
Method Article
Isolation and Fluorescence Imaging for Single-particle Reconstruction of Chlamydomonas Centrioles
In This Article
Summary
We have developed a strategy to purify and image a large number of centrioles in different orientations amenable for super-resolution microscopy and single-particle averaging.
Abstract
Centrioles are large macromolecular assemblies important for the proper execution of fundamental cell biological processes such as cell division, cell motility, or cell signaling. The green algae Chlamydomonas reinhardtii has proven to be an insightful model in the study of centriole architecture, function, and protein composition. Despite great advances toward understanding centriolar architecture, one of the current challenges is to determine the precise localization of centriolar components within structural regions of the centriole in order to better understand their role in centriole biogenesis. A major limitation lies in the resolution of fluorescence microscopy, which complicates the interpretation of protein localization in this organelle with dimensions close to the diffraction limit. To tackle this question, we are providing a method to purify and image a large number of C. reinhardtii centrioles with different orientations using super-resolution microscopy. This technique allows further processing of data through fluorescent single-particle averaging (Fluo-SPA) owing to the large number of centrioles acquired. Fluo-SPA generates averages of stained C. reinhardtii centrioles in different orientations, thus facilitating the localization of distinct proteins in centriolar sub-regions. Importantly, this method can be applied to image centrioles from other species or other large macromolecular assemblies.
Introduction
The centriole is an evolutionarily conserved organelle that lies at the core of the centrosome in animal cells and can act as a basal body (referred to as centrioles hereafter) to template cilia or flagella in many eukaryotes1,2. As such, centrioles are critical for fundamental cell biological processes ranging from spindle assembly to cell signaling. Therefore, defects in centriole assembly or function have been associated with several human pathologies including ciliopathies and cancers3.
Centrioles are nine-fold, symmetrical, microtubule triplet-based cylindrical structures that are, typically, ~450 nm long and ~250 nm wide4,5,6,7. Conventional electron microscopy and cryo-electron tomography of centrioles from different species have revealed that centrioles are polarized along their long axis with three distinct regions: a proximal region, a central core, and a distal region5,7,8,9,10,11. Importantly, each of these regions displays specific structural features. First, the lumen of the 100 nm-long proximal region contains the cartwheel structure connected to the microtubule triplet through the pinhead element12. Second, the 300–400 nm-long central region contains fibrous densities in the lumen and structural features along the inner face of the microtubules: the Y-shaped linker, the C-tubule tail, and the A-tubule stub9. Finally, the 50–100 nm distal region exhibits sub-distal and distal appendages that surround the distal part of the centriole5,13.
The last two decades have been marked by the discovery of an increasing number of centriolar proteins, leading to a current estimation of about 100 distinct proteins being part of the centriole14,15,16,17. Despite these advances, the precise localization of these proteins within the centriole remains elusive, particularly within structural sub-regions. Importantly, assigning a precise localization to structural regions of the centriole is crucial for a better understanding of their function. In this respect, C. reinhardtii centrioles have been instrumental in both aspects by first delimitating the different structural features along the cylinder9,18,19, which then allowed researchers to correlate the localization of a subset of proteins using fluorescent microscopy to a sub-structural region. This includes, for example, the proteins Bld12p and Bld10p, which localize in the proximal region, and in the cartwheel structure in particular20,21,22,23. The list of substructure localized proteins also includes POB15 and POC16, two novel proteins identified by mass spectrometry that decorate the inner central core region of C. reinhardtii centrioles17.
This paper provides a complete description of the method developed to isolate and image C. reinhardtii centrioles for subsequent super-resolution microscopy and single-particle averaging. To achieve this goal, it is important to delineate the technical limitations that need to be overcome. First, centriole purification can affect the overall architecture, with the cartwheel structure often being lost during the various steps of isolation9. Secondly, the dimensions of the centriole are very close to the diffraction limit in optical microscopy. Indeed, the lateral resolution that can be obtained in confocal microscopy is around 200 nm24, similar to the diameter of the centriole, and the resolution in the z-axis is about 2–3x lower, leading to an anisotropic volume. Thirdly, the heterogeneity of antibody labeling and centriole orientation could limit the interpretation needed to localize a protein in a specific centriolar sub-region. Finally, centrioles exist in only two copies per cell, making it difficult to acquire a large number of images and find an unambiguous centriole orientation. To circumvent these technical issues, we developed a method that relies on applying super-resolution microscopy on large numbers of isolated centrioles that adopt various orientations. We will first describe a protocol to purify C. reinhardtii centrioles that enables the purification of structurally intact centrioles and procentrioles containing the cartwheel. Then, we will describe a step-by-step protocol to concentrate the centrioles on coverslips for imaging by conventional or super-resolution fluorescent microscopy. This important step allows for increasing the number of centrioles imaged in multiple orientations. Finally, we will describe a procedure to perform single-particle averaging on data acquired on fluorescent microscopes that facilitates the detection of centrioles in different orientations. Altogether, this method can be applied to image centrioles from various species or other large macromolecular assemblies.
Protocol
1. Media Preparation for C. reinhardtii Culture and Centriole Isolation
- Preparation of media to culture C. reinhardtii cells
NOTE: Steps below describe the preparation of stock solutions for 1x TAP (Tris-acetate phosphate) medium.- Prepare a phosphate buffer (pH 7), by mixing 250 mL of 1 M K2HPO4 (174.2 g of K2HPO4 complemented to 1 L with distilled water) with ~170 mL of 1 M KH2PO4 (136.09 g of KH2PO4 in 1 L). Adjust the mixture to reach pH 7.
- Prepare solution A (40x) by mixing 96.8 g of Tris, 40 mL of phosphate buffer (pH 7), and 40 mL of acetic acid, and adjust the solution to 1 L with distilled water.
- Prepare solution B (40x) using 16 g of NH4Cl, 2 g of CaCl2, and 4 g of MgSO4. Be careful to dissolve CaCl2 in distilled water separately before adding it to the other components. Adjust the solution to 1 L with distilled water.
- Prepare Hutner's Trace Elements buffer25 as follows.
- For 1 L of buffer, dissolve each compound in the indicated volume of water: EDTA disodium salt (50 g in 250 mL), ZnSO4.7H2O (22 g in 100 mL), H3BO3 (11.4 g in 200 mL), MnCl2.4H2O (5.06 g in 50 mL), CoCl2.6H2O (1.61 g in 50 mL), CuSO4.5H2O (1.57 g in 50 mL), (NH4)6Mo7O24.4H2O (1.10 g in 50 mL), and FeSO4.7H2O (4.99 g in 50 mL).
NOTE: EDTA should be dissolved in boiling water, and the FeSO4 should be prepared last to avoid oxidation. - Mix all solutions except the EDTA together and bring to boil. Then add the EDTA, and the solution should turn green. After dissolving everything, cool the solution to 70 °C. At this temperature, add 85 mL of hot 20% KOH solution (20 g in 100 mL). Adjust the solution to 1 L with distilled water at room temperature (RT).
- Add a cotton plug to the flask and let the solution stand for 1 or 2 weeks, shaking it 1x a day. The solution should initially be green and then turn purple, leaving a rust-brown precipitate; remove the precipitate using filter paper until the solution is clear. Freeze the aliquots and store at -20 °C.
- For 1 L of buffer, dissolve each compound in the indicated volume of water: EDTA disodium salt (50 g in 250 mL), ZnSO4.7H2O (22 g in 100 mL), H3BO3 (11.4 g in 200 mL), MnCl2.4H2O (5.06 g in 50 mL), CoCl2.6H2O (1.61 g in 50 mL), CuSO4.5H2O (1.57 g in 50 mL), (NH4)6Mo7O24.4H2O (1.10 g in 50 mL), and FeSO4.7H2O (4.99 g in 50 mL).
- Prepare 1x TAP medium to grow C. reinhardtii cells by mixing the following components: 25 mL of solution A (40x) and 25 mL of solution B (40x) with 1 mL of Hutner's trace elements buffer25, and adjust the mixture to 1 L with distilled water. Sterilize the mixture using a 0.4 µm filter.
- Media preparation for centriole purification
- Prepare a deflagellation buffer using 5% sucrose in 10 mM HEPES (pH 7) at a final volume of 500 mL adjusted with distilled water.
- Prepare 250 mL of 0.5 M acetic acid.
- Prepare 250 mL of a 1 M K-PIPES stock solution (pH 7.2) by first adding 50 mL of H2O to dissolve the PIPES powder, then adding 10 N KOH until the solution starts to become clear. Titrate to pH 7.2 with 10 N and 1 N KOH and adjust the solution to a final volume of 250 mL with H2O.
- Prepare 5 sucrose solutions (w/w) as follows.
NOTE: All sucrose solutions need to be filtered after solubilization using a 0.4 µm filter plugged into a syringe. Note that the 60%- and 70%-sucrose solutions are difficult to solubilize and should be placed in a water bath pre-warmed at 60 °C to facilitate solubilization. Mix every 10 min until the sucrose is completely dissolved.- To prepare 25% sucrose, weigh 25 g of sucrose and adjust the weight to 100 g by adding 10 mM K-PIPES (pH 7.2).
- To prepare 60% sucrose, weigh 60 g of sucrose and adjust the weight to 100 g with 10 mM K-PIPES (pH 7.2).
- Prepare sucrose solutions for the sucrose gradient. Prepare 40% sucrose by weighing 40 g of sucrose and adjusting the solution to 100 g with 10 mM K-PIPES (pH 7.2). Similarly, prepare 50% (w/w) and 70% (w/w) sucrose.
- Store the solutions at -20 °C. Be careful to resuspend the sucrose solutions properly after thawing.
- Prepare 100 mL of lysis buffer by mixing 1 mM HEPES (pH 7), 0.5 mM MgCl2, and 1% NP-40, and keep it at 4 °C. Always prepare this buffer fresh on the day of the experiment. Add anti-protease tablets on the day of the centriole isolation.
- Prepare 1x phosphate buffer saline (PBS) (pH 7.4) by mixing 8 g of NaCl, 2 g of KCl, 1.44 g of Na2HPO4, and 0.24 g of KH2PO4 in 800 mL of distilled H2O. Adjust pH to 7.4 with HCl. Bring the volume of the mixture to 1 L with distilled H2O.
2. Isolation of C. reinhardtii Centrioles
NOTE: See Figure 1.
- Culture and expansion of C. reinhardtii cells
- In the evening on day 1, inoculate a cw15- strain from a solid plate into a culture Erlenmeyer flask containing 10 mL of 1x TAP. Grow the cells under white fluorescent lights (60 µE/m2s) for 2–3 days at 23 °C.
- On day 3, dilute the culture 10x (to 100 mL) in 1x TAP and grow the cells under light for 2–3 days at 23 °C.
- On day 6, dilute the culture 10x in 1x TAP to obtain 1 L of culture. Grow the cells under light at 23 °C until the culture reaches a dark green color that indicates an approximate cell density of ~1 x 107 cells/mL26 (day 9–10).
- Purification of C. reinhardtii centrioles
- Centrifuge the cw15- cells at 600 x g for 10 min in 50 mL conical tubes. Wash the pellet of cells 1x with 50 mL of 1x PBS and spin it at 600 x g for 10 min. Resuspend the pellet in 100 mL of room-temperature deflagellation buffer with a pipette.
- Deflagellate the cells with a pH shock by slowly adding drops of 0.5 M acetic acid to a final pH of 4.5–4.7 on a magnetic stirrer and incubate the cells for 2 min. Slowly add drops of 1 N KOH to restore the pH to 7.0.
- Centrifuge the cells at 600 x g for 10 min to remove any detached flagella. Remove the supernatant and store the pellet on ice. Wash the pellet 2x with 50 mL of 1x PBS precooled at 4 °C. Then, spin the pellet at 600 x g for 10 min at 4 °C.
- Resuspend the pellet in 30 mL of 1x PBS and slowly load the suspension onto a 25%-sucrose cushion of 20 mL without mixing (Figure 1B).
- Spin at 600 x g for 15 min at 4 °C to remove the remaining flagella in the supernatant; the cells are spread in the 25% sucrose (Figure 1C). Keep only the bottom-most 20 mL (red arrow, Figure 1C) by carefully aspirating the supernatant using an aspirator.
- Wash the remaining 20 mL by adding 20 mL of cold 1x PBS. Spin the sample at 600 x g for 10 min at 4 °C. Resuspend the pellet in 10 mL of cold 1x PBS (at 4 °C). Ensure that there are no clumps so that the following lysis hits all the cells at once.
- Transfer the resuspended pellet to a new 250 mL bottle. Add 100 mL of the lysis buffer supplemented with 5,000 units of DNase to the cells. It is important to add the lysis buffer to the cells, and not the other way around. Incubate the mixture for 1 h at 4 °C and mix it carefully by inverting the bottle every 15 min without forming any bubbles.
- Centrifuge the lysed cells at 600 x g for 10 min at 4 °C in a 50 mL conical tube to remove any cell debris. If the lysis has been performed correctly, the cell pellet should be white (Figure 1D). Collect the supernatant with a pipette and load it onto a 30 mL round-bottom tube containing a 60%-sucrose cushion, placed on ice. Then, spin the tube at 10,000 x g for 30 min at 4 °C.
NOTE: Several round-bottom tubes (Table of Materials) may be needed, depending on the volume of the supernatant. - Aspirate the supernatant up to 1 mL above the sucrose cushion. Note the yellow interface between the 1 mL of the remaining supernatant and the 2 mL of the sucrose cushion. Gently mix the sucrose and the remaining supernatant with a cut P1000 tip. Do not vortex at this step; otherwise, the procentrioles can be lost at this stage. Pool all sucrose cushions and store them on ice.
- Prepare a 40%- to 70%-sucrose gradient in a thin-walled 38.5 mL polypropylene tube by gently adding 3 mL of 70% sucrose (at 4 °C), followed by 3 mL of 50%, and finally, 3 mL of 40% sucrose. Load the pooled interfaces onto the 40%- to 70%-sucrose gradient; do this slowly because the cold sucrose is very viscous. Balance the tubes with the 10 mM K-PIPES buffer (pH 7.2) and centrifuge them at 68,320 x g (e.g., with an SW32Ti rotor) for 1 h and 15 min at 4 °C.
- Collect 12x 500 µL fractions at 4 °C by making a hole in the bottom of the tube with a 0.8 mm needle without disturbing the different sucrose layers. With a cut P200 pipette tip, prepare additional 10 µL aliquots from each fraction that will be used for the following immunofluorescence. Snap-freeze the fractions in liquid nitrogen.
NOTE: Isolated centrioles can be analyzed by electron microscopy to ensure that the overall ultrastructure of the centrioles is preserved.
3. Quantification of Isolated Centrioles on Coverslips: Centrifugation and Immunofluorescence
NOTE: See Figure 2.
- Preparing the tubes and coverslips
- Use 1 glass round-bottom tube (15 mL) per fraction to analyze the centriolar fractions (12 tubes in total).
- Put a custom coverslip support adaptor (called adaptor hereafter) into the round-bottom tube. Place a sterile 12 mm coverslip into the round-bottom tube.
NOTE: Here we used 12 mm coverslips, but the protocol can be adapted for 18 mm coverslips using 30 ml round-bottom tubes. - Add 5 ml of pre-cooled 10 mM K-PIPES (pH 7.2) at 4 °C. Make sure that the coverslip is not floating and stays down on the adaptor. Place the tubes on ice.
- Centrifugation of the centrioles
- Dilute each 10 µL fraction with 100 µL of cold 10 mM K-PIPES (pH 7.2). Resuspend the dilution well until the complete disappearance of the sucrose (Figure 2A). Load each diluted fraction into a round-bottom tube.
- Spin the tube at 10,000 x g for 10 min (e.g., with a JS-13.1 swinging bucket rotor) at 4 °C.
- Recover the coverslip by inserting a handmade hooked device into the hole present in the slotted edge of the adaptor and lift it up gently.
NOTE: The handmade hooked device can be made with syringe needle manually hooked and molded onto a homemade stick (Figures 2B-2D). - Upon reaching the top of the round-bottom tube, trap the edge of the adaptor with a gloved finger and remove the coverslip with tweezers. Be careful to remember which side of the coverslip contains the centrioles. Proceed to treat the coverslips for immunofluorescence.
- Immunofluorescence staining and imaging of isolated C. reinhardtii centrioles
NOTE: See Figure 2.- Prepare the material for immunofluorescence staining as follows.
- Prepare a cover-glass staining rack in a crystal polystyrene transmission lab box (60 mm in length by 50 mm in width by 43 mm in height). Fill it with 100% methanol and store it at -20 °C.
- Prepare a humid chamber. For this, assemble the humid chamber by placing a water-humidified tissue alongside the inside edges of a square Petri dish (Figure 2E). Add a piece of laboratory sealing wrap (see Table of Materials) to the middle of the Petri dish onto which the antibody mixes will be placed during the immunofluorescence procedure (steps 3.3.2–3.3.3). Cover the lid and the humid chamber with aluminum foil to protect it from light.
- Immunostain the isolated centrioles as follows.
- Fix the coverslips with the centrioles directly after the centrifugation (step 3.2.4) by incubating them for 5 min in the box filled with -20 °C methanol (step 3.3.1.1).
- Remove the coverslips with tweezers and place them in a transparent laboratory box (see Table of Materials) filled with 50 mL of 1x PBS and wash them for 5 min at room temperature.
- Pipette 60 µL of primary antibody mix [primary antibodies diluted in 1% bovine serum albumin (BSA) and 0.05% Tween-20 in PBS] on the piece of laboratory sealing wrap in the humid chamber. Carefully lay the coverslips on top of the antibody mix with the centrioles directly facing the drop. Incubate the coverslips for 45 min with primary antibodies.
NOTE: The primary antibodies that were used to generate the representative results are rabbit polyclonal Bld12 (1:300) and mouse α-tubulin (DM1A) (1:300). - Remove the coverslips and wash them for 5 min in 1x PBS, as described in step 3.3.2.2. Incubate the coverslips for 45 min with secondary antibodies in PBS containing 1% BSA and 0.05% Tween-20.
NOTE: The secondary antibodies that were used to generate the representative results are goat anti-mouse coupled to Alexa 488 (1:1,000) and goat anti-rabbit coupled to Alexa 568 (1:1,000). - Remove the coverslips and wash them for 5 min in 1x PBS, as described in step 3.3.2.2.
- Mount the coverslips on a glass slide by adding 3 µL of mounting medium on the slide and carefully placing the coverslips on top (centrioles facing the mounting medium). Seal the coverslip edge with nail polish.
- Image the isolated centrioles on a confocal microscope at a 63X oil objective with an N.A. of 1.4 while applying deconvolution27 (see Table of Materials).
NOTE: Here, the following settings were used: 500–545 nm for Alexa 488 and 580–635 nm for Alexa 568.
- Prepare the material for immunofluorescence staining as follows.
4. Concentration of Centrioles onto the Center of the Coverslips
NOTE: See Figure 3.
- Material preparation
- Prepare a 15 mL glass round-bottom tube on ice, a custom coverslip support adaptor (called adaptor hereafter, .stl file provided as Supplementary File 1), a custom concentrator (.stl file provided as Supplementary File 2), and 10 mM K-PIPES (pH 7.2) at 4 °C.
- Prepare poly-D-lysine (PDL)-coated coverslips. Dilute 10x the 1 mg/mL PDL stock solution with H2O. First, wash the coverslips with 70% EtOH, remove the ethanol, and let the coverslips dry. Coat the coverslips with PDL and incubate them for 30 min at room temperature. Wash the coverslips 3x with water and let them dry.
NOTE: Coat the coverslips with PDL to increase the number of isolated centrioles attached to the coverslips.
- Centrifugation
- Place a sterile 12 mm coverslip onto the recessed, bottom-end of the concentrator, keeping the PDL coat facedown. Cap the coverslip by placing the adaptor directly on top. Invert the round-bottom tube and place it over the concentrator, coverslip, and adaptor.
- Gently push the ensemble with tweezers until it reaches the bottom of the round-bottom tube, and invert the tube. Add 10 mM K-PIPES buffer (pH 7.2) to the round-bottom tube until it comes to the top of the concentrator. Make sure that there do not remain any bubbles in the central cylinder of the concentrator.
- Gently add 100 µL of 10 mM K-PIPES buffer (pH 7.2) to one aliquot containing enriched centriole fraction and thoroughly mix the volume.
- Remove 100 µL of 10 mM K-PIPES buffer (pH 7.2) from the hollow center of the concentrator and add 100 µL of the enriched centriolar fraction in 10 mM K-PIPES buffer (pH 7.2) to the hollow center of the concentrator, taking care that the contents remain in the hollow center.
- Centrifuge at 10,000 x g for 10 min (e.g., with a JS-13.1 swinging bucket rotor) in a pre-cooled centrifuge at 4 °C.
- Remove the concentrator with tweezers.
- Recover the coverslip by inserting a handmade hooked device into the hole present in the slotted edge of the adaptor and lift it up gently. Upon reaching the top of the round-bottom tube, trap the edge of the adaptor with a gloved finger and remove the coverslip with tweezers. Be careful to remember which side of the coverslip contain centrioles. Proceed to treat the coverslips for immunofluorescence.
NOTE: The handmade hooked device can be made with a syringe needle manually hooked and molded onto a homemade stick (Figures 2B- D). - Perform fixation and immunofluorescence staining of concentrated centrioles as done in step 3.3.2–3.3.4.
5. Single-particle Averaging
NOTE: See Figure 4.
- Imaging for Single-particle Averaging
- Mount the coverslip on a slide using a regular anti-fading mounting medium. Perform structured illumination microscopy (2D SIM) imaging using a CFI Apochromat TIRF objective (100X, NA 1.49, WD 0.12mm) and a back illuminated EM CCD camera.
NOTE: The acquisition time was set to 100 ms at a camera read out of 3 MHz. A 2.5X lens was used for SIM imaging.
NOTE: The dataset presented here was acquired on a 3-D SIM microscope (see Table of Materials). - Image the centrioles by acquiring a large stack of images comprising the total centriole signal, by setting up the top and bottom position of the Z-stack above and below the centriole signal, respectively. Proceed to project the stack and perform single-particle averaging as per steps 5.2 and 5.3.
- Mount the coverslip on a slide using a regular anti-fading mounting medium. Perform structured illumination microscopy (2D SIM) imaging using a CFI Apochromat TIRF objective (100X, NA 1.49, WD 0.12mm) and a back illuminated EM CCD camera.
- Projection of a stack
- Open the image stack with ImageJ. Then click on 'Image → Stacks → Z Project'. Set the projection type as 'Max Intensity'.
- Invert the image by clicking on 'Edit → Invert'. Save the projection that is generated (.tif format).
- Single-particle alignment with Scipion
- Create a new project in Scipion by pressing the red 'Create Project' button at the top of the page. On the left panel, double-click on 'Imports → Import micrographs'.
- Fill the 'Files directory' and 'Pattern' fields according to the data directory and names. Keep the default parameters. Click on 'Execute'.
- Double-click on 'Particles → Picking → xmipp3 - manual picking (step 1)'. Click on the magnifying glass icon close to the 'Input micrographs' field and select the imported micrographs from step 5.3.1. Click on 'Execute'.
- In the newly opened window, select particles from the different micrographs by clicking on them. When finished with every micrograph, click on the red button '+ Coordinates'.
- Double-click on 'Particles → Extract → xmipp3 - extract particles'. Click on the magnifying glass icon close to the 'Input coordinates' field and select the picked coordinates from step 5.3.4. Fill the 'Particle box size (px)' according to the particles dimensions. In the 'Preprocess' tab, set Dust Removal: No (it can generate artifacts); Invert contrast: No (black particles, white background); Phase flipping: No (linked to CTF correction); Normalize: Yes.
- Then, click on 'Execute'. When the job is done, check the extracted particles by selecting the box of the job (borders become thicker) and click on 'Analyze results' (bottom left of the main menu).
- Double-click on '2D → Align → xmipp3 - align with cl2d'. Click on the magnifying glass icon close to the 'Input particles' field and select the extracted particles from step 5.3.5. Do not use a reference image. Click on 'Execute'.
- Double-click on '2D → Align → more → xmipp3 - apply alignment 2d'. Click on the magnifying glass icon close to the 'Input particles' field and select the aligned particles from step 5.3.7.
- Click on 'Execute' as in step 5.3.6. The results can be checked by clicking on 'Analyze results' after the selection of the job box.
- Double-click on 'Particles → Mask → xmipp3 - apply 2d mask'.
- Click on the magnifying glass icon close to the 'Input particles' field and select the aligned particles from step 5.3.9. The 'Mask source' is set to 'Geometry'. Then, set the parameters of the mask as Mask type: Circular; Radius (px): looking for one particle (a centriole) without anything around it, click on the 'magic wand' icon on the left which opens a window to help find the perfect value; Shift Center: No (if the particle is perfectly centered) or Yes (if the particle is shifted); X-center offset: according to the particle position; Y-center offset: according to the particle position. Click on 'Execute'.
- Use the 'Analyze Results' button to check if the mask is correctly applied. If not, right-click on the 'Apply mask 2d' job and select 'Edit'. Modify the parameters of the mask (size and/or shifts) and run it again with the 'Execute' button.
- Double-click on '2D → Classify → xmipp3 - cl2d'.
- Click on the magnifying glass icon close to the 'Input particles' field and select the masked particles from step 5.3.11. The number of classes should be to obtain approximately 50 particles per class. Click on 'Execute'.
- Check the results by clicking on the 'Analyze Results' button. In the opened window, click on the 'eye' icon right next to "What to show".
- The new window allows the inspection of the generated classes by selecting 'Classes2D' in the 'Block' menu. Check the content of each class by selecting 'Class00N_Particles' in the same menu. Inspect each class to identify which ones contain only bad particles. Go back to the 'Classes2D' view and select the classes with good particles by clicking on each. It is possible to select several classes by keeping the 'Ctrl' key pressed during the selection.
- When all the classes with good particles have been selected, click on '+ Particles' to create a subset with these particles.
- In the same window, select a few sets of classes which represent different orientations of the particles. Create a new subset by clicking on '+ Averages'.
- For each orientation/average selected, do as follows.
- Double-click on '2D → Align → xmipp3 - align with cl2d'.
- Click on the magnifying glass icon close to the 'Input particles' field and select the good particles from step 5.3.17. Set 'Use a Reference Image' to 'Yes'. Click on the magnifying glass icon close to the 'Reference image' field and click on the white arrow left to the 'Create subset' job from step 5.3.18 and select the image to use it as a reference. Double-click on an object to open it in a separate window and check which object it is. Click on 'Execute'.
- Double-click on '2D → Align → more → xmipp3 - apply alignment 2d'. Click on the magnifying glass icon close to the 'Input particles' field and select the aligned particles from step 5.3.19.2.
- Click on 'Execute'.
- Check the result of the alignment ('Analyze Results'); it will show the aligned particles and the average of these particles.
- If the average is good and the particles are all oriented in the same way, save the average by clicking on 'Advanced → ImageJ' and save the image with ImageJ.
- If the average can be improved, select all the well-oriented images and create a new subset with the '+ Particles' button. Reiterate steps 5.3.19.1–5.3.19.4 until the subset is cleaned (all the bad particles removed). Each time the alignment is performed (step 5.3.19.2), the reference is set to the last generated average (from iteration to iteration, the average quality increases).
Results
C. reinhardtii Centriole Isolation:
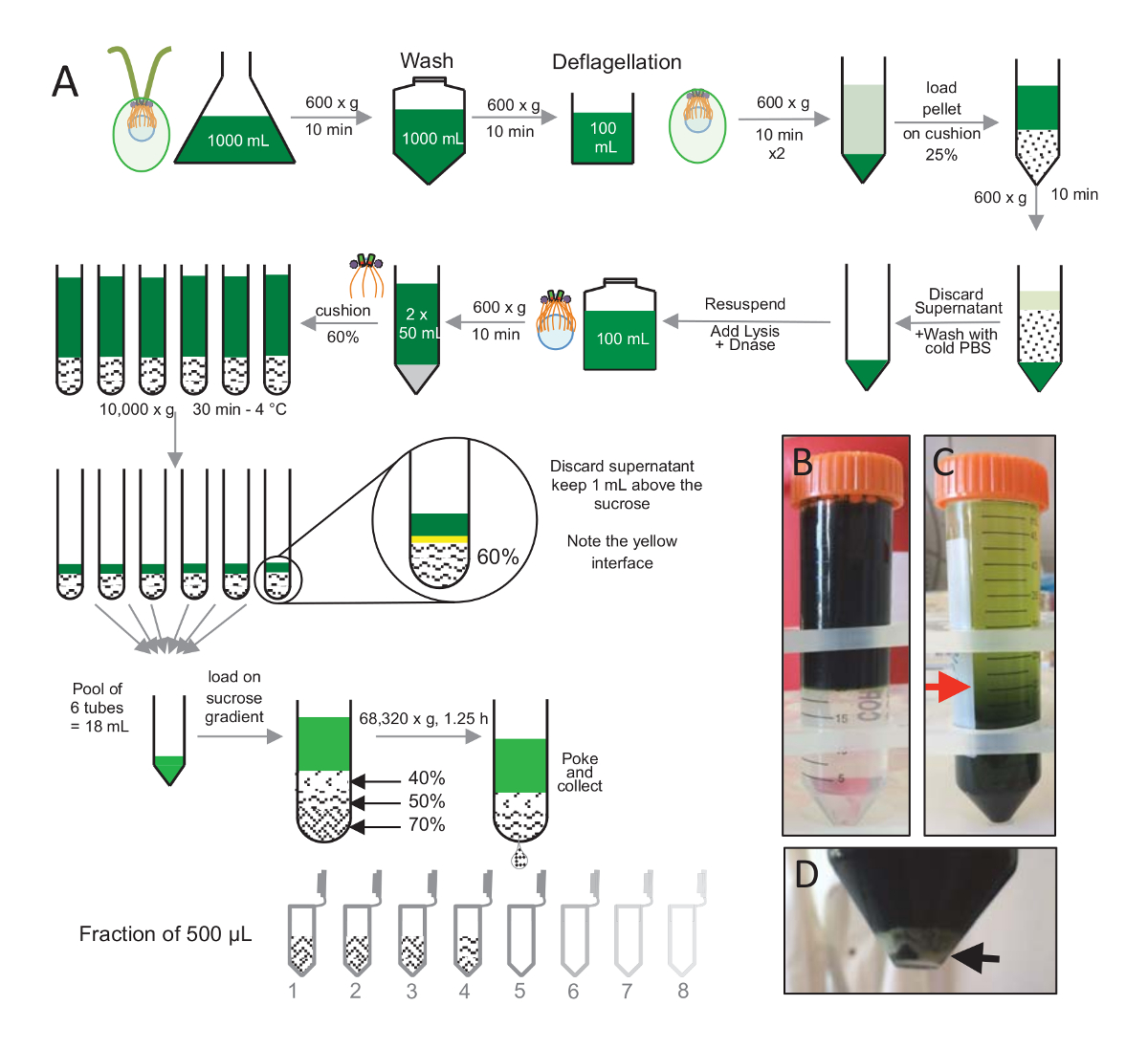
To isolate centrioles, cw15- C. reinhardtii cells were grown in liquid culture for several days under light and subsequently pelleted by centrifugation at 600 x g for 10 min. Pelleted cells were washed 1x with PBS and resuspended in a deflagellation buffer prior to deflagellation by performing a pH shock using 0.5 M acetic acid to a final pH of 4.5–4.7 for 2 min (Figure 1A). An addition of 1 N KOH was used to restore pH to 7.0. To separate detached flagella from cell bodies, deflagellated cells were first centrifuged to remove the bulk of the flagella. The pellet was then washed 2x with PBS and resuspended in 30 mL of PBS prior to loading it slowly on a 25%-sucrose cushion (Figure 1B). The tube was spun at 600 x g for 15 min at 4 °C to remove most of the detached flagella. After centrifugation, the cell bodies were spread in the sucrose cushion (Figure 1C) and recovered by removing the approximately 30 mL of supernatant (until the red arrow in Figure 1C). The resulting 20 mL of washed cells were then resuspended in 20 mL of cold PBS and centrifuged at 600 x g for 10 min. Next, the supernatant was discarded, and the cells were completely resuspended in 10 mL of PBS. The cells were transferred to a 250 mL bottle and the lysis buffer was added to the resuspended cells at once. DNase was added to the lysis and incubated for 1 h at 4 °C. After a centrifugation step to remove cell debris (see the white pellet in Figure 1D), the supernatant was collected and carefully loaded onto a 2 mL 60%-sucrose cushion prior to centrifugation at 10,000 x g for 30 min at 4 °C. Note that for 100 mL of lysis, 8 tubes of 15 mL were used to perform this step, corresponding to 12.5 mL of lysis buffer loaded on 2 mL of sucrose per tube. After centrifugation, most of the supernatant was removed up to 1 mL above the cushion. The 1 mL of remaining supernatant was then collected with the 2 mL cushion and then mixed and pooled to obtain a final volume of 24 mL. The pool was then loaded on a 40%-, 50%-, 70%-sucrose gradient and spun at 68,320 x g for 1 h and 15 min at 4 °C. Finally, the isolated centrioles were collected by making a hole in the bottom part of the centrifugation tube using a needle and the drops were collected in 12 x 500 µL fractions. Due to the high density of the different sucrose, the drop formed very slowly at the beginning (70% sucrose) and then more rapidly (40% sucrose).
Immunofluorescence of Isolated Centrioles
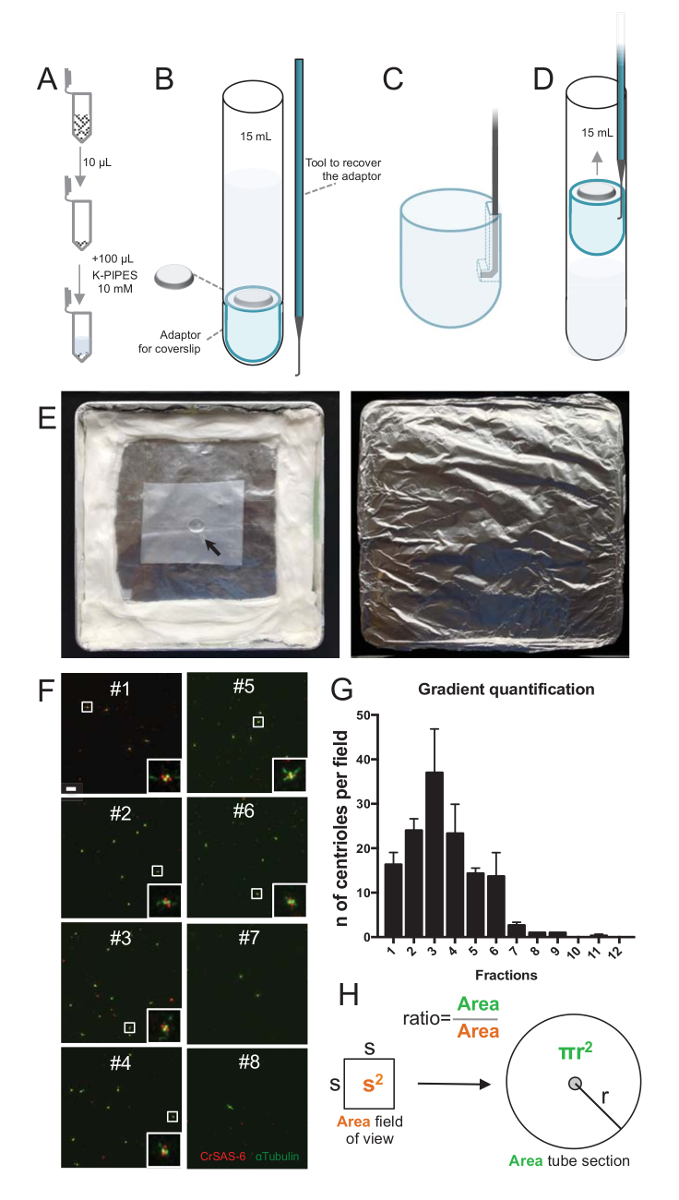
To assess the quality of the isolation procedure, 10 µL of each collected gradient fraction was then centrifuged onto a coverslip using a coverslip support adaptor (Figure 2A-2D). Importantly, to remove safely the coverslip, a custom hooked device was designed (Figure 2B). Next, the coverslips were analyzed by immunofluorescence. Antibodies against CrSAS-6(Bld12p) were used in this study to indicate the presence of the cartwheel structure and α-tubulin (DMA1) to highlight the centriolar wall. By counting the number of centrioles that were positive for both CrSAS-6 and α-tubulin per field of view and then calculating the total number of centrioles per fractions, it was possible to determine which fractions were enriched for isolated centrioles (Figure 2F-2H). Interestingly, 6 fractions were enriched for centrioles (Figure 2F and 2G, fractions #1–6), with a peak for fraction #3, while the last fractions were not, indicating that the purification worked. Note that in this particular experiment, 95% of the total centrioles were positive for CrSAS-6 and α-tubulin in fraction #3. This indicates that most isolated centrioles retained their cartwheels. If no enrichment of centrioles is observed in the first fractions, the isolation procedure did not work and should be repeated. Note that some flagellar pieces can be observed mostly in the fractions devoid of centrioles.
Next, to calculate the total number of centrioles per µL, the number of centriole per field of view should be multiplied by the ratio as presented in Figure 2H. The resulting number should then be divided by the volume of the fraction used for the immunofluorescence to obtain the number of centrioles per µL. In this particular isolation procedure, the most enriched fraction contained about 37 centrioles for an area of 0.00846 mm2 (with a field of view of 92 x 92 µm2). The surface of the tube section was 7.5 mm in radius with a total surface of 176 mm2. The corresponding ratio was then 176/0.00846 = 20,803.8, so a total of 769,740 centrioles (37x20,803.8) in 10 µL. Therefore, the number of centrioles in 1 µL was 76,974.
Concentration of Isolated Centrioles on Coverslips:
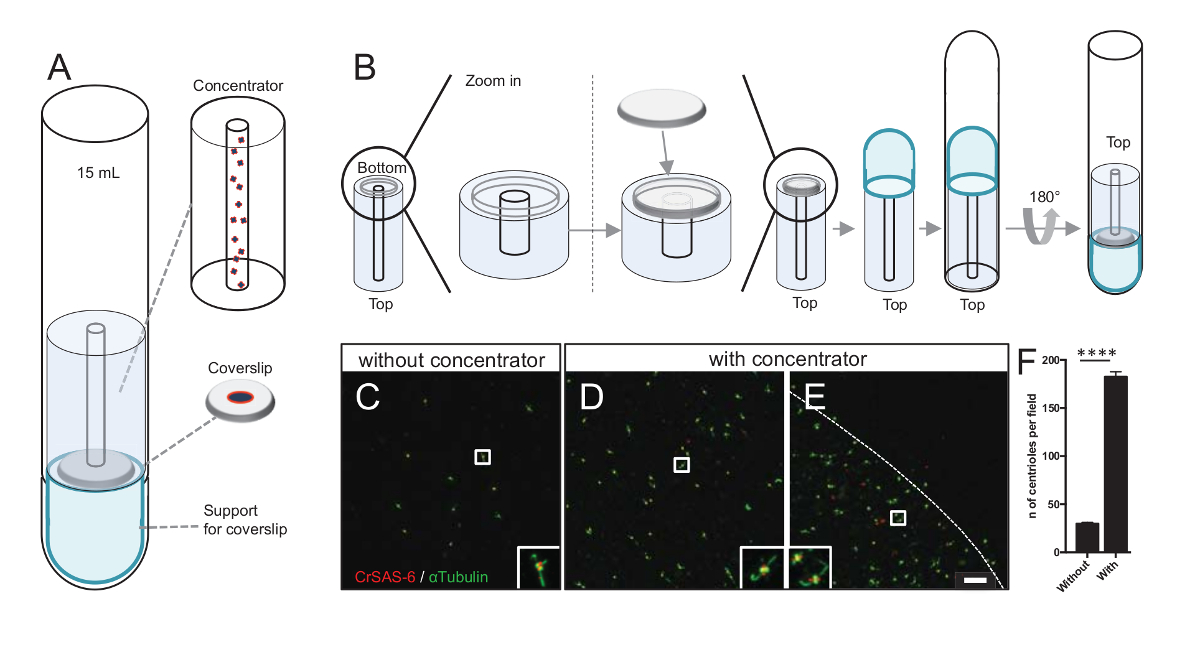
Increasing the number of centrioles per field increases the chance of detecting unambiguous centriole orientation, as well as increasing the chance of detecting similar orientations that can be used for further particle averaging procedures. As centrioles from the concentrated fractions are still sparse on the coverslip, we developed a centrifugation accessory to concentrate centrioles in the middle of the coverslip (Figure 3A) named a concentrator. Note that a .stl file with the precise measures for 3-D printing is provided with this manuscript.
First, one coverslip of 12 mm was mounted on the concentrator (Figure 3B). The adaptor was placed on top of the coverslip and the round-bottom tube was inverted and placed over the assembled adaptor and concentrator. The round-bottom tube was then gently inverted, thus allowing the loading of the sample (Protocol step 4.2). The centrioles were then centrifuged at 10,000 x g for 10 min at 4 °C. After that, the centrioles were subjected to immunofluorescence and stained for CrSAS-6/Bld12p and α-tubulin (Figure 3C-3E). Importantly, without concentrator, about 30 centrioles were seen per field of view (Figure 3C), whereas 183 centrioles were seen per field of view when the concentrator was used (Figure 3D-3F). Note that the centrioles covered only a disk of 4 mm in diameter in the middle of the coverslip. This result demonstrates that the concentration step works and allows a 6-fold enrichment of centrioles in a defined region of the coverslips, thus easing their detection and imaging.
Fluorescent Single-particle Averaging of Isolated C. reinhardtii Centrioles:
Here, using SIM microscopy that can reach a resolution of about 120 nm, centrioles stained for monoglutamylated tubulin (GT335, Figure 4), a tubulin modification present in centriolar microtubules, were imaged24. C. reinhardtii centrioles are about 500 nm long, always in pairs, and often found with associated, newly duplicated probasal bodies (referred to as procentrioles hereafter) and striated microtubule-associated fibers19. Therefore, this final assembly was about 1 µm large. For this reason, and in order to image the centrioles in their entirety, we recommend acquiring a Z-stack on the isolated centrioles.
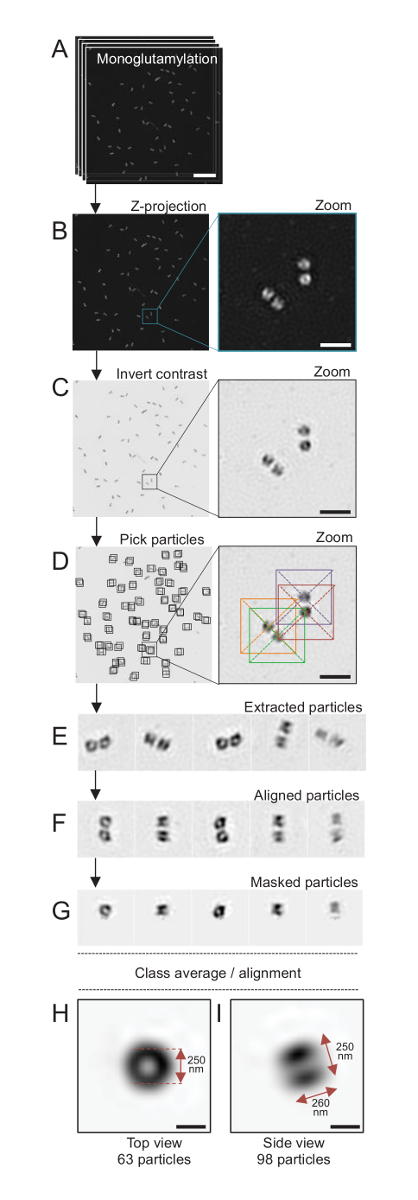
Here, after the acquisition, a final image was generated by performing a maximum intensity projection using ImageJ28 (image/Stacks/Z project/max intensity projection, Figure 4B). From such images, a single-particle analysis using cryo-electron microscopy software was performed to make classes of centrioles with similar orientations, and then averaging was performed. To do so, the image color was first inverted so as to better visualize the objects (Figure 4C). Centrioles were picked manually in a box centered over each particle using the freely available Scipion software29 that integrates several electron microscopy software programs such as Xmipp3 (Figure 4D). Note that the size of the box has to be defined by the user. Here, boxes of 50 x 50 pixels for a pixel size of 31.84 nm were used. Next, all particles were extracted (Figure 4E) and aligned using Xmipp3 (Figure 4F). Next, a circular mask of 12 pixels in radius was applied to isolate each centriole from the centriolar-pair (Figure 4G). The particles were then classified, using Xmipp3, to generate several averages. Only homogenous class averages were kept, meaning that particles that diverge from the class averages were manually excluded. This step was repeated in order to generate a near-perfect average for each chosen orientation. After five iterations, two classes of averages were generated: a top view from 63 objects (Figure 4H) and a side view from 98 particles (Figure 4I) of monoglutamylated centrioles. The dimensions of the object were determined by measuring the distance between peaks of the intensity plot profile along the monoglutamylation signal.
Importantly, the length of the side-view class average is 260 nm with a diameter of 250 nm, comparable to the measured monoglutamylated tubulin signal that was shown to localize to a region of 286 ± 33 nm in length within the core of the centriole17.

Figure 1: Purification of C. reinhardtii centrioles. (A) This is a schematic representation of each step leading to the isolation of C. reinhardtii centrioles. It includes representative steps of the protocol (B) before and (C) after centrifugation onto the 25%-sucrose gradient. The red arrow in panel C indicates the minimum volume to keep after centrifugation. (D) This panel shows the white pellet of lysed cells after centrifugation. The black arrow indicates the pellet. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Centrifugation set-up to perform immunofluorescence on isolated centrioles. (A) 10 µL of each collected fraction is diluted first in 100 µL of 10mM K-PIPES (pH 7.2). (B) This is a schematic representation of the centrifugation devices, encompassing a 15 mL round-bottom tube, a 12 mm coverslip, an adaptor for the coverslip, and a custom-made hooked device to recover the coverslip after centrifugation. Panels C and D show drawings explaining how to recover the coverslip after centrifugation. (C) Place the hooked tool into the hole present in the slotted edge of the adaptor and (D) gently pull. (E) These are pictures of the humid chamber needed to perform immunofluorescence imaging. The arrow indicates the 12 mm coverslip. (F) These are representative confocal images at 63X (zoom 2) of gradient fractions #1-8, collected during the purification procedure and stained for CrSAS-6 (red) and α-tubulin (green). The insets correspond to the region indicated by a white box in the lower magnification views. Scale bar = 10 µm. (G) This is a graph representing the number of centrioles positive for both CrSAS-6 and α-tubulin per field of view in each fraction. Note the enrichment in fractions #1-6. The average number of centrioles per field in each fraction: #1 = 16.3 ± 4.7, #2 = 24.0 ± 4.6, #3 = 37.0 ± 17.0, #4 = 23.3 ± 11.4, #5 = 14.3 ± 2.1, #6 = 13.7 ± 9.3, #7 = 2.7 ± 1.2, #8 = 1.0 ± 0.0, #9 = 1.0 ± 0.0, #10 = 0.0 ± 0.0, #11 = 0.3 ± 0.6, #12 = 0.0 ± 0.0. For each fraction, 3 random fields were imaged. Note that, when counting the most enriched fraction #3, we found that 95% of the centrioles are positive for CrSAS-6 and α-tubulin (n = 205 centrioles). (H) A ratio of the number of centrioles present in the measured area of the micrographs is used to calculate the total number of centrioles present in the area of the tube. The centriole number per µL can be calculated from the 10 µL originally centrifuged fractions. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Isolated centrioles are centrifuged using concentrators. (A) This panel shows the centrifugation devices needed to concentrate centrioles onto coverslips prior to immunofluorescence, including a 15 mL round-bottom tube, a concentrator, a 12 mm coverslip, and a support adaptor apparatus. (B) This panel shows the steps to assemble the centrifugation device. Panels C-E show confocal images of centrioles stained for CrSAS-6 (red) and α-tubulin (green) (C) without or (D-E) with the concentrator. The insets correspond to the region indicated by a white box in the lower magnification views. The scale bar is 10 µm. Note that centrioles are enriched in the middle of the coverslip (the dotted line represents the border of the concentrated area). (F) This graph represents the number of centrioles per field of view without and with the concentrator. Five random fields of view were analyzed. The average number of centrioles is, without concentrator, 29.8 ± 2.9, and with concentrator, 182.6 ± 11.5, P <0.0001. The statistical significance was assessed by an unpaired t-test. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Single-particle averaging on isolated C. reinhardtii centrioles. (A) This panel shows Z stack images of centrioles stained with GT335 and acquired using a SIM microscope. (B) These panels show a maximal intensity Z projection of the stacked images. Scale bar = 1 µm. (C) These panels show a representative image with an inverted contrast. The inset represents a zoom-in to visualize the centrioles better. (D) These panels show particle picking. The inset indicates how the particles were picked. (E) These are examples of 5 extracted particles. (F) This panel shows the particles after alignment. (G) This panel shows the particles after applying a mask. Panels H and I show two class averages: (H) a top view (63 particles) and (I) a side view (98 particles). The double arrows indicate the dimensions of the GT335 signal. Scale bar = 250 nm. Please click here to view a larger version of this figure.
{kind=link}
Supplementary File 1. Please click here to download this file.
Supplementary File 2. Please click here to download this file.
Discussion
One of the challenges in biology is to decipher the precise localization of proteins in an architectural context. The centriole is an ideal structure to apply these methods, as its architecture has been studied using cryo-electron tomography, revealing interesting ultrastructural features along its length. However, due to its dimensions close to the resolution limit in optical microscopy, it is difficult to precisely localize a protein by immunofluorescence to a structural sub-region of the centriole using conventional microscopes30.
The resolution in optical microscopy is limited by the diffraction of light that gives, roughly, a lateral maximum resolution of 200 nm in optical microscopy24. However, this limit has been by-passed by one of the major breakthroughs of the last 20 years in optical microscopy: the invention of super-resolution methods. These approaches can image beyond the diffraction limits at different resolutions: 120 nm for SIM, about 50 nm for stimulated emission depletion (STED), and 20–40 nm for single-molecule localization microscopy (SMLM)24. With these new developments of super-resolution microscopy, the structural sub-regions of the centriole are attainable. However, in practice, it is still difficult to accurately determine the localization of a protein to a structural element for the main reason that the mature centrioles exist in only 2 copies per cell and have random orientations, which makes the interpretation of localization difficult. For this reason, a protocol was set up that allows researchers to image by super-resolution a large number of centrioles, increasing the chance to observe non-ambiguous orientations. Importantly, as this method relies on the use of isolated centrioles, we are providing a method to purify intact C. reinhardtii centrioles that contain mature centrioles and procentrioles.
Finally, owing to the range of centriole orientations that can be imaged with this protocol, single-particle analysis can be applied using electron microscopy software. This results in the generation of average classes of centrioles in a particular orientation. Importantly, these resulting 2-D images can then be used to assess the localization of a specific protein along the centriole. Indeed, this method can be applied to dual-color super-resolution images, and one color can be used to reveal the centriole skeleton (e.g., tubulin), while the other color can be attributed to a specific centriolar protein. By subtracting the averages obtained with one color or two colors, it becomes easier to register a protein along the centriole (proximal, central, or distal). Note that the two channels should be accurately aligned to prevent any misleading interpretations. Moreover, averages of top views will help decipher if a protein localizes inside the centriolar lumen, along the microtubule wall, or outside the centriole.
This method has the advantage to ascertain the localization of specific proteins that could be difficult to localize otherwise due to heterogeneous labeling. Note that other methods to map proteins within the centrioles have been described in correlative 3-D SIM/SMLM with, for instance, assessing the specific orientations of the centrioles by determining the elliptical profile of a marker forming a torus around the centriole by SIM imaging. Using this parameter, it is possible to localize protein with a precision of 4–5 nm30. Note also that the method described here uses isolated centrioles with intact procentrioles, a sign that the centriole architecture is most likely largely conserved. However, we cannot exclude that some architectural features are disturbed during purification, such as the centriole diameter varying with the concentration of divalent cations as amplified with the isolation of the human centrosome5.
One of the critical steps of the protocol presented here is to obtain sufficiently concentrated isolated centrioles in different orientations amenable to Fluo-SPA. To do so, first ensure the purity and the efficiency of the centriole isolation procedure. A low concentration of isolated centrioles will prevent proper imaging and further image processing. For this purpose, we are providing a method to enrich the number of centrioles per field of view. Depending on the number of centrioles in the fraction used, the volume loaded in the concentrator should be adjusted, with a maximum volume of 250 µL.
Importantly, this method has been developed for a cell wall minus cw15- C. reinhardtii cells. In this strain, the fragility of the cell wall allows for a proper lysis of the cells and, thus, the liberation of its content. This protocol is not efficient for wild-type C. reinhardtii cells, as the cell wall prevents a proper lysis. Alternative strategies such as sonication or a pre-incubation of the cells with autolysin, an enzyme that can degrade the cell wall31, would have to be put in place to alter the cell wall prior to applying the isolation protocol presented here.
This set-up can be used with different types of microscopes, ranging from conventional confocal microscopes to high throughput dedicated super-resolution microscopes. Note that when doing SMLM, a special buffer is required for proper imaging and, thus, a chamber adapted for a 12 mm coverslip with the buffer on top of the coverslip should be used. Subsequent imaging will be performed with inverted microscopes. If the microscope set-up does not allow a 12 mm coverslip, the protocol presented here can be applied to 18 mm coverslips using a 30 mL round-bottom tube and a modified adaptor and concentrator. It is also important to note that the quality of the final reconstruction in SMLM will depend on the quality of the staining and of the primary antibody used, as well as the method of fixation.
In summary, we are providing a method that can be applied to image numerous centrioles followed by Fluo-SPA that will generate averages of centrioles in different orientations, thus helping to localize centriolar protein with precision. Importantly, this method can be applied more generally to isolated centrioles from other species, to other organelles, or to large macromolecular assemblies. Finally, the sample preparation approach presented here, combined with recent algorithm development for single-particle analysis of fluorescent SMLM data32, could open further improvement in the molecular cartography of large macromolecular assemblies.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank Pierre Gönczy and the BioImaging & Optics Platform (BIOP) at École Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland, where the SIM images of the centrioles were acquired. Nikolai Klena and Davide Gambarotto are supported by the European Research Council (ERC) Starting Grant (StG) 715289 (ACCENT) and Maeva Le Guennec, Paul Guichard, and Virginie Hamel by the Swiss National Science Foundation (SNSF) PP00P3_157517. Susanne Borgers is supported by the University of Geneva.
Materials
| Name | Company | Catalog Number | Comments |
| Mouse monoclonal anti-apha tubulin (clone DM1A) | Abcam | ab7291 | dilution 1:300 |
| DNaseI | Roche | 10104159001 | |
| 12 mm coverslips | Roth | YX03.1 | |
| 18 mm coverslips | Roth | LH23.1 | |
| K2HPO4 | Fluka | 60355 | |
| KH2PO4 | Fluka | 60230 | |
| Tris base | Biosolve Chimie SARL | 0020092391BS | |
| acetic acid | Carlo Erba Reagents | 524520 | |
| NH4Cl | Sigma | A-4514 | |
| CaCl2 | Sigma | C-7902 | |
| MgSO4 | Sigma | 63140-500G-F | |
| steritop filter | Millipore | SCGPT05RE | |
| sucrose | Sigma | S7903-1KG | |
| HEPES | AppliChem PanReac | A3724,0250 | |
| PIPES | Sigma | P6757-500G | |
| MgCl2 | ACROS ORGANICS | 197530010 | |
| NP-40 | AppliChem PanReac | A1694,0250 | |
| Round-bottom (Kimble) tubes 15 mL | Fisherscientific | 09-500-34 | |
| Round-bottom (Kimble) tubes 30 mL | Fisherscientific | 09-500-37 | |
| cover glass staining rack | Thomas scientific | 8542E40 | |
| crystal polystyrene transmission lab box | FISHERS | 11712944 | |
| Methanol | VWR | 20864.32 | |
| BSA | Roche | 10735086001 | |
| Triton X100 | Roth | 3051.3 | |
| goat anti-mouse coupled to Alexa 488 | invitrogen | A11029 | dilution 1:1,000 |
| goat anti-rabbit coupled to Alexa 568 | invitrogen | A11036 | dilution 1:1,000 |
| mounting medium | abcam | ab188804 | |
| Tube, thinwall polypropylene | Beckman Coulter | 326823 | |
| Poly-D-Lysine 1 mg/mL | SIGMA | A-003-E | |
| Mouse monoclonal anti-Polyglutamylation modification mAb (GT335) | Adipogen | AG-20B-0020 | dilution 1:1,000 |
| glycerol mounting medium with DAPI and DABCO | Abcam | ab188804 | |
| 50 mL conical tubes | Falcon | 14-432-22 | |
| Eppendorf 5810R centrifuge | Eppendorf | 5811000622 | |
| Beckman JS-13.1 swinging bucket rotor | Beckman Coulter | 346963 | |
| Beckman SW 32Ti rotor | Beckman Coulter | 369694 | |
| parafilm | Bemis | 13-374-10 | |
| Leica TCS SP8 with Hyvolution mode | Leica | ||
| OSRAM L18W/954 LUMILUX | Luxe Daylight/ OSRAM | ||
| Whatman filter paper | Sigma | WHA1001325 | |
| CrSAS-6/Bld12 antibody | dilution 1:300 (Hamel el al., 2014) | ||
| Scipion | http://scipion.i2pc.es/ | ||
| EM CCD camera (Andor iXON DU897) | Andor |
References
- Bornens, M. The centrosome in cells and organisms. Science. 335 (6067), 422-426 (2012).
- Nigg, E. A., Holland, A. J. Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nature Reviews Molecular Cell Biology. 19 (5), 297-312 (2018).
- Nigg, E. A., Raff, J. W. Centrioles, centrosomes, and cilia in health and disease. Cell. 139 (4), 663-678 (2009).
- Gönczy, P. Towards a molecular architecture of centriole assembly. Nature Reviews Molecular Cell Biology. 13 (7), 425-435 (2012).
- Paintrand, M., Moudjou, M., Delacroix, H., Bornens, M. Centrosome organization and centriole architecture: their sensitivity to divalent cations. Journal of Structural Biology. 108 (2), 107-128 (1992).
- Winey, M., O'Toole, E. Centriole structure. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 369 (1650), (2014).
- Dippell, R. V. The development of basal bodies in paramecium. Proceedings of the National Academy of Sciences of the United States of America. 61 (2), 461 (1968).
- Allen, R. D. The morphogenesis of basal bodies and accessory structures of the cortex of the ciliated protozoan Tetrahymena pyriformis. The Journal of Cell Biology. 40 (3), 716-733 (1969).
- Li, S., Fernandez, J. -. J., Marshall, W. F., Agard, D. A. Three-dimensional structure of basal body triplet revealed by electron cryo-tomography. The EMBO Journal. 31 (3), 552-562 (2012).
- Guichard, P., Chrétien, D., Marco, S., Tassin, A. -. M. Procentriole assembly revealed by cryo-electron tomography. The EMBO Journal. 29 (9), 1565-1572 (2010).
- Guichard, P., et al. Native architecture of the centriole proximal region reveals features underlying its 9-fold radial symmetry. Current Biology. 23 (17), 1620-1628 (2013).
- Hirono, M. Cartwheel assembly. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 369 (1650), (2014).
- Bornens, M., Paintrand, M., Berges, J., Marty, M. C., Karsenti, E. Structural and chemical characterization of isolated centrosomes. Cell Motility and the Cytoskeleton. 8 (3), 238-249 (1987).
- Keller, L. C., Romijn, E. P., Zamora, I., Yates, J. R., Marshall, W. F. Proteomic analysis of isolated chlamydomonas centrioles reveals orthologs of ciliary-disease genes. Current Biology. 15 (12), 1090-1098 (2005).
- Kilburn, C. L., et al. New Tetrahymena basal body protein components identify basal body domain structure. The Journal of Cell Biology. 178 (6), 905-912 (2007).
- Bauer, M., Cubizolles, F., Schmidt, A., Nigg, E. A Quantitative analysis of human centrosome architecture by targeted proteomics and fluorescence imaging. The EMBO Journal. 35 (19), 1-15 (2016).
- Hamel, V., et al. Identification of Chlamydomonas Central Core Centriolar Proteins Reveals a Role for Human WDR90 in Ciliogenesis. Current Biology. 27 (16), 2486-2498 (2017).
- Cavalier-Smith, T. basal body and flagellar development during the vegetative cell cycle and the sexual cycle of Chlamydomonas reinhardii. Journal of Cell Science. 16 (3), 529 (1974).
- Geimer, S., Melkonian, M. The ultrastructure of the Chlamydomonas reinhardtii basal apparatus: identification of an early marker of radial asymmetry inherent in the basal body. Journal of Cell Science. 117, 2663-2674 (2004).
- Nakazawa, Y., Hiraki, M., Kamiya, R., Hirono, M. SAS-6 is a cartwheel protein that establishes the 9-fold symmetry of the centriole. Current Biology. 17 (24), 2169-2174 (2007).
- Hamel, V., et al. Correlative multicolor 3D SIM and STORM microscopy. Biomedical Optics Express. 5 (10), 3326 (2014).
- Guichard, P., et al. Cell-free reconstitution reveals centriole cartwheel assembly mechanisms. Nature Communications. 8, 14813 (2017).
- Hiraki, M., Nakazawa, Y., Kamiya, R., Hirono, M. Bld10p constitutes the cartwheel-spoke tip and stabilizes the 9-fold symmetry of the centriole. Current Biology. 17 (20), 1778-1783 (2007).
- Sahl, S. J., Hell, S. W., Jakobs, S. Fluorescence nanoscopy in cell biology. Nature Reviews Molecular Cell Biology. 18 (11), 685-701 (2017).
- Hutner, S. H., Provasoli, L., Schatz, A., Haskins, C. P. Some Approaches to the Study of the Role of Metals in the Metabolism of Microorganisms. Proceedings of the American Philosophical Society. 94 (2), 152-170 (1950).
- Keller, L. C., Marshall, W. F. Isolation and proteomic analysis of chlamydomonas centrioles. Methods in Molecular Biology. 432 (1), 289-300 (2008).
- Borlinghaus, R. T., Kappel, C. HyVolution-the smart path to confocal super-resolution. Nature Methods. 13 (3), (2016).
- Schneider, C. A., Rasband, W. S., Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 9 (7), 671-675 (2012).
- de la Rosa-Trevín, J. M., et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 195 (1), 93-99 (2016).
- Gartenmann, L., et al. A combined 3D-SIM/SMLM approach allows centriole proteins to be localized with a precision of ∼4-5 nm. Current Biology. 27 (19), 1054-1055 (2017).
- Jaenicke, L., Kuhne, W., Spessert, R., Wahle, U., Waffenschmidt, S. Cell-wall lytic enzymes (autolysins) of Chlamydomonas reinhardtii are (hydroxy)proline-specific proteases. European Journal of Biochemistry. 170 (1-2), 485-491 (1987).
- Salas, D., et al. Angular reconstitution-based 3D reconstructions of nanomolecular structures from superresolution light-microscopy images. Proceedings of the National Academy of Sciences of the United States of America. 114 (35), 9273-9278 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved