Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Создание физиологической васкуляризированной модели микроопухоли человека для исследования рака
В этой статье
Резюме
Этот протокол представляет собой физиологически релевантную модель опухоли на чипе для выполнения высокопроизводительных фундаментальных и трансляционных исследований рака человека, продвижения скрининга лекарств, моделирования заболеваний и персонализированной медицины с описанием процедур загрузки, поддержания и оценки.
Аннотация
Отсутствие валидированных моделей рака, которые повторяют опухолевое микроокружение солидных раковых опухолей in vitro , остается серьезным препятствием для доклинических исследований рака и терапевтических разработок. Чтобы решить эту проблему, мы разработали васкуляризированную микроопухоль (VMT), или опухолевый чип, микрофизиологическую систему, которая реалистично моделирует сложное микроокружение опухоли человека. VMT формируется de novo в микрофлюидной платформе путем совместного культивирования нескольких типов клеток человека в динамических, физиологических условиях потока. Эта тканеинженерная микроопухолевая конструкция включает в себя живую перфузионную сосудистую сеть, которая поддерживает растущую опухолевую массу так же, как это делают новообразованные сосуды in vivo. Важно отметить, что лекарственные препараты и иммунные клетки должны пересекать эндотелиальный слой, чтобы достичь опухоли, моделируя in vivo физиологические барьеры для терапевтической доставки и эффективности. Поскольку платформа VMT оптически прозрачна, визуализация динамических процессов, таких как экстравазация иммунных клеток и метастазирование, может быть достигнута с помощью прямой визуализации флуоресцентно меченных клеток в ткани. Кроме того, VMT сохраняет гетерогенность опухоли in vivo , сигнатуры экспрессии генов и реакцию на лекарственные препараты. Практически любой тип опухоли может быть адаптирован к платформе, а первичные клетки из свежих хирургических тканей растут и реагируют на медикаментозное лечение в VMT, прокладывая путь к по-настоящему персонализированной медицине. Описаны методы создания ВМТ и его использования для онкологических исследований. Этот инновационный подход открывает новые возможности для изучения опухолей и реакции на лекарственные препараты, предоставляя исследователям мощный инструмент для продвижения исследований рака.
Введение
Рак остается серьезной проблемой здравоохранения во всем мире и является второй по значимости причиной смерти в Соединенных Штатах. Только на 2023 год Национальный центр статистики здравоохранения прогнозирует более 1,9 миллиона новых случаев заболевания раком и более 600 000 смертей от рака вСША1, что подчеркивает острую необходимость в эффективных подходах к лечению. Тем не менее, в настоящее время только 5,1% противоопухолевых препаратов, участвующих в клинических испытаниях, в конечном итоге получают одобрение FDA. Неспособность перспективных кандидатов успешно пройти клинические испытания может быть частично связана с использованием нефизиологических модельных систем, таких как 2D и сфероидные культуры, во время доклиническойразработки лекарственных средств. В этих классических моделях рака отсутствуют важнейшие компоненты микроокружения опухоли, такие как стромальная ниша, ассоциированные иммунные клетки и перфузионная сосудистая сеть, которые являются ключевыми детерминантами терапевтической резистентности и прогрессирования заболевания. Таким образом, для улучшения клинической трансляции доклинических результатов необходима новая модельная система, которая лучше имитирует микроокружение опухоли человека in vivo .
Область тканевой инженерии быстро развивается, предлагая более совершенные методы изучения заболеваний человека в лабораторных условиях. Одним из важных достижений является появление микрофизиологических систем (МПС), также известных как органные чипы или тканевые чипы, которые представляют собой функциональные, миниатюрные человеческие органы, способные воспроизводить здоровые или больныесостояния. В этом контексте для онкологических исследований были разработаны опухолевые чипы, представляющие собой трехмерные микрофлюидные модели опухолей человека in vitro 2,3,4,5,6,7,8,9,10,11,12,13 . Эти усовершенствованные модели включают биохимические и биофизические сигналы в динамическом микроокружении опухоли, что позволяет исследователям изучать поведение опухоли и реакцию на лечение в более физиологически релевантном контексте. Однако, несмотря на эти достижения, лишь немногим группам удалось успешно внедрить живую, функциональную сосудистую сеть, особенно ту, которая самоанализируется в ответ на физиологический поток 3,4,5,6. Включение функциональной сосудистой сети имеет решающее значение, поскольку позволяет моделировать физические барьеры, влияющие на доставку лекарств или клеток, наведение клеток в различные микроокружения и трансэндотелиальную миграцию опухолевых, стромальных и иммунных клеток. Благодаря этой функции опухолевый чип может лучше представить сложности, наблюдаемые в микроокружении опухоли in vivo.
Чтобы удовлетворить эту неудовлетворенную потребность, мы разработали новую платформу для скрининга лекарственных средств, которая позволяет формировать сети микрососудов внутри микрофлюидного устройства 8,9,10,11,12,13,14,15,16. Эта базовая платформа органного чипа, называемая васкуляризированным микроорганом (VMO), может быть адаптирована практически к любой системе органов для воспроизведения оригинальной физиологии тканей для моделирования заболеваний, скрининга лекарств и применения персонализированной медицины. ВМО устанавливаются путем совместного культивирования эндотелиальных колониеобразующих клеток эндотелиального происхождения (ECFC-EC), HUVEC или iPSC-EC (далее EC) и нескольких стромальных клеток в камере, включая нормальные фибробласты легких человека (NHLF), которые ремоделируют матрикс, и перициты, которые оборачивают и стабилизируют сосуды. VMO также может быть установлена в качестве системы модели рака путем совместного культивирования опухолевых клеток с ассоциированной стромой для создания васкуляризированной микроопухоли (VMT)8,9,10,11,12,13 или модели опухолевого чипа. Благодаря совместному культивированию нескольких типов клеток в среде динамического потока, перфузионные микрососудистые сети формируются de novo в тканевых камерах устройства, где васкулогенез строго регулируется скоростями интерстициального потока14,15. Среда прогоняется по микрофлюидным каналам устройства гидростатической напорной головкой, которая снабжает окружающие клетки тканевой камеры питательными веществами исключительно через микрососуды, с коэффициентом проницаемости 1,2 х 10-7 см/с, аналогично тому, что наблюдается для капилляров in vivo8.
Включение самоорганизующихся микрососудов в модель VMT представляет собой значительный прорыв, поскольку оно: 1) имитирует структуру и функции васкуляризированных опухолевых масс in vivo; 2) может моделировать ключевые этапы метастазирования, включая опухолево-эндотелиальные и стромальные клеточные взаимодействия; 3) устанавливает физиологически селективные барьеры для доставки питательных веществ и лекарственных средств, совершенствуя фармацевтический скрининг; и 4) позволяет проводить прямую оценку препаратов, обладающих антиангиогенными и антиметастатическими свойствами. Реплицируя доставку питательных веществ, лекарств и иммунных клеток in vivo в сложном 3D-микроокружении, платформа VMO/VMT является физиологически значимой моделью, которую можно использовать для проведения скрининга лекарств и изучения рака, сосудистой или органоспецифической биологии. Важно отметить, что VMT поддерживает рост различных типов опухолей, включая рак толстой кишки, меланому, рак молочной железы, глиобластому, рак легких, перитонеальный карциноматоз, рак яичников и рак поджелудочной железы 8,9,10,11,12,13. Помимо того, что микрофлюидная платформа является недорогой, легко устанавливаемой и пригодной для высокопроизводительных экспериментов, она полностью оптически совместима для анализа изображений в режиме реального времени взаимодействия опухоли и стромы и реакции на стимулы или терапевтические средства. Каждый тип клеток в системе помечен различными флуоресцентными маркерами, что позволяет напрямую визуализировать и отслеживать поведение клеток на протяжении всего эксперимента, создавая окно в динамическое микроокружение опухоли. Ранее мы показали, что VMT более точно моделирует in vivo рост опухоли, архитектуру, гетерогенность, сигнатуры экспрессии генов и реакцию на лекарственные препараты, чем стандартные методы культивирования10. Важно отметить, что VMT поддерживает рост и изучение клеток, полученных от пациентов, включая раковые клетки, что позволяет лучше моделировать патологию родительских опухолей, чем стандартные сфероидные культуры, и способствует дальнейшемуразвитию персонализированной медицины. В данной рукописи излагаются методы создания ВМТ, демонстрирующие ее полезность для изучения раковых опухолей человека.
протокол
1. Проектирование и изготовление
- Конструкция устройства
- Для изготовления микрофлюидных устройств создайте пресс-форму SU-8 с использованием слоя SU-8 толщиной 200 мкм, нанесенного на кремниевую пластину (очищенную RCA-1 и обработанную 2% фтористым водородом (HF)), с последующим этапом фотолитографии одной маски, как описано ранее 8,9.
- Отлите копию полидиметилсилоксана (PDMS) толщиной 4 мм из пресс-формы SU-8 для создания прочной полиуретановой формы для последующих этапов изготовления. Можно использовать различные итерации дизайна 8,9,10,11,12,13,14,15.
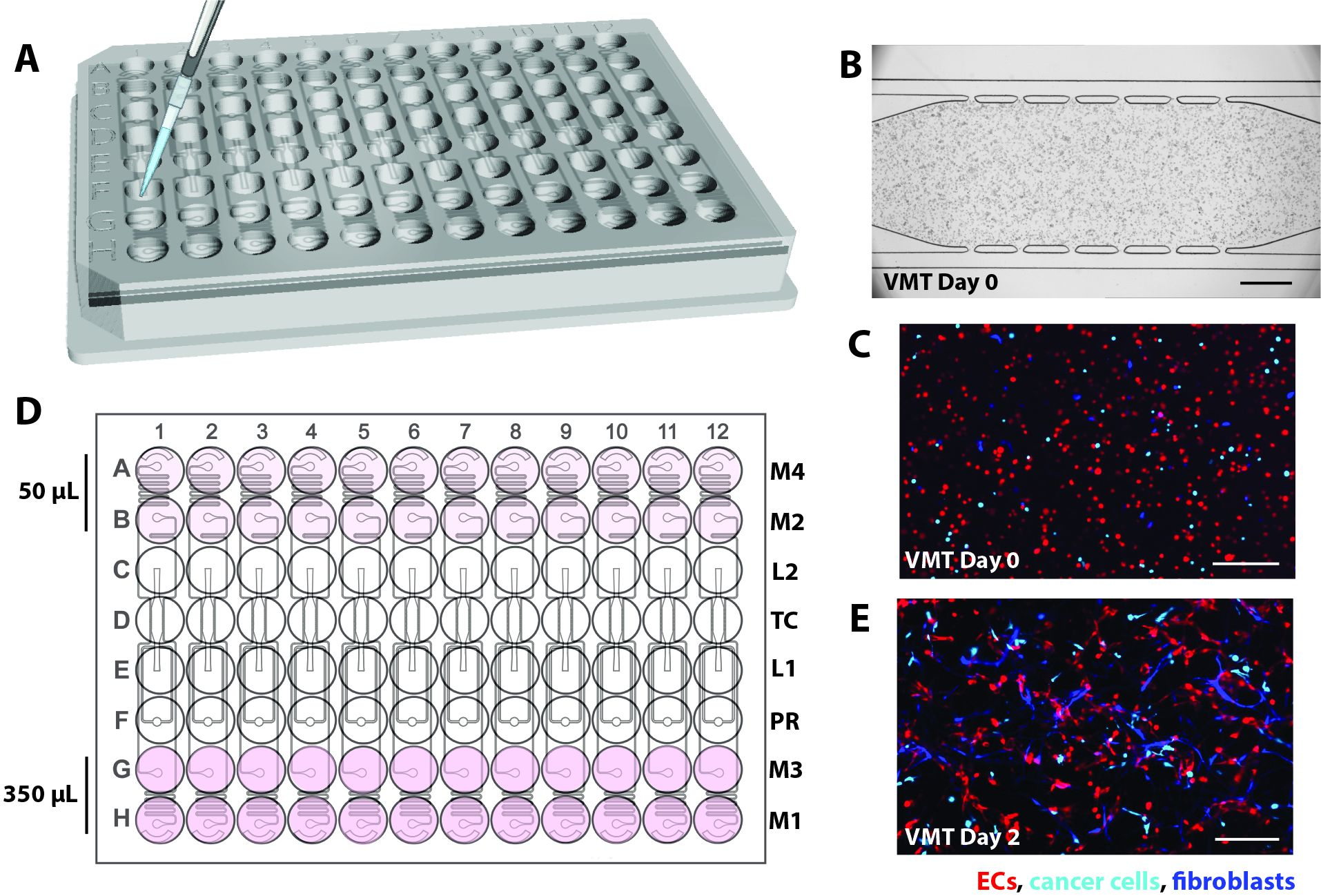
- В текущей итерации спроектируйте микрофлюидное устройство, которое будет индивидуально адаптировано к стандартному формату 96-луночного планшета и состоящему из характерного слоя PDMS толщиной 2 мм с 12 микрофлюидными устройствами, заключенными в тонкий (1/16 дюйма) прозрачный полимерный мембранный слой на дне (рис. 1A).
- Убедитесь, что отдельные тканевые блоки состоят из тканевой камеры, окруженной входным и выходным отверстиями для загрузки геля (L1) и выпускным отверстием (L2), регулятором давления (PR)16 и разъединенными микрофлюидными каналами, соединенными с 2 входами и выходами среды с каждой стороны (M1-M2, M3-M4; Рисунок 1Б).
- Расположите каждый входной и выходной отверстия в пределах одной скважины, которая служит резервуаром средней среды для создания гидростатического давления (10 ммH2O) в микрофлюидном канале. Чтобы обеспечить анастомозы сосудистой сети с наружными каналами, соедините микрофлюидные каналы с тканевой камерой через коммуникационные поры шириной 50 мкм (6 сверху, 6 снизу).
ПРИМЕЧАНИЕ: Микрофлюидные резисторы создают градиент внутритканевого давления 5 ммH2Oчерез тканевую камеру, который становится внутрипросветным, как только сосудистая сетьполностью сформирована. Последующие процедуры начинаются с полностью собранной пластины с высокой пропускной способностью.

Рисунок 1. Конструкция микрофлюидной платформы. (A) На схеме сборки платформы показан векторный слой PDMS с 12 устройствами, прикрепленными к бездонной 96-луночной пластине и запечатанными тонкой прозрачной полимерной мембраной. Каждый блок прибора занимает столбик лунок на плите. Один блок устройства, обведенный красным цветом, показан с подробными сведениями на (B). (B) На схеме одного устройства показана одна тканевая камера, расположенная в одной лунке от 96-луночной пластины, и два загрузочных отверстия с отверстиями на входе и выходе (L1-L2), пробитыми для введения клеточно-матричной смеси. Входные и выходные отверстия среды (М1-М2, М3-М4) пробиты и расположены в скважинах, которые служат резервуарами среды. Различные объемы среды создают градиент гидростатического давления в тканевой камере через несвязанные микрофлюидные каналы. Блок регулятора давления (PR) служит в качестве гелевого клапана для облегчения загрузки. Обратите внимание, что устройство имеет глубину 200 мкм, а тканевая камера составляет 2 мм x 6 мм. Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этого рисунка.
{kind=link}
2. Подготовка перед погрузкой
- Клеточная культура
- Содержат клетки согласно рекомендациям производителя в инкубаторе с влажностью 37 °C и 5%СО2 .
- Пластинчатые колбы T75 с трансдуцированными EC, NHLF или другими фибробластами/стромальными клетками и желаемыми раковыми клетками за 3-4 дня до загрузки с плотностью, указанной производителем и пользовательскими протоколами. Для этого протокола планшет 1 x 106 клеток в колбе для каждого типа клеток. Культуральный EC в полной среде для эндотелиального роста 2 (EGM2), NHLF в модифицированной среде Eagle Medium (DMEM) Dulbecco с 10% FBS и раковые клетки в соответствующих средах в зависимости от типа клеток.
- Поддерживайте клетки, подкармливая их соответствующими средами каждые 2-3 дня, и подтверждайте эффективность трансдукции или мечения, визуализируя клетки под флуоресцентным микроскопом. В день загрузки убедитесь, что EC сливается на 80%-100%, а NHLF - на 70%-80%.
- Приготовление фибриногена
- Приготовьте раствор фибриногена до желаемой концентрации (как правило, 5-8 мг/мл поддерживает образование устойчивой сосудистой сетки) с учетом процента свертываемости фибриногена. Рассчитайте необходимое количество фибриногена по следующему уравнению:
Фибриноген (мг) = (объем (мл)) x (концентрация (мг/мл))/ (% свертываемости) - Растворите фибриноген в соответствующем объеме эндотелиальной базальной среды 2 (EBM2), нагретом до 37 °C, осторожно щелкнув трубкой (не вихрить). Инкубируйте фибриноген на водяной бане с температурой 37 °C, чтобы он полностью перешел в раствор. Важно отметить, что не используйте полный носитель.
- Раствор фибриногена со стерильным фильтром с фильтром 0,22 мкм и аликвотой до требуемого объема, обычно 400 мкл на пробирку микроцентрифуги.
ПРИМЕЧАНИЕ: Другие матриксные белки (например, коллагены, фибронектин или ламинин) могут быть добавлены в смесь фибриногена.
- Приготовьте раствор фибриногена до желаемой концентрации (как правило, 5-8 мг/мл поддерживает образование устойчивой сосудистой сетки) с учетом процента свертываемости фибриногена. Рассчитайте необходимое количество фибриногена по следующему уравнению:
3. Загрузка образцов
ПРИМЕЧАНИЕ: Загрузка зависит от времени и должна быть завершена от начала (подъем ячейки) до конца (добавление среды в устройства) в течение примерно 1,5-1,75 часа для обеспечения оптимальных результатов. Каждый шаг отмечается рекомендуемым таймером, чтобы помочь пользователю не сбиться с пути.
- Подготовка материалов (День погрузки)
- Поместите на водяную баню с температурой 37 °C на 10-15 минут: сбалансированный солевой раствор Хэнка (HBSS) или фосфатно-солевой буфер (PBS) для промывки клеток, реагент для диссоциации клеток, среда (например, EGM2, DMEM)
- Храните в холодильнике при температуре 4 °C до готовности к употреблению следующие реагенты: тромбин, ламинин (размороженный в течение ночи при 4 °C).
- Разморозьте аликвоту фибриногена при комнатной температуре. Приготовьте 1,5 мкл аликвот тромбина в пробирки для микроцентрифуг по 500 мкл, по одной пробирке на единицу устройства. Убедитесь, что аликвота тромбина находится на дне каждой пробирки, чтобы облегчить загрузку.
- Поместите стерилизованные УФ-терапией пластины с высокой пропускной способностью в эксикатор не менее чем на 30 минут перед загрузкой, чтобы удалить воздух, попавший в микрофлюид.
- Подготовка клетки (Таймер старт = начало в 0 мин)
- Проверьте клетки под микроскопом при 4-кратном увеличении, чтобы подтвердить эффективность слияния и трансдукции.
- Промойте каждую колбу клеток T75 2 раза 5 мл HBSS и полностью аспирируйте. Добавьте 1 мл реагента для диссоциации в каждую колбу и инкубируйте при 37 °C, 5% CO2 в течение 1-2 мин.
- Осторожно постучите по тарелке ладонью и проверьте, все ли клетки приподняты.
- Смойте клетки с колбы 9 мл подходящей среды и соберите их в коническую колбу объемом 15 мл. Сразу же удалите маленькую аликвоту для подсчета клеток.
- Центрифугируют клетки при 300 x g в течение 3-5 мин при 4 °C. Центрифугируя ячейки, подсчитайте количество клеток. В слияющейся колбе Т75 EC или NHLF должно получиться не менее 2 x 106 ячеек.
- После центрифугирования аспирируйте среду и повторно суспендируйте гранулу в соответствующей среде в концентрации 1 x 106 клеток/мл. Держите клетки на льду.
- Приготовление смеси клеток и фибриногена (таймер = начало через 20 минут)
- Определите, сколько устройств будет загружено, добавив 1-2, чтобы учесть потери при пипетировании, и умножьте на объем смеси клеток/фибриногена, необходимый для каждого устройства. Это будет зависеть от конфигурации устройства, но для конструкции устройства, представленной в этой статье, требуется 6 мкл на устройство.
- Концентрация каждого типа клеток должна быть определена экспериментально. В качестве отправной точки загрузите EC в концентрации примерно 7 x 106 клеток/мл и NHLF в концентрации 3,5 x 106 клеток/мл. Концентрация раковых клеток может значительно варьироваться в зависимости от скорости их роста, но обычно находится в диапазоне 0,5-2 x10,6 клеток/мл. Используйте это уравнение для вычисления необходимого количества ячеек:
Необходимое количество клеток = (объем фибрина (мкл))/1000 мкл x (концентрация клеток) - Ресуспендируйте клетки в концентрации 1 x 106 клеток/мл и используйте следующее уравнение для определения необходимого объема клеток:
Необходимый объем клеток (мкл) = (необходимое количество клеток)/1000 - Смешайте соответствующие объемы EC, NHLF и раковых клеток (только для VMT) в конической пробирке и центрифуге при 300 x g в течение 3-5 мин при 4 °C.
- После отжима осторожно отсасывайте среду и удаляйте остатки среды рядом с гранулами. Аккуратно, но тщательно взвесьте гранулу в рассчитанный объем фибриногена, соблюдая особую осторожность, чтобы не попасть пузырьки воздуха. Держите на льду.
- Занесите стерилизованные планшеты и аликвоты тромбина в колпак для культивирования тканей.
- Загрузка устройств (Таймер = начало через 30-35 минут)
- С помощью пипетки P20 выпейтите 6 мкл объема из смеси клетка и фибрин. Обязательно пропитывайте смесь вверх и вниз не менее 5 раз, чтобы обеспечить однородную клеточную суспензию. Держите смесь на льду, чтобы замедлить свертывание.
- Осторожно смешайте клетку/фибрин с одной пробиркой тромбина, поместив наконечник пипетки непосредственно в аликвоту тромбина в нижней части пробирки. Сразу же проведите пипеткой вверх и вниз не менее 2 раз, стараясь не допустить появления пузырьков воздуха. Фибрин начнет сворачиваться после смешивания с тромбином, поэтому быстро, но целенаправленно выполните шаги 3.4.3. и 3.4.4. до того, как фибрин загустеет в наконечнике пипетки (~3 с).
- Поднимите пластину с высокой пропускной способностью под углом и быстро вставьте наконечник пипетки в одно из загрузочных отверстий устройства (L1 или L2). Схему см. на рисунке 2A .
- Плавным плавным плавным движением надавите на поршень пипетки до первого упора, чтобы ввести смесь клеток и фибрина в камеру ткани. Следите за тем, чтобы гель полностью прошел через камеру.
ПРИМЕЧАНИЕ: Слишком сильное давление на этом этапе может привести к разрыву геля в микрофлюидные каналы в верхней и/или нижней части камер тканей. - Осторожно положите планшет обратно в колпак для культивирования тканей, не снимая наконечник пипетки, не ослабляя поршень пипетки и не повреждая пипетку. Рукой поверните и извлеките наконечник дозатора из P20 и оставьте его в загрузочном отверстии. Не используйте кнопку выталкивателя для извлечения наконечника, так как это вызовет слишком большое давление.
- Перейдите к шагам 3.4.1-3.4.5 для остальных устройств.
- Когда загрузка будет завершена, дайте планшету спокойно постоять 2 минуты в вытяжке для культуры тканей.
- Снимите наконечники пипеток, осторожно повернув их и вытащив из загрузочных отверстий. Установите крышку на тарелку.
- Инкубируйте всю чашку в течение 15-20 минут в инкубаторе с температурой 37 °C, чтобы гель полностью полимеризовался.
- После инкубации проверьте каждый блок прибора под микроскопом. Убедитесь, что клетки равномерно распределены по всей камере без каких-либо пузырьков воздуха и что между тканевой камерой и микрофлюидными каналами есть четко видимая гелевая граница, как показано на рисунке 2B-C.
- Покрытие канала ламинином (Таймер = начало через 45-50 минут)
- После того, как гели полностью затвердеют, введите ламинин в микрофлюидные каналы, чтобы способствовать сосудистому анастомозу.
- С помощью P20 введите 4 мкл ламинина в каждый микрофлюидный канал (верхний и нижний) устройства. Вставьте наконечник пипетки в M1 или M3 и медленно вытесните ламинин, следя за тем, чтобы ламинин покрыл весь верхний канал, а затем повторите то же самое для M2 или M4, чтобы покрыть весь нижний канал.
- Определите ориентацию, пипетируя ламинин со стороны, противоположной регулятору давления, чтобы обеспечить достаточное давление для его проталкивания. Однако, если ламинин плохо перемещается с одной стороны, снимите наконечник с одной стороны и надавите на ламинин с другой стороны. Переход ко второму упору пипетки (т. е. проталкивание поршня до упора) может потребоваться для создания давления, достаточного для проталкивания ламинина через весь канал.
- Осторожно извлеките наконечник из входного/выходного отверстия для носителя. Не используйте кнопку выталкивания на P20.
- Повторите шаги 3.5.1-3.5.4 для каждого устройства и инкубируйте планшет при 37 °C, 5% CO2 в течение 10 мин.
- Добавление носителя (таймер = начало примерно через 1 час 10 минут)
- Добавьте 275 мкл полной среды EGM2 в резервуары несвязанных сред скважин в рядах A и B или G и H. Это будет высокая сторона, и ориентацию следует определить, сделав скважины на стороне, противоположной регулятору давления, большим объемом для начала. Среда будет выталкиваться с высокой стороны под действием силы тяжести.
- С помощью пипетки P200 введите 75 мкл среды на входе/выходе лунки, содержащие 275 мкл EGM2. Вставьте наконечник во впускное отверстие для носителя и медленно вытесните среду, следя за тем, чтобы среда прошла через канал и всплыла с другой стороны.
- Снимите наконечник пипетки и вытолкните оставшуюся среду из наконечника в резервуар для фильтрующего материала так, чтобы общий объем на верхней стороне составлял 350 мкл.
- Повторите шаги 3.6.1-3.6.3 для каждого устройства устройства, верхнего и нижнего каналов.
- Добавьте 50 мкл среды, чтобы полностью покрыть нижнюю сторону, лунки в рядах A и B или G и H, в зависимости от ориентации, описанной выше. Убедитесь, что дно колодца покрыто ровным слоем среды. На рисунке 2D приведена схема, показывающая объемы среды в резервуарах.
- Удаление пузырьков воздуха (после загрузки)
ПРИМЕЧАНИЕ: Удаление пузырьков является критически важным шагом для обеспечения надлежащего потока в каждом устройстве. Ко второму дню эндотелиальные клетки и фибробласты начнут растягиваться в ответ на поток (рис. 2E).- После добавления всех сред инкубируйте планшеты в течение 1-2 ч в инкубаторе с 5%CO2 при температуре 37 °C, прежде чем проверить наличие пузырьков воздуха в каналах или на входах/выходах среды.
- Визуализируйте пузырьки в каналах среды на микроскопе и вытесните их, повторно введя в каналы 75 мкл среды, чтобы вытолкнуть пузырьки наружу.
- Визуализируйте пузырьки на входе/выходе среды на глаз. Используйте пипетку P200 для удаления пузырьков воздуха, попавших на входе и выходе среды, нажимая на поршень вниз, вводя наконечник в отверстие и вытаскивая пузырь, поднимая поршень, чтобы создать отрицательное давление и всасывать пузырь.

Рисунок 2. Схема загрузки устройства. (A) С помощью пипетки P20 смесь клеток и фибрина вводится в тканевую камеру каждого устройства через одно из загрузочных отверстий. (B) На светлопольной микрофотографии видно, что микрофлюидное устройство загружает ЭК, фибробласты и раковые клетки для формирования ВМТ. Масштабная линейка = 500 мкм. (C) Флуоресцентная микрофотография прибора в B, показывающая EC красным цветом, опухоль голубым цветом и фибробласты синим цветом. (D) На схеме показано добавление среды в резервуары: 350 мкл на верхней стороне и 50 мкл на нижней стороне для создания гидростатического напора. (E) На 2-й день бактериологическое исследование ВМТ показывает, что фибробласты и ЭК начинают растягиваться, образуя сосудистую сеть. Масштабная линейка = 200 мкм. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
4. Техническое обслуживание устройства и экспериментальное применение
- Поддерживающая и медикаментозная терапия
ПРИМЕЧАНИЕ: Для поддержания потока в системе необходимо ежедневно восстанавливать гидростатическое давление, пипетируя объем среды с нижней стороны обратно на верхнюю сторону или наоборот, обеспечивая общий объем на верхней стороне на уровне 350 мкл. Направление потока меняется каждый день после 2-го дня установки VMO или VMT. Дополнительная информация о техническом обслуживании и лечении приведена ниже.- Меняйте среду через день с EGM2 до тех пор, пока сосудистая сеть полностью не установится (5-6 день). Полностью отсасывайте старые среды и заменяйте лунки высокого давления (350 мкл) и скважины низкого давления (50 мкл).
ПРИМЕЧАНИЕ: Экспериментальное определение оптимальных составов сред может быть проведено для других типов клеток, часто с использованием смеси 50:50 или добавления определенных компонентов в EGM2. - После того, как сосудистая сеть будет сформирована, а ткань полностью разовьется (4-7-й день), перед использованием приборов для экспериментов проведите тест на перфузию декстрана (шаг 4.2.1.). Используйте только те устройства, которые имеют достаточную перфузию в тканевую камеру.
- Для экспериментов с использованием терапевтических средств в день начала лечения сделайте снимки во всех флуоресцентных каналах для каждого устройства. Это послужит отправной точкой.
- Обработайте приборы желаемым терапевтическим средством, заменив среду свежей средой, содержащей разбавленный препарат в нужной концентрации. Убедитесь, что препараты разбавлены в соответствующих транспортных средствах в соответствии с рекомендациями производителя, но не превышают 0,01% ДМСО в среде.
- Подвергайте устройства воздействию препарата в течение желаемого времени (обычно 48 ч, но может быть проинформировано фармакокинетикой).
- Визуализируйте каждый канал каждого устройства через желаемый интервал времени для мониторинга ответа на лечение. Поддерживайте пластины, как указано в шаге 4.1.1, в течение всего эксперимента.
- По завершении эксперимента отбеливают пластины и помещают в контейнер с биологически опасным веществом, фиксируют 4% PFA для иммунофлуоресцентного окрашивания (шаг 4.3.) или собирают для выделения живых клеток или РНК (шаг 4.4.).
- Меняйте среду через день с EGM2 до тех пор, пока сосудистая сеть полностью не установится (5-6 день). Полностью отсасывайте старые среды и заменяйте лунки высокого давления (350 мкл) и скважины низкого давления (50 мкл).
- Перфузионные анализы
- Перфузия декстрана
ПРИМЕЧАНИЕ: Проницаемость/проходимость сосудов можно определить путем перфузии сосудистой сети флуоресцентно меченым декстрана с различной молекулярной массой (40 кДа, 70 кД или 150 кДа). В зависимости от флуоресцентной метки ЕС можно использовать FITC- или родамин-декстран.- Перед перфузией определите надлежащую экспозицию FITC или родаминового канала, добавив несколько мкл декстрана в жидкостный канал или камеру пустого устройства. Установите время экспозиции чуть ниже уровня насыщенности с помощью программного обеспечения микроскопа для отображения гистограммы интенсивности пикселей, обеспечивая динамический диапазон, характеризующийся равномерно распределенными пикселями без какой-либо заметной концентрации значений высокой интенсивности.
- Сделайте микрофотографии всех устройств в интересующих каналах, включая фоновое изображение всех устройств в канале флуоресцентного декстрана, для калибровки по фону. Используйте ту же экспозицию, что и выше, и выровняйте камеру ткани по центру рамки изображения, чтобы обеспечить согласованность изображений для количественной оценки.
- Приготовьте основной запас ФИТК-декстран или родамин-декстран в концентрации 5 мг/мл в 1x DPBS. Этот запас можно хранить при температуре 4 °C.
- Для приготовления рабочего материала разбавляют 5 мг/мл бульона до конечной концентрации 50 мкг/мл в EGM2.
- Замените среду в резервуарах разбавленным раствором декстрана в размере половины максимального объема (175 мкл в одну лунку в верхней части верхнего или нижнего канала тканевой камеры). Замените фильтрующий материал в других лунках таким образом, чтобы несвязанная верхняя сторона получала 175 мкл свежего EGM2 без декстрана, а лунки на нижней стороне имели только 50 мкл в каждой.
ПРИМЕЧАНИЕ: Декстран следует добавлять только на одну сторону микрофлюидных каналов (верхнюю или нижнюю), чтобы можно было визуализировать перемещение красителя через сторону высокого давления, в сосудистое русло и наружу со стороны низкого давления. - Под микроскопом наблюдайте за тем, как флуоресцентный декстран протекает через сосудистую сеть. Обычно это происходит в течение примерно 2 минут после добавления красителя в резервуар для фильтрующего материала.
- Начните визуализацию флуоресцентного декстранового канала (и других каналов, если это необходимо). Это точка времени T = 0. Делайте дополнительные снимки в несколько моментов времени (обычно каждые 10 минут) или одно конечное изображение.
- Перфузия клеток
ПРИМЕЧАНИЕ: Различные типы клеток могут быть перфузионированы через сосудистую сеть в зависимости от дизайна исследования, включая лимфоциты или макрофаги для иммунологических исследований рака, а также раковые клетки для исследований метастазирования. Клетки должны быть флуоресцентно помечены, чтобы облегчить отслеживание с течением времени.- По крайней мере, за 2 ч до перфузии клеток выполняйте перфузию декстрана на всех устройствах, как описано выше. Этот этап важен для определения проходимости сосудов перед добавлением клеток.
- Определите подходящую экспозицию камеры для клеток, которые необходимо перфузировать. Возьмите небольшой образец клеток для просмотра под микроскопом и установите время экспозиции для этого флуоресцентного маркера. Установите время экспозиции чуть ниже уровня насыщенности.
- Сделайте микрофотографии всех устройств в интересующих каналах, включая фоновое изображение всех устройств в канале флуоресцентного декстрана, для калибровки по фону. Используйте ту же экспозицию, что и на шаге 4.2.2.1.
- Убедитесь, что декстран полностью диффундировал из камер тканей, прежде чем собирать клетки для перфузии. Соберите и подсчитайте интересующие клетки.
- Ресуспендируйте ячейки с соответствующей плотностью в EGM2. Например, Т-клетки обычно добавляются примерно в количестве 1 x 106 клеток/мл, чтобы имитировать концентрацию в крови.
ПРИМЕЧАНИЕ: EC чувствительны к составу среды, но могут допускать смешивание до 50% с большинством других сред. Протестируйте заранее. - Добавьте 175 мкл клеточной суспензии в одну лунку на верхней стороне каждого устройства и добавьте 175 мкл EGM2 в сборе в другую лунку. На низком уровне добавьте 50 мкл полной среды EGM2 в обе лунки.
- Под микроскопом наблюдайте за тем, как флуоресцентные клетки протекают через сосудистую сеть. Обычно это происходит примерно через 2 минуты после добавления ячеек в резервуар для носителя.
- После того, как поток клеток установлен, начните визуализацию флуоресцентного клеточного канала (и других каналов, если это необходимо). Это точка времени T = 0. Сделайте дополнительные снимки в несколько временных точек или одно изображение конечной точки, в зависимости от дизайна исследования. Например, визуализация каждые 10 минут приведет к временным курсам с высоким разрешением или каждые 6-12 часов для отслеживания периодических движений клеток.
- Перфузия декстрана
- Иммунофлуоресцентное (ИФ) окрашивание
- Отсасывайте среду из скважин. Добавьте 200 мкл 4% PFA в обе лунки верхней стороны каждого устройства и 50 мкл в нижнюю сторону. Дайте PFA протекать через камеры в течение 15 минут при комнатной температуре или 30 минут при 4°C.
- Во время инкубации подготовьте 24-луночный планшет с 500 мкл 1x PBS на лунку. Рассчитайте, сколько лунок нужно для окрашивания каждого прибора.
- Полностью удалить PFA из скважин. Переверните пластину вверх дном и осторожно снимите пластиковую подложку с мембраны.
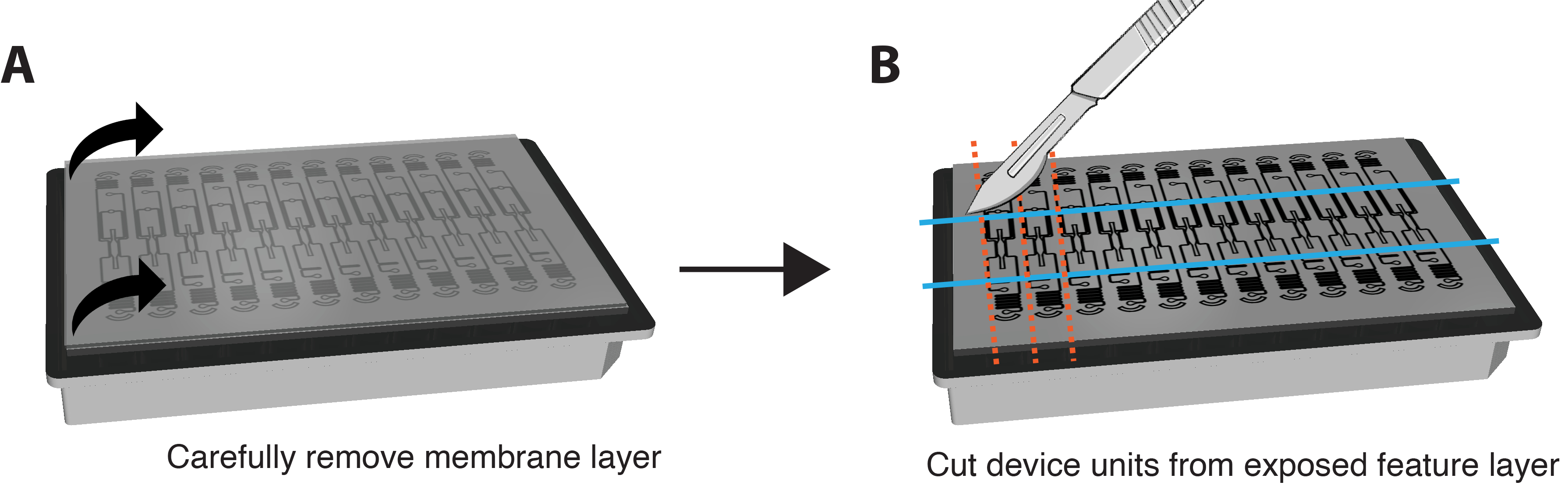
ПРИМЕЧАНИЕ: Окрашивание IF также может быть выполнено in situ без удаления мембраны и устройства. Для этого выполняйте этапы окрашивания путем перфузии реагентов через VMO/VMT и увеличивайте продолжительность каждого этапа инкубации примерно в 6 раз. - Очень осторожно и осторожно снимите нижний мембранный слой с характерного слоя устройства, чтобы обнажить камеры ткани, взявшись за оба угла и потянув вниз медленным и плавным движением. Большая часть ткани должна остаться в тканевой камере. Пожалуйста, обратитесь к рисунку 3A.
- Используйте бритвенное лезвие или скальпель, чтобы приложить достаточное усилие, чтобы полностью разрезать векторный слой и вырезать небольшой прямоугольник вокруг каждого отдельного блока устройства, как показано на рисунке 3B.
- Вклините шпатель между векторным слоем и планшетом лунки. Слегка надавите под характерный слой, чтобы осторожно удалить весь функциональный слой, содержащий тканевую камеру, с планшета лунки.
- Поместите каждый прямоугольный фрагмент векторного слоя PDMS, содержащий ткань лицевой стороной вниз, в одну лунку, содержащую PBS.
- После того, как все устройства окажутся в лунках, промойте PBS, поместив пластину на мягкое коромысло на 5 минут, аспирировав PBS из скважины и заменив его 500 мкл свежего PBS. Повторите в общей сложности 3 стирки.
- Аспирируйте PBS из каждой лунки и проникайте в ткани 500 мкл 0,5% Triton-X в PBS, 2 раза по 10 мин каждый на мягком коромысле. Удалите раствор проницаемости.
- Блокировать в 500 мкл 10% сыворотки в 0,1% Triton-X на устройство в течение 1 ч при комнатной температуре с легким покачиванием.
- Первичные антитела разводят в 3% сыворотке в 0,1% Тритоне-Х до нужной концентрации и объема. Удалите блокирующий раствор и добавьте достаточное количество раствора первичных антител, чтобы полностью покрыть дно каждой лунки и обеспечить свободное движение тканей устройства (~200 мкл). Накройте тарелку прозрачной пленкой.
- Инкубируйте пластину в течение ночи при температуре 4 °C. На следующий день доведите тарелку, содержащую салфетки устройства, до комнатной температуры (~15 мин).
- Отсасывайте раствор первичных антител из каждой лунки и промывайте камеры 500 мкл PBS, 3 раза по 5 мин каждый на мягком коромысле.
- Добавьте 200 мкл вторичного антитела в 3% сыворотке в 0,1% Triton-X в нужной концентрации. Выдерживают планшет при легком покачивании в течение 1 ч при комнатной температуре в темноте.
- Аспирировать раствор вторичных антител и промыть PBS, 3 раза по 5 мин каждый с легким покачиванием. Добавьте раствор 1x DAPI в 0,1% Triton-X на 10 мин, раскачивая в темноте.
- С помощью пинцета удалите прямоугольные вырезы устройства с испачканными тканями и положите тканевой стороной вверх на бумажное полотенце.
- Нанесите несколько мкл (~10 мкл) раствора для защиты от затухания непосредственно на каждую камеру и покровное стекло, стараясь не допустить образования пузырьков. Дайте антифейду затвердеть в камерах в течение ночи при комнатной температуре в темноте, а затем приступайте к визуализации.
- Выделение тканей и клеток для молекулярных анализов
ПРИМЕЧАНИЕ: Каждая пластина с высокой пропускной способностью будет содержать примерно 1-2 x 105 ячеек, в зависимости от времени сбора урожая. Масштабируйте количество экспериментальных репликаций, чтобы учесть общее количество клеток, а также потенциальную потерю во время сбора.- Анализ отдельных клеток
- Отсасывайте среду из каждой лунки и инвертируйте высокопроизводительную пластину так, чтобы слой устройства был обращен вверх.
- Снимите пластиковую подложку с мембраны. Очень осторожно и осторожно снимите нижнюю мембрану PDMS с характерного слоя устройства, чтобы обнажить тканевые камеры, взявшись за оба угла и потянув вниз медленным и плавным движением.
- Мембрана может быть удалена даже при правильном склеивании. Большая часть ткани должна остаться в тканевой камере после удаления мембраны; Однако, если какая-либо часть прилипла к мембране, выполните следующие действия на самой мембране.
- Промойте каждый блок устройства 500 мкл HBSS или PBS. В каждую единицу устройства добавьте 100 мкл диссоциационного реагента и оставьте его в виде капли на верхней части устройства. Поместите планшет обратно в инкубатор с температурой 37 °C на 5 минут.
- После разложения с помощью пипетки P200 пропитывайте вверх и вниз по тканевым камерам и собирайте ткани в диссоциационный реагент. Перемещайте наконечник пипетки вперед и назад по каждому устройству, чтобы обеспечить полное удаление ткани, и соберите в коническую форму объемом 15 мл с 500 мкл EGM2 для нейтрализации реагента для диссоциации.
- Для полного удаления оставшихся клеток из тканевой камеры добавьте 500 мкл EGM2 в каждый блок устройства и промойте пипеткой P200.
- Центрифугируют раствор для сбраживания, содержащий смещенные ткани, при 300 x g в течение 5 мин при 4 °C до гранулирования отдельных клеток и целых тканей.
- Осторожно аспирируйте среду и добавьте в ткани 500 мкл 1 мг/мл (200 Ед/мл) коллагеназы IV типа, 0,1 мг/мл гиалуронидазы V типа и 200 Ед/мл ДНКазы IV типа в HBSS.
- После осторожной ресуспензии дайте раствору постоять 2 минуты при комнатной температуре, прежде чем снова осторожно пипетировать, чтобы диссоциировать гель.
- Смесь для сбраживания промыть 10 мл EGM2 и центрифугировать при 300 x g в течение 5 мин при 4 °C. Ресуспендируйте ячейки в 1x DPBS с 1% BSA или HSA и пропустите через предварительно смоченный фильтр 70 мкм путем отжима при 200 x g в течение 1 минуты.
- Подсчитайте клетки и отрегулируйте объем так, чтобы конечная концентрация составляла 1000 клеток на мкл. Клеточные суспензии затем могут быть подвергнуты FACS, проточной цитометрии или секвенированию одноклеточной РНК.
- Выделение РНК из цельных тканей
- Выполните шаги 4.4.1.1 и 4.4.1.2 выше.
- Добавьте примерно 10 мкл лизисного буфера РНК в каждую открытую камеру ткани, гарантируя, что буфер накапливается непосредственно поверх тканей. Не используйте в общей сложности более 100 мкл лизисного буфера РНК.
- Выдерживать 3 мин при комнатной температуре. Используйте P20 для пипетирования вверх и вниз на каждом блоке устройства и используйте наконечник пипетки, чтобы при необходимости соскрести остатки материала из камеры для салфеток.
- Перелейте как можно больше лизисного буфера в микроцентрифужную пробирку объемом 1,5 мл. Повторите шаги 4.4.2.2.-4.4.2.3. для остальных устройств и пула образцов в пробирку объемом 1,5 мл.
- Следуйте инструкциям производителя по выделению РНК в зависимости от набора или реагентов.
- Анализ отдельных клеток

Рисунок 3. Подготовка платформы для иммуноокрашивания. (A) Схема полностью собранной платформы устройства с мембранным слоем сверху. Чтобы снять мембрану, осторожно потяните каждый угол наружного слоя вниз устойчивыми, мягкими движениями. (B) После того, как мембранный слой будет полностью удален, используйте лезвие, скальпель или нож, чтобы вырезать прямоугольники вокруг тканевой камеры каждого устройства, стараясь не порезать саму ткань. Затем под каждый прямоугольник можно просунуть шпатель, чтобы сместить его с пластины и поместить каждую единицу в одну лунку 24-луночной пластины с PBS для окрашивания. Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этого рисунка.
{kind=link}
Результаты
В соответствии с протоколами, изложенными здесь, ВМО и ВМТ были созданы с использованием коммерчески приобретенных ЭК, НХЛФ и, для ВМТ, трижды негативной клеточной линии рака молочной железы MDA-MB-231. Установленные ВМО также были перфузионированы раковыми клетками, чтобы имитировать мета...
Обсуждение
Почти каждая ткань в организме получает питательные вещества и кислород через сосудистую сеть, что делает ее критически важным компонентом для реалистичного моделирования заболеваний и скрининга лекарств in vitro. Кроме того, некоторые злокачественные новообразования и болезненны?...
Раскрытие информации
CCWH имеет долю в акционерном капитале компании Aracari Biosciences, Inc., которая занимается коммерциализацией версии технологии, описанной в этом документе. Условия этого соглашения были рассмотрены и одобрены Калифорнийским университетом в Ирвайне в соответствии с его политикой в отношении конфликта интересов. Других конфликтов интересов нет.
Благодарности
Мы благодарим сотрудников лаборатории доктора Кристофера Хьюза за их ценный вклад в описанные процедуры, а также наших сотрудников из лаборатории доктора Абрахама Ли за помощь в проектировании и изготовлении платформы. Эта работа была поддержана следующими грантами: UG3/UH3 TR002137, R61/R33 HL154307, 1R01CA244571, 1R01 HL149748, U54 CA217378 (CCWH) и TL1 TR001415 и W81XWH2110393 (SJH).
Материалы
| Name | Company | Catalog Number | Comments |
| Fabrication | |||
| (3-Mercaptopropyl)trimethoxysilane, 95% | Sigma-Aldrich | 175617-100G | |
| Greiner Bio-One μClear Bottom 96-well Polystyrene Microplates | Greiner Bio-One | 655096 | |
| Methanol ≥99.8% ACS | VWR Chemicals BDH | BDH1135-1LP | |
| MILTEX Sterile Disposable Biopsy Punch with Plunger, 1mm diameter, | Integra Miltex | 33-31AA-P/25 | |
| PDMS membrane | PAX Industries | HT-6240 | |
| Plasma Cleaner PDC-001 | Harrick Plasma | N/A | |
| Smooth-Cast 385 | Smooth-On | N/A | |
| SP Bel-Art Lab Companion Clear Polycarbonate Cabinet Style Vacuum Desiccator | Bel-Art | F42400-4031 | |
| Standard Lids with Condensation Rings, 96-well plate | VWR | 82050-827 | |
| SYLGARD 184 Silicone Elastomer Kit (PDMS) | Dow | 4019862 | |
| Cell culture/Loading | |||
| BioTek Lionheart FX Automated Microscope | Agilent | CYT5MFAW | |
| CELLvo Human Endothelial Progenitor Cells | StemBioSys | N/A | |
| Collagen I, rat tail | Enzo Life Sciences | ||
| Collagenase from Clostridium histolyticum (type 4) | Sigma-Aldrich | C5138 | |
| Corning Hank’s Balanced Salt Solution, 1X without calcium and magnesium | Corning | 21-021-CV | |
| Corning DMEM with L-Glutamine, 4.5g/L Glucose and Sodium Pyruvate | Corning | 10013CV | |
| DAPI | Sigma-Aldrich | D9542 | |
| DPBS, no calcium, no magnesium | Gibco | 14190144 | |
| EGM-2 Endothelial Cell Growth Medium-2 BulletKit | Lonza | CC-3162 | |
| Fibrinogen from bovine plasma | Neta Scientific | SIAL-341573 | |
| Fibronectin human plasma | Sigma-Aldrich | F0895 | |
| Fluorescein isothiocyanate–dextran (70kDa) | Sigma-Aldrich | FD70S-1G | |
| Gelatin from porcine skin | Sigma-Aldrich | G1890 | |
| Hyaluronidase from sheep testes (type 4) | Sigma-Aldrich | H6254 | |
| Laminin Mouse Protein | Gibco | 23017015 | |
| Leica TCS SP8 | Leica | N/A | |
| MDA-MB-231 | ATCC | HTB-26 | |
| NHLF – Normal Human Lung Fibroblasts | Lonza | CC-2512 | |
| Nikon Eclipse Ti | Nikon | N/A | |
| Paraformaldehyde 4% in 0.1M Phosphate BufferSaline, pH 7.4 | Electron Microscopy Sciences | 15735-90-1L | |
| PBMCs - Peripheral blood mononuclear cells | Lonza | CC-2702 | |
| PBS, pH 7.4 | Gibco | 10010049 | |
| Premium Grade Fetal Bovine Serum (FBS), Heat Inactivated | Avantor Seradigm | 97068-091 | |
| ProLong Gold Antifade Mountant | Invitrogen | P10144 | |
| Quick-RNA Microprep Kit | Zymo Research | R1051 | |
| Thrombin from bovine plasma | Sigma-Aldrich | T4648 | |
| Triton X-100 (Electrophoresis), | Fisher BioReagents | BP151-100 | |
| TrypLE Express Enzyme (1X), phenol red | Gibco | 12605028 | |
| Trypsin-EDTA (0.05%), phenol red | Gibco | 25300062 | |
| Vasculife | Lifeline Cell Technology | LL-0003 |
Ссылки
- Siegel, R. L., Miller, K. D., Wagle, N. S., Jemal, A. Cancer statistics, 2023. CA Cancer J Clin. 73 (1), 17-48 (2023).
- Hachey, S. J., Hughes, C. C. W. Applications of tumor chip technology. Lab Chip. 18 (19), 2893-2912 (2018).
- Ewald, M. L., Chen, Y. H., Lee, A. P., Hughes, C. C. W. The vascular niche in next generation microphysiological systems. Lab Chip. 21 (17), 3615-3616 (2021).
- Osaki, T., Sivathanu, V., Kamm, R. D. Vascularized microfluidic organ-chips for drug screening, disease models and tissue engineering. Curr Opin Biotechnol. 52, 116-123 (2018).
- Shirure, V. S., Hughes, C. C. W., George, S. C. Engineering vascularized organoid-on-a-chip models. Annu Rev Biomed Eng. 23, 141-167 (2021).
- Del Piccolo, N., et al. Tumor-on-chip modeling of organ-specific cancer and metastasis. Adv Drug Deliv Rev. 175, 113798 (2021).
- Sontheimer-Phelps, A., Hassell, B. A., Ingber, D. E. Modelling cancer in microfluidic human organs-on-chips. Nat Rev Cancer. 19 (2), 65-81 (2019).
- Sobrino, A., et al. 3D microtumors in vitro supported by perfused vascular networks. Sci Rep. 6, 31589 (2016).
- Phan, D. T. T., et al. A vascularized and perfused organ-on-a-chip platform for large-scale drug screening applications. Lab Chip. 17 (3), 511-520 (2017).
- Hachey, S. J., et al. An in vitro vascularized micro-tumor model of human colorectal cancer recapitulates in vivo responses to standard-of-care therapy. Lab Chip. 21 (7), 1333-1351 (2021).
- Hachey, S. J., et al. A Human Vascularized Micro-Tumor Model of Patient-Derived Colorectal Cancer Recapitulates Clinical Disease. Transl Res. 255, 97-108 (2023).
- Liu, Y., et al. Human in vitro vascularized micro-organ and micro-tumor models are reproducible organ-on-a-chip platforms for studies of anticancer drugs. Toxicology. 445, 152601 (2020).
- Jahid, S., et al. Structure-based Design of CDC42 Effector Interaction Inhibitors for the Treatment of Cancer. Cell Rep. 39 (4), 110760 (2022).
- Hsu, Y. H., Moya, M. L., Hughes, C. C. W., George, S. C., Lee, A. P. A microfluidic platform for generating large-scale nearly identical human microphysiological vascularized tissue arrays. Lab Chip. 13 (15), 2990-2998 (2013).
- Moya, M. L., Hsu, Y. H., Lee, A. P., Christopher, C. W. H., George, S. C. In vitro perfused human capillary networks. Tissue Eng - Part C: Methods. 19 (9), 730-737 (2013).
- Wang, X., et al. An on-chip microfluidic pressure regulator that facilitates reproducible loading of cells and hydrogels into microphysiological system platforms. Lab Chip. 16 (5), 868-876 (2016).
- Phan, D. T., et al. Blood-brain barrier-on-a-chip: Microphysiological systems that capture the complexity of the blood-central nervous system interface. Exp Biol Med. 242 (17), 1669-1678 (2017).
- Kurokawa, Y. K., et al. Human induced pluripotent stem cell-derived endothelial cells for three-dimensional microphysiological systems. Tissue Eng Part C: Methods. 23 (8), 474-484 (2017).
- Romero-López, M., et al. Recapitulating the human tumor microenvironment: Colon tumor-derived extracellular matrix promotes angiogenesis and tumor cell growth. Biomaterials. 116, 118-129 (2017).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 9 (7), 676-682 (2012).
- Carpenter, A. E., et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7 (10), R100 (2006).
- Zudaire, E., Gambardella, L., Kurcz, C., Vermeren, S. A computational tool for quantitative analysis of vascular networks. PLoS one. 6 (11), e27385 (2011).
- Corliss, B. A., et al. REAVER: A program for improved analysis of high-resolution vascular network images. Microcirculation. 27 (5), e12618 (2020).
- Urban, G., et al. Deep learning for drug discovery and cancer research: Automated analysis of vascularization images. IEEE/ACM Trans Comput Biol Bioinform. 16 (3), 1029-1035 (2019).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены