
Method Article
原発腫瘍と実験的転移発生を再現するための眼黒色腫のオルソおよび異所性ゼブラフィッシュのゼノ生着
要約
ここでは、原発腫瘍の成長速度、播種、血管外漏出、および血管周囲転移の形成とそれに対する化学的阻害の影響を評価するために、眼の黒色腫に対する汎用性の高い同所性および異所性ゼブラフィッシュ異種移植モデルを確立するためのプロトコルを紹介します。
要約
現在、転移性眼黒色腫の動物モデルはありません。転移性疾患モデルの欠如は、転移性眼黒色腫の治療のための新しい戦略の研究開発を大きく妨げています。このプロトコルでは、眼球後同所性細胞生着と血管内異所性細胞生着をそれぞれ使用して、眼球黒色腫の一次段階と播種性病期の両方に対して胚性ゼブラフィッシュモデルを生成する迅速かつ効率的な方法を説明します。これら2つの異なる生着戦略を組み合わせることで、眼の下での原発性の局所的な腫瘍増殖から尾部の血管周囲転移形成まで進行する、がんの病因を全体として要約することができます。これらのモデルにより、特定の標識、遺伝的または化学的干渉により、移植前にがん細胞を迅速かつ容易に改変することができます。また、移植された宿主を(低分子)阻害剤で治療して、腫瘍の発生を弱めることです。
ここでは、蛍光標識された安定細胞株を使用して、眼の黒色腫(結膜およびブドウ膜黒色腫)の同所性および異所性生着の両方の生成と定量化について説明します。このプロトコルは、患者の生検および患者/ PDX由来の材料(準備中の原稿)に由来する初代細胞の生着にも適用できます。生着後数時間以内に、細胞の遊走と増殖を視覚化し、定量化することができます。どちらの腫瘍病巣も、落射蛍光顕微鏡と共焦点顕微鏡の両方でイメージングに容易に利用できます。これらのモデルを用いることで、実験開始後わずか8日で化学的または遺伝的阻害戦略の活性を確認または否定することができるため、安定細胞株での高効率なスクリーニングが可能になるだけでなく、プレシジョンメディシンのアプローチに対する患者主導のスクリーニングも可能になります。
概要
転移性播種は、眼の黒色腫の主な死因と考えられています。現在、播種性眼黒色腫1,2に対する実行可能な治療法はありません。さらに、転移性疾患を反映する眼の黒色腫に利用できる動物モデルはありません。このギャップを埋めるために、原発腫瘍形成または転移性播種の初期段階を再現する2つの異なるゼブラフィッシュモデルを生成したため、通常は研究が困難なこれらのプロセスの研究を容易に行うことができます3。微小転移モデルにより、ホーミング、コロニー形成、血管外漏出など、転移性広がりの最終段階を分析できます。この段階およびそれ以降の遺伝的または化学的介入は、転移性眼黒色腫の治療に強力な手がかりを提供する可能性があります。
ゼブラフィッシュの幼生を異種移植片および同種移植片のレシピエントとして使用することは、発生の初期段階(または キャスパー 変異体全体のライフサイクル4)、高い繁殖力、 子宮外 受精5など、この種の固有の強みによって支えられています。脊椎動物における高い転写相同性は、ゼブラフィッシュとヒトとの間のコアシグナル伝達メカニズムの保持を確保し、したがって結果の潜在的な翻訳可能性が高い 6が、遺伝的アプローチは時として硬骨魚類のゲノム重複のために損なわれたり複雑になったり する7。近年の発展により、ゼブラフィッシュの異種移植モデルがヒト疾患の前臨床「アバター」8として重要であることが強調されており、単一のゼブラフィ ッシュ実験9から治療戦略の前臨床評価のための多数の個別化がん治療モデルが効果的に生まれています。
動物モデルの欠如と、転移性眼黒色腫の治療選択肢の欠如を考慮すると、当社のモデルは、遺伝子変異 (がん細胞内因性) をスクリーニングしたり、前臨床環境での化学的介入戦略を開発したりするための迅速かつ簡単なトランスレーショナル プラットフォームを提供します。同じモデル内で、ステレオ蛍光顕微鏡の低倍率を使用して、がん細胞の増殖速度、生着率/転移電位、および細胞ホーミングを動物全体で視覚化および測定し、中倍率または高倍率の共焦点顕微鏡分析を使用して同様の測定を行い、細胞内分解能 10で眼の黒色腫進行のさまざまなステップを解剖できます。
ここでは、包括的で詳細なプロトコルについて説明します:高度に最適化されたレンチウイルス形質導入11を使用した蛍光標識癌細胞の生成。その後、受精後2日間(dpf)のゼブラフィッシュ幼生にこれらの細胞を静脈内および眼窩後(RO)生着させて、それぞれ異所性モデルと同所性モデルを生成します。続いてデータの取得と分析を行います。これらの方法は、本明細書に記載のアプリケーションに対して包括的であるが、必要に応じて(注射部位または注射時間を変更することによってのみ)後脳腔、肝臓およびペリビテリン空間内の細胞を生着するように改変することができる12,13。
概念実証として、Pontes et al. 2018の知見を詳しく説明し、ゼブラフィッシュモデル 14の結膜黒色腫細胞株の用量と細胞内因性変異特異的応答を示しました。これらの知見を、転移性および原発性結膜黒色腫モデルの両方におけるBRAF V600E変異特異的阻害剤ベムラフェニブの有効性を示すことにより、詳しく説明しました。
プロトコル
すべての動物実験は、動物実験委員会(Dier Experimenten Commissie, D.E.C.)によるライセンスAVD1060020172410で承認されています。すべての動物は、標準プロトコル(www.ZFIN.org)を使用して地域のガイドラインに従って維持されました。
1. 事前準備
- 試薬
- 卵水を調製する:0.6 mg / L最終濃度海塩。
- 5 mg/mL トリカイン 25x ストックを調製する:トリカイン (3-アミノ安息香酸エチル メタンスルホン酸または MS-222) 粉末 5 g、脱塩水 900 mL、1 M トリス (pH 9) 21 mL を混合します。トリカインは、4°Cで短期間(最大6ヶ月)保存することも、日光から保護する場合は室温で1ヶ月間保存することもできます。
- 卵水に1.5%(w / v)アガロースを調製します:100 mLのDPBSに1.5 g。電子レンジで溶かします。
- 1%(w / v)低融点アガロースを卵水で調製します:100 mLのDPBSに1.5 g。電子レンジで溶かします。
- DPBS中の2%PVP40ストックを調製します:50 mLのDPBSに1 gのPVP40を。ボルテックスして37°Cでインキュベートし、溶解を促進します。室温で保存してください。
- DMSOを使用します。薬物治療の溶剤としてよく使用され、暗所で2〜8°Cで保存する必要があります。
- TrypLEは、細胞へのダメージが少なく、強く付着した細胞の穏やかな分散を可能にする合成トリプシン代替品です。
- 細胞を洗浄するために、Mg2+ およびCa2+ を含まないダルベッコのリン酸緩衝生理食塩水(DPBS)を調製します。Ca2+ の欠如は、カドヘリンを介した細胞間接着を損ないます。
- レンチウイルスプラスミドを調製します: Didier Trono から贈られた psPAX2 (プラスミド #12260) と pMD2.G (プラスミド #12259)、およびトランスファープラスミド (Addgene) をコードする GFP (プラスミド #106172) または tdTomato (プラスミド #106173) のいずれかを調製します。
- LipodD293を使用:高効率HEK293T最適化されたトランスフェクション試薬。
- アガロース皿
注:長期間保存した皿を使用する場合は、注射を開始する前に、必ず少量の卵水を皿に追加してください(これにより、魚が急速に乾燥するのを防ぎます)。- 1.5%(w / v)アガロースコーティングディッシュ(アガロースを卵水に溶解)を準備します。
- すぐに使用するか、4°Cで逆さにして保管してください。
2.針
注意: キャピラリーが使用したフィラメントで校正されていることを確認してください。フィラメントまたはキャピラリーを切り替えるときは、rを決定しますamp 使用するフィラメントのキャピラリーの値(ニードルプーラーのマニュアルを参照)。
- 1つのガラスキャピラリーは、2つのマイクロ注射針を生成します。針を作る前に、ニードルプーラーのフィラメント(2.5mmボックスフィラメント)の構造的完全性を確認してください。
- フィラメントとキャピラリーの両方が、対応するランプ値を取得するようにキャリブレーションされていることを確認してください。フィラメントの構造的完全性が損なわれた場合(つまり、不均一、穴、溶融など)、フィラメントを交換してください。
- 次のプログラムを使用します(Needle #99、Heat = ramp + 15、pull = 95、velocity = 60、time = 90)。針を指定されたペトリ皿(針を貼り付けるための粘土またはテープのいずれかを含む)に針を保管します
3. レンチウイルス粒子の生成
注:時間とリソースの浪費を防ぐために、レンチウイルス形質導入の前に迅速な腫瘍原性チェックを行うことができます。これは、使用される細胞株がゼブラフィッシュモデルにおいて十分に腫瘍形成性であることを確認するために行われ、この目的のために、Liverani et al. 2017, 15に記載されているように、細胞をCMdiI(または類似のトレーサー)で染色することができる。
- トランスフェクションの1日前にHEK 293t細胞をプレート化し、約70%のコンフルエントを達成します(通常、1日前にフルフラスコを同じ容量の培養フラスコに1:3の希釈率で分割することによって行われます)。
- トランスフェクション当日に、必要なパッケージングプラスミドであるpsPAX2およびpMD2.Gウイルスエンベロープを発現するプラスミドを、トランスファープラスミドをコードするGFP(プラスミド#106172)またはtdTomato(プラスミド#106173)のいずれかとともに同時トランスフェクションします。使用されるプラスミドの正確な量は、 表1に明記されています。
注: psPAX2 と pMD2.G はどちらも Didier Trono 氏から贈られました (それぞれ Addgene プラスミド #12260 と #12259)。

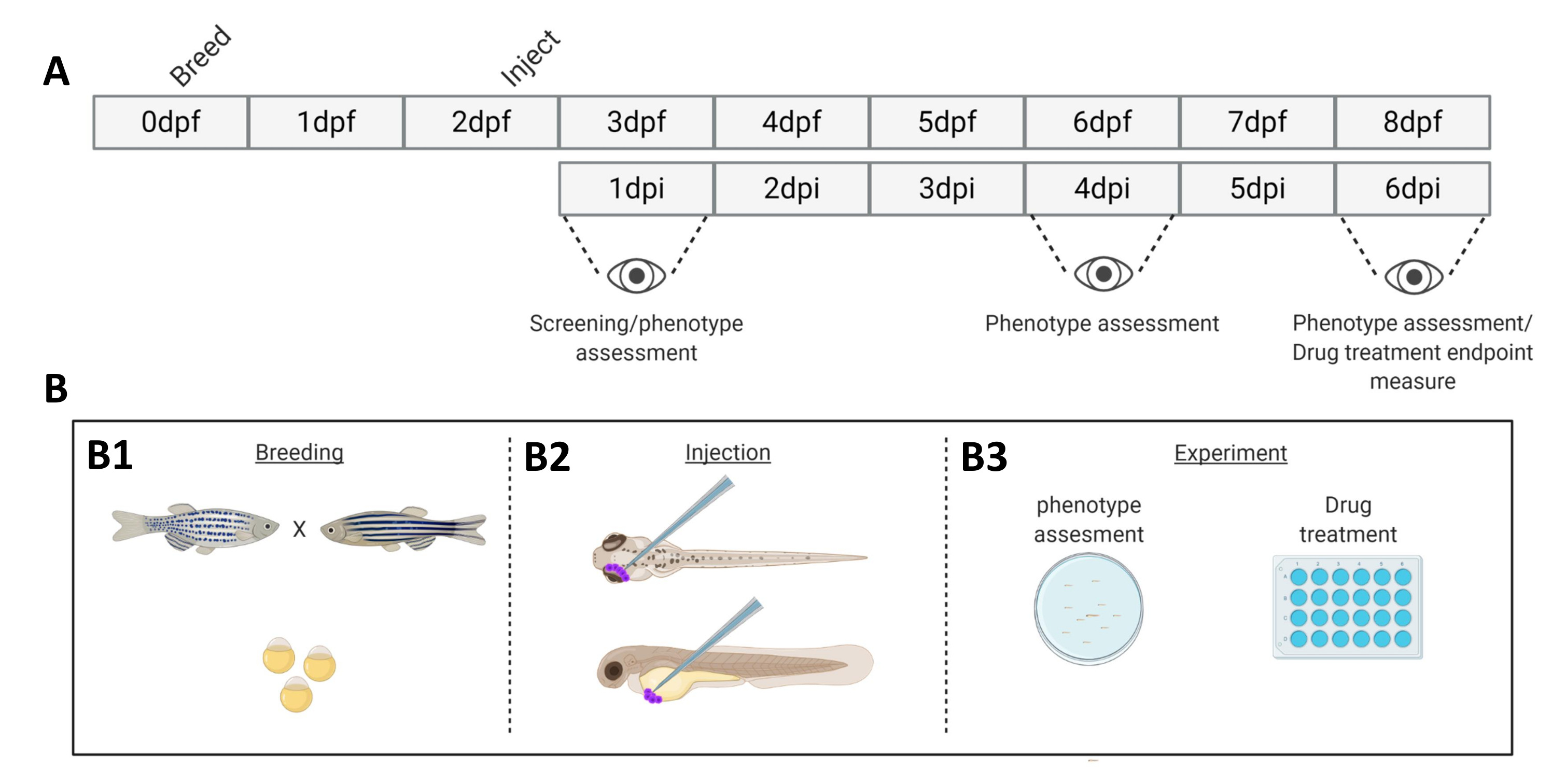
図 1.記載されたゼブラフィッシュ生着システムの概略図。 A) 0日目(B1)にゼブラフィッシュを繁殖させたアプローチのタイムライン。魚は魚を渡った後の朝(1日目)に収穫されます。48〜54時間後、魚は大部分が孵化し(絨毛膜を脱落)、絨毛膜の破片の水をきれいにした後(2日目)、魚は注入されます(眼窩後または全身、 B2)。その後、幼虫は実体蛍光顕微鏡を使用してスクリーニングされ、望ましくない表現型を示すすべての幼虫は廃棄されます(3日目)。実験の目的に応じて、幼生を経時的に画像化するか(B3、生着動態、注射後1日、4日、6日で画像化(dpi))、魚を無作為に割り付けて実験グループに入れ、薬物で治療し、ビヒクルコントロールと比較します(薬物スクリーニング、6dpiで画像化)。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
- すべてのプラスミドを500 μLの無血清培地で混合し、すべてのプラスミドを完全に混合します。32 μLのLipoD293試薬を500 μLの無血清DMEMに加え、ボルテックスして完全に混合します。両方のボリュームをよく混ぜ合わせます。プラスミドとlipoD293を20分間複合体化させます。
- 9 mLの完全培地を含む70%のコンフルエントHEK293T細胞を含む75 cm2 細胞培養フラスコに滴下します。トランスフェクション混合物を血清学的ピペット(フラスコを水平に向ける)を使用して、細胞層に直接加えます。
- トランスフェクションの16時間後に培地を20 mLの新鮮な完全DMEMと交換します。トランスフェクション後72時間後に上清を採取します。ウイルス上清を1 mLアリコートに分注し、-80°Cで保存します。 レンチウイルス上清は、-80°Cで少なくとも1年間安定です。
4. レンチウイルス形質導入
- レンチウイルス形質導入の前に、選択可能なレンチウイルスコンストラクトを使用する際にキルカーブを確立します。
- 死滅曲線については、形質導入する細胞株を12ウェルプレート(コンフルエンス約10〜20%)にプレート化します。選択薬の用量曲線を追加します(殺傷曲線のおおよその濃度:ピューロマイシン0.5-10μg/ mL、ブラストサイジン1-20μg / mL、遺伝性(G418)100-2000μg / mL、ハイグロマイシン100-2000μg / mL)。
- 選択した選択剤の安定した濃度を確保するために、3日ごとに培地を交換してください。
- 20-40%のコンフルエント細胞に最終濃度8 μg/mLのポリブレンを含む9 mLの培養培地に1 mLのレンチウイルス上清を加えます。この上清/媒体の比率を維持しながら、ボリュームを縮小できます。
- 形質導入後16-24時間で培地を交換します。必要に応じて、前のステップを繰り返して表現型の浸透度を高めます(蛍光をチェックして、別の形質導入が必要かどうかを判断します)。
- 形質導入の48時間後、レンチウイルスカセットに組み込まれた耐性マーカーに対応する抗生物質を使用して細胞を選択します。形質導入細胞集団の選択に使用する濃度は、選択剤の適用後7日以内に野生型集団を死滅させるべきである(すなわち、形質導入細胞が野生型集団よりも成長することを可能にする)。
- ウイルス上清をさまざまな感染多重度(MOI)に適用して、細胞ゲノムによって引き起こされる形質導入と遺伝的病変が細胞の生存率や腫瘍形成性に悪影響を及ぼさないようにします。
5.ゼブラフィッシュの繁殖
- がん細胞の生着の2日前の0日目に、成魚のゼブラフィッシュを室温で「ファミリークロス」方式で交配させます(図1)。
- ゼブラフィッシュの水槽をハウジングシステムから取り外します(28.5°Cに維持)。
- 魚を1:1の比率で小さな繁殖クラスターに分けます オス:メス、クラスターごとに10匹の魚。魚を小さな繁殖水槽、ハウジングシステムから汲み上げられた水、傾斜した格子の上の水に入れます(ゼブラフィッシュが自然に産卵する浅瀬を模倣するために傾斜しています)。
注:28.5°Cから室温(25°C)への温度の低下と、ダーク/ライトサイクルの次の明るい段階に入ることによって、魚は産卵します。 - その後、大人を取り外し、ハウジングタンクに移します。
- 卵を集め、ストレーナーを使用して卵水で洗います。卵を皿ごとに約75〜100個に分け、28.5°Cに保ちます。
- 収集後約6時間で、死んだ胚または奇形の胚の皿を掃除します。
- 翌朝、卵の水を交換し、死んだ胚の皿を再びきれいにします。
6.細胞の採取
注:適切な細胞調製は移植手順の鍵であり、余分な量の細胞を使用することで、下流の処理が容易になります。3番目の遠心分離ステップは、細胞ペレットのみが残り、遠心分離チューブの側面に付着した残りのPBSが最終的な再懸濁量を大幅に超えるため、重要です。
- 細胞培養に使用するすべての培地と溶液は、使用前に37°Cのウォーターバスで予温してください。
- 75 cm2 培養フラスコあたり2 mL、または25 cm2 フラスコあたり1 mLのTryplEを添加し、すべての細胞が丸くなるまでインキュベートします。ほとんどの細胞株では、2〜5分で十分です。高上皮細胞または線維芽細胞の場合、5〜10分で適切な剥離が可能になるはずです(トリプシン化が不十分な場合、下流のプロセスが妨げられ、移植中の細胞凝集が促進されます)。
- フラスコの側面を軽くたたいて、残りの細胞を取り除きます。
- 完全な培地の元の培養量に合計します。血清学的ピペットで優しく、しかし徹底的にピペットを上下させ、細胞の凝集物を単一細胞懸濁液にせん断します。このプロセスでは、泡は細胞の機械的せん断を示しているため、泡を発生させないでください。
- 滅菌済みの15 mLチューブに移し、室温で200 x g で5分間遠心分離します。上清を吸引し、滅菌PBS1mLを加えます。滅菌済みの1000μLピペットを使用して、細胞を慎重かつ完全に再懸濁します。
- 計数のために20 μLの細胞懸濁液を取り出し、残りの細胞懸濁液を遠心分離機に移します。室温で200 x gで4分間遠心分離します。
- 重要なステップ:すべてのPBSを取り外し、室温で200 x gで30秒間遠心分離し、残りのPBSを取り除きます。
- 次のように、細胞を2%ポリビニルピロリドン40(PVP40、DPBSで2%(w / v))で250細胞/ nLに希釈します。

(たとえば、
- 気泡の形成を防ぎながら、細胞を完全に再懸濁します(細胞は、腫瘍形成能を失うことなく、2%PVP40 で少なくとも2時間保持できます)。
7. 異種移植のモデリング
すべての実験は、地域の動物福祉規制に準拠して実施する必要があります。
アプリケーションに応じて、実験デザインの2つの主要なバリエーションは、表現型評価(7.1、スクリーニング前の段階)に分類され、次に、7.2、生着前に細胞が改変されたスクリーニング、または胚が化学的阻害剤で処理される7.3に分類されます。
- 腫瘍形成可能性の事前スクリーニングと決定
- 目的のゼブラフィッシュの幼生(WT、トランスジェニックまたはレポーター系統)を2 dpfで移植し、蛍光細胞の数が異なります(つまり、200、400、600±100)。
- 注入の16〜24時間後に幼虫をスクリーニングして、外れ値(異所性モデルの場合は循環中の細胞数が極端に多いまたは低い、または同所性モデルの場合は頭内の細胞)を取り除き、誤って生着した魚を取り除きます。同じ幼虫のグループ分析と速度論的分析の比較について、実験グループごとの幼虫のnrを示します。
- ゼブラフィッシュの幼生を定期的に(注射後1,2,4,6日(dpi))モニターし、±50匹の幼生のプールから20個体(ステップ9および10で説明)を画像化します。
- 一般的な表現型と疾患の進行をモニタリングし、その後ImageJ(がん細胞の蛍光色素シグナルの積分密度を測定)で定量化します。
- データをプロットして、ゼブラフィッシュ内のがん細胞の増殖速度を視覚化します(図3)。
- 細胞を先験的に改変(目的の遺伝子をノックダウンまたはノックアウト)し、ゼブラフィッシュに生着させます。
- 魚を移植し、不要な表現型をすべて取り除きます(条件ごと)。
- 1 dpi(グループあたり20匹の幼虫)で個体を画像化します。個々の個体は、設定された間隔(1、2、4、6 dpi)でイメージングできます。
- イメージング後6dpiで、トリカインを過剰投与して魚を安楽死させ(0.4 mg / mLで10倍過剰投与)、漏斗の内側に吸収紙を敷いて廃棄します。
- 生着後は魚を薬で扱います。
- 生着ゼブラフィッシュに薬剤を投与する前に、ゼブラフィッシュの最大耐量(MTD)を決定し(最大量の溶媒をネガティブコントロールとして使用して、10μM〜0.150nMから滴定します)、MTDを>80%の個体が全治療で生存する濃度として設定します。
- 注射後1日で、不要な表現型を取り除きます。
- 魚をランダムにグループ(36〜48個体/条件)に分け、1mLの卵水に井戸ごとに6匹の幼虫がいる24ウェルプレートに維持します。
- 生着後24時間で薬を塗ってください。対照として、実験グループに最大量で適用される溶媒(DMSO、EtOHなど)を同量使用します。
- 最大耐量で薬物治療を開始します。卵水を含む薬物は一日おきに交換してください。交換のたびに、卵の水と死んだ幼虫をできるだけ完全に取り除きます。
8.インジェクション
注:圧縮空気ラインに結合された空気圧パルスコントローラーを使用し、100psiの余剰圧力を供給します。これにより、注入(≈20 psi)と可能な細胞凝集体(≈100 psi)の両方を排出するのに十分な圧力が可能になります。開始圧力と時間は、20 psi で約 200 ms である必要があります。注射開始時にいずれかを50%以上減らす必要がある場合は、細胞懸 ?? 液が流動的すぎる(細胞または PVP40 濃度が低すぎる)か、針の開口部が大きすぎます。.
- キャピラリーニードルを容器から慎重に取り出します。針を折ってø20μmの開口部を作り、時計職人の細い鉗子を使用します。
- 20 μLのピペットチップを使用して、細胞を慎重かつ完全に再懸濁します。長い(マイクロローダー)チップを使用して、開いたガラスキャピラリーニードルに細胞懸濁液をピペットで固定します。針をマイクロマニピュレーターにセットします。
- 0.04 mg/mL トリカインで麻酔した ~20-40 匹の幼虫を、トランスファーピペットを使用してアガロース皿に置きます。余分な水分を取り除き、トランスファーピペットを使用して幼虫を固定します。幼虫は、まだ比較的大きな卵黄嚢が存在するため、主に横方向に向けられます。
- 異所性モデル用のキュビエ管(doC)を介して、約200、400、600個の細胞を幼虫に注入します。
- 同様に、幼虫を眼窩後(RO)に注入します。同所性モデル(100 ±50セルの注入)を生成するには、ピコポンプの空気圧パルス長を変更します(~20 psi、200 msで開始し、それに応じて調整します)。注射中は、幼虫が乾かないようにしてください。すべて(またはほとんど)の幼虫が注射されていることを確認してください。
- 注入した幼虫を新鮮な卵水で洗い流し、ラベル付きのきれいなペトリ皿に移します(皿ごとに最大150個体をプールします)。十分な幼虫が注入されるまで、このプロセスを繰り返します。
- 生着後、加湿インキュベーターで魚を34°Cに保ちます。34°Cはゼブラフィッシュが容易に許容できる最高温度であり、哺乳類の癌細胞の効率的な生着を可能にします。
注:一般に、doCとROの両方で単一細胞株を注射すると、<5%の機械的損傷によるおおよその死が観察されています(機械的損傷により、注射後1〜16時間で幼虫が死亡します)。
9. スクリーニング
- 実体蛍光顕微鏡を使用して、先験的に改変された細胞を比較する場合は、移植後1時間(または、薬物をスクリーニングする場合は、治療群にランダムに割り当てる前に、移植後1日)に魚の適切な表現型をスクリーニングします。
- DOCを通じて移植された幼虫は、着床後1時間から16時間の間に尾に細胞を持つ必要があります。注入されたプールから、異常を示す魚を含む他のすべての魚を取り除きます。
注:眼窩後部に移植された幼虫は、目の後ろの間質にのみ細胞を持つべきであり、頭や体全体に細胞が広がっている幼虫はプールから取り除かれます。 - 陽性でスクリーニングされた幼虫を洗浄し、実験グループにランダムに割り当てます。
- 生着後、加湿インキュベーターで魚を34°Cに維持し、毎日監視します。DoCを介して移植された細胞の血行性播種はほぼ瞬時に行われますが、RO腔に移植された細胞の転移性広がりは2〜4日後に広がります。
10. ゼブラフィッシュ幼生のエピ蛍光イメージング
- ゼブラフィッシュの幼生に0.2 mg / mLのトリカインを麻酔します。これは、魚の水にトリカインを追加するか、魚の亜集団をメンテナンスディッシュから0.2 mg / mLトリカインを含むディッシュに移すことによって行われます。
- ゼブラフィッシュは、側線の刺激が飛行行動を引き起こさなくなるまで、トリカインの入った皿に入れておきます。
- 魚をアガロースで覆われたペトリ皿に、皿ごとに約10個移します。皿の一方の端をゆっくりと持ち上げながら、水の大部分を取り除きます(水がペトリ皿の下端に穏やかに溜まるようにします)。慎重に行うと、すべての魚が整列し、尾が下を向くようになります。
- 皿の上から下まで、すべての魚をイメージします。次に、魚を卵水で洗い流し、トリカインを含まない皿に入れます。
- 十分な数の個体が画像化されるまで繰り返します。
- 次に、幼虫を34°Cに戻すか、トリカインを過剰投与して(6 dpiで)間引きします(つまり、0.5 mg / mLを10分間インキュベートしてから、漏斗を覆う吸収性紙に廃棄します)。
11. ゼブラフィッシュ幼生(生着)の共焦点イメージング
- 前述のように、ゼブラフィッシュを0.2 mg/mLトリカインで麻酔します。
- ガラス底の共焦点皿を実体顕微鏡の下に置き、皿の底に焦点を合わせます。5〜10匹の幼虫をガラス底の共焦点皿に移します。できるだけ多くの水を取り除きます。
- 卵水に溶かした42°C、1%の低融点アガロースで幼虫を覆います。使用する前に、アガロースが少なくとも42°Cまで冷えていることを確認してください。温度が高くなると、幼虫に害を及ぼしたり殺したりする可能性があります。
- 実体顕微鏡を使用して、トリミングされたマイクロローダーチップを使用して、幼虫を押し下げる方向をすばやく、しかし穏やかに向けます。腹側の向きが必要な場合は、時計職人の鉗子のトングで幼虫を所定の位置に保持します(胚に触れないようにします)。
- アガロースセットは幼虫の向きを微調整します。幼虫が完全に固まるのを待ってから、共焦点顕微鏡に移します。
12. 共焦点顕微鏡の設定
- 緑(488 nm)と赤(564 nm)の励起レーザーラインのスイッチを入れます。共焦点ディッシュを共焦点顕微鏡のホルダーに置きます。落射蛍光を使用して、光束を動かして最初の魚と合体させます( x と yを設定します)。接眼レンズを通して、幼虫の中心と一致するように焦点を設定します( 設定z)。
- 両方の蛍光チャンネルで700ゲイン、1〜5%のレーザー出力を設定します。レーザー出力を上げ、オフセットを小さくして、ほぼフルダイナミックレンジを実現します。信号を過度に飽和させず、信号を強調して、飽和したピクセルを数個だけ表示します。
- ステッチをキャプチャするときは、幼虫の開始と終了を1つの軸( x または y)に沿って設定し、1つの軸に沿って設定すると、胚全体を1 x 4セグメントで画像化でき、ImageJを使用して1つの画像に後処理できます。
- イメージング後、時計職人の鉗子を使用して、埋め込まれた幼虫の周りをそっと引き裂くことにより、幼虫をアガロースから取り出します。それ以外の場合は、希釈されていないトリカインを過剰摂取し、アガロースをトリカインの層で覆い、10分間インキュベートして幼虫を安楽死させます。
13. データ分析
- ImageJ/Fijiの個々のデータセット(つまり、制御、薬物A、薬物B、薬物A + B)を、車両制御から始めて個別に開きます。
- 解析マクロ (注釈付きスクリプトが利用可能) を開きます (http://doi.org/10.5281/zenodo.4290225)。
- 簡単に言うと、マクロ分析は次のことを行います:開いているすべての画像を連結します(1つの条件)。画像を画像を構成する個別のチャネルに分割します。すべてのアクセサリチャネルを閉じます(がん細胞チャネルを離れます)。連結されたシーケンス全体に対してしきい値処理アルゴリズムを実行します。個々の画像の積分密度を測定します。メジャーを Excel シートとしてルート フォルダーに保存します。
- すべての条件でマクロ分析を実行します。
- 測定値を組み合わせ(一般に、少なくともn = 2 * 20)、外れ値を削除します(グラフパッドPrism v8のQ検定)。
- 測定値を溶媒制御または 1 日目に正規化します (実験の種類によって異なり、前者は薬物阻害実験用、後者は成長速度論実験用)。図3と図4にそれぞれ示すように、時間経過または条件(x軸)に対する正規化されたがん細胞負荷(y軸)として測定値を表します。
結果
私たちは、新規細胞株からその分析まで迅速かつ簡単に進めるためのステップバイステップの手順を提供しています。まず、レンチウイルスの過剰発現カセットを使用した蛍光トレーサーの過剰発現から始めます(ステップ3および4)。これに続いて、注入中のデッドボリュームを可能な限り最小限に抑えるための細胞調製が行われ、doCと眼窩後腔の両方に高い細胞数を注入することができます(ステップ6および7)。その後、全身がん細胞播種の定性解析のために、実体蛍光顕微鏡法および高倍率共焦点顕微鏡を用いたセミハイスループットデータ取得を行います(図2 およびステップ10、11、12)。データを取得する際には、ステレオ顕微鏡イメージングと共焦点顕微鏡イメージングの両方の再現性を確保するために、一般的な設定と標準化が図られているため、注意が必要です(ステップ11および12)。データ分析(imageJ/Fijiを使用) 16と、imageJマクロを使用した標準化(ステップ13)について説明します。
ステップ3では、新しいがん細胞株の腫瘍形成可能性を評価するための迅速な事前スクリーニングを実施するための(がん)細胞の一過性標識について説明しました。重要な注意点の1つは、使いやすく長寿命であるにもかかわらず、本明細書に記載されている一過性の染色物が人工物を形成する可能性があることである(すなわち、Fiorら 9によって広く行われたように、細胞断片が全細胞と区別できるように注意しなければならない)。私たちの経験では、これらのアーティファクトの形成は、染色の極端な安定性と明るさ(細胞死後でも)に直接関連しており、細胞断片が分散して免疫細胞に取り込まれ、その後、活動性転移に由来すると誤って結論付けられる可能性があります。
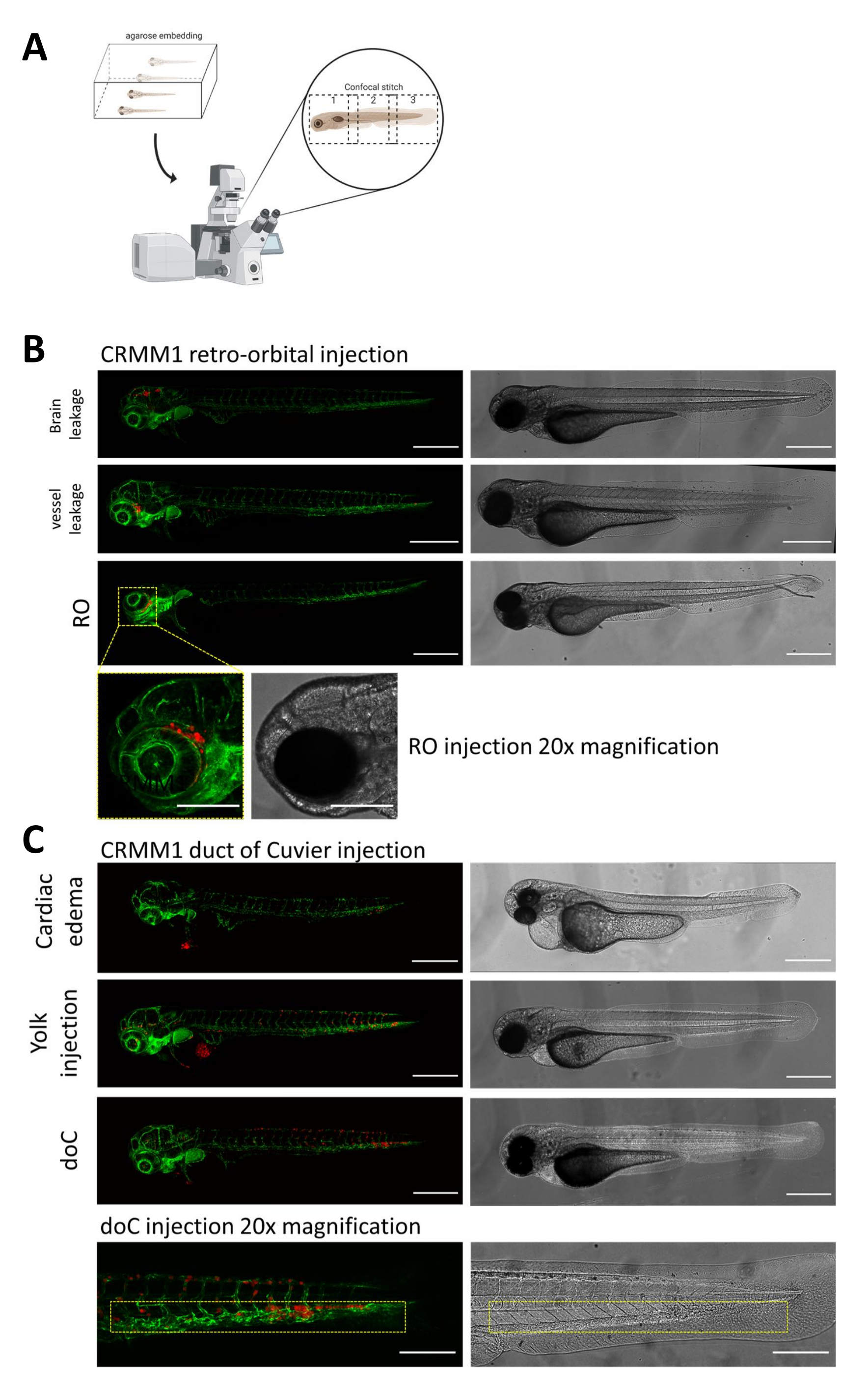
記載された両方のモデルでは、doCを介した全身の生着と後眼窩空間での局所的な生着、注射の1日後に幼虫の徹底的なスクリーニングが最も重要です。 図2B に示すように、生着した細胞が後眼窩モデルで頭部領域(眼窩後部位を越える)に機械的に変位し、卵黄嚢内の細胞を示す、または注入されたdoCプールで浮腫を示すすべての幼虫は除去する必要があります。 図2では、陰性に選択されたすべての表現型が高解像度の共焦点ステッチとして表示されていますが、実体顕微鏡観察で容易に確認および除去できます。
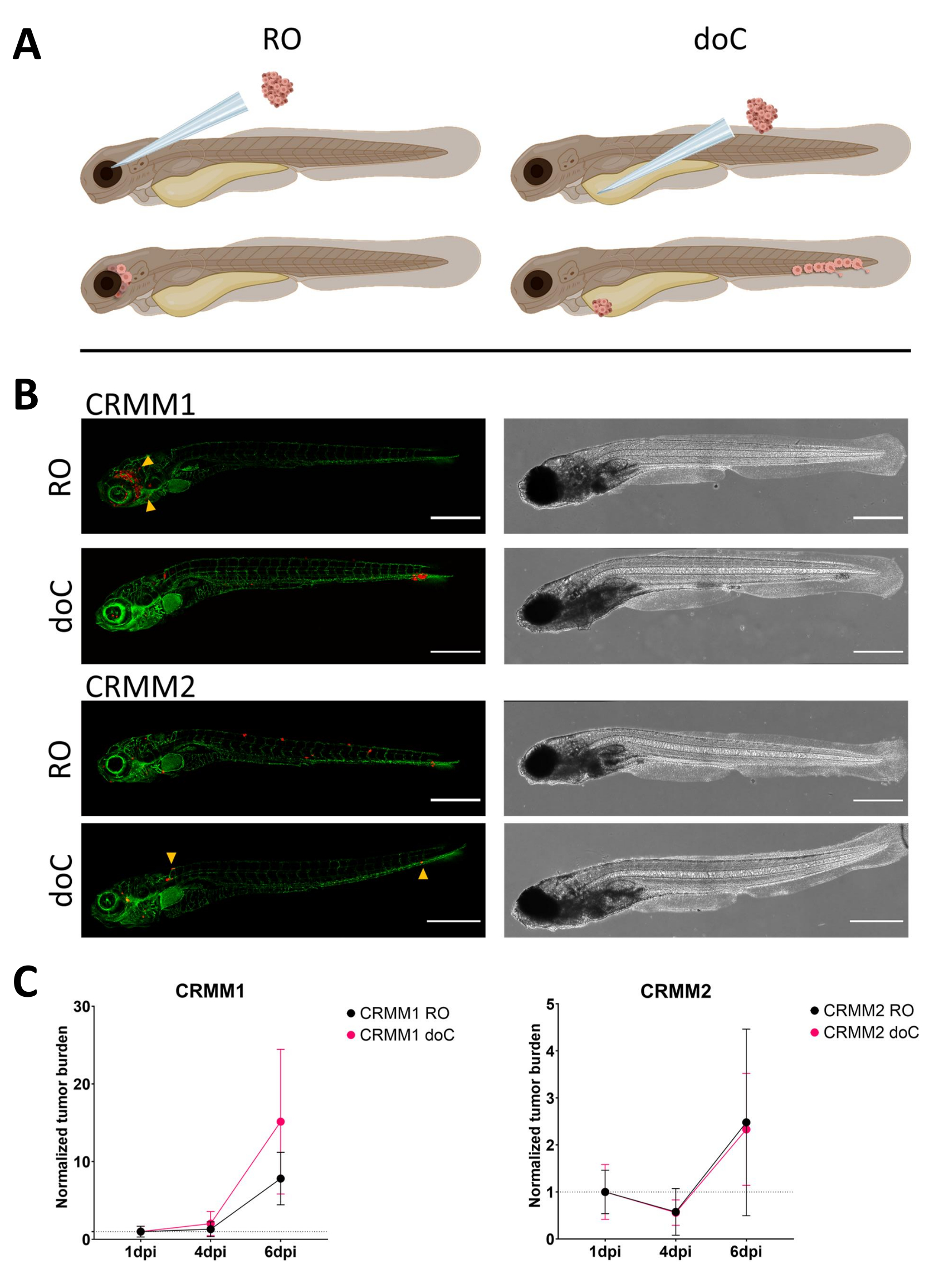
時間の経過とともに、細胞は移動と増殖の両方を行います。眼窩後モデルでは、CRMM1では隣接組織への浸潤が観察されましたが、CRMM2では増殖が少なかったことがわかりました。図 4に示すように、一部の個人(20%)では2〜4dpiの間に発生する遠隔転移が顕著に観察され、6dpiで有意差を測定しました。両方の細胞株について、両方の部位に注入した場合の増殖能をテストしました。CRMM1では、正常化された腫瘍細胞量として表示した場合、注射部位または注射部位でのがん細胞数が有意(p<0.0001)増加し、各モデルで1日目に正規化されました(7.8倍の増加、ROモデルで±3.2倍、15倍の増加±doCモデルで8.8))。CRMM2は、個々のモデルごとに1日目に正規化した場合、有意な増加を示さなかった(2.4倍の増加、±1.9倍および2.3倍の増加、±ROおよびdoCで1.14)。CRMM1は、生着後に眼窩後組織と尾側造血組織の両方で容易に増殖することがわかりました。細胞株CRMM2は、両方のモデルで増殖性が低かったが、興味深いことに、 図3B、Cに示すように、眼窩後腔に注入すると遠隔転移が可能であることがわかった。
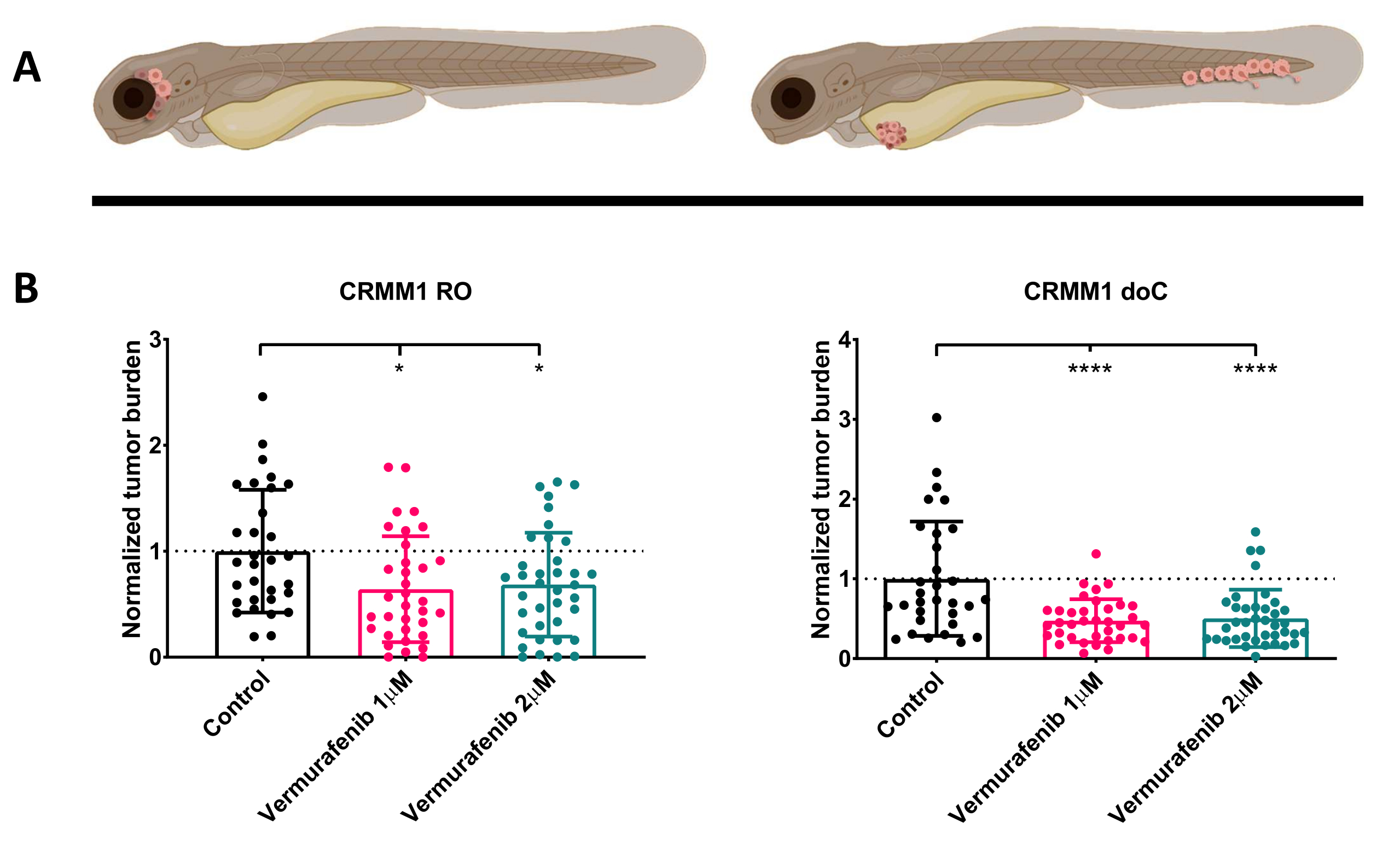
注射した幼虫を1dpiでスクリーニングし、個体を治療群または対照群のいずれかにランダムに割り当てた後、魚を6日間処理し、ベムラフェニブを含む水を交換しました(この阻害剤は、他の滴定された抗腫瘍化合物と容易に交換できます)。以前に発表された血行性結膜黒色腫播種モデル CRMM114 を詳しく説明するために、同所性 CRMM1 に対するベムラフェニブの有効性をテストすることにしました。CRMM1は、 図4に示すように、ベムラフェニブで治療された異所性生着群(P<0.0001)の強力な有意な減少と、同所性生着モデル(p<0.05)に対する発育阻害でありながら有意な反応を示しました。

図 2.注射後の表現型評価とスクリーニング。A)ゼブラフィッシュ異種共焦点ステッチ生成の概略描写、その後の共焦点投影の統合後にシームレスで高解像度の画像を生成します。ここでは、ゼブラフィッシュの異種移植片を1%低融点アガロースに埋め込み、ガラス底の共焦点皿に取り付けます(ステップ11.3で説明)。 B) キュビエ生着の後眼窩および管の可能なすべての結果は、緑色蛍光血管レポーターゼブラフィッシュ (TG:fli:GFP) に注入され、細胞は tdTomato の過剰発現でレンチウイルスで染色されて表示されます。1 dpiでの正しい生着(ROパネル)と望ましくない表現型(脳の漏出と血管の漏出の両方)を示します。後者の2つの集団は、下流の実験結果を混乱させないように削除する必要があります。 C) キュビエ管を通る血行性生着 (doC) の望ましくない表現型は、心臓浮腫性幼虫 (心臓浮腫) と卵黄嚢に細胞が漏れる幼虫 (卵黄注射) を、下流の測定への干渉を防ぐために除去する必要があるアウトラインです。正しく注入された幼虫は、ステップ7.1で説明されているように実験グループに入れられます。(共焦点顕微鏡、スケールバー200μmを使用して1dpiで取得したすべての画像黄色のボックスは、ROとdoCの両方の生着、頭部領域、尾側造血組織の転移部位をそれぞれ示しています)。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図 3.結膜黒色腫細胞株CRMM1およびCRMM2の比較解析では、転移能および増殖能の違いが示されています。A) インジェクションモデル、レトロ眼窩モデル(RO)、血行性生着モデル(doC)の模式図で、魚はTG(fli:GFP)緑色血管レポーターで、tdTomatoを過剰発現する細胞は赤色で示されています。 B)CRMM1およびCRMM2を生着させた魚の代表的な表現型であるCRMM1は、効率的な生着(ROとdoCの両方)とRO生着部位(RO、黄色の矢印)の周囲の組織への小規模な浸潤を示します。CRMM2は、両方の生着モデルで著しく低い生着効率を示しますが、眼窩後方向に注入すると遠隔転移を示します(ROで示されているように、矢印で示されています)。(すべての画像は6 dpi、共焦点顕微鏡、スケールバー200 μmで取得されました。黄色の矢印は、ROとdoCの両方の生着、頭部領域、尾側造血組織の転移部位を示しています)。 C) CRMM1 と CRMM2 の両方の動態生着プロットでは、両方の生着モデルを 1 日目 (1 日目に正規化) と比較すると、CRMM1 の正規化された腫瘍負荷 (1 dpi から 6 dpi の間) に有意 (p<0.0001) の増加があり、CRMM2 には (有意ではない) 上昇傾向があります。CRMM1は、ROとdoCの成長との間に有意差があることを明らかにしており、doCモデルでは腫瘍の拡大率が高いことが示されています(doC生着幼虫では約2倍高い)。グラフには、平均の平均と標準誤差(SEM)が表示されます。すべてのグループは、個々の条件ごとに 1 dpi に正規化されました。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図 4.BRAF V600E阻害剤であるベムラフェニブは、ROおよびdoC結膜黒色腫の生着ゼブラフィッシュの幼生を有意に阻害します。 A) ゼブラフィッシュの表現型、ROおよびdoCモデルの概略図。 B) 結膜黒色腫細胞株 CRMM1 を注射した RO および doC 生着幼虫は、正常化された腫瘍量の有意な減少を示します (それぞれ p<0.05 および P<0.001)。doC生着ゼブラフィッシュモデルは、薬物反応の増強と薬物阻害との用量に依存しない関係を示しており、阻害の飽和の可能性を示しています)。グラフは、平均値(SEM)の平均誤差と標準誤差を示しており、すべてのグループは、個々の細胞株ごとに制御するように正規化されました。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
| 試薬 | 容積 |
| psPAX2の | 1.71 pmol (12.14μg) |
| pMD2.Gの | 0.94 pmol (3.66μg) |
| トランスファープラスミド* | 1.64 pmol (正確な体積を計算) |
テーブル 1.
ディスカッション
ここでは、ゼブラフィッシュ異種移植片における原発性および転移性眼黒色腫をモデル化するための細心のアプローチを定義しました。局所的な同所性注射モデルと全身性の異所性注射モデルの両方を組み合わせることで、以前は動物モデルが利用できなかったがんの発がんの病因を再現しました。初期のゼブラフィッシュの幼生の固有の透明性により、蛍光標識されたがん細胞を動物全体で追跡することができ、潜在的な転移部位の容易な視覚化を確実にする17。さらに、高倍率の共焦点顕微鏡分析により、細胞内分解能で細胞を追跡することができます10。
私たちは、新規細胞株から異種移植片の確立とその分析まで、迅速かつ容易なアプローチで進めるためのステップバイステップの指示を提供してきました。まず、レンチウイルスの過剰発現カセットを使用した蛍光トレーサーの過剰発現(ステップ3および4)から始め、続いて細胞調製を行い、注入中のデッドボリュームを可能な限り最小限に抑えます。これにより、doC空間と眼窩後空間の両方に高い細胞数を注入することができます(ステップ7および8)。次に、全身がん細胞播種の定性分析のために、実体蛍光顕微鏡法と高倍率共焦点顕微鏡を使用したセミハイスループットデータ取得を行います(図2 、ステップ9および10)。データを取得する際には、ステレオ顕微鏡イメージングと共焦点顕微鏡イメージングの両方の再現性を確保するために、一般的な設定と標準化が図られているため、注意が必要です(ステップ11および12)。データ分析(imageJ/Fijiを使用) 16と、ImageJマクロを使用した標準化(ステップ13)について説明します。
ステップ3では、新しいがん細胞株の腫瘍形成可能性を評価するための迅速な事前スクリーニングを実施するための(がん)細胞の一過性標識について言及します。重要な注意点の1つは、使いやすく長寿命であるにもかかわらず、本明細書に記載されている一過性の染色は人工物を形成する可能性があることです(例えば、Fiorら 9によって広範に行われたように、細胞断片を全細胞と区別できるように注意を払う必要があります)。私たちの経験では、これらのアーティファクトの形成は、染色の極端な安定性と明るさ(細胞死後でも)に直接関連しており、細胞断片が分散して免疫細胞に取り込まれ、その後、活動性転移に由来すると誤って結論付けられる可能性があります。
これらのモデルを使用して、生着した細胞を眼窩後間隙内に物理的に閉じ込めることにより、原発腫瘍の発生をシミュレートしました。その後、生着後1日で徹底的なスクリーニングを行うことで、実験の後半で離れた場所で見つかった細胞が活発に転移していることが確認されます(血管内および播種し、最終的には転移性ニッチで血管外に放出されます)。胚の共通枢機卿静脈であるDoCを介した生着は、細胞の大量移植を容易かつ高度に再現性の高いものにし(適切に濃縮すると600個の細胞の余剰)、転移カスケードの一次段階を効果的に回避し(血管内)、転移カスケードの後期段階(接着、血管外漏出、伸長)に焦点を当てることができます。適切に使用すれば強力なツールですが、実験の後半の段階で偽陽性の結論が引き出されないように、生着後の最初の日に両方のモデルを広範囲に監視する必要があります。
以前の出版物に沿って、結膜黒色腫線は、ゼブラフィッシュの血液循環系全体に播種した後、転移性コロニーを容易に形成することを示しました14。本稿では、同所性モデルとして眼窩後注入による生着レパートリーの拡大と、その後のCRMM2細胞株の尾側造血組織への活性転移について報告する。続いて、ゼブラフィッシュの幼生をモデル化した場合、BRAF V600E特異的阻害剤であるベムラフェニブが結膜黒色腫の主要形態にも有効であることを報告します。
前述の方法を使用して、熟練した研究者は、提案されたどちらのモデルでも、1日あたり数百匹(1時間あたり約200匹)を超える生着幼虫を生成することができます。2週間のタイムスケールで、薬物を最大耐量まで滴定し、確立された異種移植モデルでスクリーニングすることができます。形質導入されていない細胞株の使用開始から終了まで、ゼブラフィッシュモデルでの薬剤感受性プロファイルの取得まで、1か月以内に達成できます(注入された細胞株がゼブラフィッシュモデル内で腫瘍形成されている場合)。私たちの手では、実験ごとにわずか20匹の幼虫と2つの生物学的反復が再現性よく強力な薬物阻害をもたらしましたが、2つの個々の実験が競合する(または統計的に有意な成長阻害をもたらさない)場合、3番目の生物学的反復を行うことができます。
これらのモデルは、わずかな調整により、神経膠芽腫(後脳腔注射)、乳がん(doC注射)、骨肉腫(doC)などに対してこれらの移植戦略を迅速に適応させることができました18,19,20,21。これらのモデルは、その後、単剤戦略と併用薬戦略の両方の基礎研究と前臨床スクリーニングの両方に活用できます。最近、これらのモデル13を使用して、薬物のさまざまな投与体制とその光活性化について説明しました。
開示事項
何一つ。
謝辞
この研究は、助成金契約No 667787(UM Cure 2020プロジェクト、www.umcure2020.org)に基づく欧州連合のHorizon 2020研究およびイノベーションプログラムからの資金提供によって支援されました。中国奨学金評議会は、J.Y.への博士号の助成金について親切に認められています。
資料
| Name | Company | Catalog Number | Comments |
| 2.5mm box filament | Science products | FB255B | for pulling micro injection needles using a Sutter P97 or P1000 |
| 3mL transfer pipettes | Merck | Z350796 | for transfer and selection of zebrafish embryos |
| Agarose | Milipore | 2120 | 1.5% (w/v) in eggwater, 1.5 g in 100 mL DPBS, microwave to dissolve, for injecting and stereofluorescence imaging of zebrafish larvae |
| Capillaries: borosilicate glass outer | World precision instruments | BF100-78-10 | Borosilicate glass capillaries used for needle preparation |
| DMSO | Sigma | D8418 | Often used as solvent in drug treatments, should be stored at 2-8°C the dark. |
| DPBS | Thermo Fischer Scientific | 14190144 | Dulbecco’s phosphate buffered saline, without Mg2+ and Ca2+ for washing the cells, lack of Ca2+ impairs cell-cell adhesion through cadherins and prevents cell aggregation during injection |
| Egg water | Instant ocean | SS15-10 | 0.6 mg/L final concentration sea salt in demineralized water |
| GFP encoding lentiviral transfer plasmid | Addgene | Plasmid #106172 | Generated in Snaar lab, available at Addgene |
| Hek293T | ATCC | CRL-3216 | Stable cell line for generating lentiviral particles, contains SV40-T antigen required for the generation of lentiviral particles |
| Leica sp8 confocal | Leica | Leica TCS SP8 | automated stage confocal microscope with 405/488/514/635nm lasers |
| LipodD293 | Signagen | SL100668 | Highly efficient HEK293t optimized transfection reagent |
| Low-melting agarose | Milipore | 2070 | 1% (w/v) in eggwater 1.5 g in 100 mL DPBS, microwave to dissolve, for embedding zebrafish larvae for confocal imaging |
| Micro loader tips | Fischer scientific | 10289651 | flexible microloader tips |
| Micro manipulator | World precision instruments | M3301R | x/y/z manual micro manipulator for microinjection |
| Needle puller: P-97 or P-1000 | Sutter | P-97 | needle puller used for generating standardized micro engraftment needles |
| Nr.5 watchmakers forceps | VWR | HAMMHSC818-11 | fine watchmakers forceps used for breaking back needles |
| Picopump | World precision instruments | SYS-PV820 | pulse controller supplying pressure for microinjection |
| pMD2.G | Addgene | plasmid #12259 | Gifted by Didier Trono, 2nd generation lentiviral virulence plasmid |
| psPAX2 | Addgene | plasmid #12260 | Gifted by Didier Trono, 2nd generation lentiviral packaging plasmid |
| PVP40 | Sigma-Aldrich | PVP40 | Polyvinylpyrrolidone average mol wt 40,000) PVP40 2% (w/v) in DPBS, 1 g PVP40 in 50 mL DPBS. Vortex and incubate at 37°C to facilitate dissolving. Store at room temperature. |
| tdTomato encoding lentiviral transfer plasmid | Addgene | Plasmid #106173 | Generated in Snaar lab, available at Addgene |
| transmitted light stereo microscope | Leica | leica M50 with (MDG33 base) | leica transmitted light microscope with mirror adjustable illumination. |
| Tricaine | Sigma-Aldrich | E10521 | Ethyl 3-aminobenzoate methanesulfonate or MS-222 |
| TryplE | Thermo Fischer Scientific | 12604-01 | Synthetic trypsine replacement, less damaging to the cells and allows for the gentle dispersion of strongly adherent cells. (Thermo- |
| willco dish | WillCo wells | GWST-5040 | 50mm glass bottom dishes, allow for the embedding of up to 20 zebrafish larvae, enabling the imaging of multiple conditions in one dish due to its large optical glass surfac |
参考文献
- Yang, J., Manson, D. K., Marr, B. P., Carvajal, R. D. Treatment of uveal melanoma: where are we now. Therapeutic Advances in Medical Oncology. 10, (2018).
- Wong, J. R., Nanji, A. A., Galor, A., Karp, C. L. Management of conjunctival malignant melanoma: A review and update. Expert Review of Ophthalmology. 9, 185-204 (2014).
- Nguyen, D. X., Bos, P. D., Massagué, J. Metastasis: from dissemination to organ-specific colonization. Nature Reviews Cancer. 9, 274-284 (2009).
- White, R. M., et al. Transparent Adult Zebrafish as a Tool for In Vivo Transplantation Analysis. Cell Stem Cell. 2, 183-189 (2008).
- Zon, L. I., Peterson, R. T. In vivo drug discovery in the zebrafish. Nature Reviews Drug Discovery. 4, 35-44 (2005).
- Howe, K., et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 496, 498-503 (2013).
- Palmblad, M., et al. Parallel deep transcriptome and proteome analysis of zebrafish larvae. BMC Research Notes. 6, 428(2013).
- Yan, C., et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell. 177, 1903-1914 (2019).
- Fior, R., et al. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proceedings of the National Academy of Sciences of the United States of America. 114, 8234-8243 (2017).
- Campbell, P. D., Chao, J. A., Singer, R. H., Marlow, F. L. Dynamic visualization of transcription and RNA subcellular localization in zebrafish. Development. 142, Cambridge. 1368-1374 (2015).
- Campeau, E., et al. A Versatile Viral System for Expression and Depletion of Proteins in Mammalian Cells. PLoS One. 4, 6529(2009).
- vander Helm, D., et al. Mesenchymal stromal cells prevent progression of liver fibrosis in a novel zebrafish embryo model. Scientific Reports. 8, 16005(2018).
- Chen, Q., et al. TLD1433 photosensitizer inhibits conjunctival melanoma cells in zebrafish ectopic and orthotopic tumour models. Cancers. 12, (2020).
- Pontes, K. C. deS., et al. Evaluation of ( fli:GFP ) Casper Zebrafish Embryos as a Model for Human Conjunctival Melanoma. Investigative Opthalmology & Visual Science. 58, 6065(2017).
- Liverani, C., et al. Innovative approaches to establish and characterize primary cultures: an ex vivo 3D system and the zebrafish model. Biology Open. 6, 133-140 (2017).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9, 676-682 (2012).
- White, R. M., et al. Transparent Adult Zebrafish as a Tool for In Vivo Transplantation Analysis. Cell Stem Cell. 2, 183-189 (2008).
- Mercatali, L., et al. Development of a patient-derived xenograft (PDX) of breast cancer bone metastasis in a Zebrafish model. International Journal of Molecular Sciences. 17, (2016).
- Tulotta, C., et al. Imaging cancer angiogenesis and metastasis in a zebrafish embryo model. Advances in Experimental Medicine and Biology. 916, Springer New York LLC. 239-263 (2016).
- Paauwe, M., et al. Endoglin expression on cancer-associated fibroblasts regulates invasion and stimulates colorectal cancer metastasis. Clinical Cancer Research. 24, 6331-6344 (2018).
- Cao, J., et al. Overexpression of EZH2 in conjunctival melanoma offers a new therapeutic target. Journal of Pathology. 245, (2018).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved