
Method Article
Ortho- und ektopische Zebrafisch-Xeno-Transplantation eines okulären Melanoms zur Rekapitulation des Primärtumors und experimentelle Metastasenbildung
In diesem Artikel
Zusammenfassung
In dieser Arbeit stellen wir ein Protokoll zur Etablierung vielseitiger orthotoper und ektopischer Zebrafisch-Xenotransplantatmodelle für okuläres Melanom vor, um die Wachstumskinetik des Primärtumors, der Dissemination, der Extravasation und der entfernten, perivaskulären Metastasenbildung und der Wirkung chemischer Hemmung darauf zu beurteilen.
Zusammenfassung
Für das metastasierende okuläre Melanom gibt es derzeit keine Tiermodelle. Der Mangel an metastasierenden Krankheitsmodellen hat die Erforschung und Entwicklung neuartiger Strategien zur Behandlung des metastasierten okulären Melanoms stark behindert. In diesem Protokoll beschreiben wir einen schnellen und effizienten Weg zur Generierung embryonaler Zebrafischmodelle sowohl für das primäre als auch für das disseminierte Stadium des okulären Melanoms, wobei retroorbitale orthotope bzw. intravaskuläre ektopische Zelltransplantationen verwendet werden. Durch die Kombination dieser beiden unterschiedlichen Transplantationsstrategien können wir die Ätiologie des Krebses in seiner Gesamtheit rekapitulieren, beginnend mit dem primären, lokalisierten Tumorwachstum unter dem Auge bis hin zu einer perivaskulären Metastasenbildung im Schwanz. Diese Modelle ermöglichen es uns, die Krebszellen vor der Implantation schnell und einfach mit spezifischer Markierung, genetischer oder chemischer Interferenz zu modifizieren; und die transplantierten Wirte mit (kleinen molekularen) Inhibitoren zu behandeln, um die Tumorentwicklung zu verlangsamen.
In dieser Arbeit beschreiben wir die Generierung und Quantifizierung sowohl der orthotopen als auch der ektopischen Transplantation von okulären Melanomen (Bindehaut- und Aderhautmelanom) unter Verwendung fluoreszenzmarkierter stabiler Zelllinien. Dieses Protokoll ist auch anwendbar für die Transplantation von Primärzellen, die aus der Patientenbiopsie und von Patienten/PDX abgeleitetem Material stammen (Manuskript in Vorbereitung). Innerhalb weniger Stunden nach der Transplantation können die Zellmigration und -proliferation visualisiert und quantifiziert werden. Beide Tumorherde stehen sowohl mit der Epifluoreszenzmikroskopie als auch mit der konfokalen Mikroskopie für die Bildgebung zur Verfügung. Mit diesen Modellen können wir die Aktivität chemischer oder genetischer Hemmungsstrategien innerhalb von nur 8 Tagen nach Beginn des Experiments bestätigen oder widerlegen, was nicht nur ein hocheffizientes Screening an stabilen Zelllinien ermöglicht, sondern auch ein patientenorientiertes Screening für präzisionsmedizinische Ansätze.
Einleitung
Die metastasierende Ausbreitung gilt als die Haupttodesursache des okulären Melanoms. Derzeit gibt es kein praktikables Behandlungsschema für das disseminierte okuläre Melanom 1,2. Darüber hinaus gibt es keine Tiermodelle für das okuläre Melanom, das die metastasierende Erkrankung widerspiegelt. Um diese Lücke zu schließen, haben wir zwei unterschiedliche Zebrafischmodelle generiert, die entweder die Primärtumorbildung oder die frühen Stadien der metastasierenden Ausbreitung rekapitulieren und so die Untersuchung dieser normalerweise schwer zu untersuchenden Prozesse ermöglichen 3. Die Mikrometastasenmodelle ermöglichen die Analyse der letzten Phasen der Metastasierung, einschließlich Homing, Kolonisation und Extravasation. Genetische oder chemische Eingriffe in diesem Stadium und darüber hinaus könnten möglicherweise eine starke Unterstützung bei der Behandlung des metastasierten okulären Melanoms bieten.
Die Verwendung der Zebrafischlarven als Empfänger von Xeno- und Allotransplantaten wird durch die intrinsischen Stärken dieser Art unterstützt, wie z.B. ihre optische Transparenz in den frühen Entwicklungsstadien (oder ihren gesamten Lebenszyklus für Casper-Mutanten 4), ihre hohe Fruchtbarkeit und Ex-utero-Befruchtung 5. Eine hohe transkriptionelle Homologie bei Wirbeltieren gewährleistet die Beibehaltung der Kernsignalmechanismen zwischen dem Zebrafisch und dem Menschen und damit eine hohe potenzielle Übertragbarkeit der Ergebnisse 6, obwohl genetische Ansätze aufgrund der Teleost-Genomduplikation manchmal beeinträchtigt oder kompliziert sind 7. Jüngste Entwicklungen haben die Bedeutung von Zebrafisch-Xenotransplantatmodellen als präklinische "Avatare" menschlicher Krankheiten unterstrichen8 und effektiv eine Vielzahl personalisierter Krebstherapiemodelle für die präklinische Bewertung von Behandlungsstrategien aus einem einzigen Zebrafischexperiment hervorgebracht 9.
In Anbetracht des Mangels an Tiermodellen und des gleichzeitigen Mangels an Behandlungsmöglichkeiten für metastasiertes okuläres Melanom bieten unsere Modelle eine schnelle und einfache translationale Plattform, um sowohl genetische Veränderungen (intrinsische Krebszellen) zu screenen als auch chemische Interventionsstrategien in einem präklinischen Umfeld zu entwickeln. Innerhalb desselben Modells können wir die Kinetik des Wachstums von Krebszellen, die Transplantationsrate/das Metastasierungspotenzial und das Zellhoming auf Ganztierebene mit geringer Vergrößerung in einem Stereofluoreszenzmikroskop visualisieren und messen und ähnliche Messungen mit konfokaler mikroskopischer Analyse mit mittlerer oder hoher Vergrößerung durchführen, um verschiedene Schritte der Progression des okulären Melanoms mit subzellulärer Auflösung zu analysieren 10.
Hier beschreiben wir umfassende und detaillierte Protokolle für: die Erzeugung fluoreszenzmarkierter Krebszellen mittels hochoptimierter lentiviraler Transduktion11; nachfolgende intravenöse und retroorbitale (RO) Transplantationen dieser Zellen in Zebrafischlarven 2 Tage nach der Befruchtung (DPF), um ektopische bzw. orthotope Modelle zu erzeugen; gefolgt von der Datenerfassung und -analyse. Obwohl diese Verfahren für die hierin beschriebenen Anwendungen umfassend sind, können sie modifiziert werden, um Zellen in der Hinterhirnhöhle, der Leber und dem Perivitellinraum zu transplantieren, wenn dies erforderlich ist (ausschließlich durch Ändern der Injektionsstelle oder des Zeitpunkts der Injektion)12,13.
Als Proof-of-Concept haben wir die Ergebnisse von Pontes et al. 2018 weiterentwickelt, in denen wir eine dosis- und zellintrinsische mutationsspezifische Reaktion von konjunktivalen Melanom-Zelllinien im Zebrafischmodell zeigten 14. Wir haben diese Ergebnisse vertieft, indem wir die Wirksamkeit des BRAF V600E-Mutations-spezifischen Inhibitors Vemurafenib sowohl in metastasierten als auch in primären konjunktivalen Melanommodellen gezeigt haben.
Protokoll
Alle Tierversuche wurden vom Tierversuchsausschuss (Dier Experimenten Commissie, D.E.C.) unter Lizenz AVD1060020172410 genehmigt. Alle Tiere wurden in Übereinstimmung mit den lokalen Richtlinien unter Verwendung von Standardprotokollen (www.ZFIN.org) gehalten.
1. Vorbereitung
- Reagenzien
- Bereiten Sie Eiwasser vor: 0,6 mg/L Endkonzentration Meersalz.
- Bereiten Sie 5 mg/ml Tricain 25x Brühe vor: Mischen Sie 5 g Tricainpulver (Ethyl-3-aminobenzoat-Methansulfonat oder MS-222), 900 ml demineralisiertes Wasser und 21 ml 1 M Tris (pH 9). Auf pH 7 einstellen und bis zu 1 l füllen. Tricain kann kurzfristig (bis zu sechs Monate) bei 4 °C gelagert werden oder einen Monat lang bei Raumtemperatur bei Raumtemperatur gelagert werden, wenn es vor Sonnenlicht geschützt ist.
- Bereiten Sie 1,5 % (w/v) Agarose in Eiwasser zu: 1,5 g in 100 ml DPBS. Mikrowelle zum Auflösen.
- Bereiten Sie 1 % (w/v) niedrigschmelzende Agarose in Eiwasser zu: 1,5 g in 100 ml DPBS. Mikrowelle zum Auflösen.
- Bereiten Sie 2 % (w/v) PVP40-Brühe in DPBS vor: 1 g PVP40 in 50 ml DPBS. Vortexen und bei 37 °C inkubieren, um das Auflösen zu erleichtern. Bei Raumtemperatur lagern.
- Verwenden Sie DMSO. Es wird häufig als Lösungsmittel in medikamentösen Behandlungen verwendet und sollte bei 2-8 °C dunkel gelagert werden.
- Verwenden Sie TrypLE, einen synthetischen Trypsin-Ersatz, der die Zellen weniger schädigt und eine sanfte Dispergierung stark anhaftender Zellen ermöglicht.
- Bereiten Sie die phosphatgepufferte Kochsalzlösung (DPBS) von Dulbecco ohne Mg2+ und Ca2+ zum Waschen der Zellen vor. Der Mangel an Ca2+ beeinträchtigt die Zell-Zell-Adhäsion durch Cadherine.
- Bereiten Sie lentivirale Plasmide vor: psPAX2 (Plasmid #12260) und pMD2.G (Plasmid #12259), geschenkt von Didier Trono und entweder ein GFP (Plasmid #106172) oder tdTomato (Plasmid #106173), das für das Transferplasmid (Addgen) kodiert.
- Verwenden Sie LipodD293: Hocheffizientes HEK293T optimiertes Transfektionsreagenz.
- Agarose-Gericht
HINWEIS: Wenn Sie Geschirr verwenden, das längere Zeit gelagert wurde, achten Sie darauf, dass Sie vor Beginn der Injektion eine kleine Menge Eiwasser in das Geschirr geben (dies verhindert, dass der Fisch zu schnell austrocknet).- Bereiten Sie 1,5 % (w/v) mit Agarose überzogene Gerichte zu (Agarose in Eiwasser aufgelöst).
- Sofort verwenden oder bei 4 °C in umgekehrter Position lagern.
2. Nadeln
HINWEIS: Stellen Sie sicher, dass die Kapillaren auf dem verwendeten Filament kalibriert wurden. Bestimmen Sie beim Wechsel des Filaments oder der Kapillare den Rampenwert der Kapillaren am verwendeten Filament (siehe Handbuch Nadelabzieher).
- Eine Glaskapillare ergibt zwei Mikroinjektionsnadeln. Überprüfen Sie vor der Herstellung von Nadeln die strukturelle Integrität des Filaments (2,5-mm-Box-Filament) des Nadelabziehers.
- Stellen Sie sicher, dass sowohl das Filament als auch die Kapillare kalibriert sind, um den entsprechenden Rampenwert zu erhalten. Wenn die strukturelle Integrität des Filaments beeinträchtigt ist (z. B. uneben, Löcher, geschmolzen usw.), wechseln Sie das Filament.
- Verwenden Sie das folgende Programm (Nadel #99, Hitze = Rampe + 15, Zug = 95, Geschwindigkeit = 60, Zeit = 90). Bewahren Sie die Nadeln in einer dafür vorgesehenen Petrischale auf (die entweder Ton oder Klebeband enthält, um die Nadeln zu kleben)
3. Erzeugung von lentiviralen Partikeln
HINWEIS: Um eine Verschwendung von Zeit und Ressourcen zu vermeiden, kann vor der lentiviralen Transduktion eine schnelle Tumorigenitätskontrolle durchgeführt werden. Dies geschieht, um sicherzustellen, dass die zu verwendende Zelllinie im Zebrafischmodell ausreichend tumorigen ist, zu diesem Zweck können die Zellen mit einem CMdiI (oder analogen Tracer) gefärbt werden, wie in Liverani et al. 2017 15 beschrieben.
- HEK 293t-Zellen einen Tag vor der Transfektion abplatten, um eine Konfluenz von ca. 70 % zu erreichen (routinemäßig durch Aufteilen eines vollen Kolbens in denselben Volumenkulturkolben in einer Verdünnung von 1:3 einen Tag zuvor).
- Am Tag der Transfektion co-transfizieren Sie die erforderlichen Verpackungsplasmide psPAX2 und pMD2.G aus der viralen Hüllhülle, die das Plasmid exprimieren, zusammen mit entweder einem GFP (Plasmid #106172) oder tdTomato (Plasmid #106173), das für das Transferplasmid kodiert. Die genaue Menge des verwendeten Plasmids ist in Tabelle 1 angegeben.
HINWEIS: Sowohl psPAX2 als auch pMD2.G wurden von Didier Trono geschenkt (Addgene Plasmid #12260 bzw. #12259).

Abbildung 1. Schematische Darstellung des beschriebenen Zebrafisch-Transplantationssystems. A) Der zeitliche Ablauf des Ansatzes mit der Zucht des Zebrafisches am Tag 0 (B1). Die Fische werden am Morgen nach der Kreuzung der Fische (Tag 1) geerntet. Nach 48-54 Stunden sind die Fische weitgehend geschlüpft (und haben ihr Chorion abgestoßen) und die Fische werden nach der Reinigung des Wassers von den Chorionresten (Tag 2) injiziert (retroorbital oder systemisch, B2). Anschließend werden die Larven mit einem Stereofluoreszenzmikroskop gescreent und alle Larven mit unerwünschten Phänotypen verworfen (Tag 3). Je nach Ziel des Experiments werden entweder die Larven über die Zeit abgebildet (B3, Transplantationskinetik, 1, 4 und 6 Tage nach der Injektion (dpi)) oder die Fische werden randomisiert und in Versuchsgruppen eingeteilt, mit Medikamenten behandelt und mit der Vehikelkontrolle verglichen (Wirkstoffscreening, abgebildet bei 6 dpi). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
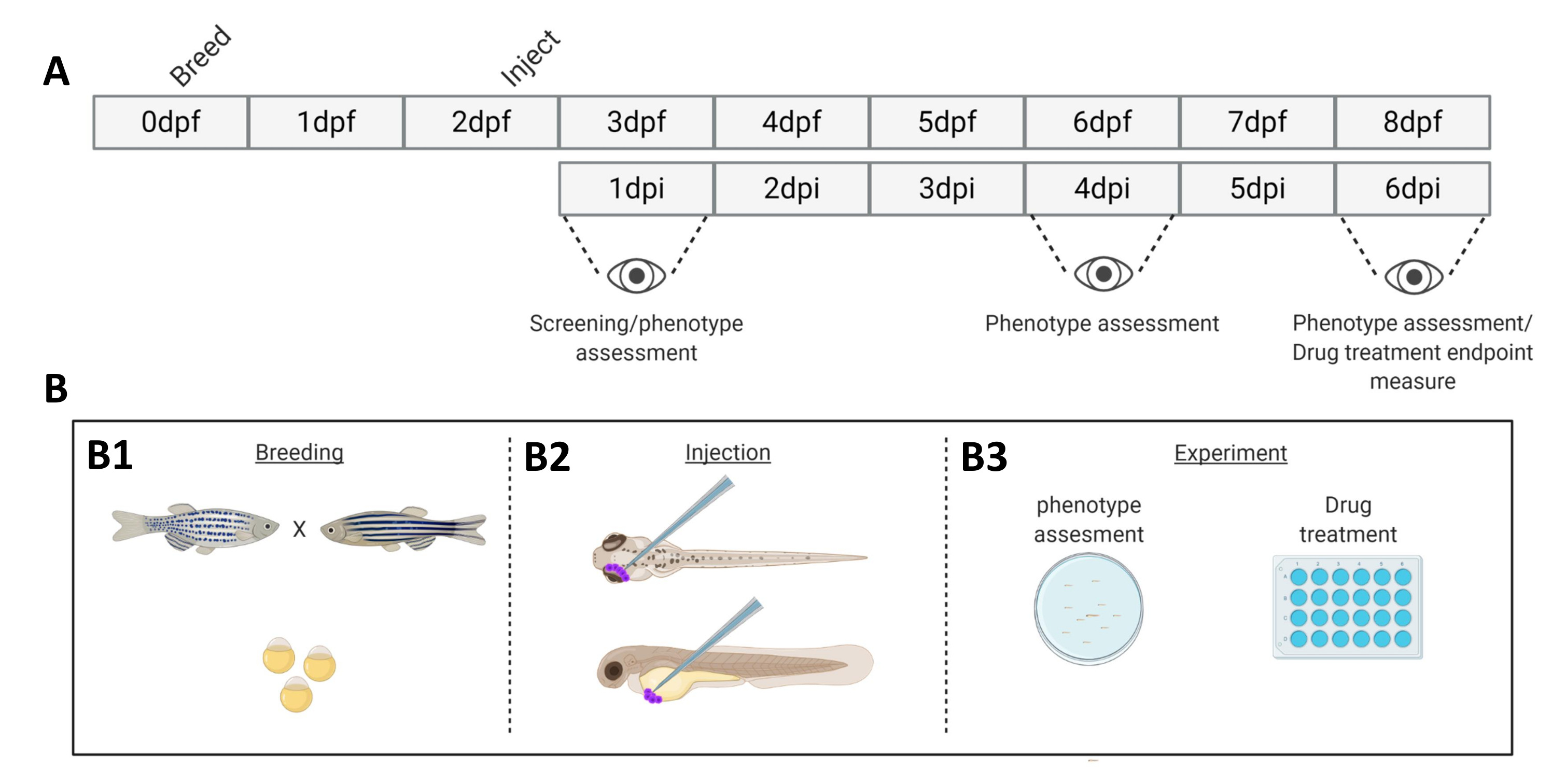{kind=link}
- Mischen Sie alle Plasmide in 500 μl serumfreiem Medium, um ein vollständiges Mischen aller Plasmide zu ermöglichen. Fügen Sie 32 μl LipoD293-Reagenz zu 500 μl serumfreiem DMEM hinzu und vermischen Sie sich vollständig mit einem Vortex. Mischen Sie beide Volumina gründlich miteinander. Lassen Sie die Plasmide und das lipoD293 20 Minuten lang komplexieren.
- Tröpfchenweise in einen 75cm großen 2-Zell-Kulturkolben geben, der 70 % konfluente HEK293T Zellen enthält, die 9 ml vollständiges Kulturmedium enthalten. Geben Sie das Transfektionsgemisch mit einer serologischen Pipette (Kolben in horizontaler Ausrichtung) direkt in die Zellschicht.
- Ersetzen Sie das Medium 16 Stunden nach der Transfektion durch 20 ml frisches, vollständiges DMEM. Ernte des Überstands 72 Stunden nach der Transfektion. Aliquoter Virusüberstand in 1 mL Aliquots und bei -80 °C lagern. Der lentivirale Überstand ist bei -80 °C mindestens 1 Jahr stabil.
4. Lentivirale Transduktion
- Legen Sie vor der lentiviralen Transduktion eine Kill-Kurve fest, wenn Sie ein selektierbares lentivirales Konstrukt verwenden.
- Für die Kill-Kurve wird die zu transduzierende Zelllinie in einer 12-Well-Platte (Konfluenz ca. 10-20%) plattiert. Fügen Sie eine Dosiskurve des Selektorats hinzu (ungefähre Konzentrationen für Tötungskurven: Puromycin 0,5-10 μg/ml, Blasticidin 1-20 μg/ml, Genetikin (G418) 100-2000 μg/ml, Hygromycin 100-2000 μg/ml).
- Wechseln Sie das Medium alle drei Tage, um eine stabile Konzentration des gewählten Selektors zu gewährleisten.
- 1 ml lentiviraler Überstand wird zu 9 ml Kulturmedium mit einer Endkonzentration von 8 μg/ml Polybren auf 20-40 % konfluenten Zellen gegeben. Die Volumina können verkleinert werden, wobei dieses Verhältnis von Überstand/Medium beibehalten wird.
- 16-24 Stunden nach der Transduktion, Tauschen Sie das Medium aus. Wiederholen Sie bei Bedarf den vorherigen Schritt, um die Penetranz des Phänotyps zu erhöhen (überprüfen Sie die Fluoreszenz, um zu entscheiden, ob eine weitere Transduktion erforderlich ist).
- 48 Stunden nach der Transduktion wählen Sie die Zellen mit dem Antibiotikum aus, das dem in die lentivirale Kassette eingebauten Resistenzmarker entspricht. Die Konzentration, die für die Selektion der transduzierten Zellpopulation verwendet werden soll, sollte die Wildtyp-Population innerhalb von 7 Tagen nach Anwendung des Selektors abtöten (d. h. den transduzierten Zellen ermöglichen, aus der Wildtyp-Population herauszuwachsen).
- Wenden Sie den Virusüberstand in verschiedenen Infektionsmultiplizitäten (MOIs) an, um sicherzustellen, dass die Transduktion und die genetischen Läsionen, die durch das zelluläre Genom verursacht werden, die Lebensfähigkeit oder Tumorigenität der Zellen nicht negativ beeinflussen.
5. Zebrafische züchten
- Am Tag 0, 2 Tage vor der Transplantation von Krebszellen, paaren sich adulte Zebrafische in "Family Cross"-Manier bei Raumtemperatur (Abbildung 1).
- Nehmen Sie das Zebrafischbecken aus dem Gehäusesystem (bei 28,5 °C gehalten).
- Teilen Sie die Fische in kleine Zuchtgruppen im Verhältnis 1:1 Männchen: Weibchen, mit 10 Fischen pro Gruppe. Setzen Sie die Fische in kleine Aufzuchtbecken, in Wasser, das aus dem Stallsystem entnommen wird, über einem schrägen Gitter (schräg, um die Untiefen nachzuahmen, in denen Zebrafische von Natur aus laichen).
HINWEIS: Induziert durch den Temperaturabfall von 28,5°C auf Raumtemperatur (25°C) und den Eintritt in die nächste helle Phase des Dunkel-Hell-Zyklus laichen die Fische. - Entfernen Sie anschließend die erwachsenen Tiere und setzen Sie sie in ihren Haltungstank um.
- Die Eier auffangen und mit Eiwasser und einem Sieb waschen. Die Eier auf ca. 75-100 pro Teller verteilen und bei 28,5°C aufbewahren.
- Reinigen Sie ca. 6 Stunden nach der Entnahme das Geschirr von toten oder missgebildeten Embryonen.
- Tauschen Sie am nächsten Morgen das Eiwasser aus und reinigen Sie das Geschirr erneut von toten Embryonen.
6. Ernte von Zellen
HINWEIS: Die richtige Zellvorbereitung ist der Schlüssel zum Implantationsverfahren, die Verwendung einer überflüssigen Menge an Zellen ermöglicht eine einfachere nachgelagerte Verarbeitung. Der dritte Zentrifugationsschritt ist entscheidend, da Sie nur das Küvettenpellet übrig haben, während das verbleibende PBS, das an den Seiten des Zentrifugenröhrchens haftet, das endgültige Resuspensionsvolumen bei weitem übersteigt.
- Alle Medien und Lösungen, die in der Zellkultur verwendet werden, vor der Verwendung in einem 37 °C warmen Wasserbad erwärmen.
- 2 ml TryplE pro 75 cm2 Kulturkolben oder 1 ml pro 25 cm2 Kolben zugeben und inkubieren, bis alle Zellen abgerundet sind. Für die meisten Zelllinien sollten 2-5 Minuten ausreichend sein. Bei hochgradig epithelialen Zellen oder fibroblastischen Zellen sollten 5-10 Minuten eine ordnungsgemäße Ablösung ermöglichen (eine unzureichende Trypsinisierung behindert nachgelagerte Prozesse und erleichtert die Zellaggregation während der Implantation).
- Klopfen Sie vorsichtig auf die Seite des Kolbens, um die verbleibenden Zellen zu entfernen.
- Addieren Sie das ursprüngliche Kulturvolumen des gesamten Mediums. Pipettieren Sie vorsichtig, aber gründlich mit einer serologischen Pipette auf und ab, um die Zellklumpen in Einzelzellsuspension zu scheren. Erzeugen Sie während dieses Prozesses keinen Schaum, da Schaum auf eine mechanische Scherung der Zellen hinweist.
- In ein steriles 15-ml-Röhrchen umfüllen und 5 Minuten bei 200 x g bei Raumtemperatur zentrifugieren. Überstand aspirieren und 1 ml steriles PBS hinzufügen. Resuspendieren Sie die Zellen vorsichtig und gründlich mit einer sterilen 1000-μl-Pipette.
- Entfernen Sie 20 μl Zellsuspension zum Zählen und geben Sie die restliche Zellsuspension in die Zentrifuge. 4 Minuten bei 200 x g bei Raumtemperatur zentrifugieren.
- KRITISCHER SCHRITT: Entfernen Sie alle PBS, zentrifugieren Sie 30 s lang bei 200 x g bei Raumtemperatur und entfernen Sie die restlichen PBS.
- Die Zellen werden wie folgt auf 250 Zellen/nL in 2 % Polyvinylpyrrolidon 40 (PVP40, 2 % w/v) in DPBS) verdünnt:

(z. B.
- Resuspendieren Sie die Zellen gründlich und verhindern Sie gleichzeitig die Bildung von Luftblasen (die Zellen können mindestens 2 Stunden lang in 2% PVP40 gehalten werden, ohne das tumorigene Potenzial zu verlieren).
7. Modellierung von Xenotransplantaten
Alle Versuche sollten in Übereinstimmung mit den örtlichen Tierschutzbestimmungen durchgeführt werden.
Je nach Anwendung werden zwei Hauptvarianten im Versuchsdesign klassifiziert: eine Phänotyp-Bewertung (7.1 die Vor-Screening-Phase) und zweitens 7.2 ein Screening, bei dem entweder die Zellen vor der Transplantation modifiziert wurden, oder 7.3, bei dem die Embryonen mit einem chemischen Inhibitor behandelt werden.
- Vorscreening und Bestimmung des tumorigenen Potenzials
- Transplantieren Sie Zebrafischlarven von Interesse (WT, transgene oder Reporterlinie) bei 2 dpf mit einer unterschiedlichen Anzahl fluoreszierender Zellen (d. h. 200, 400, 600 ±100).
- Untersuchen Sie die Larven 16-24 Stunden nach der Injektion, um Ausreißer (extrem hohe oder niedrige Zellzahlen im Umlauf für das ektope Modell oder Zellen im Kopf für das orthotope Modell) zu entfernen und falsch transplantierte Fische zu entfernen. Geben Sie die Anzahl der Larven pro Versuchsgruppe für die Gruppenanalyse im Vergleich zur kinetischen Analyse derselben Larven an.
- Überwachen Sie die Zebrafischlarven in regelmäßigen Abständen (1,2,4,6 Tage nach der Injektion (dpi)) und bilden Sie 20 Individuen (wie in den Schritten 9 und 10 beschrieben) aus einem Pool von ±50 Larven ab.
- Überwachen Sie den allgemeinen Phänotyp und das Fortschreiten der Erkrankung und quantifizieren Sie anschließend mit ImageJ (Messung der integrierten Dichte des Fluorophorsignals in den Krebszellen).
- Plotten Sie die Daten, um die Wachstumskinetik der Krebszellen im Zebrafisch zu visualisieren (Abbildung 3).
- Modifizieren Sie Zellen a priori (Knock-Down oder Knock-out eines Gens von Interesse) und transplantieren Sie sie in den Zebrafisch.
- Transplantieren Sie Fische und entfernen Sie alle unerwünschten Phänotypen (pro Bedingung).
- Stellen Sie sich die Individuen bei 1 dpi (20 Larven pro Gruppe) vor. Individuen können in festgelegten Intervallen (1,2,4 und 6 dpi) abgebildet werden.
- Bei 6 dpi nach der Bildgebung wird der Fisch durch Überdosierung mit Tricain eingeschläfert (10-fache Überdosierung bei 0,4 mg/ml) und auf saugfähigem Papier, das einen Trichter auskleidet, entsorgt.
- Behandeln Sie Fische nach der Transplantation mit Medikamenten.
- Bestimmen Sie vor der Anwendung des Medikaments auf transplantierten Zebrafischen die maximal verträgliche Dosis (MTD) auf Zebrafischen (Titrat von 10 μM auf 0,150 nM, unter Verwendung des höchsten Lösungsmittelvolumens als Negativkontrolle). Wir haben die MTD als die Konzentration festgelegt, bei der >80 % der Individuen die gesamte Behandlung überleben.
- Entfernen Sie einen Tag nach der Injektion die unerwünschten Phänotypen.
- Teilen Sie die Fische nach dem Zufallsprinzip in Gruppen (36-48 Individuen/Zustand) ein und halten Sie in einer 24-Well-Platte mit 6 Larven pro Well in 1 mL Eiwasser.
- Tragen Sie Medikamente 24 Stunden nach der Transplantation auf. Als Kontrolle ist die gleiche Menge Lösungsmittel (DMSO, EtOH usw.) in der höchsten Menge zu verwenden, die für eine Versuchsgruppe aufgetragen wurde.
- Beginnen Sie die medikamentöse Behandlung mit der maximal verträglichen Dosis. Wechseln Sie das Eiwasser, das das Medikament enthält, jeden zweiten Tag. Entfernen Sie bei jedem Wechsel Eiwasser und abgestorbene Larven so vollständig wie möglich.
8. Injektion
HINWEIS: Verwenden Sie einen pneumatischen Impulsregler, der an eine Druckluftleitung gekoppelt ist und einen Druck von mehr als 100 psi liefert. Dies ermöglicht einen ausreichenden Druck, um sowohl zu injizieren (≈20 psi) als auch mögliche Zellaggregate auszustoßen (≈100 psi). Der Startdruck und die Startzeit sollten ca. 200 ms bei 20 psi betragen. Wenn eine der beiden Injektionen zu Beginn der Injektion um mehr als 50 % verringert werden muss, ist entweder die Zellsuspension zu flüssig (Zell- oder PVP40-Konzentration zu niedrig) oder die Nadelöffnung ist zu groß.
- Entferne vorsichtig eine Kapillarnadel aus ihrem Behälter. Brechen Sie die Nadel mit einer feinen Uhrmacherzange zu einer Öffnung von ø20 μm.
- Resuspendieren Sie die Zellen vorsichtig und gründlich mit einer 20-μl-Pipettenspitze. Pipettieren Sie die Zellsuspension mit einer langen Spitze (Mikrolader) in die offene Glaskapillarnadel. Legen Sie die Nadel in den Mikromanipulator ein.
- Legen Sie ~20-40 Larven, die in 0,04 mg/ml Tricain betäubt sind, mit einer Transferpipette auf eine Agaroseschale. Entfernen Sie überschüssige Feuchtigkeit, um die Larven mit einer Transferpipette zu immobilisieren. Die Larven werden aufgrund des Vorhandenseins eines noch relativ großen Dottersacks meist seitlich ausgerichtet.
- Injizieren Sie den Larven ca. 200, 400 und 600 Zellen über den Ductus of Cuvier (doC) für das ektopische Modell.
- Injizieren Sie auf ähnliche Weise Larven retroorbital (RO). Um das orthotope Modell (Injektion von 100 ±50 Zellen) zu erhalten, ändern Sie die pneumatische Impulslänge an der Pikopumpe (beginnen Sie bei ~20 psi, 200 ms und passen Sie sie entsprechend an). Achten Sie bei der Injektion darauf, dass die Larven nicht austrocknen. Stellen Sie sicher, dass alle (oder die meisten) Larven injiziert werden.
- Spülen Sie die injizierten Larven mit frischem Eiwasser ab und geben Sie sie in eine beschriftete, saubere Petrischale (mit bis zu 150 Individuen pro Schale). Wiederholen Sie diesen Vorgang, bis genügend Larven injiziert sind.
- Halten Sie den Fisch nach der Transplantation bei 34 °C in einem befeuchteten Inkubator, wo 34 °C die höchste Temperatur ist, die von Zebrafischen leicht toleriert wird und eine effiziente Transplantation von Säugetierkrebszellen ermöglicht.
HINWEIS: Im Allgemeinen haben wir bei der Injektion einzelner Zelllinien sowohl in doC als auch in RO einen ungefähren Tod aufgrund mechanischer Schädigung von <5% beobachtet (mechanische Schädigung tötet die Larven zwischen 1 und 16 Stunden nach der Injektion).
9. Vorführung
- Untersuchen Sie die Fische mit einem Stereofluoreszenzmikroskop 1 Stunde nach der Implantation auf den entsprechenden Phänotyp, wenn Sie a priori veränderte Zellen vergleichen (oder 1 Tag nach der Implantation, wenn Sie Medikamente screenen, vor der zufälligen Einteilung in Behandlungsgruppen).
- Larven, die durch den doC implantiert wurden, sollten zwischen 1 Stunde und 16 Stunden nach der Implantation Zellen im Schwanz haben. Entfernen Sie alle anderen Fische, einschließlich Fische, die Anomalien aufweisen, aus dem injizierten Pool.
HINWEIS: Larven, die retroorbital implantiert werden, sollten Zellen nur im Interstitium hinter dem Auge haben, Larven, deren Zellen über den Kopf oder Körper verteilt sind, werden aus dem Pool entfernt. - Positiv gescreente Larven reinigen und nach dem Zufallsprinzip in Versuchsgruppen einteilen.
- Halten Sie die Fische nach der Transplantation bei 34 °C in einem befeuchteten Inkubator und überwachen Sie sie täglich. Die hämatogene Ausbreitung von Zellen, die durch den doC implantiert werden, erfolgt fast augenblicklich, während sich die metastatische Ausbreitung von Zellen, die in die RO-Höhle implantiert werden, nach 2-4 Tagen ausbreitet.
10. Epifluoreszierende Bildgebung von Zebrafischlarven
- Betäuben Sie Zebrafischlarven mit 0,2 mg/ml Tricain, entweder durch Zugabe von Tricain zum Wasser des Fisches oder durch Umsetzen einer Teilpopulation von Fischen aus der Erhaltungsschale in eine Schale mit 0,2 mg/ml Tricain.
- Halten Sie Zebrafische in einer Schale mit Tricain, bis sie stationär bleiben, bis die Stimulation der Seitenlinie kein Flugverhalten mehr induziert.
- Übertragen Sie den Fisch in eine mit Agarose bedeckte Petrischale, ca. 10 pro Schale. Entfernen Sie den größten Teil des Wassers, indem Sie ein Ende der Schale vorsichtig anheben (damit sich das Wasser sanft am unteren Ende der Petrischale sammelt). Wenn es vorsichtig gemacht wird, richten sich alle Fische mit dem Schwanz nach unten aus.
- Stellen Sie sich alle Fische von oben nach unten vor. Dann den Fisch mit Eiwasser in eine Schüssel ohne Tricain abwaschen.
- Wiederholen Sie diesen Vorgang, bis genügend Individuen abgebildet sind.
- Dann werden die Larven entweder wieder auf 34 °C gebracht oder durch Überdosierung mit Tricain (d. h. 0,5 mg/ml, 10 Minuten inkubiert, bevor sie auf saugfähigem Papier, das einen Trichter auskleidet) entsorgt werden.
11. Konfokale Bildgebung von (transplantierten) Zebrafischlarven
- Betäuben Sie den Zebrafisch mit 0,2 mg/ml Tricain, wie zuvor beschrieben.
- Stellen Sie eine konfokale Schale mit Glasboden unter ein Stereomikroskop und fokussieren Sie auf den Boden der Schale. Übertragen Sie 5-10 Larven in eine konfokale Schale mit Glasboden. Entferne so viel Wasser wie möglich.
- Bedecken Sie die Larven mit 42 °C, 1 % niedrigschmelzender Agarose, die in Eiwasser aufgelöst ist. Stellen Sie sicher, dass die Agarose vor der Verwendung auf mindestens 42 °C abgekühlt ist; Höhere Temperaturen können die Larven schädigen oder töten.
- Richten Sie die Larven mit dem Stereomikroskop schnell, aber vorsichtig aus, indem Sie es mit einer abgekürzten Mikroladerspitze nach unten drücken. Wenn eine ventrale Ausrichtung erforderlich ist, halten Sie die Larven mit der Zange einer Uhrmacherzange an Ort und Stelle (ohne den Embryo zu berühren).
- Während die Agarose-Sets feine Anpassungen an die Ausrichtung der Larven vornehmen. Lassen Sie die Larven vollständig aushärten, bevor Sie sie in das Konfokalmikroskop übertragen.
12. Einstellen des konfokalen Mikroskops
- Schalten Sie die grünen (488 nm) und roten (564 nm) Anregungslaserlinien ein. Stellen Sie die Konfokalschale in die Halterung des Konfokalmikroskops. Bewegen Sie das Lichtbündel mithilfe der Epifluoreszenz so, dass es mit dem ersten Fisch verschmilzt (Einstellung x und y). Stellen Sie mit dem Okular den Fokus so ein, dass er mit dem Zentrum der Larven übereinstimmt (Einstellung z).
- Stellen Sie 700 Verstärkung auf beiden Fluoreszenzkanälen ein, 1-5 % Laserleistung. Erhöhen Sie die Laserleistung und verringern Sie den Offset, um sich dem vollen Dynamikbereich anzunähern. Übersättigen Sie das Signal nicht, sondern verstärken Sie das Signal, um nur einige gesättigte Pixel anzuzeigen.
- Wenn Sie einen Stich aufnehmen, legen Sie den Anfang und das Ende der Larven entlang einer Achse (entweder x oder y) fest, wenn Sie sie entlang einer Achse setzen, kann ein ganzer Embryo in 1 x 4 Segmenten abgebildet und mit ImageJ zu einem Bild nachbearbeitet werden.
- Entfernen Sie nach der Bildgebung die Larven aus der Agarose, indem Sie sie mit einer Uhrmacherzange vorsichtig um die eingebetteten Larven herum reißen. Andernfalls euthanasieren Sie die Larven durch Überdosierung mit unverdünntem Tricain, bedecken Sie die Agarose mit einer Schicht Tricain und inkubieren Sie sie 10 Minuten.
13. Datenanalyse
- Öffnen Sie die einzelnen Datensätze in ImageJ/Fiji (d.h. Kontrolle, Medikament A, Medikament B, Medikament A+B) separat, beginnend mit Fahrzeugsteuerung.
- Öffnen Sie das Analysemakro (annotiertes Skript verfügbar) (http://doi.org/10.5281/zenodo.4290225).
- Kurz gesagt, die Makroanalyse macht folgendes: verkettet alle geöffneten Bilder (eine Bedingung); teilt die Bilder in die separaten Kanäle auf, aus denen das Bild besteht; schließt alle akzessorischen Kanäle (verlässt den Krebszellkanal); führt einen Schwellenwertalgorithmus für die gesamte verkettete Sequenz aus; misst die integrierte Dichte jedes einzelnen Bildes; und speichert die Kennzahlen als Excel-Tabelle im Stammordner.
- Führen Sie die Makroanalyse für alle Bedingungen aus.
- Kombinieren Sie Messungen (in der Regel mindestens n=2*20) und entfernen Sie Ausreißer (Q-Test in Graph Pad Prism v8).
- Normalisieren Sie die Messungen entweder auf die Lösungsmittelkontrolle oder auf Tag 1 (abhängig von der Art des Experiments, ersteres für ein Experiment zur Medikamentenhemmung und letzteres für ein Wachstumskinetik-Experiment). Drücken Sie Messungen als normalisierte Krebszelllast (y-Achse) über die Zeit oder den Zustand (x-Achse ) aus, wie in Abbildung 3 bzw. Abbildung 4 gezeigt.
Ergebnisse
Wir haben eine Schritt-für-Schritt-Anleitung für einen schnellen und einfachen Ansatz bereitgestellt, um von einer neuartigen Zelllinie zu ihrer Analyse zu gelangen. Wir beginnen mit der Überexpression eines fluoreszierenden Tracers unter Verwendung einer lentiviralen Überexpressionskassette (Schritte 3 und 4). Darauf folgt die Zellvorbereitung, um ein möglichst geringes Totvolumen während der Injektion zu gewährleisten, was die Injektion hoher Zellzahlen sowohl in den doC- als auch in den retroorbitalen Raum ermöglicht (Schritte 6 und 7). Anschließend führen wir eine Semi-Hochdurchsatz-Datenerfassung mittels Stereofluoreszenzmikroskopie und konfokaler Mikroskopie mit höherer Vergrößerung zur qualitativen Analyse der Ganzkörper-Krebszellverbreitung durch (Abbildung 2 und Schritte 10, 11 und 12). Bei der Datenerfassung ist darauf zu achten, dass zur Gewährleistung der Reproduzierbarkeit sowohl für die stereo- als auch für die konfokalmikroskopische Bildgebung die generischen Einstellungen und die Standardisierung abgegrenzt werden (Schritte 11 und 12). Die Datenanalyse (mit imageJ/Fiji) 16 wird zusammen mit der Standardisierung mit imageJ-Makros (Schritt 13) diskutiert.
In Schritt 3 haben wir die transiente Markierung von (Krebs-)Zellen erwähnt, um ein schnelles Pre-Screening durchzuführen, um das tumorigene Potenzial einer neuen Krebszelllinie zu bewerten. Ein wichtiger Vorbehalt ist, dass der hier beschriebene transiente Farbstoff, obwohl er einfach zu verwenden und langlebig ist, die Möglichkeit hat, Artefakte zu bilden (d.h. es muss darauf geachtet werden, dass Zellfragmente von ganzen Zellen unterschieden werden können, wie dies von Fior und Kollegen ausführlich durchgeführt wurde 9). Unserer Erfahrung nach steht die Bildung dieser Artefakte in direktem Zusammenhang mit der extremen Stabilität der Färbung und der Helligkeit (auch nach dem Zelltod), bei der Zellfragmente dispergiert und von Immunzellen aufgenommen werden, die anschließend fälschlicherweise auf aktive Metastasen zurückgeführt werden könnten.
In beiden beschriebenen Modellen, der systemischen Transplantation durch den doC und der lokalisierten Transplantation im retroorbitalen Raum, ist ein gründliches Screening der Larven einen Tag nach der Injektion von größter Bedeutung. Wie in Abbildung 2B gezeigt, sollten alle Larven entfernt werden, die im retroorbitalen Modell eine mechanische Verschiebung der transplantierten Zellen in den Kopfbereich (über die retroorbitale Stelle hinaus) und Zellen im Dottersack oder ein Ödem im doC-injizierten Pool aufweisen. Alle negativ selektierten Phänotypen sind in Abbildung 2 als hochauflösende konfokale Stiche dargestellt, können aber durch stereomikroskopische Betrachtung leicht gesehen und entfernt werden.
Im Laufe der Zeit wandern die Zellen und vermehren sich. Für das retroorbitale Modell beobachteten wir für CRMM1 eine Infiltration in benachbarte Gewebe, für CRMM2 jedoch eine geringere Proliferation. Auffallend ist, dass bei einigen Individuen (20%) Fernmetastasen zwischen 2 und 4 dpi auftraten, wobei wir einen signifikanten Unterschied bei 6 dpi maßen, wie in Abbildung 4 gezeigt. Für beide Zelllinien haben wir das proliferative Potenzial getestet, wenn es an beiden Stellen injiziert wird. Für CRMM1 gab es einen signifikanten (p<0,0001) Anstieg der Krebszellzahl für oder an den Injektionsstellen, wenn sie als normalisierte Tumorzelllast dargestellt wurde, die sich für jedes Modell bis zum ersten Tag normalisierte (7,8-facher Anstieg, ±3,2 für das RO-Modell und ein 15-facher Anstieg ±8,8 für das doC-Modell). CRMM2 zeigte kein signifikantes Wachstum, wenn es für jedes einzelne Modell auf den ersten Tag normalisiert wurde (2,4-facher Anstieg, ±1,9- und 2,3-facher Anstieg, ±1,14 für RO und doC). Es wurde festgestellt, dass CRMM1 sowohl im retroorbitalen Gewebe als auch im kaudalen hämatopoetischen Gewebe nach der Transplantation leicht proliferiert. Die Zelllinie CRMM2 war in beiden Modellen weniger proliferativ, aber interessanterweise wurde festgestellt, dass sie in der Lage ist, Fernmetastasen zu bilden, wenn sie in den retroorbitalen Raum injiziert wird, wie in Abbildung3B,C gezeigt.
Nach dem Screening der injizierten Larven bei 1 dpi und der zufälligen Zuordnung der Individuen zu Behandlungs- oder Kontrollgruppen wurden die Fische 6 Tage lang behandelt, wobei das Vemurafenib enthaltende Wasser gewechselt wurde (dieser Inhibitor kann leicht gegen jede andere titrierte Antitumorverbindung ausgetauscht werden). Wir haben uns entschieden, das zuvor veröffentlichte hämatogene konjunktivale Melanom-Verbreitungsmodell CRMM114 weiterzuentwickeln, indem wir die Wirksamkeit von Vemurafenib bei orthotopisch transplantiertem CRMM1 getestet haben. CRMM1 zeigte eine starke signifikante Reduktion der mit Vemurafenib behandelten ektopisch transplantierten Gruppe (P<0,0001) und ein verkümmertes, aber signifikantes Ansprechen für das orthotopisch transplantierte Modell (p<0,05), wie in Abbildung 4 gezeigt.

Abbildung 2. Phänotypische Beurteilung und Screening nach der Injektion. A) Schematische Darstellung der konfokalen Stichgenerierung von Zebrafisch-Xenotransplantaten, die nach Integration der anschließenden konfokalen Projektion nahtlose, hochauflösende Bilder liefert. Hier werden Zebrafisch-Xenotransplantate in 1% niedrigschmelzende Agarose eingebettet und auf eine konfokale Glasbodenschale montiert (wie in Schritt 11.3 beschrieben). B) Alle möglichen Ergebnisse der retroorbitalen und des Cuvier-Gang-Transplantats werden in grün fluoreszierenden Blutgefäßreporter-Zebrafischen (TG:fli:GFP) injiziert, wobei die Zellen durch lentivirale Überexpression von tdTomato gefärbt wurden. Wir bezeichnen die korrekte Transplantation bei 1 dpi (RO-Panel) und die unerwünschten Phänotypen (sowohl Hirnleckage als auch Blutgefäßleckage). Die beiden letztgenannten Populationen müssen entfernt werden, um sicherzustellen, dass sie die nachgelagerten experimentellen Ergebnisse nicht verfälschen. C) Die unerwünschten Phänotypen für die hämatogene Transplantation durch den Cuvier-Ductus (doC) sind Umrisse, bei denen kardiale ödematöse Larven (Herzödem) und Larven mit Zellen, die in den Dottersack austreten (Dotterinjektion), entfernt werden müssen, um eine Interferenz mit nachgeschalteten Messungen zu vermeiden. Die korrekt injizierten Larven werden wie in Schritt 7.1 beschrieben in Versuchsgruppen eingeteilt. (Alle Bilder wurden bei 1 dpi mit einem konfokalen Mikroskop aufgenommen, Maßstabsbalken 200 μm. Gelbe Kästchen zeigen Metastasierungsstellen sowohl für RO- als auch für doC-Transplantate, für die Kopfregion bzw. für kaudales hämatopoetisches Gewebe an). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
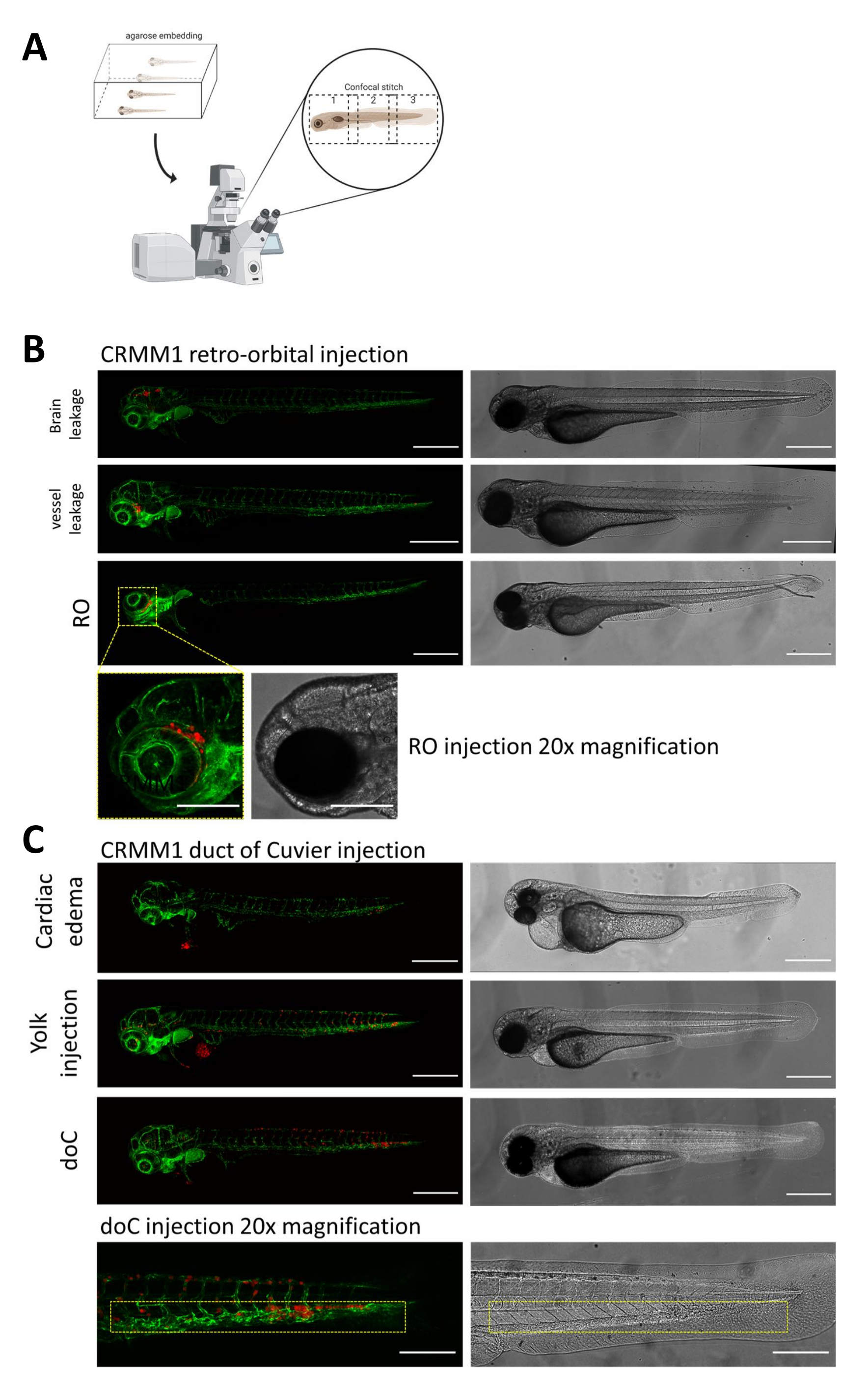{kind=link}

Abbildung 3. Vergleichende Analysen der konjunktivalen Melanomzelllinien CRMM1 und CRMM2 zeigen eine unterschiedliche Metastasierungs- und Wachstumskapazität. A) Schematische Darstellung der Injektionsmodelle, des retroorbitalen Modells (RO) und des hämatogenen Transplantationsmodells (doC): Bei den verwendeten Fischen handelt es sich um TG(fli:GFP) grüne Blutgefäßreporter, wobei die Zellen, die tdTomate überexprimieren, rot dargestellt sind. B) Repräsentative Phänotypen von Fischen, die mit CRMM1 und CRMM2 transplantiert wurden, CRMM1 zeigt eine effiziente Transplantation (sowohl RO als auch doC) und eine kleinskalige Invasion in das Gewebe, das die RO-Transplantationsstelle umgibt (RO, gelbe Pfeilspitzen). CRMM2 weist bei beiden Transplantationsmodellen eine bemerkenswert geringere Transplantationseffizienz auf, zeigt jedoch bei retroorbitaler Injektion Fernmetastasen (wie in RO gezeigt, gekennzeichnet durch die Pfeilspitzen). (Alle Bilder wurden bei 6 dpi mit einem konfokalen Mikroskop aufgenommen, Maßstabsbalken 200 μm. Gelbe Pfeilspitzen zeigen Metastasen sowohl für RO- als auch für doC-Transplantate, für die Kopfregion bzw. für kaudales hämatopoetisches Gewebe an). C) Kinetische Transplantationsdiagramme sowohl für CRMM1 als auch für CRMM2 beim Vergleich beider Transplantationsmodelle mit Tag 1 (Normalisierung bis Tag 1) ergab sich ein signifikanter (p<0,0001) Anstieg der normalisierten Tumorlast für Zelllinien-CRMM1 (zwischen 1 dpi und 6 dpi), wobei es einen (nicht signifikanten) Aufwärtstrend für CRMM2 gibt. CRMM1 zeigt einen signifikanten Unterschied zwischen RO- und doC-Wachstum, wobei das doC-Modell eine höhere Tumorexpansionsrate zeigt (etwa 2-fach höher für die doC-transplantierten Larven). Diagramme zeigen den Mittelwert und den Standardfehler des Mittelwerts (SEM) an. Alle Gruppen wurden für jede einzelne Bedingung auf 1 dpi normiert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
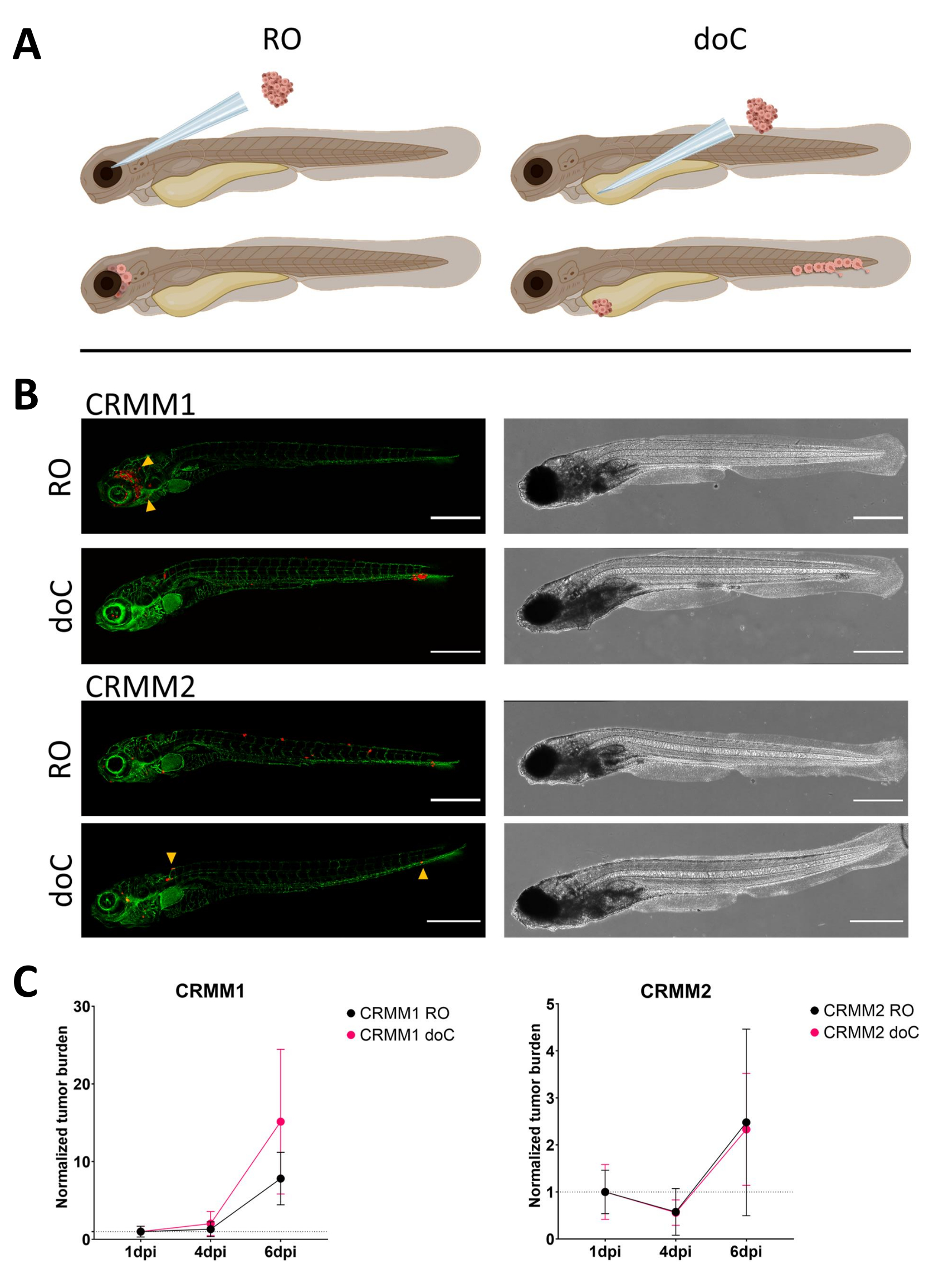{kind=link}

Abbildung 4. Der BRAF-V600E-Inhibitor Vemurafenib hemmt signifikant sowohl RO- als auch doC-Transplantatlarven aus konjunktivalem Melanom. A) Schematische Darstellung von Zebrafisch-Phänotypen, RO- und doC-Modellen. B) Sowohl RO- als auch doC-transplantierte Larven, denen die konjunktivale Melanomzelllinie CRMM1 injiziert wurde, zeigen eine signifikante Verringerung der normalisierten Tumorlast (p<0,05 bzw. P<0,001). Die doC-transplantierten Zebrafischmodelle deuten auf ein erhöhtes Wirkstoffansprechen und eine dosisunabhängige Beziehung zur Arzneimittelhemmung hin, was auf eine mögliche Sättigung der Hemmung hinweist. Die Diagramme zeigen den Mittelwert und den Standardfehler des Mittelwerts (SEM), Alle Gruppen wurden normalisiert, um jede einzelne Zelllinie zu kontrollieren. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
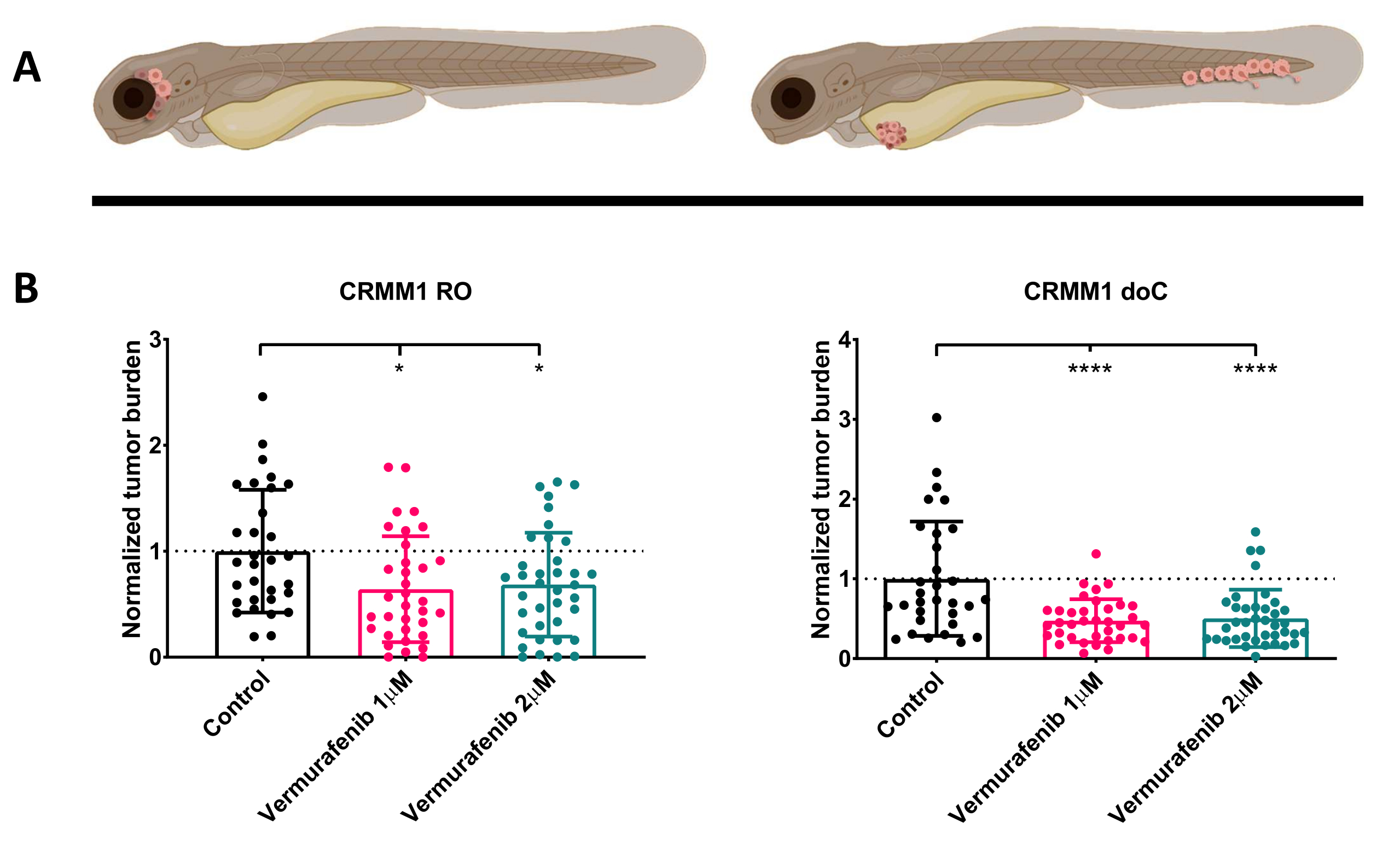{kind=link}
| Reagenz | Volumen |
| psPAX2 | 1,71 pmol (12,14 μg) |
| pMD2.G | 0,94 pmol (3,66 μg) |
| Plasmid übertragen* | 1.64 pmol (exaktes Volumen berechnen) |
Tabelle 1.
Diskussion
In dieser Arbeit haben wir einen sorgfältigen Ansatz zur Modellierung des primären und metastasierten okulären Melanoms in Zebrafisch-Xenotransplantaten definiert. Durch die Kombination eines lokalisierten, orthotopen Injektionsmodells und eines systemischen, ektopischen Injektionsmodells konnten wir die Ätiologie der Karzinogenese für eine Krebserkrankung rekapitulieren, für die bisher keine Tiermodelle zur Verfügung standen. Die inhärente Transparenz der frühen Zebrafischlarve ermöglicht die Verfolgung von fluoreszenzmarkierten Krebszellen auf der Ebene des gesamten Tieres und gewährleistet so eine einfache Visualisierung potenzieller Metastasierungsstellen17. Darüber hinaus ermöglicht uns die konfokale mikroskopische Analyse mit hoher Vergrößerung, Zellen mit einer subzellulären Auflösungzu verfolgen 10.
Wir haben eine Schritt-für-Schritt-Anleitung für einen schnellen und einfachen Ansatz bereitgestellt, um von einer neuen Zelllinie zur Etablierung des Xenotransplantats und seiner Analyse zu gelangen. Wir beginnen mit der Überexpression eines fluoreszierenden Tracers unter Verwendung einer lentiviralen Überexpressionskassette (Schritt 3 und 4), gefolgt von einer Zellvorbereitung, um ein möglichst geringes Totvolumen während der Injektion zu gewährleisten. Dies ermöglicht die Injektion hoher Zellzahlen sowohl in den doC- als auch in den retroorbitalen Raum (Schritt 7 und 8). Dann führen wir eine Semi-Hochdurchsatz-Datenerfassung mit Stereofluoreszenzmikroskopie und konfokaler Mikroskopie mit höherer Vergrößerung durch, um die Ausbreitung von Ganzkörperkrebszellen zu analysieren (Abbildung 2 und Schritt 9 und 10). Bei der Datenerfassung ist darauf zu achten, dass zur Gewährleistung der Reproduzierbarkeit sowohl für die stereo- als auch für die konfokalmikroskopische Bildgebung die generischen Einstellungen und die Standardisierung abgegrenzt werden (Schritte 11 und 12). Die Datenanalyse (mit imageJ/Fiji) 16 wird zusammen mit der Standardisierung mit ImageJ-Makros (Schritt 13) diskutiert.
In Schritt 3 erwähnen wir die transiente Markierung von (Krebs-)Zellen, um ein schnelles Pre-Screening durchzuführen, um das tumorigene Potenzial einer neuen Krebszelllinie zu beurteilen. Ein wichtiger Vorbehalt besteht darin, dass der hier beschriebene transiente Farbstoff, obwohl er einfach zu verwenden und langlebig ist, die Möglichkeit hat, Artefakte zu bilden (z. B. muss darauf geachtet werden, dass Zellfragmente von ganzen Zellen unterschieden werden können, wie dies von Fior und Kollegen ausführlich durchgeführt wurde 9). Unserer Erfahrung nach steht die Bildung dieser Artefakte in direktem Zusammenhang mit der extremen Stabilität der Färbung und der Helligkeit (auch nach dem Zelltod), bei der Zellfragmente dispergiert und von Immunzellen aufgenommen werden, die anschließend fälschlicherweise auf aktive Metastasen zurückgeführt werden könnten.
Mit diesen Modellen simulierten wir die Entwicklung von Primärtumoren, indem wir die transplantierten Zellen physisch innerhalb des retroorbitalen Interstizes einschlossen. Ein anschließendes gründliches Screening 1 Tag nach der Transplantation stellt sicher, dass Zellen, die später im Experiment an einer entfernten Stelle gefunden wurden, aktiv metastasiert haben (intravasiert und disseminiert, um schließlich an der Metastasierungsnische zu extravasieren). Die Transplantation durch die doC, die embryonale Vena cardinal communis, ermöglicht eine einfache und hochgradig reproduzierbare Implantation großer Zellmengen (mit einem Überschuss von 600 Zellen bei richtiger Konzentration), wodurch die primären Stadien der Metastasierungskaskade (Intravasation) effektiv umgangen werden und wir uns auf die späteren Stadien der Metastasierungskaskade (Adhäsion, Extravasation und Auswachstum) konzentrieren können. Obwohl beide Modelle bei richtiger Anwendung leistungsfähige Werkzeuge sind, sollten sie am ersten Tag nach der Transplantation umfassend überwacht werden, um sicherzustellen, dass in den späteren Phasen des Experiments keine falsch positiven Schlussfolgerungen gezogen werden.
In Übereinstimmung mit früheren Veröffentlichungen haben wir gezeigt, dass konjunktivale Melanomlinien nach der Verbreitung im Blutkreislaufsystem von Zebrafischen leicht metastasierende Kolonien bilden14. Hier berichten wir über die Erweiterung des Transplantationsrepertoires mit der retroorbitalen Injektion als orthotopes Modell und die anschließende aktive Metastasierung in das kaudale hämatopoetische Gewebe der Zelllinie CRMM2. Anschließend berichten wir über die Wirksamkeit des BRAF V600E-spezifischen Inhibitors Vemurafenib auch auf die primäre Form des konjunktivalen Melanoms, wenn er in Zebrafischlarven modelliert wurde.
Unter Verwendung der oben genannten Methoden ist ein erfahrener Forscher in der Lage, mehr als Hunderte von transplantierten Larven pro Tag (ungefähr 200 pro Stunde) eines der vorgeschlagenen Modelle zu erzeugen. In einem Zeitraum von zwei Wochen kann ein Medikament sowohl auf die maximal verträgliche Dosis titriert als auch auf ein etabliertes Xenotransplantatmodell gescreent werden. Von Anfang bis Ende kann unter Verwendung einer nicht transduzierten Zelllinie innerhalb eines Monats ein Arzneimittelsensitivitätsprofil im Zebrafischmodell erreicht werden (vorausgesetzt, die injizierte Zelllinie ist im Zebrafischmodell tumorigen). In unseren Händen haben bereits 20 Larven pro Versuch und zwei biologische Wiederholungen reproduzierbar zu einer robusten Wirkstoffhemmung geführt, wenn zwei Einzelversuche in Konflikt stehen (oder keine statistisch signifikante Wachstumshemmung ergeben), kann eine dritte biologische Wiederholung durchgeführt werden.
Durch geringfügige Anpassungen haben diese Modelle es uns ermöglicht, diese Implantationsstrategien unter anderem für Glioblastome (Injektion der Hinterhirnhöhle), Brustkrebs (doC-Injektion) und Osteosarkome (doC) schnell anzupassen 18,19,20,21. Diese Modelle können anschließend sowohl für die Grundlagenforschung als auch für das präklinische Screening von Einzelmedikamenten und kombinatorischen Wirkstoffstrategien verwendet werden. Kürzlich haben wir verschiedene Verabreichungsschemata von Medikamenten und deren Photoaktivierung anhand dieser Modelle beschrieben 13.
Offenlegungen
Nichts.
Danksagungen
Diese Arbeit wurde durch Mittel aus dem Forschungs- und Innovationsprogramm Horizon 2020 der Europäischen Union im Rahmen der Finanzhilfevereinbarung Nr. 667787 (UM Cure 2020 project, www.umcure2020.org) unterstützt. Der Chinese Scholarship Council dankt uns für ein PhD-Stipendium an J.Y.
Materialien
| Name | Company | Catalog Number | Comments |
| 2.5mm box filament | Science products | FB255B | for pulling micro injection needles using a Sutter P97 or P1000 |
| 3mL transfer pipettes | Merck | Z350796 | for transfer and selection of zebrafish embryos |
| Agarose | Milipore | 2120 | 1.5% (w/v) in eggwater, 1.5 g in 100 mL DPBS, microwave to dissolve, for injecting and stereofluorescence imaging of zebrafish larvae |
| Capillaries: borosilicate glass outer | World precision instruments | BF100-78-10 | Borosilicate glass capillaries used for needle preparation |
| DMSO | Sigma | D8418 | Often used as solvent in drug treatments, should be stored at 2-8°C the dark. |
| DPBS | Thermo Fischer Scientific | 14190144 | Dulbecco’s phosphate buffered saline, without Mg2+ and Ca2+ for washing the cells, lack of Ca2+ impairs cell-cell adhesion through cadherins and prevents cell aggregation during injection |
| Egg water | Instant ocean | SS15-10 | 0.6 mg/L final concentration sea salt in demineralized water |
| GFP encoding lentiviral transfer plasmid | Addgene | Plasmid #106172 | Generated in Snaar lab, available at Addgene |
| Hek293T | ATCC | CRL-3216 | Stable cell line for generating lentiviral particles, contains SV40-T antigen required for the generation of lentiviral particles |
| Leica sp8 confocal | Leica | Leica TCS SP8 | automated stage confocal microscope with 405/488/514/635nm lasers |
| LipodD293 | Signagen | SL100668 | Highly efficient HEK293t optimized transfection reagent |
| Low-melting agarose | Milipore | 2070 | 1% (w/v) in eggwater 1.5 g in 100 mL DPBS, microwave to dissolve, for embedding zebrafish larvae for confocal imaging |
| Micro loader tips | Fischer scientific | 10289651 | flexible microloader tips |
| Micro manipulator | World precision instruments | M3301R | x/y/z manual micro manipulator for microinjection |
| Needle puller: P-97 or P-1000 | Sutter | P-97 | needle puller used for generating standardized micro engraftment needles |
| Nr.5 watchmakers forceps | VWR | HAMMHSC818-11 | fine watchmakers forceps used for breaking back needles |
| Picopump | World precision instruments | SYS-PV820 | pulse controller supplying pressure for microinjection |
| pMD2.G | Addgene | plasmid #12259 | Gifted by Didier Trono, 2nd generation lentiviral virulence plasmid |
| psPAX2 | Addgene | plasmid #12260 | Gifted by Didier Trono, 2nd generation lentiviral packaging plasmid |
| PVP40 | Sigma-Aldrich | PVP40 | Polyvinylpyrrolidone average mol wt 40,000) PVP40 2% (w/v) in DPBS, 1 g PVP40 in 50 mL DPBS. Vortex and incubate at 37°C to facilitate dissolving. Store at room temperature. |
| tdTomato encoding lentiviral transfer plasmid | Addgene | Plasmid #106173 | Generated in Snaar lab, available at Addgene |
| transmitted light stereo microscope | Leica | leica M50 with (MDG33 base) | leica transmitted light microscope with mirror adjustable illumination. |
| Tricaine | Sigma-Aldrich | E10521 | Ethyl 3-aminobenzoate methanesulfonate or MS-222 |
| TryplE | Thermo Fischer Scientific | 12604-01 | Synthetic trypsine replacement, less damaging to the cells and allows for the gentle dispersion of strongly adherent cells. (Thermo- |
| willco dish | WillCo wells | GWST-5040 | 50mm glass bottom dishes, allow for the embedding of up to 20 zebrafish larvae, enabling the imaging of multiple conditions in one dish due to its large optical glass surfac |
Referenzen
- Yang, J., Manson, D. K., Marr, B. P., Carvajal, R. D. Treatment of uveal melanoma: where are we now. Therapeutic Advances in Medical Oncology. 10, (2018).
- Wong, J. R., Nanji, A. A., Galor, A., Karp, C. L. Management of conjunctival malignant melanoma: A review and update. Expert Review of Ophthalmology. 9, 185-204 (2014).
- Nguyen, D. X., Bos, P. D., Massagué, J. Metastasis: from dissemination to organ-specific colonization. Nature Reviews Cancer. 9, 274-284 (2009).
- White, R. M., et al. Transparent Adult Zebrafish as a Tool for In Vivo Transplantation Analysis. Cell Stem Cell. 2, 183-189 (2008).
- Zon, L. I., Peterson, R. T. In vivo drug discovery in the zebrafish. Nature Reviews Drug Discovery. 4, 35-44 (2005).
- Howe, K., et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 496, 498-503 (2013).
- Palmblad, M., et al. Parallel deep transcriptome and proteome analysis of zebrafish larvae. BMC Research Notes. 6, 428 (2013).
- Yan, C., et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell. 177, 1903-1914 (2019).
- Fior, R., et al. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proceedings of the National Academy of Sciences of the United States of America. 114, 8234-8243 (2017).
- Campbell, P. D., Chao, J. A., Singer, R. H., Marlow, F. L. Dynamic visualization of transcription and RNA subcellular localization in zebrafish. Development. 142, 1368-1374 (2015).
- Campeau, E., et al. A Versatile Viral System for Expression and Depletion of Proteins in Mammalian Cells. PLoS One. 4, 6529 (2009).
- vander Helm, D., et al. Mesenchymal stromal cells prevent progression of liver fibrosis in a novel zebrafish embryo model. Scientific Reports. 8, 16005 (2018).
- Chen, Q., et al. TLD1433 photosensitizer inhibits conjunctival melanoma cells in zebrafish ectopic and orthotopic tumour models. Cancers. 12, (2020).
- Pontes, K. C. d. e. S., et al. Evaluation of ( fli:GFP ) Casper Zebrafish Embryos as a Model for Human Conjunctival Melanoma. Investigative Opthalmology & Visual Science. 58, 6065 (2017).
- Liverani, C., et al. Innovative approaches to establish and characterize primary cultures: an ex vivo 3D system and the zebrafish model. Biology Open. 6, 133-140 (2017).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9, 676-682 (2012).
- White, R. M., et al. Transparent Adult Zebrafish as a Tool for In Vivo Transplantation Analysis. Cell Stem Cell. 2, 183-189 (2008).
- Mercatali, L., et al. Development of a patient-derived xenograft (PDX) of breast cancer bone metastasis in a Zebrafish model. International Journal of Molecular Sciences. 17, (2016).
- Tulotta, C., et al. Imaging cancer angiogenesis and metastasis in a zebrafish embryo model. Advances in Experimental Medicine and Biology. 916, 239-263 (2016).
- Paauwe, M., et al. Endoglin expression on cancer-associated fibroblasts regulates invasion and stimulates colorectal cancer metastasis. Clinical Cancer Research. 24, 6331-6344 (2018).
- Cao, J., et al. Overexpression of EZH2 in conjunctival melanoma offers a new therapeutic target. Journal of Pathology. 245, (2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten