, יש לדגור על צלחות במשך 1-2 שעות באינקובטור של 37°C, 5% CO2 לפני בדיקת בועות אוויר בתעלות או בכניסות / שקעים בינוניים.
- דמיינו בועות בערוצי המדיה במיקרוסקופ וגרשו על ידי החדרה מחדש של 75 מיקרוליטר מדיה לערוצים כדי לדחוף את הבועות החוצה.
- דמיינו בועות בכניסות / שקעים בינוניים לפי העין. השתמשו בפיפטת P200 כדי להסיר בועות אוויר שנלכדו בכניסות ובשקעים הבינוניים על ידי דחיפת הבוכנה כלפי מטה, החדרת הקצה לתוך החור ומשיכת הבועה החוצה על ידי הרמת הבוכנה כדי להפעיל לחץ שלילי ולשאוב את הבועה.

איור 2. סכמטי של טעינת המכשיר. (A) באמצעות פיפטה P20, תערובת תאים/פיברין מוחדרת לתא הרקמה של כל יחידת התקן דרך אחת מיציאות הטעינה. (B) מיקרוגרף ברייטפילד מראה מכשיר מיקרופלואידי טעון EC, פיברובלסטים ותאים סרטניים ליצירת VMT. סרגל קנה מידה = 500 מיקרומטר. (C) מיקרוגרף פלואורסצנטי של המכשיר ב-B המציג EC באדום, גידול בציאן ופיברובלסטים בכחול. (D) הסכימה מראה תוספת של תווך למאגרים, עם 350 μL בצד הגבוה ו-50 μL בצד הנמוך כדי ליצור את ראש הלחץ ההידרוסטטי. (E) יום 2 של תרבית VMT מראה פיברובלסטים וEC מתחילים להימתח וליצור את רשת כלי הדם. סרגל קנה מידה = 200 מיקרומטר. לחץ כאן כדי להציג גרסה גדולה יותר של איור זה.
{kind=link}
4. תחזוקת מכשירים ויישומים ניסיוניים
- תחזוקה וטיפול תרופתי
הערה: כדי לשמור על זרימה במערכת, יש ליצור מחדש לחץ הידרוסטטי מדי יום על ידי צנרת נפח המדיה מהצד הנמוך בחזרה לצד הגבוה או להיפך, להבטיח שהנפח הכולל בצד הגבוה יישאר על 350 מיקרוליטר. כיוון הזרימה משתנה כל יום לאחר יום 2 של הקמת VMO או VMT. פרטי תחזוקה וטיפול נוספים מסופקים להלן.- החליפו מדיה אחת ליומיים עם EGM2 מלא עד להתבססות מלאה של כלי הדם (יום 5-6). לשאוף מדיה ישנה לחלוטין ולהחליף בארות בלחץ גבוה (350 μL) ובארות בלחץ נמוך (50 μL).
הערה: ניתן לבצע קביעה ניסיונית של פורמולציות מדיה ממוטבות עבור סוגי תאים אחרים, לעתים קרובות באמצעות תערובת של 50:50 או הוספה של רכיבים ספציפיים ל-EGM2. - לאחר יצירת רשת כלי הדם והתפתחות מלאה של הרקמה (יום 4-7), בצע בדיקת זילוח דקסטרן לפני השימוש במכשירים לניסויים (שלב 4.2.1). השתמש רק במכשירים שיש להם זילוח מספיק לתוך תא הרקמה.
- עבור ניסויים באמצעות טיפולים, ביום תחילת הטיפול, צלם תמונות בכל ערוצי הפלואורסצנט עבור כל מכשיר. זה ישמש כנקודת בסיס.
- יש לטפל במכשירים בעלי הטיפול הרצוי על ידי החלפת המדיום בתווך טרי המכיל את התרופה המדוללת בריכוז הרצוי. ודא שהתרופות מדוללות בכלי רכב מתאימים בהתאם להמלצת היצרן, אך לא יעלו על 0.01% DMSO בתקשורת.
- חשוף מכשירים לתרופה למשך פרק הזמן הרצוי (בדרך כלל 48 שעות, אך ניתן ליידע אותו על ידי פרמקוקינטיקה ).
- צלם כל ערוץ של כל מכשיר במרווח הזמן הרצוי כדי לעקוב אחר תגובת הטיפול. שמור על לוחות כפי שמצוין בשלב 4.1.1 למשך הניסוי.
- בסיום הניסוי, יש לתלות את לוחות האקונומיקה ולהניח במיכל מסוכן ביולוגית, לתקן עם 4% PFA עבור צביעה אימונופלואורסצנטית (שלב 4.3.), או לקצור עבור בידוד תאים חיים או RNA (שלב 4.4).
- החליפו מדיה אחת ליומיים עם EGM2 מלא עד להתבססות מלאה של כלי הדם (יום 5-6). לשאוף מדיה ישנה לחלוטין ולהחליף בארות בלחץ גבוה (350 μL) ובארות בלחץ נמוך (50 μL).
- מבחני זילוח
- זילוח של דקסטרן
הערה: ניתן לקבוע חדירות/פטנט של כלי הדם על ידי ערבוב רשת כלי הדם עם דקסטרן בעל תווית פלואורסצנטית במשקלים מולקולריים משתנים (40 kD, 70 kD או 150 kD). ניתן להשתמש ב- FITC- או רודמין-דקסטרן בהתאם לתווית הפלואורסצנטית של האיחוד האירופי.- לפני הזלוף, קבע את החשיפה המתאימה של תעלת FITC או רודמין על-ידי הוספת כמה μL של דקסטרן בתוך התעלה הנוזלית או התא של מכשיר ריק. הגדר את זמן החשיפה מעט מתחת לרמת הרוויה באמצעות שימוש בתוכנת מיקרוסקופ כדי להציג היסטוגרמה של עוצמות פיקסלים, ולהבטיח טווח דינמי המאופיין בפיקסלים מפוזרים באופן אחיד ללא ריכוז בולט של ערכים בעוצמות גבוהות.
- צלם מיקרוגרפים של כל המכשירים בערוצים מעניינים, כולל תמונת רקע של כל המכשירים בערוץ דקסטרן פלואורסצנטי, כדי לכייל על הרקע. השתמשו באותה חשיפה שנקבעה לעיל ויישרו את תא הרקמה במרכז מסגרת התמונה כדי להבטיח תמונות עקביות לכימות.
- הכינו מלאי עיקרי של FITC-דקסטרן או רודמין-דקסטרן בריכוז של 5 מ"ג/מ"ל ב-1x DPBS. מלאי זה יכול להישמר ב 4 °C (75 °F).
- להכנת מלאי תקין, יש לדלל מלאי של 5 מ"ג/מ"ל לריכוז סופי של 50 מיקרוגרם/מ"ל ב-EGM2.
- החלף את המדיה במאגרים בתמיסת דקסטרן מדוללת כחצי נפח מקסימלי (175 μL לבאר אחת ולצד הגבוה בתעלה העליונה או התחתונה של תא הרקמה). החלף את המדיה בבארות האחרות כך שהצד הגבוה הלא מצומד יקבל 175 μL של EGM2 טרי ללא דקסטרן, והבארות בצד הנמוך יכילו רק 50 μL בכל אחת.
הערה: יש להוסיף דקסטרן רק לצד אחד של התעלות המיקרופלואידיות (עליונות או תחתונות) כדי לאפשר הדמיה של צבע העובר דרך הצד בלחץ גבוה, לתוך מיטת כלי הדם, והחוצה מהצד בלחץ נמוך. - מתחת למיקרוסקופ, צפו בדקסטרן הפלואורסצנטי שזורם דרך רשת כלי הדם. זה יקרה בדרך כלל תוך כ -2 דקות לאחר הוספת הצבע למאגר המדיה.
- התחל לצלם את תעלת דקסטרן פלואורסצנטית (וערוצים אחרים, אם תרצה). זוהי נקודת זמן T = 0. צלם תמונות נוספות במספר נקודות זמן (בדרך כלל כל 10 דקות) או בתמונה אחת של נקודת קצה.
- זילוח תאים
הערה: סוגי תאים שונים יכולים להיות מחוררים דרך כלי הדם בהתאם לתכנון המחקר, כולל לימפוציטים או מקרופאגים למחקרי אימונולוגיה של סרטן, כמו גם תאים סרטניים למחקרי גרורות. תאים חייבים להיות מסומנים באופן פלואורסצנטי כדי להקל על המעקב לאורך זמן.- לפחות שעתיים לפני ניקוז התאים, יש לבצע זילוח דקסטרן בכל המכשירים כמפורט לעיל. שלב זה חשוב כדי לקבוע את פטנטיות כלי הדם לפני הוספת תאים.
- קבע את חשיפת המצלמה המתאימה לניקוב התאים. קח דגימה קטנה של תאים כדי לצפות מתחת למיקרוסקופ ולהגדיר את זמן החשיפה עבור סמן פלואורסצנטי זה. הגדירו את זמן החשיפה ממש מתחת לרמת הרוויה.
- צלם מיקרוגרפים של כל המכשירים בערוצים מעניינים, כולל תמונת רקע של כל המכשירים בערוץ דקסטרן פלואורסצנטי, כדי לכייל על הרקע. השתמש באותה חשיפה שנקבעה בשלב 4.2.2.1.
- אשרו כי דקסטרן התפזר לחלוטין מחוץ לתאי הרקמה לפני קצירת התאים לזילוח. קצור וספור את התאים המעניינים.
- השהה מחדש תאים בצפיפות מתאימה לתוך EGM2. לדוגמה, תאי T מתווספים בדרך כלל בערך 1 x 106 תאים / מ"ל כדי לחקות את הריכוז בדם.
הערה: EC רגישים להרכב מדיה אך יכולים לסבול עד 50% ערבוב עם רוב המדיה האחרת. בדוק מראש. - הוסף 175 μL של תרחיף תאים לבאר אחת בצד הגבוה של כל מכשיר והוסף 175 μL של EGM2 שלם לבאר השנייה. בצד הנמוך, הוסף 50 μL של מדיה מלאה EGM2 לשתי הבארות.
- מתחת למיקרוסקופ, שימו לב שהתאים הפלואורסצנטיים יזרמו דרך רשת כלי הדם. זה יתרחש בדרך כלל תוך כ -2 דקות לאחר הוספת התאים למאגר המדיה.
- לאחר שזרימת התא נוצרת, התחל לצלם את תעלת התא הפלואורסצנטית (וערוצים אחרים, אם תרצה). זוהי נקודת זמן T = 0. צלם תמונות נוספות במספר נקודות זמן או תמונה אחת של נקודת קצה, בהתאם לעיצוב המחקר. לדוגמה, הדמיה כל 10 דקות תגרום למסלולי זמן ברזולוציה גבוהה או כל 6-12 שעות כדי לעקוב אחר תנועות תאים תקופתיות.
- זילוח של דקסטרן
- צביעה אימונופלואורסצנטית (IF)
- שאפו מדיה מבארות. הוסף 200 μL של 4% PFA לשתי הבארות של הצד הגבוה של כל יחידת התקן ו- 50 μL לצד הנמוך. אפשר ל-PFA לזרום דרך התאים במשך 15 דקות בטמפרטורת החדר או 30 דקות ב-4°C.
- במהלך הדגירה, להכין צלחת 24 באר עם 500 μL של 1x PBS לכל באר. חשב כמה בארות דרושות כדי להכתים כל מכשיר.
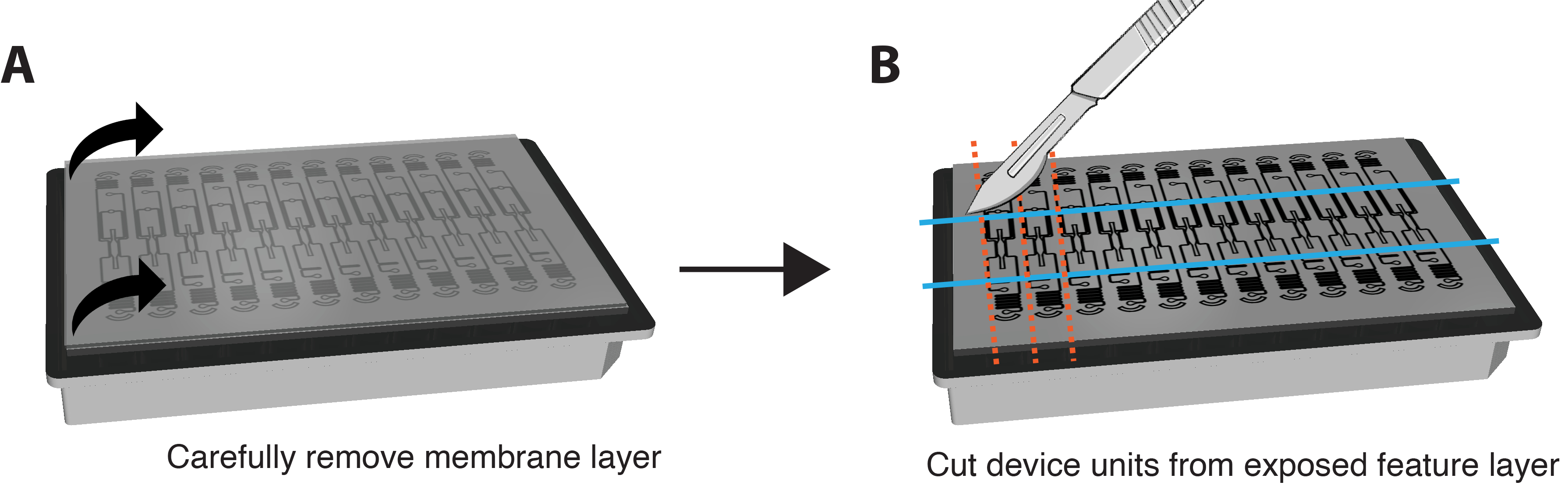
- הסר את PFA לחלוטין מבארות. הופכים את הצלחת ומסירים בזהירות את גב הפלסטיק על הממברנה.
הערה: אם ניתן לבצע צביעה גם באתרה מבלי להסיר את הממברנה וההתקן. לשם כך, בצע שלבי צביעה על ידי החדרת ריאגנטים דרך VMO/VMT והגדל את משך הזמן של כל שלב דגירה בערך פי 6. - בעדינות רבה ובזהירות לקלף את שכבת הקרום התחתונה משכבת התכונה של המכשיר כדי לחשוף את תאי הרקמה על ידי אחיזה בשתי הפינות ומשיכה מטה בתנועה איטית וחלקה. רוב הרקמה צריכה להישאר בתא הרקמה. עיין באיור 3A.
- השתמשו בסכין גילוח או באזמל כדי להפעיל מספיק כוח כדי לחתוך באופן מלא את שכבת התכונה ולחתוך מלבן קטן סביב כל יחידת התקן בנפרד, כפי שמוצג באיור 3B.
- תקעו מרית בין שכבת התכונה לבין צלחת הבאר. הפעילו לחץ עדין מתחת לשכבת התכונה כדי להסיר בזהירות את כל שכבת התכונה המכילה את תא הרקמה מלוח הבאר.
- הניחו כל פיסת שכבת PDMS מלבנית המכילה את הרקמה עם הפנים כלפי מטה לבאר אחת המכילה PBS.
- לאחר שכל היחידות נמצאות בבארות, שטפו עם PBS על ידי הנחת הצלחת על נדנדה עדינה למשך 5 דקות, שאיפת PBS מהבאר והחלפתה ב-500 מיקרוליטר PBS טרי. חזרו על הפעולה במשך 3 כביסות בסך הכל.
- שאפו PBS מכל באר וחדרו רקמות עם 500 μL של 0.5% Triton-X ב-PBS, 2x למשך 10 דקות כל אחת על נדנדה עדינה. הסר תמיסת חלחול.
- יש לחסום ב-500 מיקרוליטר של 10% סרום ב-0.1% Triton-X לכל מכשיר למשך שעה אחת בטמפרטורת החדר עם נדנוד עדין.
- לדלל נוגדנים ראשוניים בסרום 3% ב-0.1% טריטון-X לריכוז ולנפח הרצויים. הסר את תמיסת החסימה והוסף מספיק תמיסת נוגדנים ראשונית כדי לכסות באופן מלא את תחתית כל באר ולאפשר תנועה חופשית של רקמות המכשיר (~ 200 μL). מכסים את הצלחת בסרט שקוף.
- לדגור את הצלחת מתנדנד לילה ב 4 מעלות צלזיוס. למחרת, החזירו את הצלחת המכילה רקמות המכשיר לטמפרטורת החדר (~ 15 דקות).
- יש לשאוף תמיסת נוגדנים ראשונית מכל באר ולשטוף תאים עם 500 μL של PBS, 3x למשך 5 דקות כל אחד על נדנדה עדינה.
- הוסף 200 μL של נוגדן משני בסרום 3% ב 0.1% Triton-X בריכוז הרצוי. דוגרים על הצלחת בנדנוד עדין במשך שעה בטמפרטורת החדר בחושך.
- יש לשאוף תמיסת נוגדנים משנית ולשטוף עם PBS, 3 פעמים למשך 5 דקות כל אחת עם נדנוד עדין. הוסף תמיסה של 1x DAPI ב-0.1% Triton-X למשך 10 דקות תוך כדי נדנוד בחושך.
- הסר מהצלחת מגזרות התקן מלבניות המכילות רקמות מוכתמות באמצעות פינצטה והנח את צד הרקמה כלפי מעלה על מגבת נייר.
- פיפטה כמה μL (~ 10 μL) של תמיסת אנטי-דהייה ישירות על כל תא וכיסוי, תוך הקפדה לא להכניס בועות. יש לאפשר לאנטי-דהייה להירפא על תאים למשך הלילה בטמפרטורת החדר בחושך ולאחר מכן להמשיך בהדמיה.
- בידוד רקמות ותאים לבדיקות מולקולריות
הערה: כל צלחת תפוקה גבוהה תכיל בערך 1-2 x 105 תאים, בהתאם לנקודת הזמן של הקציר. שנה את קנה המידה של מספר העותקים המשוכפלים הניסיוניים כדי לקחת בחשבון את סך התאים וכן אובדן פוטנציאלי במהלך הקציר.- אנליזות של תא בודד
- שאפו מדיה מכל באר והפכו את הצלחת בעלת התפוקה הגבוהה כך ששכבת המכשיר פונה כלפי מעלה.
- הסר את גב הפלסטיק על הממברנה. קלף בעדינות ובזהירות רבה את הקרום התחתון של PDMS משכבת התכונות של המכשיר כדי לחשוף את תאי הרקמה על ידי אחיזה בשתי הפינות ומשיכה מטה בתנועה איטית וחלקה.
- ניתן להסיר את הממברנה גם עם מליטה נכונה. רוב הרקמה צריכה להישאר בתא הרקמה לאחר הסרת הממברנה; עם זאת, אם חלק כלשהו דבוק לממברנה, בצע את השלבים הבאים על הממברנה עצמה.
- שטפו כל יחידת מכשיר עם 500 μL של HBSS או PBS. לכל יחידת מכשיר, הוסף 100 μL של מגיב דיסוציאציה ואפשר לו לשבת כטיפה על גבי המכשיר. מחזירים את הצלחת לאינקובטור של 37 מעלות צלזיוס למשך 5 דקות.
- לאחר העיכול, השתמש פיפטה P200 כדי צינורות למעלה ולמטה על פני חדרי הרקמה ולאסוף את הרקמות מגיב דיסוציאציה. הזז את קצה הצינור קדימה ואחורה על פני כל מכשיר כדי להבטיח הסרה מלאה של הרקמה ולאסוף לתוך חרוט 15 מ"ל עם 500 μL של EGM2 כדי לנטרל את מגיב הדיסוציאציה.
- להסרה מלאה של התאים הנותרים מתא הרקמה, יש להוסיף 500 μL של EGM2 לכל יחידת התקן ולשטוף עם פיפטה P200.
- צנטריפוגה את תמיסת העיכול המכילה את הרקמות שנעקרו ב 300 x גרם במשך 5 דקות ב 4 ° C כדי גלולה תאים בודדים ורקמות שלמות.
- יש לשאוף בזהירות מדיה ולהוסיף 500 μL של 1 מ"ג/מ"ל (200 U/mL) collagenase סוג IV, 0.1 mg/mL hyaluronidase type V ו-200 U/mL DNAse type IV ב-HBSS.
- לאחר השעיה עדינה, הניחו לתמיסה לשבת במשך 2 דקות בטמפרטורת החדר לפני שתפיצו שוב בעדינות כדי לנתק את הג'ל.
- לשטוף את העיכול לערבב עם 10 מ"ל של EGM2 וצנטריפוגה ב 300 x גרם במשך 5 דקות ב 4 ° C. יש להשהות מחדש תאים ב-DPBS אחד עם 1% BSA או HSA ולעבור דרך מסנן 70 מיקרומטר שהורטב מראש על ידי סיבוב במהירות של 200 x גרם למשך דקה אחת.
- ספרו את התאים וכווננו את עוצמת הקול כך שהריכוז הסופי יהיה 1000 תאים למיקרוליטר. לאחר מכן ניתן להעביר תרחיפים תאיים ל-FACS, ציטומטריית זרימה או ריצוף RNA חד-תאי.
- בידוד RNA מרקמות שלמות
- בצע את שלבים 4.4.1.1 ו- 4.4.1.2 לעיל.
- הוסף כ -10 μL של חיץ RNA lysis על כל תא רקמה חשוף, להבטיח כי מאגר מאגר ישירות על גבי הרקמות. אין להשתמש ביותר מ-100 μL של מאגר RNA lysis בסך הכל.
- יש לדגור במשך 3 דקות בטמפרטורת החדר. השתמש ב- P20 כדי לצנרת למעלה ולמטה בכל יחידת התקן והשתמש בקצה הצינור כדי לגרד כל חומר שאריות מתא הרקמה במידת הצורך.
- העבר חיץ ליזיס רב ככל האפשר לתוך צינור מיקרוצנטריפוגה 1.5 מ"ל. חזור על שלבים 4.4.2.2.-4.4.2.3. עבור שאר המכשירים ודגימות מאגר לתוך צינור 1.5 מ"ל.
- עקוב אחר הוראות היצרן לבידוד RNA, בהתאם לערכה או לריאגנטים.
- אנליזות של תא בודד

איור 3. הכנת מצע לצביעת חיסון. (A) סכמה של פלטפורמת התקן מורכבת במלואה עם שכבת קרום למעלה. כדי להסיר את הממברנה, משכו בזהירות כל פינה של השכבה החיצונית כלפי מטה בתנועה יציבה ועדינה. (B) לאחר הסרת שכבת הממברנה לחלוטין, השתמש בלהב, אזמל או סכין כדי לחתוך מלבנים סביב תא הרקמה של כל יחידת מכשיר, תוך הקפדה שלא לחתוך לתוך הרקמה עצמה. לאחר מכן ניתן לתקוע מרית מתחת לכל מלבן כדי לעקור אותו מהצלחת ולהכניס כל יחידה לבאר אחת של צלחת בת 24 בארות עם PBS להכתמה. אנא לחץ כאן כדי להציג גרסה גדולה יותר של איור זה.
{kind=link}
תוצאות
בעקבות הפרוטוקולים המתוארים כאן, VMOs ו- VMTs הוקמו באמצעות EC, NHLF, שנרכשו באופן מסחרי, ועבור VMT, קו תאי סרטן השד טריפל נגטיב MDA-MB-231. VMOs מבוססים היו גם מחוררים עם תאים סרטניים כדי לחקות גרורות. בכל מודל, ביום החמישי של התרבית המשותפת, רשת כלי דם מתכנסת בעצמה בתגובה לזרימה המונעת על ידי כוח הכבידה על ?...
Discussion
כמעט כל רקמה בגוף מקבלת חומרים מזינים וחמצן דרך כלי הדם, מה שהופך אותה למרכיב קריטי למידול מחלות מציאותי ולבדיקת תרופות במבחנה. יתר על כן, מספר ממאירויות ומצבי מחלה מוגדרים על ידי תפקוד לקוי של אנדותל כלי הדם וחדירות יתר3. יש לציין כי בסרטן, כלי הדם הקשורים לגידול הם לעתים ?...
Disclosures
ל-CCWH יש עניין במניות בחברת Aracari Biosciences, Inc., אשר ממסחרת גרסה של הטכנולוגיה המתוארת במאמר זה. תנאי הסדר זה נבדקו ואושרו על ידי אוניברסיטת קליפורניה, אירווין, בהתאם למדיניות ניגוד העניינים שלה. אין ניגודי עניינים אחרים.
Acknowledgements
אנו מודים לחברי מעבדתו של ד"ר כריסטופר יוז על תרומתם המוערכת להליכים המתוארים, כמו גם לשותפינו במעבדתו של ד"ר אברהם לי על עזרתם בעיצוב וייצור פלטפורמה. עבודה זו נתמכה על ידי המענקים הבאים: UG3/UH3 TR002137, R61/R33 HL154307, 1R01CA244571, 1R01 HL149748, U54 CA217378 (CCWH) ו-TL1 TR001415 and W81XWH2110393 (SJH).
Materials
| Name | Company | Catalog Number | Comments |
| Fabrication | |||
| (3-Mercaptopropyl)trimethoxysilane, 95% | Sigma-Aldrich | 175617-100G | |
| Greiner Bio-One μClear Bottom 96-well Polystyrene Microplates | Greiner Bio-One | 655096 | |
| Methanol ≥99.8% ACS | VWR Chemicals BDH | BDH1135-1LP | |
| MILTEX Sterile Disposable Biopsy Punch with Plunger, 1mm diameter, | Integra Miltex | 33-31AA-P/25 | |
| PDMS membrane | PAX Industries | HT-6240 | |
| Plasma Cleaner PDC-001 | Harrick Plasma | N/A | |
| Smooth-Cast 385 | Smooth-On | N/A | |
| SP Bel-Art Lab Companion Clear Polycarbonate Cabinet Style Vacuum Desiccator | Bel-Art | F42400-4031 | |
| Standard Lids with Condensation Rings, 96-well plate | VWR | 82050-827 | |
| SYLGARD 184 Silicone Elastomer Kit (PDMS) | Dow | 4019862 | |
| Cell culture/Loading | |||
| BioTek Lionheart FX Automated Microscope | Agilent | CYT5MFAW | |
| CELLvo Human Endothelial Progenitor Cells | StemBioSys | N/A | |
| Collagen I, rat tail | Enzo Life Sciences | ||
| Collagenase from Clostridium histolyticum (type 4) | Sigma-Aldrich | C5138 | |
| Corning Hank’s Balanced Salt Solution, 1X without calcium and magnesium | Corning | 21-021-CV | |
| Corning DMEM with L-Glutamine, 4.5g/L Glucose and Sodium Pyruvate | Corning | 10013CV | |
| DAPI | Sigma-Aldrich | D9542 | |
| DPBS, no calcium, no magnesium | Gibco | 14190144 | |
| EGM-2 Endothelial Cell Growth Medium-2 BulletKit | Lonza | CC-3162 | |
| Fibrinogen from bovine plasma | Neta Scientific | SIAL-341573 | |
| Fibronectin human plasma | Sigma-Aldrich | F0895 | |
| Fluorescein isothiocyanate–dextran (70kDa) | Sigma-Aldrich | FD70S-1G | |
| Gelatin from porcine skin | Sigma-Aldrich | G1890 | |
| Hyaluronidase from sheep testes (type 4) | Sigma-Aldrich | H6254 | |
| Laminin Mouse Protein | Gibco | 23017015 | |
| Leica TCS SP8 | Leica | N/A | |
| MDA-MB-231 | ATCC | HTB-26 | |
| NHLF – Normal Human Lung Fibroblasts | Lonza | CC-2512 | |
| Nikon Eclipse Ti | Nikon | N/A | |
| Paraformaldehyde 4% in 0.1M Phosphate BufferSaline, pH 7.4 | Electron Microscopy Sciences | 15735-90-1L | |
| PBMCs - Peripheral blood mononuclear cells | Lonza | CC-2702 | |
| PBS, pH 7.4 | Gibco | 10010049 | |
| Premium Grade Fetal Bovine Serum (FBS), Heat Inactivated | Avantor Seradigm | 97068-091 | |
| ProLong Gold Antifade Mountant | Invitrogen | P10144 | |
| Quick-RNA Microprep Kit | Zymo Research | R1051 | |
| Thrombin from bovine plasma | Sigma-Aldrich | T4648 | |
| Triton X-100 (Electrophoresis), | Fisher BioReagents | BP151-100 | |
| TrypLE Express Enzyme (1X), phenol red | Gibco | 12605028 | |
| Trypsin-EDTA (0.05%), phenol red | Gibco | 25300062 | |
| Vasculife | Lifeline Cell Technology | LL-0003 |
References
- Siegel, R. L., Miller, K. D., Wagle, N. S., Jemal, A. Cancer statistics, 2023. CA Cancer J Clin. 73 (1), 17-48 (2023).
- Hachey, S. J., Hughes, C. C. W. Applications of tumor chip technology. Lab Chip. 18 (19), 2893-2912 (2018).
- Ewald, M. L., Chen, Y. H., Lee, A. P., Hughes, C. C. W. The vascular niche in next generation microphysiological systems. Lab Chip. 21 (17), 3615-3616 (2021).
- Osaki, T., Sivathanu, V., Kamm, R. D. Vascularized microfluidic organ-chips for drug screening, disease models and tissue engineering. Curr Opin Biotechnol. 52, 116-123 (2018).
- Shirure, V. S., Hughes, C. C. W., George, S. C. Engineering vascularized organoid-on-a-chip models. Annu Rev Biomed Eng. 23, 141-167 (2021).
- Del Piccolo, N., et al. Tumor-on-chip modeling of organ-specific cancer and metastasis. Adv Drug Deliv Rev. 175, 113798 (2021).
- Sontheimer-Phelps, A., Hassell, B. A., Ingber, D. E. Modelling cancer in microfluidic human organs-on-chips. Nat Rev Cancer. 19 (2), 65-81 (2019).
- Sobrino, A., et al. 3D microtumors in vitro supported by perfused vascular networks. Sci Rep. 6, 31589 (2016).
- Phan, D. T. T., et al. A vascularized and perfused organ-on-a-chip platform for large-scale drug screening applications. Lab Chip. 17 (3), 511-520 (2017).
- Hachey, S. J., et al. An in vitro vascularized micro-tumor model of human colorectal cancer recapitulates in vivo responses to standard-of-care therapy. Lab Chip. 21 (7), 1333-1351 (2021).
- Hachey, S. J., et al. A Human Vascularized Micro-Tumor Model of Patient-Derived Colorectal Cancer Recapitulates Clinical Disease. Transl Res. 255, 97-108 (2023).
- Liu, Y., et al. Human in vitro vascularized micro-organ and micro-tumor models are reproducible organ-on-a-chip platforms for studies of anticancer drugs. Toxicology. 445, 152601 (2020).
- Jahid, S., et al. Structure-based Design of CDC42 Effector Interaction Inhibitors for the Treatment of Cancer. Cell Rep. 39 (4), 110760 (2022).
- Hsu, Y. H., Moya, M. L., Hughes, C. C. W., George, S. C., Lee, A. P. A microfluidic platform for generating large-scale nearly identical human microphysiological vascularized tissue arrays. Lab Chip. 13 (15), 2990-2998 (2013).
- Moya, M. L., Hsu, Y. H., Lee, A. P., Christopher, C. W. H., George, S. C. In vitro perfused human capillary networks. Tissue Eng - Part C: Methods. 19 (9), 730-737 (2013).
- Wang, X., et al. An on-chip microfluidic pressure regulator that facilitates reproducible loading of cells and hydrogels into microphysiological system platforms. Lab Chip. 16 (5), 868-876 (2016).
- Phan, D. T., et al. Blood-brain barrier-on-a-chip: Microphysiological systems that capture the complexity of the blood-central nervous system interface. Exp Biol Med. 242 (17), 1669-1678 (2017).
- Kurokawa, Y. K., et al. Human induced pluripotent stem cell-derived endothelial cells for three-dimensional microphysiological systems. Tissue Eng Part C: Methods. 23 (8), 474-484 (2017).
- Romero-López, M., et al. Recapitulating the human tumor microenvironment: Colon tumor-derived extracellular matrix promotes angiogenesis and tumor cell growth. Biomaterials. 116, 118-129 (2017).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 9 (7), 676-682 (2012).
- Carpenter, A. E., et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7 (10), R100 (2006).
- Zudaire, E., Gambardella, L., Kurcz, C., Vermeren, S. A computational tool for quantitative analysis of vascular networks. PLoS one. 6 (11), e27385 (2011).
- Corliss, B. A., et al. REAVER: A program for improved analysis of high-resolution vascular network images. Microcirculation. 27 (5), e12618 (2020).
- Urban, G., et al. Deep learning for drug discovery and cancer research: Automated analysis of vascularization images. IEEE/ACM Trans Comput Biol Bioinform. 16 (3), 1029-1035 (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved