
Method Article
分離とクラミドモナス由来の単一粒子再構成のための蛍光イメージング
要約
浄化し、多数の超解像顕微鏡法と平均単一粒子の影響を受けやすい向きが異なる由来のイメージ戦略を開発しました。
要約
由来は、大規模な高分子アセンブリ細胞分裂、細胞運動、シグナル伝達などの基礎的な細胞生物学的プロセスの適切な実行のために重要です。緑藻クラミドモナスは中心小体のアーキテクチャ、機能、およびタンパク質合成の研究で洞察力に富んだモデルであると証明しました。セントリ アーキテクチャについての偉大な進歩にもかかわらず、現在の課題の 1 つはで彼らの役割を理解するために小体の構造領域内でセントリ コンポーネントの正確な局在を決定することです。中心小体の形成。主な制限は、回折限界に近い寸法でこのオルガネラにおけるタンパク質局在の解釈を複雑にする蛍光顕微鏡の分解能にあります。この質問を取り組むためには、我々 は浄化し、超解像顕微鏡を用いたさまざまな方向とC. クラミドモナス由来の多数のイメージ法を提供しています。この手法は、さらに蛍光単一粒子由来の買収の数が多いため (蛍光 SPA) の平均を使用してデータの処理が可能します。蛍光スパ生成の平均の向きが異なるためセントリ サブ領域の明瞭な蛋白質のローカリゼーションを容易C. クラミドモナス由来の染色します。重要なは、このメソッドは、他の種や他の大規模な高分子アセンブリからイメージ由来に適用できます。
概要
小体は進化的に保存された細胞小器官動物細胞における中心体の中核に位置しテンプレート繊毛または多くの真核生物1,2鞭毛基底ボディ (以下由来といいます) として使用できます。など、由来は基礎的な細胞生物学的プロセスのシグナルを細胞に紡錘アセンブリに至る重要です。したがって、中心小体アセンブリまたは機能の欠陥に関連付けられていくつか人間キレる ciliopathies と癌の3。
由来は、nine-fold、対称、トリプレット ベース構造が円筒形微小管は、通常、〜 450 nm 長いと ~ 250 nm ワイド4,5,6,7。従来の電子顕微鏡と異なる種から由来の低温電子トモグラフィーは由来が 3 つの領域に彼らの長い軸に沿って偏光は明らかにした: 近位部、中央のコアおよび遠位領域5,7,8,9,10,11します。 重要なのは、これらの領域のそれぞれは特定の構造特徴を表示します。まず、100 nm の長さの近位部の内腔には、ピンヘッド要素12を介して微小管トリプレットに接続されている車輪の構造が含まれています。第二に、300-400 nm の長さの中央部を含む線維密度微小管の内面に沿ってルーメンと構造機能: Y 字リンカー、C 管尾、A 管スタブ9。最後に、50-100 nm の遠位領域中心小体5,13の遠位部分を囲むサブ遠位部と遠位肢を展示します。
最後の二十年は、中心体タンパク質、中心小体14,15,16,の一部である約 100 の異なる蛋白質の現在の推定数の増加の発見によってマークされています。17. これらの進歩にもかかわらずこれらのタンパク質小体内の正確な局在ままとらえどころのない、特に構造のサブ領域内で。重要なは、小体の構造領域に正確な局在の割り当ては、その機能の理解を深めるために重要です。この点で、 C. クラミドモナス由来尽力している両方の面で最初の delimitating シリンダー9,18,19、それから許可に沿ってさまざまな構造機能でサブ構造領域に蛍光顕微鏡を用いたタンパク質のサブセットのローカリゼーションを関連付ける研究者。これは、たとえば、含まれています、タンパク質 Bld12p と Bld10p、近位部と車輪の構造を特にローカライズ20,報21,22,23。ローカライズされた部分構造蛋白質のリストは、POB15、POC16、 C. クラミドモナス由来17の内側中心領域を飾る 2 つの新規タンパク質質量分析法による識別にも含まれます。
このペーパーでは、開発を分離し、その後超解像顕微鏡の単一粒子平均C. クラミドモナス由来のイメージ方法の完全な説明を提供します。この目標を達成するために克服しなければならない技術的な制限を記述することが重要です。まず、中心小体の浄化はしばしば分離9の様々 なステップの中に失われる車輪構造と全体的なアーキテクチャを及ぼします。第二に、小体の寸法が非常に回折限界に近い光学顕微鏡検査です。確かに、共焦点顕微鏡で得ることができる横方向の分解能は約 200 nm24と同様、中心小体とzの解像度の直径-軸は 2-3 倍低い、異方性体積に 。第三に、抗体の標識と中心小体の向きの不均一性は特定セントリ サブ領域でタンパク質をローカライズするために必要な解釈を制限できます。最後に、由来はイメージの大規模な数を取得し、明確な中心小体方向を見つけるが難しく、セル当たりの 2 つのコピーに存在します。これらの技術的な問題を回避するには、多数のさまざまな方向を採用する分離の由来に超解像顕微鏡法を適用することに依存するメソッドを開発しました。まず構造的にそのまま由来と、車輪を含む procentrioles の精製を可能にするc. クラミドモナス由来を浄化するプロトコルについて述べる。その後、中心体を従来のイメージングや超解像蛍光顕微鏡 coverslips に集中するステップバイ ステップ プロトコルについて述べる。この重要なステップは、由来の複数の向きでイメージの数を増やします。最後に、単一粒子の向きが異なる由来の検出を容易に蛍光顕微鏡で取得したデータを平均化を実行する手順を説明します。全体では、このメソッドは、様々 な種やその他の大規模な高分子アセンブリからイメージ由来に適用できます。
プロトコル
1. C. クラミドモナスの文化の中心小体分離メディアの準備
-
C. クラミドモナス細胞培養するメディアの準備
注: 以下の手順は、1 タップ (リン酸トリス酢酸) 中 x ストック溶液の調製をについて説明します。- 1 M KH2PO4 (KH2PO4 1 L 136.09 g) ~ 170 mL で 1 M K2HPO4 (K2HPO4蒸留水 1 L に混じって 174.2 g) 250 mL を混合することによってリン酸バッファー (pH 7) を準備します。PH 7 に到達する混合気を調整します。
- トリスの 96.8 グラム、40 mL のリン酸バッファー (pH 7)、酢酸 40 mL を混合することによってソリューション (40 x) を準備し、蒸留水 1 L にソリューションを調整します。
- ソリューション B の準備 (40 x) NH4Cl の 16 g、CaCl2, 2 g と MgSO4の 4 g を使用しています。その他のコンポーネントを追加する前に個別に CaCl2蒸留水を溶解する注意してください。蒸留水 1 L にソリューションを調整します。
- とおり25 Hutner のトレース要素のバッファーを準備します。
- バッファーの 1 L の溶解水の示された量の各化合物: EDTA 二ナトリウム塩 (250 mL に 50 g)、ZnSO4.7H2O (100 mL で 22 g) H3ボー3 (200 mL で 11.4 g)、MnCl2.4H2O (50 mL の 5.06 g)、CoCl2.6H FeSO4.7H2O (50 mL の 4.99 g) (NH4) 6 ヶ月頃7O24.4H2O (50 ml 1.10 g)、CuSO4.5H2O (50 mL の 1.57 g)2O (50 mL の 1.61 g)。
注: EDTA は、水を沸騰で解体されるべき、FeSO4は、酸化を避けるために最後用意する必要があります。 - EDTA を除くすべてのソリューションを一緒に混合し、沸騰させます。EDTA を追加し、ソリューションが緑色に変わります。すべてを溶解後クール 70 ° C への解決策この温度でホット 20 %koh 溶液 (100 mL に 20 g) 85 mL を追加します。常温 (RT) 蒸留水 1 L にソリューションを調整します。
- フラスコに綿のプラグを追加し、それを揺する 1 または 2 週間立つソリューション日 x 1。ソリューション必要があります最初は緑と紫、錆茶色の沈殿物; を残して電源を入れますソリューションがクリアされるまで、ろ紙を使用して沈殿物を削除します。試料を凍結し、-20 ° C で保存
- バッファーの 1 L の溶解水の示された量の各化合物: EDTA 二ナトリウム塩 (250 mL に 50 g)、ZnSO4.7H2O (100 mL で 22 g) H3ボー3 (200 mL で 11.4 g)、MnCl2.4H2O (50 mL の 5.06 g)、CoCl2.6H FeSO4.7H2O (50 mL の 4.99 g) (NH4) 6 ヶ月頃7O24.4H2O (50 ml 1.10 g)、CuSO4.5H2O (50 mL の 1.57 g)2O (50 mL の 1.61 g)。
- 次のコンポーネントを混合することによってc. クラミドモナス細胞を成長するタップ中 × 1 を準備: 溶液 (40 x) 25 mL、25 mL の溶液 B (40 倍) 1 mL Hutner の微量元素のバッファーの25、蒸留水 1 L に混合気を調整と。0.4 μ m フィルターを使用して混合物を滅菌します。
-
中心小体の浄化のためのメディアの準備
- 500 mL の蒸留水で調整の最終巻には 10 mM HEPES (pH 7) で 5% ショ糖を使用して deflagellation バッファーを準備します。
- 0.5 M 酢酸の 250 mL を準備します。
- H2O パイプ粉体を溶解するための最初の追加の 50 mL の 1 M K パイプ ストック溶液 (pH 7.2) の 250 mL を準備し、解決するまで N KOH をクリアになる開始 10 を追加します。10 N 1 N 島と pH 7.2 に滴定し、H2O を 250 mL の最終巻にソリューションを調整
- 5 のショ糖液 (w/w) を次のとおり準備します。
注: すべてのショ糖液 0.4 μ m フィルターを用いた可溶化は注射器に接続後、フィルターを適用する必要があります。60% と 70% ショ糖ソリューションが可溶化しにくいし、可溶化を容易にする 60 ° C に加温水槽に配置する必要がありますに注意してください。サッカロースは完全にまでは、10 分ごとをミックスします。- 25% ショ糖を準備、ショ糖の 25 グラムの重量を量る、10 mM K パイプ (pH 7.2) を追加することにより 100 g 重量を調整します。
- 60% ショ糖を準備、ショ糖の 60 グラムの重量を量る 10 ミリメートル K パイプ (pH 7.2) 100 g に重量を調整します。
- ショ糖密度勾配のショ糖液を準備します。ショ糖の 40 グラムの重量を量る 10 ミリメートル K パイプ (pH 7.2) 100 g ソリューションを調整することによって 40% ショ糖を準備します。同様に、50% (w/w) と 70% (w/w) ショ糖を準備します。
- -20 ° C でソリューションを格納します。注意解凍後ショ糖ソリューションを正しく再懸濁します。
- 100 ml 混合 1 mm HEPES (pH 7) 換散バッファー、0.5 mM MgCl2、および 1 np-40、および維持の 4 ° C でそれ常に、実験の日にこのバッファーの新鮮なを準備します。中心小体分離日抗プロテアーゼ タブレットを追加します。
- 1 混合物のボリューム塩酸持参で KH2PO4蒸留 H2o. 調整 pH 7.4 に 800 mL の 0.24 g Na2HPO4、1.44 g、KCl の 2 g の塩化ナトリウム 8 g を混合することによってリン酸バッファー生理食塩水 (PBS) (pH 7.4) x 1 を準備します。蒸留 H2o ・ L
2. C. クラミドモナス由来の分離
注: は、図 1を参照してください。
-
文化とC. クラミドモナス細胞の拡大
- 1 日目の夕方、文化 1 10 ml 三角フラスコに固体板からcw15 -ひずみを接種するタップ x。23 ° C で 2-3 日間の白色蛍光灯 (60 Με/m2s) の下のセルを成長します。
- 3 日目、希釈文化 10 (100 mL) x 1 x をタップし、23 ° C で 2-3 日の光の下で細胞の成長
- 6 日に希釈文化 10 x 1 x でタップすると文化の 1 L を入手します。文化 〜 107セル/mL26 (日 9-10) x 1 のおおよその細胞密度を示す暗い緑の色に到達するまで 23 ° c の光の下で細胞を成長します。
-
C. クラミドモナス由来の精製
- 50 mL の円錐管で 10 分間 600 x gでcw15 -細胞を遠心します。ペレットを洗浄細胞の 50 mL の 1x PBS で 1 x と 100 mL のピペットで室温 deflagellation バッファーのペレット 600 x gで 10 分間再懸濁しますでそれをスピンします。
- PH ショックで細胞をゆっくりと磁性攪拌器で 4.5-4.7 の最終的な pH に 0.5 M 酢酸の滴を追加することによって deflagellate し、2 分ゆっくりと pH を 7.0 に復元する 1 の N 島の滴の追加セルを孵化させなさい。
- 600 × g 10 分の戸建の鞭毛を削除するために細胞を遠心します。上澄みを除去し、氷の上にペレットを格納します。ペレット 2 x は 50 mL の 1x PBS 4 ° C で冷却のその後、4 ° C で 10 分間 600 x gでペレットをスピンします。
- 30 mL の 1x PBS でペレットを再懸濁し、(図 1B) を混合せずに 20 mL の 25% ショ糖クッションの上に懸濁液をゆっくりとロードします。
- 清; 残りの鞭毛を削除する 4 ° C で 15 分間 600 x gでスピンします。セルは 25% ショ糖 (図 1C) に広がっています。慎重に、吸引器を使用して上清を吸引によってのみ、一番 20 mL (赤矢印、図 1C) を保ちます。
- 20 mL の冷 1x PBS を追加することによって残りの 20 mL を洗浄します。4 ° C で 10 分間 600 x gでサンプルをスピンします。10 mL の (4 ° C) で冷 1x PBS でペレットを再懸濁します。次の換散は、一度にすべてのセルを当るように塊がないことを確認します。
- 新しい 250 ml にペレットを再懸濁を転送します。100 mL のセルに DNase の 5,000 単位を補完換散バッファーを追加します。セル、および他の方法で周りに換散バッファーを追加することが重要です。4 ° C で 1 h の混合物を孵化し、任意の気泡を形成することがなくボトル 15 分毎の反転によって慎重に混ぜます。
- 任意細胞の残骸を削除する 50 mL のコニカル チューブの 4 ° C で 10 分間の 600 x gで分離細胞を遠心します。細胞ペレットを白にする必要があります、溶解が正しく実行された場合 (図 1D)。ピペットで上澄みを収集し、氷上に置いた 60% ショ糖クッションを含む 30 mL 丸底チューブにそれをロードします。その後、4 ° C で 30 分間 10,000 x gでチューブをスピンします。
注: いくつかの丸底チューブ (材料表) が必要で、培養上清の量に応じて。 - ショ糖クッションの上 1 mL までの培養上清を吸引します。残りの上清 1 mL、2 mL ショ糖クッションの間黄色のインターフェイスに注意してください。優しく P1000 カットとショ糖および残りの上清をミックスします。この手順でボルテックスにかけないでください。それ以外の場合、procentrioles はこの段階で失われます。プールのすべてのショ糖クッション、氷の上に保管します。
- 薄肉 38.5 mL ポリプロピレン チューブに 40%-70% ショ糖密度勾配を準備するには、軽く 70% ショ糖 (4 ° C) で続いて 3 mL の 50% と 40% のショ糖の最後に、3 mL 3 mL を追加します。40%-70% ショ糖グラデーションにプールされたインターフェイスを読み込む冷たいショ糖は非常に粘性のため、ゆっくりとこれを行います。10 mM K パイプ バッファー (pH 7.2) と管のバランス、4 ° C で 1 時間 15 分の 68,320 x g (例えば、SW32Ti 回転体を持つ) でそれらを遠心力場
- 4 ° C で 12 x 500 μ L 分画を収集するには、異なるショ糖層を乱すことがなくチューブの底に 0.8 mm の針で穴を作るします。カット P200 ピペット チップで次の蛍光抗体法に使用される各画分から追加の 10 μ L の因数を準備します。液体窒素で分数をスナップ-凍結。
注: 孤立した由来は、中心体の全体的な微細構造が保持されることを確保するための電子顕微鏡による分析できます。
3. Coverslips に分離由来の定量化: 遠心分離と蛍光抗体法
注: は、図 2を参照してください。
-
チューブと coverslips 準備
- 分数あたり 1 ガラス丸底チューブ (15 mL) を使用して、中心体画分 (合計 12 管) を分析します。
- サポートのカスタム coverslip アダプター (アダプターを以下と呼ばれる) を丸底チューブに入れます。丸底チューブ滅菌 12 mm coverslip の場所。
注意: 12 mm coverslips を使用してここで、18 mm coverslips 30 ml 丸底チューブを使用してのプロトコルを合わせることができます。 - 4 ° C で予冷 10 mm K パイプ (pH 7.2) 5 ml を追加します。Coverslip フローティングではないアダプターをご利用いただけますことを確認します。氷の上には、チューブを配置します。
-
中心体の遠心分離
- コールド 10 mM K パイプ (pH 7.2) の 100 μ L で各 10 μ L 分画を希釈します。スクロース (図 2A) の完全な消失まで希釈を再懸濁します。丸底チューブにそれぞれの希釈割合をロードします。
- 4 ° C で 10,000 x gで 10 分間 (例えば、JS 13.1 スイング バケツ ローター) で管をスピンします。
- アダプターのスロットの端にある穴に手作りフック デバイスを挿入して、coverslip を回復し、そっと持ち上げます。
注: 手動でフックし、自家製棒 (図 2B 2D) に成形の注射針で自作フック装置が可能です。 - 丸底チューブの先頭に達すると、時に手袋をはめた指でアダプターの端をトラップし、ピンセットで、coverslip を削除します。Coverslip のどちら側に中心体が含まれていることに留意します。蛍光抗体法の coverslips の治療に進みます。
-
蛍光染色とイメージング分離C. クラミドモナス由来
注: は、図 2を参照してください。- 蛍光抗体法により次のように染色のための材料を準備します。
- 結晶ポリスチレン伝送ラボ ボックス (高さ 43 mm、幅 50 mm の長さは 60 mm) でカバー ガラス染色ラックを準備します。100% メタノールでそれを埋めるし、-20 ° C で保存
- 湿気の多い部屋を準備します。このため、水加湿の組織内部の横に配置することによって湿気のある商工会議所を組み立てるペトリ皿 (図 2E) の正方形のエッジ。シーリング ラップ研究室の一部を追加 (材料表参照) 蛍光抗体法の手順 (手順 3.3.2–3.3.3) の間に配置されます抗体ミックスをシャーレの中央に。ふたをし、光から保護するためにアルミ箔で湿気のある商工会議所をカバーしてください。
- Immunostain として分離の由来に従います。
- -20 ° C のメタノール (ステップ 3.3.1.1) にそれらをインキュベート ボックスで 5 分で遠心分離 (ステップ 3.2.4) 入力後中心体と直接、coverslips を修正します。
- ピンセットと場所研究室の透明なボックスに 50 mL の 1x PBS でいっぱい (材料の表を参照してください)、室温で 5 分間洗って coverslips を削除します。
- 一次抗体のミックスの 60 μ L をピペット [0.05% と 1% ウシ血清アルブミン (BSA) で希釈した一次抗体 PBS でトゥイーン 20] 研究室の湿気のある部屋でラップをシールの部分に。ドロップに直接直面している中心体と抗体のミックスの上に coverslips をゆっくりと寝かせます。一次抗体と 45 分のため coverslips を孵化させなさい。
注: 代表的な結果の生成に使用された一次抗体がウサギのポリクローナル Bld12 (1: 300) とマウス α-チューブリン (DM1A) (1: 300)。 - Coverslips を削除し、1 × PBS で 5 分間洗って 3.3.2.2 の手順で説明するようします。1 %bsa および 0.05% を含む PBS で二次抗体と 45 分のため coverslips をインキュベート トゥイーン 20。
注: 代表的な結果の生成に使用された二次抗体、アレクサ 488 (1:1, 000) と結合したヤギ抗マウスとヤギ抗うさぎ Alexa 568 (1:1, 000) に結合します。 - Coverslips を削除し、1 × PBS で 5 分間洗って 3.3.2.2 の手順で説明するようします。
- スライド ガラス上のスライドに媒体をマウントおよび上 (メディアをマウントに直面して由来)、coverslips を注意深く配置 3 μ L を追加することによって、coverslips をマウントします。マニキュアと coverslip エッジをシールします。
- 画像のデコンボリューション27を適用中 1.4 の n. a. と石油目的 X 63 共焦点顕微鏡の分離の由来 (材料の表を参照してください)。
注: ここでは、次の設定が使用された: アレクサ 488 と Alexa 568 580-635 nm の 500-545 nm。
- 蛍光抗体法により次のように染色のための材料を準備します。
4、Coverslips の中央に由来の濃度
注: は、図 3を参照してください。
-
材料の準備
- 氷、(呼ばれるアダプター以下、.stl ファイル補足のファイル 1として提供) カスタム coverslip サポート アダプター、カスタム コンセントレーター (.stl ファイル補足のファイル 2として提供)、10 mM K パイプ (pH 15 mL ガラス丸底チューブを準備します。7.2) 4 ° C で
- ポリ-D-リジン (PDL) を準備-coverslips をコーティングします。10 x H2O を 1 mg/mL PDL の貯蔵液を希釈します。まず、70 %etoh、削除、エタノール、coverslips 乾燥させると coverslips を洗います。PDL を coverslips をコートし、室温で 30 分間インキュベートします。Coverslips 3 を洗って水で x と乾燥させます。
注: は、分離の由来、coverslips に接続の数を増やすための PDL を coverslips をコートします。
-
遠心分離
- 凹に滅菌 12 mm coverslip の場所、PDL を維持、コンセントレーターの下端が下向きをコートします。上に直接アダプターを配置することによって、coverslip をキャップします。丸底チューブを反転し、コンセントレーター、coverslip、アダプターの上に置きます。
- 軽く丸底チューブの底部に達するまでピンセットでアンサンブルを押して、チューブを反転します。コンセントレーターの一番上に来るまで、丸底チューブ 10 mM K パイプ バッファー (pH 7.2) を追加します。コンセントレーターの中央のシリンダーで任意の気泡そこに残らないことを確認してください。
- 優しく豊かな中心小体分数を含む 1 つの因数に 10 ミリメートル K パイプ バッファー (pH 7.2) の 100 μ L を追加し、ボリュームを徹底的にミックスします。
- コンセントレーターの中空部から 10 mM K パイプ バッファー (pH 7.2) の 100 μ L を削除し、内容が中空の中心に残ることを世話、コンセントレーターの中空部に 10 mM K パイプ バッファー (pH 7.2) の豊かな中心体分画の 100 μ L を追加します。
- 遠心 10,000 × gの 4 ° C で事前冷却遠心機 (例えば、JS 13.1 スイング バケツ ローター) 10 分
- ピンセットで、コンセントレーターを削除します。
- アダプターのスロットの端にある穴に手作りフック デバイスを挿入して、coverslip を回復し、そっと持ち上げます。丸底チューブの先頭に達すると、時に手袋をはめた指でアダプターの端をトラップし、ピンセットで、coverslip を削除します。Coverslip のどちら側に由来が含まれていることに留意します。蛍光抗体法の coverslips の治療に進みます。
注: 手動でフックし、自家製棒 (図 2 b D) に成形の注射針で自作フック装置が可能です。 - 固定および濃縮由来の免疫蛍光染色手順 3.3.2–3.3.4 で実行します。
5. 単一粒子の平均
注: は、図 4を参照してください。
- 単一粒子の平均のためのイメージング
- 正規の耐フェード マウント媒体を使用してスライド上の coverslip をマウントします。CFI アポクロマート全反射の目的を使用して構造化照明顕微鏡 (2 D SIM) イメージングを実行 (100 X、NA 1.49 WD 0.12 mm) とバック点灯 EM CCD カメラ。
注: 同期捕捉時間は 3 MHz から読み取るカメラで 100 ms に設定されました。2.5 X レンズが使用された SIM 用。
注: ここに表示されているデータセットは、3-D SIM 顕微鏡に買収された (材料の表を参照してください)。 - 画像総中心小体を構成する画像の大規模なスタックを取得することによって中心体信号、中心小体信号の上下に上記の Z スタックの上部と下部の位置をそれぞれ設定。プロジェクトのスタックと 5.2 と 5.3 の手順に従って単一粒子の平均を実行に進みます。
- 正規の耐フェード マウント媒体を使用してスライド上の coverslip をマウントします。CFI アポクロマート全反射の目的を使用して構造化照明顕微鏡 (2 D SIM) イメージングを実行 (100 X、NA 1.49 WD 0.12 mm) とバック点灯 EM CCD カメラ。
- スタックの投影
- ImageJ で画像のスタックを開きます。をクリックして '→ スタックをイメージ → Z プロジェクト」。投影の種類を設定として最大強度'。
- クリックして、イメージの反転 '編集 → 反転'。生成される投影を保存 (.tif 形式)。
- Scipion 単一粒子整列
- 赤を押すと Scipion で新しいプロジェクトを作成」ページの上部に'のプロジェクトの作成] ボタン。左側のパネルでダブルクリック '輸入 → 輸入顕微鏡'。
- 入力、'ファイルのディレクトリ'と'パターン 'フィールド データ ディレクトリおよび名前によると。既定のパラメーターを保持します。'実行'をクリックします。
- '粒子 → ピッキング → xmipp3 - マニュアル (ステップ 1) を選ぶ'をダブルクリックします。'入力顕微鏡'フィールドの近くに虫眼鏡アイコンをクリックし、手順 5.3.1 から輸入された顕微鏡を選択します。'実行'をクリックします。
- 新しく開いたウィンドウでそれらをクリックして別の顕微鏡写真から粒子を選択します。すべての顕微鏡写真を完了したら、赤いボタン「+ 調整」をクリックしてします。
- '粒子 → 抽出 → xmipp3 - 抽出粒子」をダブルクリックします。'入力座標'フィールドの近くに虫眼鏡アイコンをクリックし、手順 5.3.4 から選んだ座標を選択します。粒子寸法によると「粒子ボックス サイズ (px)」をご記入ください。「前処理」タブでダスト除去を設定: なし (それは成果物を生成することができます)。コントラストの反転: (黒い粒子、白背景)。位相反転: なし (CTF 補正にリンク);ノーマライズ: はい。
- 「実行」をクリックします。仕事が終わったらをクリックしジョブ (厚くなるボーダー) のボックスを選択して抽出された粒子をチェック '分析結果(メイン メニューの左下)。
- '2 D → 整列 → xmipp3 - cl2d に合わせて'をダブルクリックします。'入力粒子'フィールドの近くに虫眼鏡アイコンをクリックし、手順 5.3.5 から抽出された粒子を選択します。参照イメージを使用しません。'実行'をクリックします。
- ダブルクリックして、 '2 D → 整列 → もっと → xmipp3 - 配置を適用 2 d'。'入力粒子'フィールドの近くに虫眼鏡アイコンをクリックし、手順 5.3.7 から一直線に並べられた粒子を選択します。
- 5.3.6 の手順のように「実行」をクリックします。ジョブ ボックスの選択後「分析結果」をクリックしてして結果を確認できます。 されます
- '粒子 → マスク → xmipp3 - 2d のマスクを適用する」をダブルクリックします。
- '入力粒子'フィールドの近くに虫眼鏡アイコンをクリックし、手順 5.3.9 から一直線に並べられた粒子を選択します。「マスク ソース」は、 「ジオメトリ」に設定されます。その後、マスク型としてマスクのパラメーターを設定: 円形;半径 (ピクセル): をクリックして、その周りに何もない 1 つの粒子 (中心小体) を探して、'魔法の杖 ' 、最適な値を見つけるためにウィンドウを開く左のアイコンシフト センター: なし (粒子は完全に中央に) 場合や (パーティクルが移動) する場合は Yes です。X 中心のオフセット: 粒子の位置によるとY センター オフセット: 粒子の位置によると。'実行'をクリックします。
- 「分析結果」ボタンを使用して、マスクが正しく適用されます。そうでない場合は、 '2 d をマスク [適用]ジョブを右クリックし、 '編集'を選択します。それもう一度「実行」ボタンを使用して (サイズおよび/またはシフト) マスクと実行パラメーターを変更します。
- '2 D → 分類 → xmipp3 - cl2d'をダブルクリックします。
- '入力粒子'フィールドの近くに虫眼鏡アイコンをクリックし、手順 5.3.11 から仮面の粒子を選択します。クラスの数は、1 クラス約 50 粒子を取得するはずです。'実行'をクリックします。
- 「分析結果」ボタンをクリックして、結果確認してください。開いたウィンドウで「何を」のすぐ隣に「目」アイコンをクリックします。
- 新しいウィンドウは、 'ブロック'メニューの'Classes2D'を選択することによって生成されたクラスの検査をできます。各クラスの内容をチェックするには、同じメニューの'Class00N_Particles'を選択します。唯一の悪い粒子が含まれているかを識別するために各クラスを検査します。'Classes2D'ビューに戻るし、それぞれをクリックして良い粒子を使用してクラスを選択します。選択時に押される「Ctrl」キーを保つことによっていくつかのクラスを選択することが可能です。
- 良い粒子のすべてのクラスが選択されている場合は、 '+ 粒子'これらの粒子にサブセットを作成するをクリックします。
- 同じウィンドウで粒子のさまざまな方向を表すクラスのいくつかのセットを選択します。新しいサブセットを作成するには、 '+ 平均'をクリックします。
- 各方向/平均の選択、次の手順に従います。
- '2 D → 整列 → xmipp3 - cl2d に合わせて'をダブルクリックします。
- '入力粒子'フィールドの近くに虫眼鏡アイコンをクリックし、手順 5.3.17 から良い粒子を選択します。'Yes'に'Use の参照イメージ'を設定します。'参照イメージ'フィールドの近くに虫眼鏡アイコンをクリックしてステップ 5.3.18 から'サブセットを作成ジョブに白い矢印左側クリックし、参照として使用するイメージを選択します。別のウィンドウで開き、どのオブジェクトをチェックするオブジェクトをダブルクリックします。'実行'をクリックします。
- ダブルクリックして、 '2 D → 整列 → もっと → xmipp3 - 配置を適用 2 d'。'入力粒子'フィールドの近くに虫眼鏡アイコンをクリックし、5.3.19.2 のステップから一直線に並べられた粒子を選択します。
- '実行'をクリックします。
- アライメントの結果をチェック ('分析結果);一直線に並べられた粒子は、これらの粒子の平均が表示されます。
- 平均は得意と粒子はすべて同じ方法で指向された場合をクリックして平均を保存' 詳細 → ImageJ'と ImageJ で画像を保存します。
- 平均を改善することができます、よく指向のすべての画像を選択し、 '+ 粒子'ボタンで新しいサブセットを作成します。サブセットは (すべて悪い粒子を削除) きれいになるまでは、手順 5.3.19.1–5.3.19.4 を繰り返します。配置するたびに実行 (ステップ 5.3.19.2)、(平均品質向上のイテレーションにイテレーション) からは最後の生成のなかに参照が設定されます。
結果
C. クラミドモナス小体分離:
C. クラミドモナス細胞由来、 cw15- を分離する光の下で数日間液体文化で育つ, 10 分 Pelleted 細胞に 1x PBS で洗浄したためその後 600 × gで遠心分離によってペレットの再懸濁とは2 分(図 1)の最終 pH 4.5-4.7 0.5 M 酢酸を使用して pH ショックを実行することによって deflagellation する前にバッファーを deflagellation します。1 N KOH 添加 pH を 7.0 に戻すに使用されました。戸建鞭毛を細胞体に区別、deflagellated 細胞は鞭毛の大部分を削除する最初遠心分離しました。ペレットは 2 倍の PBS で洗浄し、25% ショ糖クッション (図 1B) にゆっくりとそれをロードする前に 30 mL の PBS で再停止されます。戸建の鞭毛の大部分を削除する 4 ° C で 15 分間 600 x gでチューブを分離されました。遠心分離後、細胞体がショ糖クッション (図 1C) で広がり、削除して回復、上清 (図 1Cの赤矢印) までの約 30 mL。洗浄のセルの結果 20 mL、20 mL の冷 PBS で再停止されるされ 600 x gで 10 分間遠心分離します。次に、清が破棄され、セルが完全に 10 mL の PBS で再停止されます。セルは、250 mL のボトルに移された、換散バッファーは一度に再懸濁のセルに追加されました。DNase、換散を添加し、4 ° C で 1 時間インキュベート細胞の残骸を削除する遠心分離のステップ後 (図 1Dの白いペレットを参照)、上清を集めた後、慎重に 4 ° C で 30 分間、10,000 × gで遠心分離前に 2 mL 60% ショ糖クッションにロード換散の 100 ml は、8 チューブ 15 ml 12.5 mL のショ糖チューブあたり 2 mL に読み込まれる換散バッファーに対応する、この手順を実行する使用されました。遠心分離の後上澄みのほとんどが削除されたクッションの上 1 mL まで。残りの上清 1 mL、2 mL のクッションを使用して収集、混合し、プール 24 mL の最終的なボリュームを取得します。プールをロードし、40%、50%、70% ショ糖勾配、4 ° C で 1 時間 15 分の 68,320 x gでスピン最後に、分離の由来は針を使用して遠心分離の管の底の部分に穴を作るによって収集された、12 x 500 μ L 分画の滴を集めた。異なるショ糖の高密度のためドロップ形成 (70% ショ糖) 初めしより多くの非常にゆっくりと急速に (40% ショ糖)。
分離由来の蛍光
分離のプロシージャの品質を評価するために各勾配フラクションの 10 μ L coverslip サポート アダプター (図 2 a-2 D) を使用して観察に次いで遠心分離。重要なは、安全に、coverslip、削除するカスタム フック デバイス設計されました (図 2B)。次に、coverslips は蛍光抗体法により分析した.CrSAS-6(Bld12p) 抗体は、車輪構造と α-チューブリン セントリ壁を強調する (DMA1) の存在を示すため、本研究で使用されました。CrSAS 6 と α-チューブリンの視野あたり陽性由来の数をカウントし、分数あたり由来の合計数を計算して、それはどの分画の濃縮された分離由来 (を決定することが可能図 2 階-2 H)。興味深いことに、6 分画はあった最後の分数はなかったが、浄化が働いたことを示す分数 #3 のピークを持つ (図 2 fと2 G分数 #1-6) 由来の濃縮します。この特定の実験で合計由来の 95% CrSAS 6 と α-チューブリン分数 #3 で陽性であったことに注意してください。これは、最も孤立した由来が、側転を保持されることを示します。最初の分数は由来の濃縮を認め、分離のプロシージャは働かなかったし、繰り返す必要があります。由来を欠いている分数でほとんどいくつかのべん毛の作品を観察できることに注意してください。
次に、μ L あたり由来の合計数を計算するため視野あたり中心小体の数は図 2Hに記載されている比かける必要があります。結果の数は、螢光抗体の μ L あたり由来の番号の取得に使用する分数のボリュームで、分けする必要があります。この特定の分離手順で豊かな分数 (92 x 92 μ m2のビューのフィールド) を 0.00846 mm2の面積に約 37 由来に含まれます。チューブのセクションの表面 176 mm2の総表面の半径 7.5 mm であった。対応する比率は当時 176/0.00846、20,803.8 を = 10 μ L の 769,740 中心体 (37 x 20、803.8) の合計。したがって、1 μ L の中心体の数は、76,974 でした。
Coverslips に分離由来の濃度:
さらに粒子平均プロシージャで使用することができますと同様の方向を検出する機会の増加し同様、明確な中心小体の向きの検出のチャンスを増加フィールドごと由来の数を増やします。集中の一部分からの由来はまだまばら、coverslip、集中という名前のコンセントレーター coverslip (図 3A) の真ん中に由来する遠心分離アクセサリーを開発しました。本稿では、3次元印刷の正確な対策と .stl ファイルが提供されることに注意してください。
最初に、12 mm の 1 つ coverslip コンセントレーター (図 3B) にマウントされていた。アダプターは、coverslip の上に置かれた、丸底チューブは逆さにして組み立てられたアダプターとコンセントレーター上置します。丸底チューブは、軽く反転サンプル (プロトコル手順 4.2) の読み込みをできるように。中心体は、4 ° C で 10 分間、10,000 × gで遠心分離しました。その後、中心体が蛍光抗体法を受けるし、CrSAS-6/Bld12p と α-チューブリン (図 3-3 e) のステンド グラスします。重要なは、コンセントレーター、なし 183 の由来は、コンセントレーターの使用 (図 3 D-3F) 頃視野あたり見られたに対し、約 30 の由来は各フィールドのビュー (図 3C) 見られました。中心体カバー、coverslip の真ん中に直径 4 mm のディスクのみであることに注意してください。この結果は、濃度ステップ動作でき、coverslips、従って緩和その検出とイメージングの定義済みの領域の中心体の 6 濃縮を示します。
単一粒子を分離の平均蛍光C. クラミドモナス由来:
ここでは、約 120 の解像度に到達することができます SIM 顕微鏡を用いた nm、由来 monoglutamylated チューブリン (GT335、図 4)、中心体の微小管にチューブリン変更のため染色されたイメージ24。C. クラミドモナス由来が約 500 nm ペアで常に、長く、しばしば見つけ関連付けられている新しく複製 (以下 procentrioles) probasal 体、微小管関連繊維19紋します。したがって、この最終組み立て約 1 μ m の大きさであった。このため、および全体の中心体をイメージするために、分離の由来に Z スタックを取得をお勧めします。
ここでは、買収後最終的なイメージは、ImageJ28 (画像/スタック/Z プロジェクト/最大強度投影図 4B) を使用して最大強度投影を実行によって生成されました。このようなイメージからクライオ電子顕微鏡のソフトウェアを使用して単一粒子分析由来と同様の方向でのクラスの作成を行い、平均を行った。これを行うには、画像の色は最初オブジェクト (図 4C) を見やすくために逆になった。由来は、Xmipp3 などのいくつかの電子顕微鏡ソフトウェア プログラムを統合する自由に利用できる Scipion ソフトウェア29を使用して各粒子の中央にボックスで手動で選ばれた (図 4D)。ボックスのサイズは、ユーザーによって定義されることに注意してください。ここでは、31.84 のピクセル サイズ 50 × 50 ピクセルのボックス nm が使用されました。次に、すべての粒子が (図 4E) を抽出し、Xmipp3 を使用して、配置 (図 4F)。次に、12 ピクセル半径の円形のマスクはセントリ ペア (図 4G) から各中心小体を分離に適用されました。その後、Xmipp3 を使用して複数回の平均を生成する、粒子分類されました。のみクラス平均から分岐する粒子が手動で除外されたことを意味均等クラス平均は保たれました。この手順は、選択した各方向の完璧に近い平均を生成するために繰り返されました。5 つの繰り返しの後の平均の 2 つのクラスが生成された: monoglutamylated 由来の 98 粒子 (図 4私) から 63 オブジェクト (図 4H) から上面と側面を表示します。オブジェクトの寸法をに沿って monoglutamylation 信号強度プロット プロファイルのピーク間の距離を測定することによって求めた。
重要な側面図クラスの平均の長さは、260 nm 直径 250 nm、中心小体17のコア内長さ 286 ± 33 nm の領域にローカライズする示された測定 monoglutamylated チューブリン信号に匹敵します。

図 1:精製C. クラミドモナス由来。(A) これはC. クラミドモナス由来の分離に至る各ステップの概略図。上に 25% ショ糖密度勾配遠心分離後の前にプロトコル (B) と (C) の代表的な手順が含まれています。パネルCの赤矢印は、遠心分離後も保持するボリュームの最小値を示します。(D) このパネルでは、遠心分離後分離細胞の白いペレットを示しています。黒い矢印は、ペレットを示します。この図の拡大版を表示するのにはここをクリックしてください。
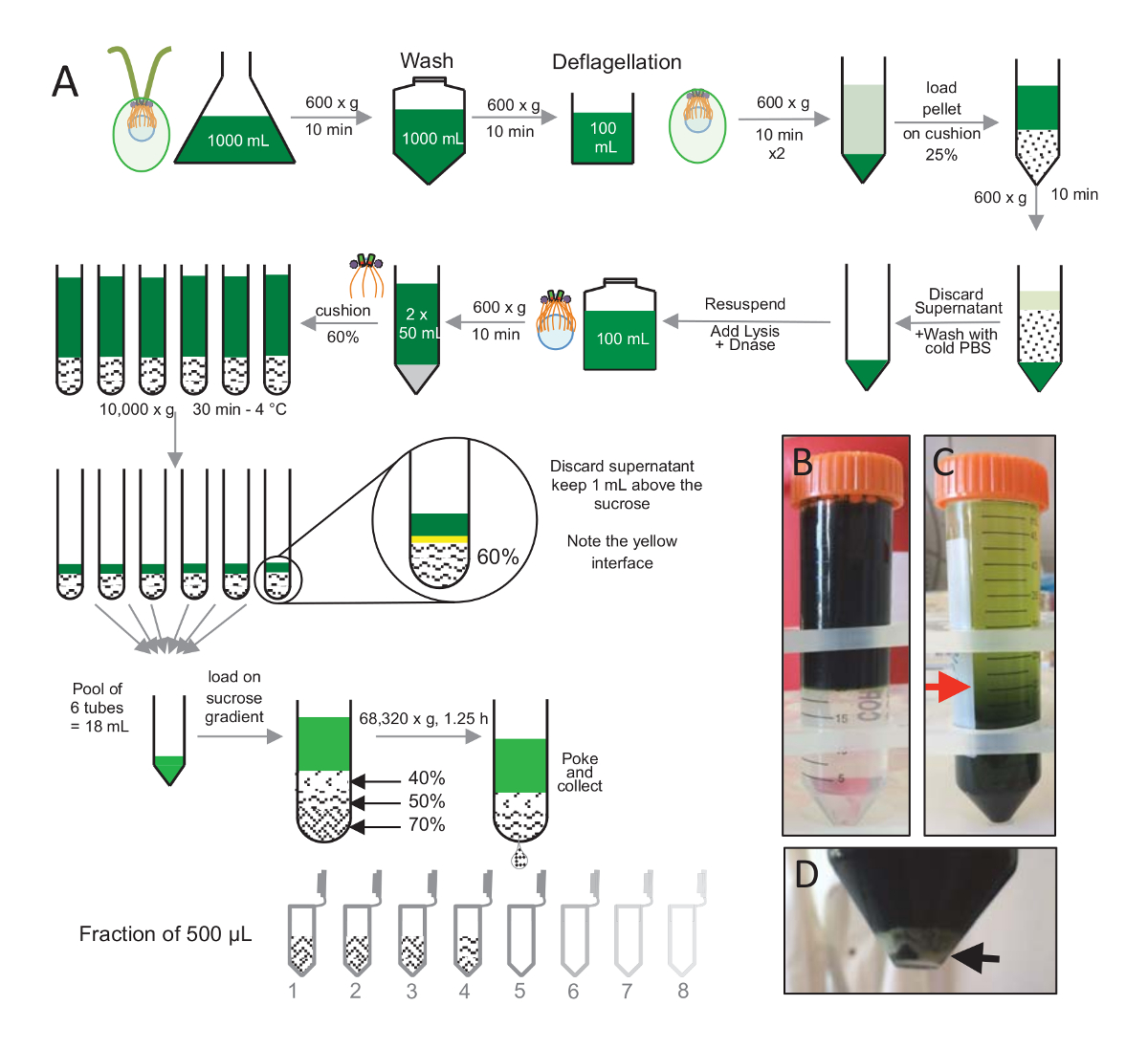{kind=link}

図 2:遠心分離で蛍光抗体法を実行するセットアップ分離由来。各フラクションの (A) 10 μ L を最初 100 μ L の 10 ミリメートル K パイプ (pH 7.2) で希釈します。(B) これは、観察用のアダプター、12 mm coverslip 15 mL 丸底チューブを包括的な遠心分離装置と遠心分離後、coverslip を回復する特注フック デバイスの概略図です。CとDのパネルは、遠心分離後、coverslip を回復する方法を説明する図面を表示します。(C) 場所アダプターのスロットの端にある穴にフック ツールおよび (D) 慎重に引き出します。(E) 蛍光イメージングを実行に必要な湿気のある商工会議所の写真です。矢印は、12 mm coverslip を示します。(F) 63 X (ズーム 2) グラデーション分数 #1-8、浄化手順中に収集されたので代表的な共焦点画像と CrSAS-6 (赤) と α-チューブリン (緑) のステンド グラスします。インセットは、低い倍率ビューの白いボックスで示される領域に対応しています。スケール バー = 10 μ m. (G) CrSAS-6 と α-チューブリン各画分の視野あたりの由来の正の数を表すグラフです。分数 #1-6 にエンリッチメントに注意してください。由来各画分のフィールドごとの平均数: #1 = 16.3 ± 4.7、#2 = 24.0 ± 4.6, #3 = 37.0 ± 17.0, #4 = 23.3 ± 11.4、#5 = 14.3 ± 2.1 #6 = 13.7 ± 9.3、#7 = 2.7 ± 1.2 #8 = 1.0 ± 0.0 #9 = 1.0 ± 0.0 #10 = 0.0 ± 0.0 #11 = 0.3 ± 0.6、#12 = 0.0 ± 0.0。各分画の 3 つのランダムなフィールドをイメージしました。#3 の最も豊かの分数をカウントするとき私たち中心体の 95% が CrSAS 6 と α-チューブリンの肯定的なことを見、(n = 205 由来)。由来、顕微鏡の測定領域内に存在数の (H) の比率を使用して、チューブの領域に存在由来の合計数を計算します。中心小体の数 μ L は、10 μ L もともと遠心分画から計算できます。この図の拡大版を表示するのにはここをクリックしてください。
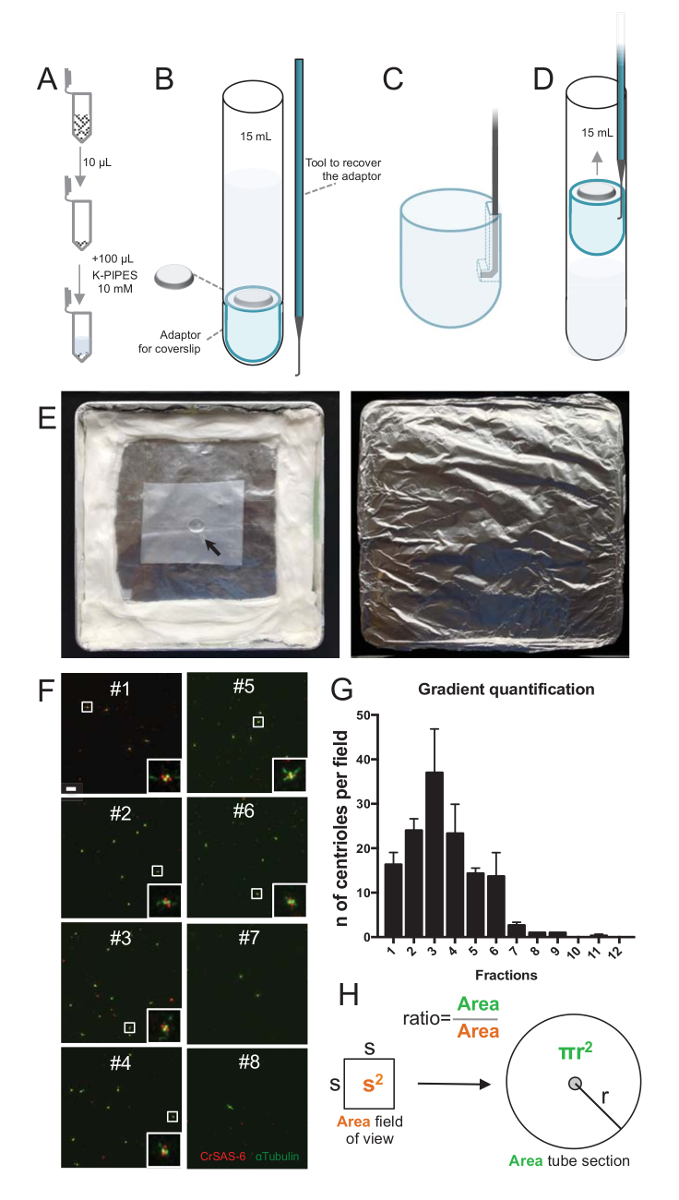{kind=link}

図 3: コンセントレーターを使用して分離された由来は、遠心分離します。(A) このパネル由来蛍光、15 mL 丸底チューブ、コンセントレーター、12 mm coverslip サポート アダプター装置など前のカバーガラス上に集中するために必要なの遠心分離デバイスを示しています。(B) このパネルは、遠心分離装置を組み立て手順を示しています。パネルC-Eは、コンセントレーターに染まった CrSAS 6 (赤) と α-チューブリン (緑) (C) なしまたは (のD-E) 由来の共焦点画像を表示します。インセットは、低い倍率ビューの白いボックスで示される領域に対応しています。スケール バーは、10 μ m. 由来 (点線集中領域の境界を表す) coverslip の真ん中に濃縮されていることに注意してください。(F) このグラフでは、ビューのフィールドなしとコンセントレーターあたり由来の数を表します。5 つのランダムなフィールドの表示を行った。由来の平均数は、コンセントレーター、29.8 ± 2.9 とコンセントレーター、182.6 ± 11.5、 P < 0.0001。統計的有意性は、対になっていないtによって評価された-をテストします。この図の拡大版を表示するのにはここをクリックしてください。
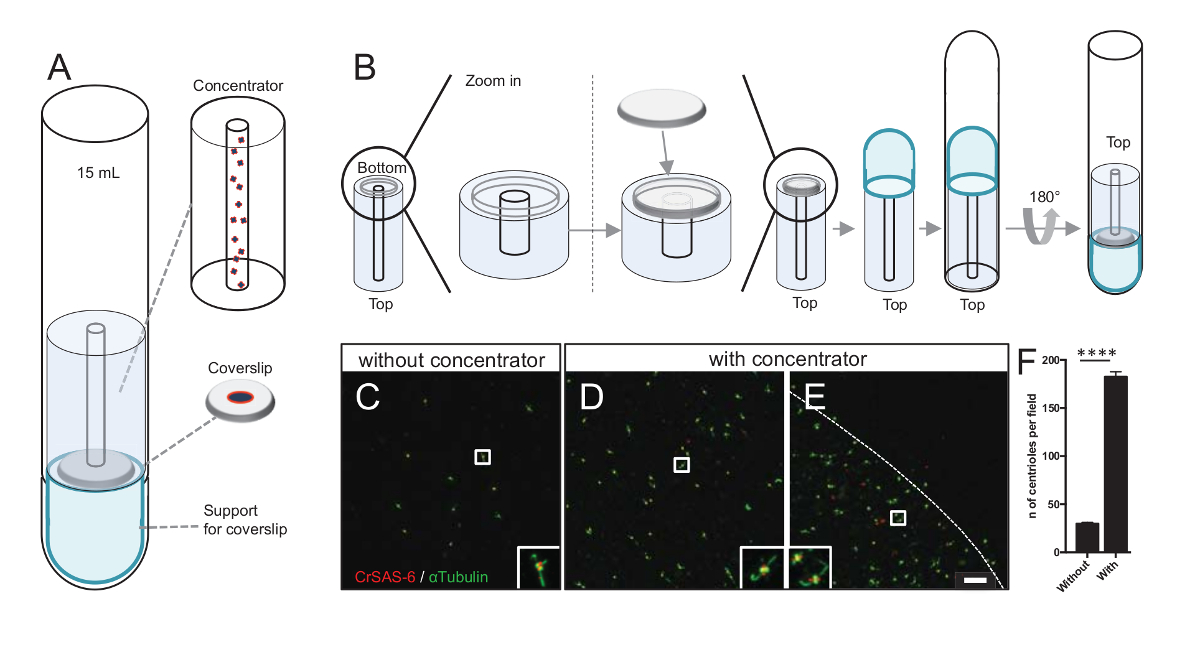{kind=link}

図 4:単一粒子の平均分離C. クラミドモナス由来。(A) このパネル ショー Z は、由来 GT335 で染色し、SIM 顕微鏡を用いての画像をスタック。(B) これらのパネル スタックのイメージの最大強度 Z 投影を表示します。スケール バー = 1 μ m. (C) これらのパネルを見る代表的な画像コントラストが反転します。良い由来を視覚化するためのズームのはめ込みを表します。(D) これらのパネルを粒子のピッキング。挿入図は、粒子が選ばれた方法を示します。(E) これらは、抽出された 5 例の粒子。このパネル (F) は、配置後の粒子を示しています。(G) このパネルでは、マスクを適用した後、粒子を示しています。パネルH 、私は、2 つのクラス平均を表示: (H) トップ ビュー (63 粒子) と (私) サイドビュー (98 粒子)。二重の矢印は、GT335 信号の大きさを示しています。スケール バー = 250 nm。この図の拡大版を表示するのにはここをクリックしてください。
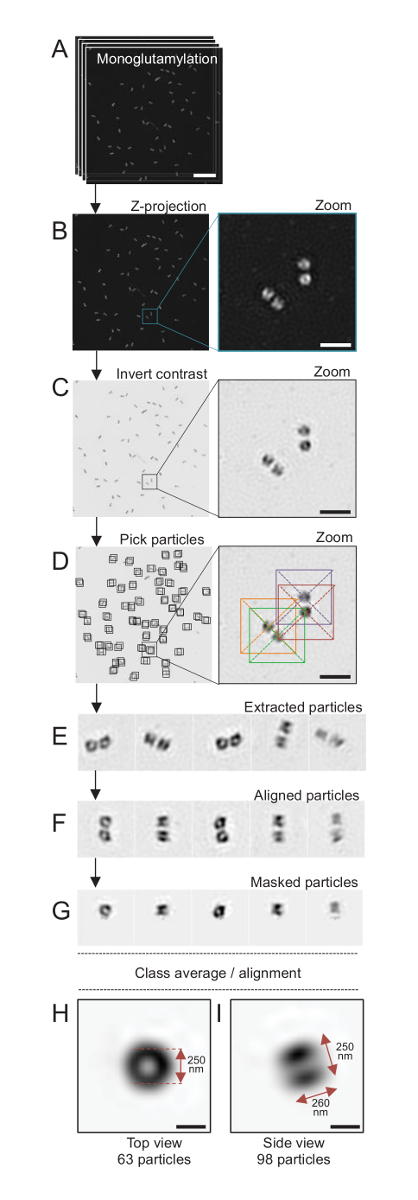{kind=link}
1 の補足ファイル。このファイルをダウンロードするここをクリックしてください。
2 の補足ファイル。このファイルをダウンロードするここをクリックしてください。
ディスカッション
建築におけるタンパク質の正確な局在を解読する生物学の課題の一つです。小体は、そのアーキテクチャは、その長さに沿って微細構造の特徴を明らかに低温電子トモグラフィーを用いた研究されているこれらの方法を適用する理想的な構造です。ただし、光学顕微鏡の解像限界に近いその寸法のため従来の顕微鏡30を使用して中心小体の構造サブ領域に蛍光タンパク質を正確にローカライズすることは困難です。
光学顕微鏡の解像度は、約、200 の横方向の最大解像度を与える光の回折によって限られた光学顕微鏡24nm。ただし、この制限にされている最後の 20 年の主要なブレークスルーの 1 つによってバイパス光学顕微鏡: 超解像法の発明。これらのアプローチは、異なる解像度での回折限界を超えてイメージできる: 120 nm の SIM、約 50 の誘導放出の枯渇 (STED) の nm と単一分子局在顕微鏡 (SMLM)24の 20-40 nm。超解像顕微鏡法のこれらの新しい開発、小体の地域構造が達成可能であります。しかし、実際には、まだ成熟した由来がセルあたりのみ 2 つのコピーし、の解釈になるランダムな方向性があります主な理由の構造要素にタンパク質の局在を正確に判断することは困難です。ローカライズが困難です。このため、画像超解像による多数の由来、非あいまいな方向を観察する機会を増やすことを研究者を許可するプロトコルは設定。重要なは、このメソッドは、分離由来の使用に依存する、我々 は、そのままc. クラミドモナスを浄化する方法を提供して成熟由来と procentrioles を含む由来。
最後に、このプロトコルでイメージを作成することができます中心小体向きの範囲により単一粒子解析適用できる電子顕微鏡のソフトウェアを使用しています。これは、結果、特定の向きの中心体の平均クラスの生成。重要なは、これらの結果の 2 D 画像は小体に沿って特定の蛋白質の局在化を評価するために、使用できます。確かに、この方法は、デュアル色超解像画像に適用できるし、特定セントリ タンパク質に起因する他の色、中心小体スケルトン (例えばチューブリン)、明らかにする 1 つの色を使用できます。1 色または 2 色で得られた平均値を引いて、中心小体 (近位、中央、または遠位) に沿ってタンパク質を登録しやすくなります。任意の誤った解釈を防ぐために 2 つのチャネルする必要があります正確に位置合わせされることに注意してください。また、トップ ビューの平均はセントリ ルーメン、小体の内外微小管壁に沿って内部タンパク質にローカライズする場合に役立ちます。
このメソッドには、異機種混在環境ラベリングのためそれ以外の場合をローカライズすることは困難かもしれない特定の蛋白質の局在を確認するために利点があります。中心粒内の蛋白質をマップする他の方法に記載されている相関の 3-D SIM/SMLM、例えば、周辺のトーラスを形成マーカーの楕円形のプロファイルを決定することによって中心体の特定の方向を評価するに注意してください、SIM イメージングによる中心小体。このパラメーターを使用すると、4-5 nm30の精度をもつタンパク質をローカライズすることが可能です。説明する方法がここでそのまま procentrioles、中心小体アーキテクチャは、大部分は保存される可能性が最も高い記号で分離由来を使用するにも注意してください。しかし、我々 はいくつかのアーキテクチャ上の機能が人間体5絶縁増幅の二価陽イオンの濃度変化中心小体の直径などの浄化中に邪魔を除外できません。
ここで提示されたプロトコルの重要な手順の 1 つは蛍光スパを受けやすいさまざまな方向に十分に集中して孤立した由来を得ることです。これを行うには、最初、純度、中心小体分離処理の効率性を確認します。分離由来の低濃度は、適切な画像と更なるイメージ処理できなくなります。この目的のため、我々 は由来の視野あたりの数を豊かにするためのメソッドを提供しています。使用する分数の中心体の数、に応じて、コンセントレーターに読み込まれているボリューム調整してください、250 μ L の最大音量で。
重要なは、マイナスcw15 C. クラミドモナス細胞細胞壁のこの法を開発しました。この株の細胞壁の脆弱性は細胞の適切な換散とは、したがって、その内容の解放ことができます。このプロトコルが野生型c. クラミドモナスの効率的な細胞、細胞壁は適切な換散を防ぐようです。ここで紹介の隔離のプロトコルを適用する前に細胞壁を変更する場所に置かれる超音波処理や溶解酵素、細胞壁31、低下することが酵素で細胞の前培養などの代替戦略でしょう。
この設定は、高スループット専用超解像顕微鏡まで従来の共焦点顕微鏡から、顕微鏡の種類で使用できます。SMLM の際に、特別なバッファーが適切なイメージングのため必要であるに注意してください、したがって、商工会議所、coverslip の上にバッファーを持つ 12 ミリメートル coverslip の適応を使用する必要があります。その後画像は倒立顕微鏡で実行されます。顕微鏡のセットアップが 12 mm coverslip を許可しない場合、ここで示されるプロトコルは 30 mL 丸底チューブと変更されたアダプターとコンセントレーターを使用して 18 mm coverslips に適用できます。また、固定の方法と同様に、SMLM で最終的な再建の品質が染色の使用、一次抗体の品質に依存することに注意してくださいすることが重要です。
要約すると、蛍光-スパの向きが異なる由来の平均を生成する精度でセントリ タンパク質をローカライズすることで多数の由来が続いてイメージに適用することができますメソッドを提供しています。重要なは、このメソッドより一般的に他の種からの分離の由来、その他の細胞内小器官、または大規模な高分子アセンブリに適用できます。最後に、サンプル準備方法紹介、蛍光 SMLM データ32の単一粒子解析の最近のアルゴリズム開発と組み合わせて、大規模な高分子の分子の地図の更なる改善を開くことができません。アセンブリ。
開示事項
著者が明らかに何もありません。
謝辞
我々 は、エコール連邦工科ローザンヌ (EPFL)、中心体のシムの画像を取得したスイス、ローザンヌでピエール ・ Gönczy ・ バイオ イメージング光学系プラットフォーム (BIOP) を感謝します。ニコライ ・ クレナとダビデ ・ Gambarotto は、欧州研究会議 (ERC) 開始助成金 (StG) 715289 (アクセント) とマエバ ル Guennec、ポール ・ ギシャール、スイス科学財団 (SNSF) PP00P3_157517 によってヴィルジニ ・ ハメルによってサポートされます。スザンヌは、ジュネーブ大学でサポートします。
資料
| Name | Company | Catalog Number | Comments |
| Mouse monoclonal anti-apha tubulin (clone DM1A) | Abcam | ab7291 | dilution 1:300 |
| DNaseI | Roche | 10104159001 | |
| 12 mm coverslips | Roth | YX03.1 | |
| 18 mm coverslips | Roth | LH23.1 | |
| K2HPO4 | Fluka | 60355 | |
| KH2PO4 | Fluka | 60230 | |
| Tris base | Biosolve Chimie SARL | 0020092391BS | |
| acetic acid | Carlo Erba Reagents | 524520 | |
| NH4Cl | Sigma | A-4514 | |
| CaCl2 | Sigma | C-7902 | |
| MgSO4 | Sigma | 63140-500G-F | |
| steritop filter | Millipore | SCGPT05RE | |
| sucrose | Sigma | S7903-1KG | |
| HEPES | AppliChem PanReac | A3724,0250 | |
| PIPES | Sigma | P6757-500G | |
| MgCl2 | ACROS ORGANICS | 197530010 | |
| NP-40 | AppliChem PanReac | A1694,0250 | |
| Round-bottom (Kimble) tubes 15 mL | Fisherscientific | 09-500-34 | |
| Round-bottom (Kimble) tubes 30 mL | Fisherscientific | 09-500-37 | |
| cover glass staining rack | Thomas scientific | 8542E40 | |
| crystal polystyrene transmission lab box | FISHERS | 11712944 | |
| Methanol | VWR | 20864.32 | |
| BSA | Roche | 10735086001 | |
| Triton X100 | Roth | 3051.3 | |
| goat anti-mouse coupled to Alexa 488 | invitrogen | A11029 | dilution 1:1,000 |
| goat anti-rabbit coupled to Alexa 568 | invitrogen | A11036 | dilution 1:1,000 |
| mounting medium | abcam | ab188804 | |
| Tube, thinwall polypropylene | Beckman Coulter | 326823 | |
| Poly-D-Lysine 1 mg/mL | SIGMA | A-003-E | |
| Mouse monoclonal anti-Polyglutamylation modification mAb (GT335) | Adipogen | AG-20B-0020 | dilution 1:1,000 |
| glycerol mounting medium with DAPI and DABCO | Abcam | ab188804 | |
| 50 mL conical tubes | Falcon | 14-432-22 | |
| Eppendorf 5810R centrifuge | Eppendorf | 5811000622 | |
| Beckman JS-13.1 swinging bucket rotor | Beckman Coulter | 346963 | |
| Beckman SW 32Ti rotor | Beckman Coulter | 369694 | |
| parafilm | Bemis | 13-374-10 | |
| Leica TCS SP8 with Hyvolution mode | Leica | ||
| OSRAM L18W/954 LUMILUX | Luxe Daylight/ OSRAM | ||
| Whatman filter paper | Sigma | WHA1001325 | |
| CrSAS-6/Bld12 antibody | dilution 1:300 (Hamel el al., 2014) | ||
| Scipion | http://scipion.i2pc.es/ | ||
| EM CCD camera (Andor iXON DU897) | Andor |
参考文献
- Bornens, M. The centrosome in cells and organisms. Science. 335 (6067), 422-426 (2012).
- Nigg, E. A., Holland, A. J. Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nature Reviews Molecular Cell Biology. 19 (5), 297-312 (2018).
- Nigg, E. A., Raff, J. W. Centrioles, centrosomes, and cilia in health and disease. Cell. 139 (4), 663-678 (2009).
- Gönczy, P. Towards a molecular architecture of centriole assembly. Nature Reviews Molecular Cell Biology. 13 (7), 425-435 (2012).
- Paintrand, M., Moudjou, M., Delacroix, H., Bornens, M. Centrosome organization and centriole architecture: their sensitivity to divalent cations. Journal of Structural Biology. 108 (2), 107-128 (1992).
- Winey, M., O'Toole, E. Centriole structure. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 369 (1650), (2014).
- Dippell, R. V. The development of basal bodies in paramecium. Proceedings of the National Academy of Sciences of the United States of America. 61 (2), 461 (1968).
- Allen, R. D. The morphogenesis of basal bodies and accessory structures of the cortex of the ciliated protozoan Tetrahymena pyriformis. The Journal of Cell Biology. 40 (3), 716-733 (1969).
- Li, S., Fernandez, J. -. J., Marshall, W. F., Agard, D. A. Three-dimensional structure of basal body triplet revealed by electron cryo-tomography. The EMBO Journal. 31 (3), 552-562 (2012).
- Guichard, P., Chrétien, D., Marco, S., Tassin, A. -. M. Procentriole assembly revealed by cryo-electron tomography. The EMBO Journal. 29 (9), 1565-1572 (2010).
- Guichard, P., et al. Native architecture of the centriole proximal region reveals features underlying its 9-fold radial symmetry. Current Biology. 23 (17), 1620-1628 (2013).
- Hirono, M. Cartwheel assembly. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 369 (1650), (2014).
- Bornens, M., Paintrand, M., Berges, J., Marty, M. C., Karsenti, E. Structural and chemical characterization of isolated centrosomes. Cell Motility and the Cytoskeleton. 8 (3), 238-249 (1987).
- Keller, L. C., Romijn, E. P., Zamora, I., Yates, J. R., Marshall, W. F. Proteomic analysis of isolated chlamydomonas centrioles reveals orthologs of ciliary-disease genes. Current Biology. 15 (12), 1090-1098 (2005).
- Kilburn, C. L., et al. New Tetrahymena basal body protein components identify basal body domain structure. The Journal of Cell Biology. 178 (6), 905-912 (2007).
- Bauer, M., Cubizolles, F., Schmidt, A., Nigg, E. A Quantitative analysis of human centrosome architecture by targeted proteomics and fluorescence imaging. The EMBO Journal. 35 (19), 1-15 (2016).
- Hamel, V., et al. Identification of Chlamydomonas Central Core Centriolar Proteins Reveals a Role for Human WDR90 in Ciliogenesis. Current Biology. 27 (16), 2486-2498 (2017).
- Cavalier-Smith, T. basal body and flagellar development during the vegetative cell cycle and the sexual cycle of Chlamydomonas reinhardii. Journal of Cell Science. 16 (3), 529 (1974).
- Geimer, S., Melkonian, M. The ultrastructure of the Chlamydomonas reinhardtii basal apparatus: identification of an early marker of radial asymmetry inherent in the basal body. Journal of Cell Science. 117, 2663-2674 (2004).
- Nakazawa, Y., Hiraki, M., Kamiya, R., Hirono, M. SAS-6 is a cartwheel protein that establishes the 9-fold symmetry of the centriole. Current Biology. 17 (24), 2169-2174 (2007).
- Hamel, V., et al. Correlative multicolor 3D SIM and STORM microscopy. Biomedical Optics Express. 5 (10), 3326 (2014).
- Guichard, P., et al. Cell-free reconstitution reveals centriole cartwheel assembly mechanisms. Nature Communications. 8, 14813 (2017).
- Hiraki, M., Nakazawa, Y., Kamiya, R., Hirono, M. Bld10p constitutes the cartwheel-spoke tip and stabilizes the 9-fold symmetry of the centriole. Current Biology. 17 (20), 1778-1783 (2007).
- Sahl, S. J., Hell, S. W., Jakobs, S. Fluorescence nanoscopy in cell biology. Nature Reviews Molecular Cell Biology. 18 (11), 685-701 (2017).
- Hutner, S. H., Provasoli, L., Schatz, A., Haskins, C. P. Some Approaches to the Study of the Role of Metals in the Metabolism of Microorganisms. Proceedings of the American Philosophical Society. 94 (2), 152-170 (1950).
- Keller, L. C., Marshall, W. F. Isolation and proteomic analysis of chlamydomonas centrioles. Methods in Molecular Biology. 432 (1), 289-300 (2008).
- Borlinghaus, R. T., Kappel, C. HyVolution-the smart path to confocal super-resolution. Nature Methods. 13 (3), (2016).
- Schneider, C. A., Rasband, W. S., Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 9 (7), 671-675 (2012).
- de la Rosa-Trevín, J. M., et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 195 (1), 93-99 (2016).
- Gartenmann, L., et al. A combined 3D-SIM/SMLM approach allows centriole proteins to be localized with a precision of ∼4-5 nm. Current Biology. 27 (19), 1054-1055 (2017).
- Jaenicke, L., Kuhne, W., Spessert, R., Wahle, U., Waffenschmidt, S. Cell-wall lytic enzymes (autolysins) of Chlamydomonas reinhardtii are (hydroxy)proline-specific proteases. European Journal of Biochemistry. 170 (1-2), 485-491 (1987).
- Salas, D., et al. Angular reconstitution-based 3D reconstructions of nanomolecular structures from superresolution light-microscopy images. Proceedings of the National Academy of Sciences of the United States of America. 114 (35), 9273-9278 (2017).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved