Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Etablierung eines physiologischen humanen vaskularisierten Mikrotumormodells für die Krebsforschung
In diesem Artikel
Zusammenfassung
Dieses Protokoll stellt ein physiologisch relevantes Tumor-on-a-Chip-Modell vor, um Hochdurchsatz-Grundlagen- und translationale Krebsforschung am Menschen durchzuführen und das Arzneimittel-Screening, die Krankheitsmodellierung und Ansätze der personalisierten Medizin mit einer Beschreibung von Lade-, Wartungs- und Bewertungsverfahren voranzutreiben.
Zusammenfassung
Der Mangel an validierten Krebsmodellen, die die Tumormikroumgebung solider Krebsarten in vitro rekapitulieren, stellt nach wie vor einen erheblichen Engpass für die präklinische Krebsforschung und die therapeutische Entwicklung dar. Um dieses Problem zu lösen, haben wir den vaskularisierten Mikrotumor (VMT) oder Tumorchip entwickelt, ein mikrophysiologisches System, das die komplexe Mikroumgebung des menschlichen Tumors realistisch modelliert. Die VMT bildet sich de novo innerhalb einer mikrofluidischen Plattform durch Co-Kultur mehrerer humaner Zelltypen unter dynamischen, physiologischen Strömungsbedingungen. Dieses Gewebe-Engineering-Mikrotumor-Konstrukt enthält ein lebendes, durchblutetes Gefäßnetzwerk, das die wachsende Tumormasse genauso unterstützt, wie es neu gebildete Gefäße in vivo tun. Wichtig ist, dass Medikamente und Immunzellen die Endothelschicht überwinden müssen, um den Tumor zu erreichen, wodurch physiologische Barrieren für die therapeutische Verabreichung und Wirksamkeit in vivo modelliert werden. Da die VMT-Plattform optisch transparent ist, kann eine hochauflösende Bildgebung dynamischer Prozesse wie Immunzellextravasation und Metastasierung mit direkter Visualisierung von fluoreszenzmarkierten Zellen innerhalb des Gewebes erreicht werden. Darüber hinaus behält die VMT in vivo die Tumorheterogenität, die Genexpressionssignaturen und das Ansprechen auf Medikamente bei. Praktisch jeder Tumortyp kann an die Plattform angepasst werden, und Primärzellen aus frischem chirurgischem Gewebe wachsen und sprechen auf die medikamentöse Behandlung in der VMT an, was den Weg zu einer wirklich personalisierten Medizin ebnet. Hier werden die Methoden zur Etablierung des VMT und dessen Nutzung für die onkologische Forschung skizziert. Dieser innovative Ansatz eröffnet neue Möglichkeiten für die Untersuchung von Tumoren und Arzneimittelreaktionen und gibt Forschern ein leistungsfähiges Werkzeug an die Hand, um die Krebsforschung voranzutreiben.
Einleitung
Krebs ist nach wie vor ein großes Gesundheitsproblem weltweit und die zweithäufigste Todesursache in den Vereinigten Staaten. Allein für das Jahr 2023 rechnet das National Center for Health Statistics mit mehr als 1,9 Millionen neuen Krebsfällen und über 600.000 Krebstodesfällen in den USA1, was den dringenden Bedarf an wirksamen Behandlungsansätzen unterstreicht. Derzeit erhalten jedoch nur 5,1 % der Krebstherapeutika, die sich in klinischen Studien befinden, letztendlich eine FDA-Zulassung. Das Scheitern vielversprechender Kandidaten, klinische Studien erfolgreich zu durchlaufen, kann teilweise auf die Verwendung nicht-physiologischer Modellsysteme wie 2D- und Sphäroidkulturen während der präklinischen Arzneimittelentwicklung zurückgeführt werden2. Diesen klassischen Krebsmodellen fehlen wesentliche Komponenten der Tumormikroumgebung, wie z. B. eine stromale Nische, assoziierte Immunzellen und durchblutete Gefäße, die wichtige Determinanten der therapeutischen Resistenz und des Fortschreitens der Krankheit sind. Daher ist ein neues Modellsystem notwendig, das die menschliche in vivo Tumormikroumgebung besser nachahmt, um die klinische Translation präklinischer Befunde zu verbessern.
Das Gebiet des Tissue Engineering schreitet rasant voran und bietet verbesserte Methoden zur Untersuchung menschlicher Krankheiten im Labor. Eine wichtige Entwicklung ist das Aufkommen mikrophysiologischer Systeme (MPS), auch bekannt als Organchips oder Gewebechips, bei denen es sich um funktionelle, miniaturisierte menschliche Organe handelt, die in der Lage sind, gesunde oder kranke Zustände zu replizieren 3,4,5. In diesem Zusammenhang wurden für die onkologische Forschung Tumorchips, d.h. dreidimensionale mikrofluidikbasierte in vitro humane Tumormodelle, entwickelt 2,3,4,5,6,7,8,9,10,11,12,13 . Diese fortschrittlichen Modelle enthalten biochemische und biophysikalische Hinweise in einer dynamischen Tumormikroumgebung und ermöglichen es den Forschern, das Verhalten von Tumoren und das Ansprechen auf Behandlungen in einem physiologisch relevanteren Kontext zu untersuchen. Trotz dieser Fortschritte haben jedoch nur wenige Gruppen erfolgreich ein lebendes, funktionelles Gefäßsystem integriert, insbesondere eines, das sich als Reaktion auf den physiologischen Fluss selbst mustert 3,4,5,6. Die Einbeziehung eines funktionellen vaskulären Netzwerks ist von entscheidender Bedeutung, da es die Modellierung physikalischer Barrieren ermöglicht, die die Verabreichung von Medikamenten oder Zellen, das Homing von Zellen in bestimmte Mikroumgebungen und die transendotheliale Migration von Tumor-, Stroma- und Immunzellen beeinflussen. Durch die Einbeziehung dieses Merkmals kann der Tumorchip die Komplexität, die in der In-vivo-Tumormikroumgebung beobachtet wird, besser darstellen.
Um diesen ungedeckten Bedarf zu decken, haben wir eine neuartige Wirkstoff-Screening-Plattform entwickelt, die es ermöglicht, Mikrogefäßnetzwerke innerhalb eines mikrofluidischen Gerätszu bilden 8,9,10,11,12,13,14,15,16. Diese Basis-Organchip-Plattform, die als vaskularisiertes Mikroorgan (VMO) bezeichnet wird, kann an praktisch jedes Organsystem angepasst werden, um die ursprüngliche Gewebephysiologie für die Krankheitsmodellierung, das Arzneimittelscreening und Anwendungen der personalisierten Medizin zu replizieren. VMOs werden durch die Co-Kultivierung von endothelialen koloniebildenden zellabgeleiteten Endothelzellen (ECFC-EC), HUVEC oder iPSC-EC (im Folgenden EC) und mehreren Stromazellen in der Kammer etabliert, einschließlich normaler menschlicher Lungenfibroblasten (NHLF), die die Matrix umbauen, und Perizyten, die die Gefäße umhüllen und stabilisieren. Das VMO kann auch als Krebsmodellsystem etabliert werden, indem Tumorzellen mit dem zugehörigen Stroma kokultiviert werden, um einen vaskularisierten Mikrotumor (VMT)8,9,10,11,12,13 oder ein Tumorchip-Modell zu erstellen. Durch die Co-Kultur mehrerer Zelltypen in einer dynamischen Flussumgebung bilden perfundierte mikrovaskuläre Netzwerke de novo in den Gewebekammern des Geräts, wo die Vaskulogenese durch interstitielle Flussraten eng reguliert wird14,15. Das Medium wird durch einen hydrostatischen Druckkopf durch die mikrofluidischen Kanäle des Geräts getrieben, der die umgebenden Zellen der Gewebekammer ausschließlich über die Mikrogefäße mit Nährstoffen versorgt, mit einem Permeabilitätskoeffizienten von 1,2 x 10-7 cm/s, ähnlich wie bei Kapillaren in vivo8.
Die Integration von selbstorganisierenden Mikrogefäßen in das VMT-Modell stellt einen bedeutenden Durchbruch dar, da es: 1) die Struktur und Funktion vaskularisierter Tumormassen in vivo nachahmt; 2) kann Schlüsselschritte der Metastasierung modellieren, einschließlich Tumor-Endothel- und Stromazell-Interaktionen; 3) etabliert physiologisch selektive Barrieren für die Nährstoff- und Arzneimittelabgabe und verbessert so das pharmazeutische Screening; und 4) ermöglicht die direkte Bewertung von Medikamenten mit anti-angiogenen und anti-metastasierenden Eigenschaften. Durch die Replikation der In-vivo-Verabreichung von Nährstoffen, Medikamenten und Immunzellen in einer komplexen 3D-Mikroumgebung ist die VMO/VMT-Plattform ein physiologisch relevantes Modell, das zur Durchführung von Wirkstoff-Screenings und zur Untersuchung der Krebs-, Gefäß- oder organspezifischen Biologie verwendet werden kann. Wichtig ist, dass die VMT das Wachstum verschiedener Arten von Tumoren unterstützt, darunter Dickdarmkrebs, Melanom, Brustkrebs, Glioblastom, Lungenkrebs, Peritonealkarzinose, Eierstockkrebs und Bauchspeicheldrüsenkrebs 8,9,10,11,12,13. Die mikrofluidische Plattform ist nicht nur kostengünstig, einfach einzurichten und für Hochdurchsatzexperimente geeignet, sondern auch optisch vollständig kompatibel für die Echtzeit-Bildanalyse von Tumor-Stroma-Interaktionen und die Reaktion auf Stimuli oder Therapeutika. Jeder Zelltyp im System ist mit einem anderen Fluoreszenzmarker markiert, um eine direkte Visualisierung und Verfolgung des Zellverhaltens während des gesamten Experiments zu ermöglichen und ein Fenster in die dynamische Tumormikroumgebung zu schaffen. Wir haben bereits gezeigt, dass die VMT das Wachstum, die Architektur, die Heterogenität, die Genexpressionssignaturen und das Ansprechen von Medikamenten in vivo genauer modelliert als Standardkulturmodalitäten10. Wichtig ist, dass die VMT das Wachstum und die Untersuchung von Patientenzellen, einschließlich Krebszellen, unterstützt, die die Pathologie der Elterntumoren besser modellieren als Standard-Sphäroidkulturen und die Bemühungen um personalisierte Medizin weiter vorantreiben11. Dieses Manuskript skizziert die Methoden zur Etablierung des VMT und zeigt seinen Nutzen für die Untersuchung menschlicher Krebserkrankungen.
Protokoll
1. Design und Herstellung
- Geräte-Design
- Für die Herstellung von mikrofluidischen Geräten wird eine SU-8-Form mit einer 200-μm-Schicht aus SU-8 hergestellt, die auf einen Si-Wafer aufgetragen wird (RCA-1 gereinigt und mit 2 % Fluorwasserstoff (HF) behandelt), gefolgt von einem Photolithographieschritt mit einer einzigen Maske, wie zuvor beschrieben 8,9.
- Gießen Sie eine 4 mm dicke Polydimethylsiloxan (PDMS)-Replik aus der SU-8-Form, um eine langlebige Polyurethanform für nachgelagerte Fertigungsschritte zu erzeugen. Es können verschiedene Design-Iterationen verwendet werden 8,9,10,11,12,13,14,15.
- In der aktuellen Iteration wird das mikrofluidische Gerät so entworfen, dass es individuell in ein standardmäßiges 96-Well-Plattenformat eingepasst wird und aus einer 2 mm dicken PDMS-Feature-Schicht mit 12 mikrofluidischen Geräteeinheiten besteht, die von einer dünnen (1/16 Zoll) transparenten Polymermembranschicht auf der Unterseite umschlossen sind (Abbildung 1A).
- Stellen Sie sicher, dass die einzelnen Gewebeeinheiten aus einer Gewebekammer bestehen, die von einem Gelladeeinlass (L1) und -auslass (L2), einem Druckregler (PR)16 und entkoppelten mikrofluidischen Kanälen flankiert wird, die mit 2 Medienein- und -auslässen auf jeder Seite verbunden sind (M1-M2, M3-M4; Abbildung 1B).
- Positionieren Sie jeden Ein- und Auslass in einer einzigen Vertiefung, die als Mediumreservoir dient, um einen hydrostatischen Druck (10 mmH2O) über den mikrofluidischen Kanal zu erzeugen. Um Anastomosen des Gefäßnetzwerks mit den äußeren Kanälen zu ermöglichen, verbinden Sie mikrofluidische Kanäle über 50 μm breite Kommunikationsporen (6 oben, 6 unten) mit der Gewebekammer.
HINWEIS: Mikrofluidische Widerstände erzeugen einen interstitiellen Druckgradienten von 5 mm H2O über die Gewebekammer, der intraluminal wird, sobald das Gefäßnetz vollständig ausgebildet ist 8,10. Die nachfolgenden Verfahren beginnen mit einer fertig montierten Hochdurchsatzplatte.

Abbildung 1. Design der mikrofluidischen Plattform. (A) Das Schema der Plattformanordnung zeigt die PDMS-Feature-Schicht mit 12 Bauteileinheiten, die mit einer bodenlosen 96-Well-Platte verbunden und mit einer dünnen transparenten Polymermembran versiegelt sind. Jede Geräteeinheit belegt eine Säule von Vertiefungen auf der Platte. Die rot umrandete einzelne Geräteeinheit ist mit Details in (B) dargestellt. (B) Das Schema einer Geräteeinheit zeigt eine einzelne Gewebekammer, die sich in einer Vertiefung der 96-Well-Platte befindet, und zwei Ladeöffnungen mit Einlass- und Auslasslöchern (L1-L2), die gestanzt sind, um die Einführung einer Zellmatrixmischung zu ermöglichen. Die Ein- und Auslässe des Mediums (M1-M2, M3-M4) werden gelocht und in Vertiefungen positioniert, die als Medienreservoirs dienen. Unterschiedliche Medienvolumina erzeugen über entkoppelte mikrofluidische Kanäle einen hydrostatischen Druckgradienten über die Gewebekammer. Die Druckreglereinheit (PR) dient als Gel-Berstventil, um die Beladung zu erleichtern. Beachten Sie, dass das Gerät 200 μm tief ist und die Gewebekammer 2 mm x 6 mm groß ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
2. Vorbereitungen vor dem Verladen
- Zellkultur
- Halten Sie die Zellen gemäß den Empfehlungen des Herstellers in einem befeuchteten Inkubator mit 37 °C und 5 % CO2 auf .
- Platten-T75-Kolben mit transduzierten EC-, NHLF- oder anderen Fibroblasten-/Stromazellen und gewünschten Krebszellen 3-4 Tage vor der Beladung mit einer Dichte, die vom Hersteller und den Benutzerprotokollen angegeben wird. Für dieses Protokoll wird 1 x 106 Zellen pro Kolben für jeden Zelltyp aufgetragen. Kultur EC in Endothelial Growth Media 2 (EGM2) kompletten Medien, NHLF in Dulbecco's Modified Eagle Medium (DMEM) mit 10% FBS und Krebszellen in geeigneten Medien je nach Zelltyp.
- Pflegen Sie die Zellen, indem Sie alle 2-3 Tage mit den entsprechenden Medien füttern, und bestätigen Sie die Transduktions- oder Markierungseffizienz erneut, indem Sie die Zellen unter einem Fluoreszenzmikroskop visualisieren. Stellen Sie am Tag der Beladung sicher, dass der EC-Wert zu 80 % bis 100 % konfluent ist, während der NHLF-Wert bei 70 % bis 80 % subkonfluent ist.
- Herstellung von Fibrinogen
- Bereiten Sie die Fibrinogenlösung auf die gewünschte Konzentration vor (typischerweise unterstützen 5-8 mg/ml die Bildung eines robusten Gefäßnetzwerks), wobei die prozentuale Gerinnung von Fibrinogen berücksichtigt wird. Berechnen Sie die benötigte Menge an Fibrinogen mit der folgenden Gleichung:
Fibrinogen (mg) = (Volumen (ml)) x (Konzentration (mg/ml))/ (Gerinnung %) - Lösen Sie das Fibrinogen in einem geeigneten Volumen des endothelialen Basalmediums 2 (EBM2), das auf 37 °C erwärmt wird, durch vorsichtiges Schnippen des Röhrchens auf (nicht wirbeln). Fibrinogen in einem 37 °C warmen Wasserbad inkubieren, damit es vollständig in Lösung gehen kann. Wichtig ist, dass Sie kein vollständiges Medium verwenden.
- Sterilfilter-Fibrinogenlösung mit 0,22-μm-Filter und Aliquot auf das gewünschte Volumen, typischerweise 400 μl pro Mikrozentrifugenröhrchen.
HINWEIS: Andere Matrixproteine (z. B. Kollagene, Fibronektin oder Laminin) können in die Fibrinogenmischung eingebracht werden.
- Bereiten Sie die Fibrinogenlösung auf die gewünschte Konzentration vor (typischerweise unterstützen 5-8 mg/ml die Bildung eines robusten Gefäßnetzwerks), wobei die prozentuale Gerinnung von Fibrinogen berücksichtigt wird. Berechnen Sie die benötigte Menge an Fibrinogen mit der folgenden Gleichung:
3. Laden von Proben
HINWEIS: Das Laden ist zeitkritisch und sollte vom Start (Anheben der Zelle) bis zum Ende (Hinzufügen von Medien zu den Geräten) innerhalb von etwa 1,5-1,75 h abgeschlossen sein, um optimale Ergebnisse zu gewährleisten. Jeder Schritt wird mit einem vorgeschlagenen Timer notiert, um den Benutzer auf Kurs zu halten.
- Vorbereitung der Materialien (Tag der Verladung)
- In ein 37 °C warmes Wasserbad für 10-15 min geben: Hank's Balanced Salt Solution (HBSS) oder phosphatgepufferte Kochsalzlösung (PBS) zum Waschen von Zellen, Zelldissoziationsreagenz, Medien (z.B. EGM2, DMEM)
- Bewahren Sie die folgenden Reagenzien bis zur Verwendung im Kühlschrank bei 4 °C auf: Thrombin, Laminin (über Nacht bei 4 °C aufgetaut).
- Fibrinogen-Aliquot bei Raumtemperatur auftauen. Bereiten Sie 1,5 μl Aliquotes Thrombin in 500-μl-Mikrozentrifugenröhrchen mit einem Röhrchen pro Geräteeinheit vor. Stellen Sie sicher, dass sich das Thrombinaliquot am Boden jedes Röhrchens befindet, um das Laden zu erleichtern.
- Legen Sie UV-sterilisierte Hochdurchsatzplatten vor dem Laden mindestens 30 Minuten lang in einen Exsikkator, um die in der Mikrofluidik eingeschlossene Luft zu entfernen.
- Zellvorbereitung (Timer-Start = Start bei 0 min)
- Überprüfen Sie die Zellen unter dem Mikroskop bei 4-facher Vergrößerung, um die Konfluenz und Transduktionseffizienz zu bestätigen.
- Jeder T75-Kolben mit Zellen 2x mit 5 ml HBSS waschen und vollständig absaugen. In jeden Kolben wird 1 ml Dissoziationsreagenz gegeben und bei 37 °C und 5 % CO2 1-2 min inkubiert.
- Klopfen Sie vorsichtig mit der Handfläche auf die Platte und überprüfen Sie, ob alle Zellen angehoben wurden.
- Waschen Sie die Zellen mit 9 ml geeignetem Medium vom Kolben ab und sammeln Sie sie in einem 15-ml-Konus. Entfernen Sie sofort ein kleines Aliquot für die Zellzählung.
- Die Zellen werden bei 300 x g für 3-5 min bei 4 °C zentrifugiert. Zählen Sie beim Zentrifugieren der Zellen die Zellen. Ein konfluenter T75-Kolben EC oder NHLF sollte mindestens 2 x 106 Zellen ergeben.
- Nach der Zentrifugation werden die Medien abgesaugt und das Pellet in geeigneten Medien in einer Konzentration von 1 x 106 Zellen/ml resuspendiert. Halten Sie die Zellen auf Eis.
- Zubereitung von Zell-Fibrinogen-Gemisch (Timer = Beginn bei 20 min)
- Bestimmen Sie, wie viele Geräte geladen werden, addieren Sie 1-2, um den Pipettierverlust zu berücksichtigen, und multiplizieren Sie es mit dem Volumen der Zell-/Fibrinogenmischung, die pro Gerät benötigt wird. Dies hängt von der Gerätekonfiguration ab, aber für das in diesem Artikel vorgestellte Gerätedesign sind 6 μl pro Gerät erforderlich.
- Die Konzentration jedes Zelltyps sollte experimentell bestimmt werden. Als Ausgangspunkt ist EC mit einer Konzentration von ca. 7 × 106 Zellen/ml und NHLF mit einer Konzentration von 3,5 × 106 Zellen/ml zu laden. Die Konzentration von Krebszellen kann je nach Wachstumsrate erheblich variieren, liegt aber typischerweise im Bereich von 0,5-2 x 106 Zellen/ml. Verwenden Sie diese Gleichung, um die Anzahl der benötigten Zellen zu berechnen:
Anzahl der benötigten Zellen = (Volumen des Fibrins (μl))/1000 μl x (Konzentration der Zellen) - Resuspendieren Sie die Zellen bei einer Konzentration von 1 x 106 Zellen/ml und verwenden Sie die folgende Gleichung, um das benötigte Zellvolumen zu bestimmen:
Volumen der benötigten Zellen (μl) = (Anzahl der benötigten Zellen)/1000 - Mischen Sie die entsprechenden Volumina von EC-, NHLF- und Krebszellen (nur für VMT) in einem konischen Röhrchen und zentrifugieren Sie bei 300 x g für 3-5 Minuten bei 4 °C.
- Nach dem Schleudern das Medium vorsichtig ansaugen und alle Restmedien in der Nähe des Pellets abpipettieren. Resuspendieren Sie das Pellet vorsichtig, aber gründlich in das berechnete Fibrinogenvolumen und achten Sie besonders darauf, keine Luftblasen einzuführen. Auf Eis bleiben.
- Sterilisierte Platten und Thrombin-Aliquots in die Gewebekulturhaube bringen.
- Ladegeräte (Timer = Beginn bei 30-35 min)
- Pipettieren Sie mit einer P20-Pipette 6 μl Volumen aus dem Zell-Fibrin-Gemisch. Achten Sie darauf, die Mischung mindestens 5x nach oben und unten zu pipettieren, um eine gleichmäßige Zellsuspension zu gewährleisten. Bewahren Sie die Mischung auf Eis auf, um die Gerinnung zu verlangsamen.
- Mischen Sie die Zelle/das Fibrin vorsichtig in ein Röhrchen Thrombin, indem Sie die Pipettenspitze direkt in das Thrombin-Aliquot am Boden des Röhrchens stecken. Sofort mindestens 2x auf- und abpipeutieren, dabei darauf achten, dass keine Luftblasen entstehen. Das Fibrin beginnt zu gerinnen, sobald es mit Thrombin vermischt ist, also führen Sie die Schritte 3.4.3 schnell, aber absichtlich aus. und 3.4.4. bevor das Fibrin in der Pipettenspitze geliert (~3 s).
- Heben Sie die Platte mit hohem Durchsatz schräg an und führen Sie die Pipettenspitze schnell in eine der Geräteladeöffnungen (L1 oder L2) ein. Siehe Abbildung 2A für einen Schaltplan.
- Drücken Sie den Pipettenkolben mit einer sanften, flüssigen Bewegung bis zum ersten Anschlag, um das Zell-Fibrin-Gemisch in die Gewebekammer zu injizieren. Achte darauf, dass das Gel die Kammer vollständig durchquert.
HINWEIS: Wenn Sie während dieses Schritts zu viel Druck ausüben, kann dies dazu führen, dass das Gel in die mikrofluidischen Kanäle oben und/oder unten der Gewebekammern platzt. - Legen Sie die Platte vorsichtig wieder flach in die Gewebekulturhaube, ohne die Pipettenspitze zu entfernen, den Pipettenkolben loszulassen oder die Pipette zu stören. Drehen und entfernen Sie die Pipettenspitze mit der Hand vom P20 und lassen Sie sie im Ladeloch. Verwenden Sie nicht die Auswerfertaste, um die Spitze zu entfernen, da dies zu viel Druck verursacht.
- Fahren Sie mit den Schritten 3.4.1 bis 3.4.5 für die restlichen Geräte fort.
- Wenn das Laden abgeschlossen ist, lassen Sie die Platte 2 Minuten lang ungestört in der Gewebekulturhaube stehen.
- Entfernen Sie die Pipettenspitzen, indem Sie sie vorsichtig drehen und von den Ladeöffnungen entfernen. Setzen Sie den Deckel wieder auf die Platte.
- Inkubieren Sie die gesamte Platte 15-20 Minuten lang in einem 37 °C warmen Inkubator, damit das Gel vollständig polymerisieren kann.
- Überprüfen Sie nach der Inkubation jede Geräteeinheit unter dem Mikroskop. Prüfen Sie, ob die Zellen gleichmäßig in der Kammer verteilt sind, ohne dass Luftblasen entstehen, und dass eine deutlich sichtbare Gelgrenzfläche zwischen der Gewebekammer und den mikrofluidischen Kanälen vorhanden ist, wie in Abbildung 2B-C dargestellt.
- Kanalbeschichtung mit Laminin (Timer = Beginn bei 45-50 min)
- Nachdem die Gele vollständig fest sind, führen Sie Laminin in die mikrofluidischen Kanäle ein, um die vaskuläre Anastomose zu fördern.
- Führen Sie mit einem P20 4 μl Laminin in jeden mikrofluidischen Kanal (oben und unten) des Geräts ein. Führen Sie die Pipettenspitze in M1 oder M3 ein und stoßen Sie das Laminin langsam aus, wobei Sie darauf achten müssen, dass das Laminin den gesamten oberen Kanal bedeckt, und wiederholen Sie dann den Vorgang für M2 oder M4, um den gesamten unteren Kanal zu beschichten.
- Bestimmen Sie die Ausrichtung, indem Sie das Laminin von der dem Druckregler gegenüberliegenden Seite pipettieren, damit genügend Druck vorhanden ist, um es durchzudrücken. Wenn sich das Laminin jedoch nicht leicht von einer Seite bewegt, entfernen Sie die Spitze von einer Seite und drücken Sie das Laminin von der anderen Seite. Es kann erforderlich sein, zum zweiten Anschlag der Pipette zu gehen (d. h. den Kolben ganz nach unten zu drücken), um genügend Druck zu erzeugen, um das Laminin durch den gesamten Kanal zu drücken.
- Entfernen Sie die Spitze vorsichtig vom Einlass/Auslass des Mediums. Verwenden Sie nicht die Auswerfertaste am P20.
- Wiederholen Sie die Schritte 3.5.1-3.5.4 für jedes Gerät und inkubieren Sie die Platte bei 37 °C und 5 % CO2 für 10 Minuten.
- Medienzugabe (Timer = Beginn bei ca. 1 h 10 min)
- Geben Sie 275 μl des vollständigen EGM2-Mediums in die entkoppelten Medienreservoirs der Wells in den Reihen A und B oder G und H. Dies wird die hohe Seite sein, und die Ausrichtung sollte bestimmt werden, indem die Vertiefungen auf der dem Druckregler gegenüberliegenden Seite zu Beginn ein hohes Volumen haben. Die Medien werden durch die Schwerkraft von der hohen Seite gedrückt.
- Geben Sie mit einer P200-Pipette 75 μl Medien in die Medieneinlässe/-auslässe der Vertiefungen, die die 275 μl EGM2 enthalten. Führen Sie die Spitze in das Einlassloch des Mediums ein und stoßen Sie das Medium langsam aus, wobei Sie darauf achten, dass das Medium durch den Kanal fließt und auf der anderen Seite Blasen aufwirft.
- Entfernen Sie die Pipettenspitze und schieben Sie das verbleibende Medium von der Spitze in das Medienreservoir, so dass das Gesamtvolumen auf der oberen Seite 350 μl beträgt.
- Wiederholen Sie die Schritte 3.6.1 bis 3.6.3 für jede Geräteeinheit, obere und untere Kanäle.
- Fügen Sie je nach oben beschriebener Ausrichtung 50 μl Medien hinzu, um die untere Seite, die Vertiefungen in den Reihen A und B oder G und H, vollständig zu bedecken. Stellen Sie sicher, dass der Boden des Wells mit einer gleichmäßigen Medienschicht bedeckt ist. In Abbildung 2D finden Sie ein Schema, das die Medienvolumina in den Reservoirs zeigt.
- Entfernen von Luftblasen (Nachladen)
HINWEIS: Das Entfernen von Blasen ist ein wichtiger Schritt, um einen ordnungsgemäßen Durchfluss in jedem Gerät zu gewährleisten. An Tag 2 beginnen sich Endothelzellen und Fibroblasten als Reaktion auf den Fluss zu dehnen (Abbildung 2E).- Sobald alle Medien zugegeben sind, inkubieren Sie die Platten 1-2 h lang in einem 37 °C heißen Inkubator mit 5 % CO2 , bevor Sie die Kanäle oder die Ein-/Auslässe des Mediums auf Luftblasen prüfen.
- Visualisieren Sie Blasen in den Medienkanälen auf dem Mikroskop und stoßen Sie sie aus, indem Sie 75 μl Medien wieder in die Kanäle einführen, um die Blasen herauszudrücken.
- Visualisieren Sie Blasen in den Ein- und Auslässen des Mediums mit dem Auge. Verwenden Sie eine P200-Pipette, um Luftblasen zu entfernen, die an den Ein- und Auslässen des Mediums eingeschlossen sind, indem Sie den Kolben nach unten drücken, die Spitze in das Loch einführen und die Blase herausziehen, indem Sie den Kolben anheben, um Unterdruck auszuüben und die Blase aufzusaugen.

Abbildung 2. Schematische Darstellung der Gerätebelastung. (A) Mit einer P20-Pipette wird das Zell-/Fibringemisch über einen der Ladeanschluss in die Gewebekammer jeder Geräteeinheit eingeführt. (B) Die hellfeldmikroskopische Aufnahme zeigt ein mikrofluidisches Gerät, das EC, Fibroblasten und Krebszellen belädt, um eine VMT zu bilden. Maßstabsbalken = 500 μm. (C) Fluoreszenzmikroskopische Aufnahme des Geräts in B, die EC in Rot, Tumor in Cyan und Fibroblasten in Blau zeigt. (D) Das Schema zeigt die Zugabe von Medium in die Behälter mit 350 μl auf der oberen Seite und 50 μl auf der unteren Seite, um die hydrostatische Druckhöhe zu erzeugen. (E) Tag 2 der VMT-Kultur zeigt, dass Fibroblasten und EC beginnen, sich auszudehnen, um das vaskuläre Netzwerk zu bilden. Maßstabsleiste = 200 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
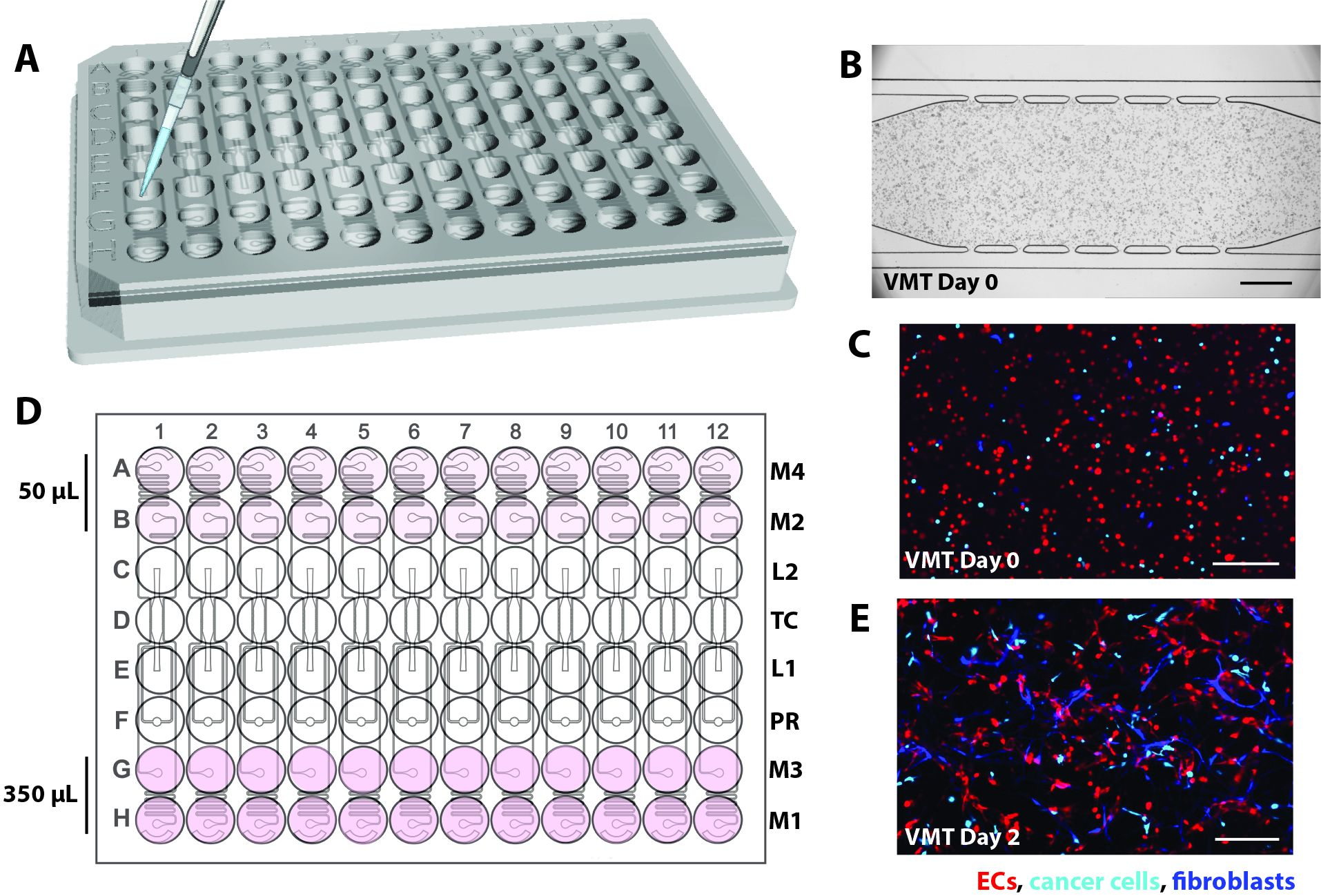{kind=link}
4. Gerätewartung und experimentelle Anwendungen
- Erhaltungstherapie und medikamentöse Behandlung
HINWEIS: Um den Durchfluss im System aufrechtzuerhalten, muss der hydrostatische Druck täglich wieder hergestellt werden, indem das Medienvolumen von der unteren Seite zurück zur hohen Seite oder umgekehrt pipettiert wird, wobei sichergestellt wird, dass das Gesamtvolumen auf der hohen Seite bei 350 μl bleibt. Die Durchflussrichtung wird jeden Tag nach Tag 2 der VMO- oder VMT-Etablierung geändert. Weitere Wartungs- und Behandlungsdetails finden Sie weiter unten.- Wechseln Sie das Medium jeden zweiten Tag mit EGM2, bis sich das Gefäßsystem vollständig etabliert hat (Tag 5-6). Altmedien vollständig absaugen und Hochdruckbrunnen (350 μL) und Niederdruckbrunnen (50 μL) ersetzen.
HINWEIS: Die experimentelle Bestimmung optimierter Medienformulierungen kann für andere Zelltypen durchgeführt werden, oft mit einer 50:50-Mischung oder der Zugabe spezifischer Komponenten zu EGM2. - Sobald sich das Gefäßnetz gebildet hat und das Gewebe vollständig entwickelt ist (Tag 4-7), ist ein Dextransfusionstest durchzuführen, bevor die Geräte für Experimente verwendet werden (Schritt 4.2.1.). Verwenden Sie nur Geräte, die über eine ausreichende Durchblutung der Gewebekammer verfügen.
- Bei Experimenten mit Therapeutika werden am Tag des Behandlungsbeginns Bilder in allen Fluoreszenzkanälen für jedes Gerät aufgenommen. Dies dient als Grundlage.
- Behandeln Sie Geräte mit dem gewünschten Therapeutikum, indem Sie das Medium durch ein frisches Medium ersetzen, das das verdünnte Arzneimittel in der gewünschten Konzentration enthält. Stellen Sie sicher, dass Arzneimittel je nach Empfehlung des Herstellers in geeigneten Vehikeln verdünnt werden, aber 0,01 % DMSO in den Medien nicht überschreiten.
- Setzen Sie die Geräte dem Arzneimittel für die gewünschte Zeitspanne aus (in der Regel 48 Stunden, kann aber durch Pharmakokinetik informiert werden).
- Bilden Sie jeden Kanal jedes Geräts im gewünschten Zeitintervall ab, um das Ansprechen auf die Behandlung zu überwachen. Die Platten sind wie in Schritt 4.1.1 beschrieben für die Dauer des Versuchs aufzubewahren.
- Nach Beendigung des Versuchs werden Bleichplatten aufgetragen und in einen Biohazard-Behälter gegeben, mit 4 % PFA für die Immunfluoreszenzfärbung fixiert (Schritt 4.3.) oder zur Isolierung von lebenden Zellen oder RNA geerntet (Schritt 4.4.).
- Wechseln Sie das Medium jeden zweiten Tag mit EGM2, bis sich das Gefäßsystem vollständig etabliert hat (Tag 5-6). Altmedien vollständig absaugen und Hochdruckbrunnen (350 μL) und Niederdruckbrunnen (50 μL) ersetzen.
- Perfusions-Assays
- Perfusion von Dextran
HINWEIS: Die vaskuläre Permeabilität/Durchgängigkeit kann durch Perfusion des Gefäßnetzwerks mit fluoreszenzmarkiertem Dextran mit unterschiedlichem Molekulargewicht (40 kD, 70 kD oder 150 kD) bestimmt werden. FITC- oder Rhodamin-Dextran können je nach Fluoreszenzmarkierung des EC verwendet werden.- Bestimmen Sie vor der Perfusion die angemessene Exposition des FITC- oder Rhodaminkanals, indem Sie einige μl Dextran in den fluidischen Kanal oder die Kammer eines leeren Geräts geben. Stellen Sie die Belichtungszeit knapp unter der Sättigungsstufe ein, indem Sie eine Mikroskopsoftware verwenden, um ein Histogramm der Pixelintensitäten anzuzeigen und einen Dynamikbereich zu gewährleisten, der durch gleichmäßig verteilte Pixel ohne nennenswerte Konzentration von Werten mit hoher Intensität gekennzeichnet ist.
- Machen Sie Mikroskopaufnahmen von allen Geräten in den interessierenden Kanälen, einschließlich eines Hintergrundbilds aller Geräte im fluoreszierenden Dextrankanal, um sie vor dem Hintergrund zu kalibrieren. Verwenden Sie die gleiche Belichtung wie oben bestimmt und richten Sie die Gewebekammer in der Mitte des Bildrahmens aus, um konsistente Bilder für die Quantifizierung zu gewährleisten.
- Bereiten Sie einen Hauptvorrat an FITC-Dextran oder Rhodamin-Dextran in einer Konzentration von 5 mg/ml in 1x DPBS vor. Dieser Vorrat kann bei 4 °C gelagert werden.
- Zur Herstellung eines Arbeitsstoffs werden 5 mg/ml Stammstoff in EGM2 auf eine Endkonzentration von 50 μg/ml verdünnt.
- Ersetzen Sie das Medium in den Reservoirs durch die verdünnte Dextranlösung als halbes maximales Volumen (175 μl in eine Vertiefung sowie die obere Seite auf dem oberen oder unteren Kanal der Gewebekammer). Ersetzen Sie die Medien in den anderen Vertiefungen, so dass die entkoppelte obere Seite 175 μl frisches EGM2 ohne Dextran erhält und die Vertiefungen auf der unteren Seite jeweils nur 50 μl enthalten.
HINWEIS: Dextran sollte nur auf einer Seite der mikrofluidischen Kanäle (oben oder unten) hinzugefügt werden, um die Visualisierung des Farbstoffs zu ermöglichen, der durch die Hochdruckseite, in das Gefäßbett und aus der Niederdruckseite fließt. - Achten Sie unter dem Mikroskop darauf, dass das fluoreszierende Dextran durch das Gefäßnetz fließt. Dies geschieht in der Regel innerhalb von ca. 2 Minuten nach Zugabe des Farbstoffs in das Medienreservoir.
- Beginnen Sie mit der Abbildung des fluoreszierenden Dextrankanals (und anderer Kanäle, falls gewünscht). Dies ist der Zeitpunkt T = 0. Nehmen Sie zusätzliche Bilder zu mehreren Zeitpunkten (in der Regel alle 10 Minuten) oder ein einzelnes Endpunktbild auf.
- Durchblutung von Zellen
HINWEIS: Je nach Studiendesign können verschiedene Zelltypen durch das Gefäßsystem perfundiert werden, darunter Lymphozyten oder Makrophagen für Krebsimmunologiestudien sowie Krebszellen für Metastasierungsstudien. Die Zellen müssen fluoreszenzmarkiert werden, um die Nachverfolgung im Laufe der Zeit zu erleichtern.- Führen Sie mindestens 2 Stunden vor der Durchblutung der Zellen eine Dextranperfusion an allen Geräten wie oben beschrieben durch. Dieser Schritt ist wichtig, um die Durchgängigkeit der Gefäße vor dem Hinzufügen von Zellen zu bestimmen.
- Bestimmen Sie die geeignete Kamerabelichtung für die zu perfundierten Zellen. Nehmen Sie eine kleine Probe von Zellen, um sie unter dem Mikroskop zu betrachten, und stellen Sie die Belichtungszeit für diesen fluoreszierenden Marker ein. Stellen Sie die Belichtungszeit knapp unter der Sättigungsstufe ein.
- Machen Sie Mikroskopaufnahmen von allen Geräten in den interessierenden Kanälen, einschließlich eines Hintergrundbilds aller Geräte im fluoreszierenden Dextrankanal, um sie vor dem Hintergrund zu kalibrieren. Verwenden Sie die gleiche Exposition wie in Schritt 4.2.2.1 bestimmt.
- Vergewissern Sie sich, dass Dextran vollständig aus den Gewebekammern diffundiert ist, bevor die Zellen für die Perfusion entnommen werden. Ernten und zählen Sie die Zellen, die Sie interessieren.
- Resuspendieren Sie Zellen mit entsprechender Dichte in EGM2. Zum Beispiel werden T-Zellen typischerweise mit etwa 1 x 106 Zellen/ml hinzugefügt, um die Konzentration im Blut nachzuahmen.
HINWEIS: EC reagieren empfindlich auf die Zusammensetzung des Mediums, können aber bis zu 50 % Mischung mit den meisten anderen Medien tolerieren. Testen Sie vorher. - Geben Sie 175 μl Zellsuspension in eine Vertiefung auf der oberen Seite jedes Geräts und geben Sie 175 μl EGM2 vollständig in die andere Vertiefung. Auf der niedrigen Seite geben Sie 50 μl des vollständigen EGM2-Mediums in beide Vertiefungen.
- Achten Sie unter dem Mikroskop darauf, dass die fluoreszierenden Zellen durch das Gefäßnetz fließen. Dies geschieht in der Regel innerhalb von ca. 2 Minuten nach dem Hinzufügen der Zellen zum Medienreservoir.
- Sobald der Zellfluss hergestellt ist, beginnen Sie mit der Abbildung des fluoreszierenden Zellkanals (und anderer Kanäle, falls gewünscht). Dies ist der Zeitpunkt T = 0. Nehmen Sie je nach Studiendesign zusätzliche Bilder zu mehreren Zeitpunkten oder ein einzelnes Endpunktbild auf. Zum Beispiel führt die Bildgebung alle 10 Minuten zu hochauflösenden Zeitverläufen oder alle 6-12 Stunden, um periodische Zellbewegungen zu verfolgen.
- Perfusion von Dextran
- Immunfluoreszierende (IF) Färbung
- Saugen Sie Medien aus Brunnen ab. Geben Sie 200 μl 4 % PFA in beide Vertiefungen der oberen Seite jeder Geräteeinheit und 50 μl in die untere Seite. Lassen Sie PFA 15 Minuten bei Raumtemperatur oder 30 Minuten bei 4 °C durch die Kammern fließen.
- Bereiten Sie während der Inkubation eine 24-Well-Platte mit 500 μl 1x PBS pro Well vor. Berechnen Sie, wie viele Wells benötigt werden, um jedes Gerät zu färben.
- PFA vollständig aus den Vertiefungen entfernen. Drehen Sie die Platte auf den Kopf und entfernen Sie vorsichtig die Kunststoffrückseite der Membran.
HINWEIS: Die IF-Färbung kann auch in situ durchgeführt werden, ohne die Membran und das Gerät zu entfernen. Führen Sie dazu Färbeschritte durch, indem Sie Reagenzien durch das VMO/VMT perfundiert und die Dauer jedes Inkubationsschritts um etwa das 6-fache erhöhen. - Ziehen Sie die untere Membranschicht sehr vorsichtig und vorsichtig von der Funktionsschicht des Geräts ab, um die Gewebekammern freizulegen, indem Sie beide Ecken greifen und in einer langsamen und gleichmäßigen Bewegung nach unten ziehen. Der größte Teil des Gewebes sollte in der Gewebekammer verbleiben. Siehe Abbildung 3A.
- Verwenden Sie eine Rasierklinge oder ein Skalpell, um genügend Kraft aufzuwenden, um die Feature-Ebene vollständig zu durchtrennen, und schneiden Sie ein kleines Rechteck um jede einzelne Geräteeinheit, wie in Abbildung 3B dargestellt.
- Klemmen Sie einen Spachtel zwischen die Feature-Schicht und die Well-Platte. Üben Sie sanften Druck unter der Feature-Schicht aus, um die gesamte Feature-Schicht, die die Gewebekammer enthält, vorsichtig von der Well-Platte zu entfernen.
- Legen Sie jedes rechteckige PDMS-Feature-Layer-Stück, das das Gewebe enthält, mit der Vorderseite nach unten in eine einzelne Vertiefung, die PBS enthält.
- Sobald sich alle Einheiten in Vertiefungen befinden, waschen Sie mit PBS, indem Sie die Platte 5 Minuten lang auf eine sanfte Wippe stellen, PBS aus der Vertiefung ansaugen und durch 500 μl frisches PBS ersetzen. Wiederholen Sie dies für insgesamt 3 Wäschen.
- PBS aus jeder Vertiefung absaugen und Gewebe mit 500 μl 0,5% Triton-X in PBS permeabilisieren, 2x für jeweils 10 min auf einer sanften Wippe. Permeabilisierungslösung entfernen.
- Blockieren Sie 500 μl 10%iges Serum in 0,1% Triton-X pro Gerät für 1 h bei Raumtemperatur mit leichtem Schaukeln.
- Verdünnen Sie die Primärantikörper in 3 % Serum in 0,1 % Triton-X auf die gewünschte Konzentration und das gewünschte Volumen. Entfernen Sie die blockierende Lösung und fügen Sie genügend primäre Antikörperlösung hinzu, um den Boden jeder Vertiefung vollständig zu bedecken und eine freie Bewegung des Gerätegewebes (~200 μl) zu ermöglichen. Decken Sie die Platte mit einer transparenten Folie ab.
- Die Platte über Nacht bei 4 °C schaukeln lassen. Am nächsten Tag bringen Sie die Platte mit den Gerätetaschentüchern wieder auf Raumtemperatur (~15 min).
- Primäre Antikörperlösung aus jeder Vertiefung und Waschkammer mit 500 μL PBS absaugen, 3x für jeweils 5 min auf einer sanften Wippe.
- 200 μl sekundärer Antikörper in 3 % Serum in 0,1 % Triton-X in der gewünschten Konzentration hinzufügen. Die Platte mit leichtem Schaukeln 1 h bei Raumtemperatur im Dunkeln inkubieren.
- Sekundäre Antikörperlösung absaugen und mit PBS waschen, 3x für jeweils 5 min unter leichtem Schaukeln. Fügen Sie eine Lösung von 1x DAPI in 0,1% Triton-X für 10 Minuten hinzu, während Sie im Dunkeln schaukeln.
- Entfernen Sie rechteckige Geräteausschnitte mit verfärbtem Gewebe mit einer Pinzette von der Platte und legen Sie sie mit der Taschentuchseite nach oben auf ein Papiertuch.
- Pipettieren Sie einige μl (~10 μl) Antifade-Lösung direkt auf jede Kammer und jedes Deckglas und achten Sie darauf, dass keine Blasen entstehen. Lassen Sie das Antifade über Nacht bei Raumtemperatur im Dunkeln in den Kammern aushärten und fahren Sie dann mit der Bildgebung fort.
- Gewebe- und Zellisolierung für molekulare Assays
HINWEIS: Jede Platte mit hohem Durchsatz enthält je nach Erntezeitpunkt ca. 1-2 x 105 Zellen. Skalieren Sie die Anzahl der experimentellen Replikationen, um die Gesamtzahl der Zellen sowie den potenziellen Verlust während der Ernte zu berücksichtigen.- Einzelzell-Analysen
- Saugen Sie Medien aus jeder Vertiefung an und drehen Sie die Platte mit hohem Durchsatz um, so dass die Geräteschicht nach oben zeigt.
- Entfernen Sie die Kunststoffrückseite auf der Membran. Ziehen Sie die PDMS-Bodenmembran sehr vorsichtig und vorsichtig von der Funktionsschicht des Geräts ab, um die Gewebekammern freizulegen, indem Sie beide Ecken greifen und in einer langsamen und gleichmäßigen Bewegung nach unten ziehen.
- Die Membran kann auch bei ordnungsgemäßer Verklebung entfernt werden. Der größte Teil des Gewebes sollte nach dem Entfernen der Membran in der Gewebekammer verbleiben; Wenn jedoch ein Teil an der Membran klebt, befolgen Sie die folgenden Schritte auf der Membran selbst.
- Waschen Sie jede Geräteeinheit mit 500 μl HBSS oder PBS. Geben Sie 100 μl Dissoziationsreagenz in jede Geräteeinheit und lassen Sie es als Tröpfchen auf dem Gerät sitzen. Stellen Sie die Platte für 5 Minuten zurück in den 37 °C warmen Inkubator.
- Verwenden Sie nach dem Aufschluss eine P200-Pipette, um die Gewebekammern auf und ab zu pipepsieren und das Gewebe im Dissoziationsreagenz zu sammeln. Bewegen Sie die Pipettenspitze über jedes Gerät hin und her, um sicherzustellen, dass das Gewebe vollständig entfernt wird, und sammeln Sie es in einem 15-ml-Konus mit 500 μl EGM2, um das Dissoziationsreagenz zu neutralisieren.
- Um die verbleibenden Zellen vollständig aus der Gewebekammer zu entfernen, geben Sie 500 μl EGM2 in jede Geräteeinheit und waschen Sie sie mit einer P200-Pipette.
- Die Aufschlusslösung, die das gelöste Gewebe enthält, wird bei 300 x g für 5 Minuten bei 4 °C zentrifugiert, um einzelne Zellen und ganze Gewebe zu pelletieren.
- Saugen Sie das Medium vorsichtig ab und geben Sie 500 μl 1 mg/ml (200 U/ml) Kollagenase Typ IV, 0,1 mg/ml Hyaluronidase Typ V und 200 U/ml DNAse Typ IV in HBSS in das Gewebe hinzu.
- Lassen Sie die Lösung nach der sanften Resuspension 2 Minuten lang bei Raumtemperatur einwirken, bevor Sie sie erneut vorsichtig pipettieren, um das Gel zu dissoziieren.
- Die Aufschlussmischung mit 10 ml EGM2 wird gewaschen und bei 300 x g 5 min bei 4 °C zentrifugiert. Die Zellen werden in 1x DPBS mit 1 % BSA oder HSA resuspendiert und durch einen vorgetränkten 70-μm-Filter geleitet, indem sie 1 Minute lang bei 200 x g geschleudert werden.
- Zählen Sie die Zellen und passen Sie das Volumen so an, dass die endgültige Konzentration 1000 Zellen pro μl beträgt. Zelluläre Suspensionen können dann einer FACS, Durchflusszytometrie oder Einzelzell-RNA-Sequenzierung unterzogen werden.
- RNA-Isolierung aus ganzen Geweben
- Befolgen Sie die Schritte 4.4.1.1 und 4.4.1.2 oben.
- Geben Sie ca. 10 μl RNA-Lysepuffer in jede exponierte Gewebekammer und stellen Sie sicher, dass sich die Puffer direkt auf dem Gewebe ansammeln. Verwenden Sie insgesamt nicht mehr als 100 μl RNA-Lysepuffer.
- 3 Minuten bei Raumtemperatur inkubieren. Verwenden Sie den P20, um auf jeder Geräteeinheit auf und ab zu pipettieren, und verwenden Sie die Pipettenspitze, um bei Bedarf Restmaterial aus der Gewebekammer zu kratzen.
- Geben Sie so viel Lysepuffer wie möglich in ein 1,5-ml-Mikrozentrifugenröhrchen. Wiederholen Sie die Schritte 4.4.2.2.-4.4.2.3. für restliche Geräte und Poolproben in das 1,5-ml-Röhrchen.
- Befolgen Sie die Anweisungen des Herstellers zur Isolierung von RNA, je nach Kit oder Reagenzien.
- Einzelzell-Analysen

Abbildung 3. Vorbereitung der Plattform für die Immunfärbung. (A) Schematische Darstellung einer vollständig montierten Geräteplattform mit Membranschicht auf der Oberseite. Um die Membran zu entfernen, ziehen Sie vorsichtig jede Ecke der äußeren Schicht in einer gleichmäßigen, sanften Bewegung nach unten. (B) Sobald die Membranschicht vollständig entfernt ist, schneiden Sie mit einer Klinge, einem Skalpell oder einem Messer Rechtecke um die Gewebekammer jeder Geräteeinheit herum und achten Sie darauf, nicht in das Gewebe selbst zu schneiden. Ein Spatel kann dann unter jedes Rechteck geklemmt werden, um es von der Platte zu lösen, und jede Einheit in eine einzelne Vertiefung einer 24-Well-Platte mit PBS zum Färben zu legen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
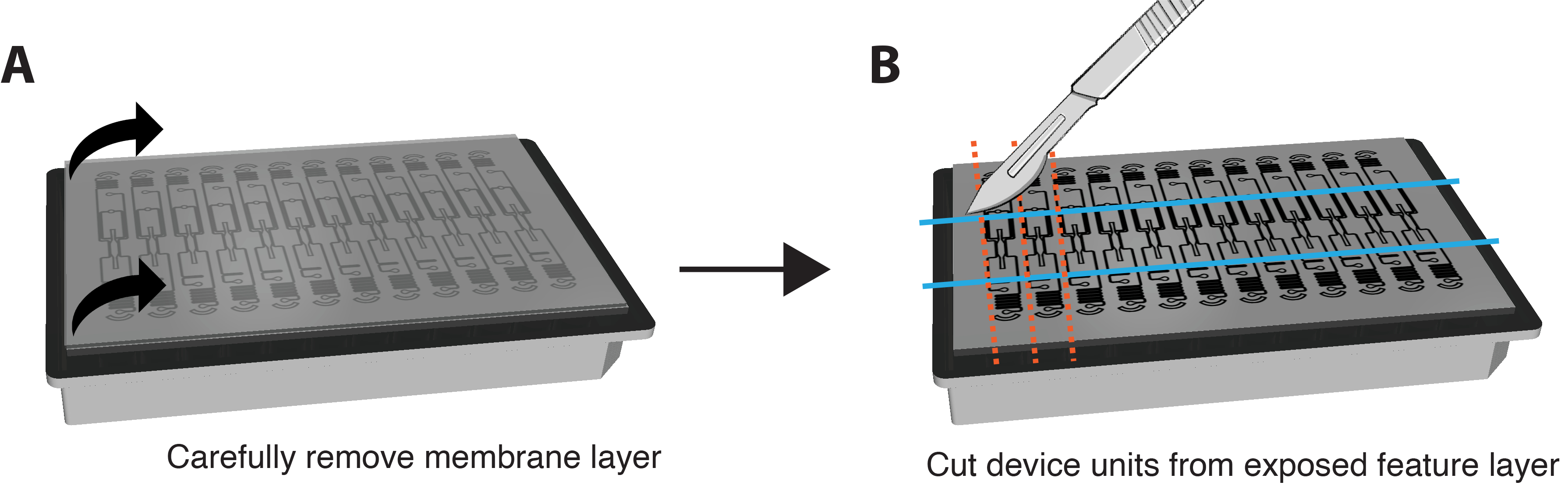{kind=link}
Ergebnisse
Gemäß den hier beschriebenen Protokollen wurden VMOs und VMTs unter Verwendung von kommerziell erworbenen EC-, NHLF- und für VMT der triple-negativen Brustkrebszelllinie MDA-MB-231 etabliert. Etablierte VMOs wurden auch mit Krebszellen durchblutet, um die Metastasierung nachzuahmen. In jedem Modell bildet sich an Tag 5 der Co-Kultur ein vaskuläres Netzwerk als Reaktion auf die schwerkraftgetriebene Strömung durch die Gewebekammer selbst und dient als Kanal für die In-vivo-artige Abgabe von Nährstoffen, Th...
Diskussion
Fast jedes Gewebe im Körper erhält Nährstoffe und Sauerstoff über das Gefäßsystem, was es zu einer kritischen Komponente für eine realistische Krankheitsmodellierung und ein Arzneimittelscreening in vitro macht. Darüber hinaus werden verschiedene Malignome und Krankheitszustände durch vaskuläre endotheliale Dysfunktion und Hyperpermeabilität definiert3. Bemerkenswert ist, dass bei Krebs das tumorassoziierte Gefäßsystem oft schlecht durchblutet, gestört und undicht ist, was a...
Offenlegungen
CCWH ist an Aracari Biosciences, Inc. beteiligt, das eine Version der in diesem Artikel beschriebenen Technologie vermarktet. Die Bedingungen dieser Vereinbarung wurden von der University of California, Irvine, in Übereinstimmung mit ihren Richtlinien zu Interessenkonflikten geprüft und genehmigt. Es bestehen keine weiteren Interessenkonflikte.
Danksagungen
Wir danken den Mitgliedern des Labors von Dr. Christopher Hughes für ihren wertvollen Beitrag zu den beschriebenen Verfahren sowie unseren Mitarbeitern im Labor von Dr. Abraham Lee für ihre Unterstützung bei der Entwicklung und Herstellung der Plattform. Diese Arbeit wurde durch folgende Zuschüsse unterstützt: UG3/UH3 TR002137, R61/R33 HL154307, 1R01CA244571, 1R01 HL149748, U54 CA217378 (CCWH) und TL1 TR001415 und W81XWH2110393 (SJH).
Materialien
| Name | Company | Catalog Number | Comments |
| Fabrication | |||
| (3-Mercaptopropyl)trimethoxysilane, 95% | Sigma-Aldrich | 175617-100G | |
| Greiner Bio-One μClear Bottom 96-well Polystyrene Microplates | Greiner Bio-One | 655096 | |
| Methanol ≥99.8% ACS | VWR Chemicals BDH | BDH1135-1LP | |
| MILTEX Sterile Disposable Biopsy Punch with Plunger, 1mm diameter, | Integra Miltex | 33-31AA-P/25 | |
| PDMS membrane | PAX Industries | HT-6240 | |
| Plasma Cleaner PDC-001 | Harrick Plasma | N/A | |
| Smooth-Cast 385 | Smooth-On | N/A | |
| SP Bel-Art Lab Companion Clear Polycarbonate Cabinet Style Vacuum Desiccator | Bel-Art | F42400-4031 | |
| Standard Lids with Condensation Rings, 96-well plate | VWR | 82050-827 | |
| SYLGARD 184 Silicone Elastomer Kit (PDMS) | Dow | 4019862 | |
| Cell culture/Loading | |||
| BioTek Lionheart FX Automated Microscope | Agilent | CYT5MFAW | |
| CELLvo Human Endothelial Progenitor Cells | StemBioSys | N/A | |
| Collagen I, rat tail | Enzo Life Sciences | ||
| Collagenase from Clostridium histolyticum (type 4) | Sigma-Aldrich | C5138 | |
| Corning Hank’s Balanced Salt Solution, 1X without calcium and magnesium | Corning | 21-021-CV | |
| Corning DMEM with L-Glutamine, 4.5g/L Glucose and Sodium Pyruvate | Corning | 10013CV | |
| DAPI | Sigma-Aldrich | D9542 | |
| DPBS, no calcium, no magnesium | Gibco | 14190144 | |
| EGM-2 Endothelial Cell Growth Medium-2 BulletKit | Lonza | CC-3162 | |
| Fibrinogen from bovine plasma | Neta Scientific | SIAL-341573 | |
| Fibronectin human plasma | Sigma-Aldrich | F0895 | |
| Fluorescein isothiocyanate–dextran (70kDa) | Sigma-Aldrich | FD70S-1G | |
| Gelatin from porcine skin | Sigma-Aldrich | G1890 | |
| Hyaluronidase from sheep testes (type 4) | Sigma-Aldrich | H6254 | |
| Laminin Mouse Protein | Gibco | 23017015 | |
| Leica TCS SP8 | Leica | N/A | |
| MDA-MB-231 | ATCC | HTB-26 | |
| NHLF – Normal Human Lung Fibroblasts | Lonza | CC-2512 | |
| Nikon Eclipse Ti | Nikon | N/A | |
| Paraformaldehyde 4% in 0.1M Phosphate BufferSaline, pH 7.4 | Electron Microscopy Sciences | 15735-90-1L | |
| PBMCs - Peripheral blood mononuclear cells | Lonza | CC-2702 | |
| PBS, pH 7.4 | Gibco | 10010049 | |
| Premium Grade Fetal Bovine Serum (FBS), Heat Inactivated | Avantor Seradigm | 97068-091 | |
| ProLong Gold Antifade Mountant | Invitrogen | P10144 | |
| Quick-RNA Microprep Kit | Zymo Research | R1051 | |
| Thrombin from bovine plasma | Sigma-Aldrich | T4648 | |
| Triton X-100 (Electrophoresis), | Fisher BioReagents | BP151-100 | |
| TrypLE Express Enzyme (1X), phenol red | Gibco | 12605028 | |
| Trypsin-EDTA (0.05%), phenol red | Gibco | 25300062 | |
| Vasculife | Lifeline Cell Technology | LL-0003 |
Referenzen
- Siegel, R. L., Miller, K. D., Wagle, N. S., Jemal, A. Cancer statistics, 2023. CA Cancer J Clin. 73 (1), 17-48 (2023).
- Hachey, S. J., Hughes, C. C. W. Applications of tumor chip technology. Lab Chip. 18 (19), 2893-2912 (2018).
- Ewald, M. L., Chen, Y. H., Lee, A. P., Hughes, C. C. W. The vascular niche in next generation microphysiological systems. Lab Chip. 21 (17), 3615-3616 (2021).
- Osaki, T., Sivathanu, V., Kamm, R. D. Vascularized microfluidic organ-chips for drug screening, disease models and tissue engineering. Curr Opin Biotechnol. 52, 116-123 (2018).
- Shirure, V. S., Hughes, C. C. W., George, S. C. Engineering vascularized organoid-on-a-chip models. Annu Rev Biomed Eng. 23, 141-167 (2021).
- Del Piccolo, N., et al. Tumor-on-chip modeling of organ-specific cancer and metastasis. Adv Drug Deliv Rev. 175, 113798 (2021).
- Sontheimer-Phelps, A., Hassell, B. A., Ingber, D. E. Modelling cancer in microfluidic human organs-on-chips. Nat Rev Cancer. 19 (2), 65-81 (2019).
- Sobrino, A., et al. 3D microtumors in vitro supported by perfused vascular networks. Sci Rep. 6, 31589 (2016).
- Phan, D. T. T., et al. A vascularized and perfused organ-on-a-chip platform for large-scale drug screening applications. Lab Chip. 17 (3), 511-520 (2017).
- Hachey, S. J., et al. An in vitro vascularized micro-tumor model of human colorectal cancer recapitulates in vivo responses to standard-of-care therapy. Lab Chip. 21 (7), 1333-1351 (2021).
- Hachey, S. J., et al. A Human Vascularized Micro-Tumor Model of Patient-Derived Colorectal Cancer Recapitulates Clinical Disease. Transl Res. 255, 97-108 (2023).
- Liu, Y., et al. Human in vitro vascularized micro-organ and micro-tumor models are reproducible organ-on-a-chip platforms for studies of anticancer drugs. Toxicology. 445, 152601 (2020).
- Jahid, S., et al. Structure-based Design of CDC42 Effector Interaction Inhibitors for the Treatment of Cancer. Cell Rep. 39 (4), 110760 (2022).
- Hsu, Y. H., Moya, M. L., Hughes, C. C. W., George, S. C., Lee, A. P. A microfluidic platform for generating large-scale nearly identical human microphysiological vascularized tissue arrays. Lab Chip. 13 (15), 2990-2998 (2013).
- Moya, M. L., Hsu, Y. H., Lee, A. P., Christopher, C. W. H., George, S. C. In vitro perfused human capillary networks. Tissue Eng - Part C: Methods. 19 (9), 730-737 (2013).
- Wang, X., et al. An on-chip microfluidic pressure regulator that facilitates reproducible loading of cells and hydrogels into microphysiological system platforms. Lab Chip. 16 (5), 868-876 (2016).
- Phan, D. T., et al. Blood-brain barrier-on-a-chip: Microphysiological systems that capture the complexity of the blood-central nervous system interface. Exp Biol Med. 242 (17), 1669-1678 (2017).
- Kurokawa, Y. K., et al. Human induced pluripotent stem cell-derived endothelial cells for three-dimensional microphysiological systems. Tissue Eng Part C: Methods. 23 (8), 474-484 (2017).
- Romero-López, M., et al. Recapitulating the human tumor microenvironment: Colon tumor-derived extracellular matrix promotes angiogenesis and tumor cell growth. Biomaterials. 116, 118-129 (2017).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 9 (7), 676-682 (2012).
- Carpenter, A. E., et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7 (10), R100 (2006).
- Zudaire, E., Gambardella, L., Kurcz, C., Vermeren, S. A computational tool for quantitative analysis of vascular networks. PLoS one. 6 (11), e27385 (2011).
- Corliss, B. A., et al. REAVER: A program for improved analysis of high-resolution vascular network images. Microcirculation. 27 (5), e12618 (2020).
- Urban, G., et al. Deep learning for drug discovery and cancer research: Automated analysis of vascularization images. IEEE/ACM Trans Comput Biol Bioinform. 16 (3), 1029-1035 (2019).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten