
Method Article
Xeno-attecchimento orto- ed ectopico di zebrafish del melanoma oculare per ricapitolare il tumore primario e lo sviluppo di metastasi sperimentali
In questo articolo
Riepilogo
Qui, presentiamo un protocollo per stabilire modelli versatili di xenotrapianto ortotopico ed ectopico di zebrafish per il melanoma oculare per valutare la cinetica di crescita del tumore primario, la disseminazione, lo stravaso e la formazione di metastasi perivascolari a distanza e l'effetto dell'inibizione chimica su di esso.
Abstract
Attualmente non esistono modelli animali per il melanoma oculare metastatico. La mancanza di modelli di malattia metastatica ha notevolmente ostacolato la ricerca e lo sviluppo di nuove strategie per il trattamento del melanoma oculare metastatico. In questo protocollo delineiamo un modo rapido ed efficiente per generare modelli embrionali di zebrafish sia per lo stadio primario che disseminato del melanoma oculare, utilizzando rispettivamente l'attecchimento di cellule ectopiche retro-orbitali e intravascolari. Combinando queste due diverse strategie di attecchimento possiamo ricapitolare l'eziologia del cancro nella sua totalità, progredendo dalla crescita primaria e localizzata del tumore sotto l'occhio a una formazione di metastasi peri-vascolari nella coda. Questi modelli ci permettono di modificare rapidamente e facilmente le cellule tumorali prima dell'impianto con specifiche marcature, interferenze genetiche o chimiche; e trattare gli ospiti impiantati con inibitori (piccoli molecolari) per attenuare lo sviluppo del tumore.
Qui, descriviamo la generazione e la quantificazione dell'attecchimento ortotopico ed ectopico dei melanomi oculari (melanoma congiuntivale e uveale) utilizzando linee cellulari stabili marcate in fluorescenza. Questo protocollo è applicabile anche per l'attecchimento di cellule primarie derivate da biopsia del paziente e materiale derivato da paziente/PDX (manoscritto in preparazione). Entro poche ore dall'attecchimento, la migrazione e la proliferazione cellulare possono essere visualizzate e quantificate. Entrambi i focolai tumorali sono prontamente disponibili per l'imaging sia con la microscopia a epifluorescenza che con la microscopia confocale. Utilizzando questi modelli, possiamo confermare o confutare l'attività di strategie di inibizione chimica o genetica entro soli 8 giorni dall'inizio dell'esperimento, consentendo non solo uno screening altamente efficiente su linee cellulari stabili, ma anche uno screening diretto al paziente per approcci di medicina di precisione.
Introduzione
La disseminazione metastatica è considerata la principale causa di morte del melanoma oculare; Attualmente non esiste un regime di trattamento praticabile per il melanoma oculare disseminato 1,2. Inoltre, non sono disponibili modelli animali per il melanoma oculare che riflette la malattia metastatica. Per colmare questa lacuna, abbiamo generato due distinti modelli di zebrafish che ricapitolano la formazione del tumore primario o le prime fasi della disseminazione metastatica, consentendo così prontamente lo studio di questi processi normalmente difficili da studiare 3. I modelli di micro-metastasi consentono l'analisi delle ultime fasi di diffusione metastatica, tra cui homing, colonizzazione e stravaso. Gli interventi genetici o chimici in questa fase e oltre potrebbero potenzialmente fornire un potente appiglio nel trattamento del melanoma oculare metastatico.
L'uso delle larve di zebrafish come recipiente di xeno e allotrapianti è supportato dai punti di forza intrinseci di questa specie, come la sua trasparenza ottica nelle prime fasi di sviluppo (o il suo intero ciclo di vita per i mutanti casper 4), l'elevata fecondità e la fecondazione ex utero 5. L'elevata omologia trascrizionale nei vertebrati garantisce il mantenimento dei meccanismi di segnalazione principali tra il pesce zebra e l'uomo e quindi un'elevata traducibilità potenziale dei risultati 6, sebbene gli approcci genetici siano talvolta rovinati o complicati a causa della duplicazione del genoma del teleosteo 7. Recenti sviluppi hanno sottolineato l'importanza dei modelli di xenotrapianto di zebrafish come "avatar" preclinici della malattia umana8, producendo efficacemente una moltitudine di modelli di terapia del cancro personalizzati per la valutazione preclinica delle strategie di trattamento da un singolo esperimento di zebrafish 9.
Considerando la mancanza di modelli animali e la concorde mancanza di opzioni di trattamento per il melanoma oculare metastatico, i nostri modelli forniscono una piattaforma traslazionale rapida e semplice per lo screening di alterazioni genetiche (cellule tumorali intrinseche) o per lo sviluppo di strategie di intervento chimico in un contesto preclinico. All'interno dello stesso modello possiamo visualizzare e misurare la cinetica di crescita delle cellule tumorali, il tasso di attecchimento/potenziale metastatico e l'homing cellulare su un intero livello di animale utilizzando un ingrandimento a basso livello in un microscopio a stereofluorescenza e fare misurazioni simili utilizzando l'analisi microscopica confocale a medio o alto ingrandimento per sezionare diverse fasi della progressione del melanoma oculare alla risoluzione subcellulare 10.
Qui, descriviamo protocolli completi e dettagliati per: la generazione di cellule tumorali marcate in fluorescenza utilizzando la trasduzione lentivirale altamente ottimizzata11; successivi attecchimenti endovenosi e retro-orbitali (RO) di queste cellule in larve di zebrafish 2 giorni dopo la fecondazione (DPF) per generare rispettivamente modelli ectopici e ortotopici; seguito dall'acquisizione e dall'analisi dei dati. Questi metodi, sebbene completi per le applicazioni qui descritte, possono essere modificati per innestare cellule nella cavità cerebrale posteriore, nel fegato e nello spazio delle perivitelline quando necessario (cambiando esclusivamente il sito di iniezione o il tempo di iniezione)12,13.
Come prova di concetto abbiamo elaborato i risultati di Pontes et al. 2018, dove abbiamo mostrato una risposta specifica alla dose e alla mutazione intrinseca cellulare delle linee cellulari di melanoma congiuntivale nel modello 14 di zebrafish. Abbiamo elaborato questi risultati mostrando l'efficacia dell'inibitore specifico della mutazione BRAF V600E vemurafenib in modelli di melanoma congiuntivale sia metastatico che primario.
Protocollo
Tutti gli esperimenti sugli animali sono stati approvati dal Comitato per gli esperimenti sugli animali (Dier Experimenten Commissie, D.E.C.) su licenza AVD1060020172410. Tutti gli animali sono stati mantenuti in conformità con le linee guida locali utilizzando protocolli standard (www.ZFIN.org).
1. Preparazione
- Reagenti
- Preparare l'acqua d'uovo: 0,6 mg/L di sale marino alla concentrazione finale.
- Preparare 5 mg/mL di Tricaina 25x brodo: Mescolare 5 g di Tricaina (etil-3-amminobenzoato metansolfonato o MS-222) in polvere, 900 mL di acqua demineralizzata e 21 mL di 1 M Tris (pH 9). Regolare a pH 7 e riempire fino a 1 L. La tricaina può essere conservata a 4 °C per un breve periodo (fino a sei mesi) o può essere conservata a temperatura ambiente per un mese a temperatura ambiente se protetta dalla luce solare.
- Preparare l'agarosio all'1,5% (p/v) in acqua d'uovo: 1,5 g in 100 ml di DPBS. Microonde per sciogliere.
- Preparare l'agarosio a basso punto di fusione all'1% (p/v) in acqua d'uovo: 1,5 g in 100 ml di DPBS. Microonde per sciogliere.
- Preparare il 2% (p/v) di PVP40 in DPBS: 1 g di PVP40 in 50 mL di DPBS. Agitare e incubare a 37 °C per facilitare la dissoluzione. Conservare a temperatura ambiente.
- Usa il DMSO. Viene spesso utilizzato come solvente nei trattamenti farmacologici e deve essere conservato a 2-8 °C al buio.
- Utilizzare TrypLE, un sostituto sintetico della tripsina che è meno dannoso per le cellule e consente la dispersione delicata delle cellule fortemente aderenti.
- Preparare la soluzione salina tamponata con fosfato di Dulbco (DPBS) senza Mg2+ e Ca2+ per il lavaggio delle cellule. La mancanza di Ca2+ compromette l'adesione cellula-cellula attraverso le caderine.
- Preparare plasmidi lentivirali: psPAX2 (plasmide #12260) e pMD2.G (plasmide #12259) donati da Didier Trono e un plasmide di trasferimento codificante GFP (Plasmide #106172) o tdTomato (Plasmide #106173) (Addgene).
- Utilizzare LipodD293: reagente di trasfezione altamente efficiente HEK293T ottimizzato.
- Piatto di agarosio
NOTA: Quando si utilizzano stoviglie che sono state conservate per molto tempo, assicurarsi di aggiungere un piccolo volume di acqua d'uovo alle stoviglie prima di iniziare l'iniezione (questo eviterà che il pesce si asciughi troppo velocemente).- Preparare piatti ricoperti di agarosio all'1,5% (p/v) (agarosio sciolto in acqua d'uovo).
- Utilizzare immediatamente o conservare a 4 °C in posizione capovolta.
2. Aghi
NOTA: Assicurarsi che i capillari siano stati calibrati sul filamento utilizzato. Quando si commuta il filamento o il capillare, determinare il valore di rampa dei capillari sul filamento utilizzato (vedere il manuale dell'estrattore dell'ago).
- Un capillare di vetro produrrà due micro aghi per iniezione. Prima di realizzare gli aghi, verificare l'integrità strutturale del filamento (filamento a scatola da 2,5 mm) dell'estrattore dell'ago.
- Assicurarsi che sia il filamento che il capillare siano calibrati per ottenere il valore di rampa corrispondente. Quando l'integrità strutturale dei filamenti è compromessa (ad esempio, irregolare, fori, fuso, ecc.), sostituire il filamento.
- Utilizzare il seguente programma (Ago #99, Calore=rampa+15, trazione=95, velocità=60, tempo=90). Conservare gli aghi in una capsula di Petri designata (contenente argilla o nastro adesivo su cui attaccare gli aghi)
3. Generazione di particelle lentivirali
NOTA: Per evitare uno spreco di tempo e risorse, è possibile eseguire un rapido controllo della tumorigenicità prima della trasduzione lentivirale. Questo viene fatto per garantire che la linea cellulare da utilizzare sia sufficientemente tumorigenica nel modello di zebrafish, a tal fine le cellule possono essere colorate con un CMdiI (o tracciante analogo) come descritto in Liverani et al. 2017 15.
- Piastra di cellule HEK 293t un giorno prima della trasfezione per ottenere una confluenza di circa il 70% (di routine facendo dividere un pallone pieno nello stesso pallone di coltura dello stesso volume a una diluizione 1:3 il giorno prima).
- Al giorno della trasfezione, co-trasfettare i plasmidi di imballaggio richiesti psPAX2 e pMD2.G che esprimono l'involucro virale insieme a un GFP (Plasmide #106172) o tdTomato (Plasmide #106173) che codifica per il plasmide di trasferimento. La quantità esatta di plasmide utilizzata è specificata nella Tabella 1.
NOTA: Sia psPAX2 che pMD2.G sono stati donati da Didier Trono (plasmide Addgene #12260 e #12259, rispettivamente).

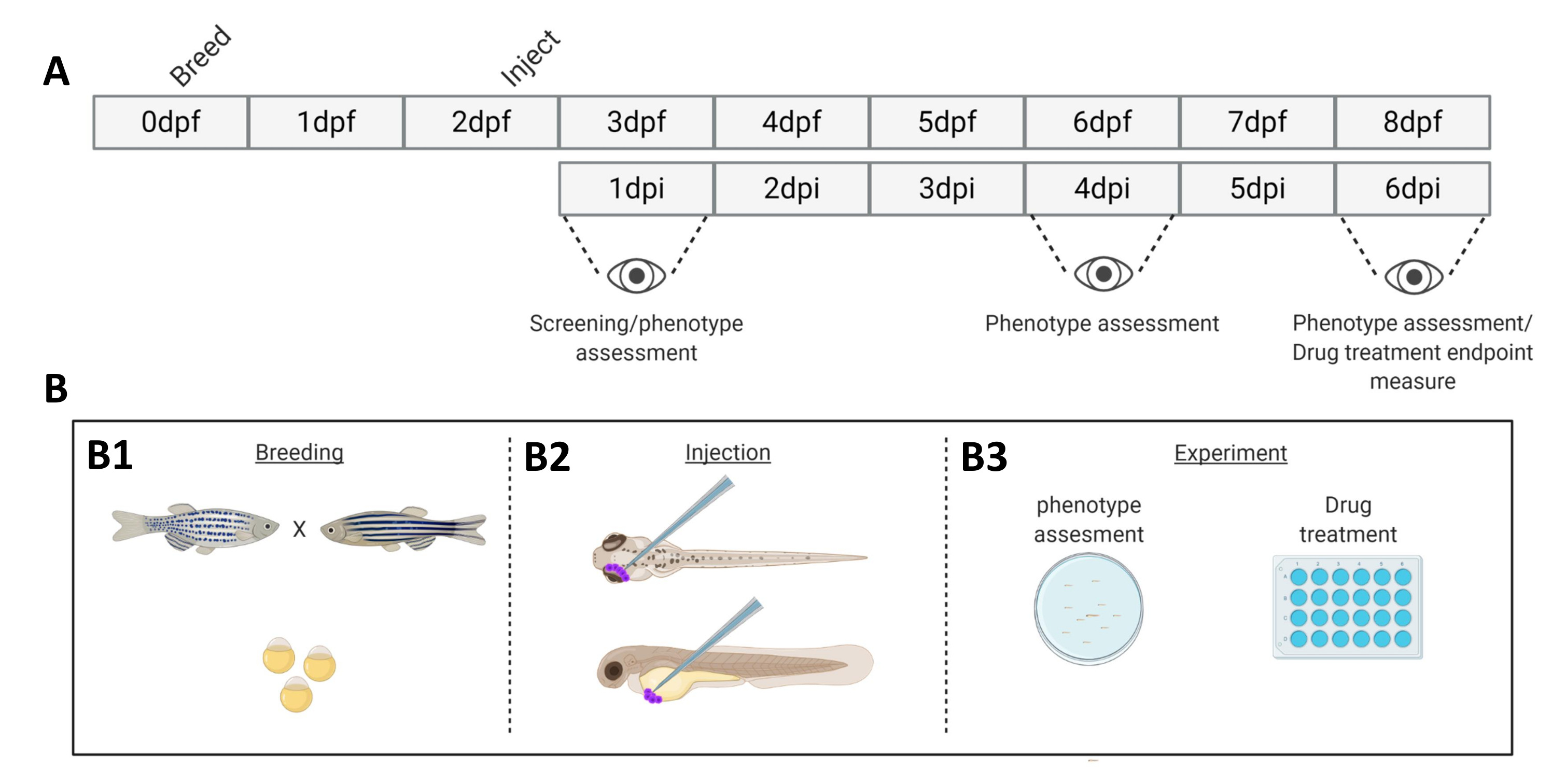
Figura 1. Rappresentazione schematica del sistema di attecchimento del pesce zebra descritto. A) La tempistica dell'avvicinamento, con l'allevamento del pesce zebra al giorno 0 (B1). Il pesce viene raccolto al mattino dopo l'incrocio (giorno 1). Dopo 48-54 ore i pesci si sono in gran parte schiusi (perdendo il loro corion) e i pesci vengono iniettati (retro-orbitalmente o sistemicamente, B2) dopo aver ripulito l'acqua dai detriti del corion (giorno 2). Le larve vengono successivamente sottoposte a screening utilizzando un microscopio a stereofluorescenza e tutte le larve che mostrano fenotipi indesiderati vengono scartate (giorno 3). A seconda dell'obiettivo dell'esperimento, le larve vengono visualizzate nel tempo (B3, cinetica di attecchimento, visualizzate a 1, 4 e 6 giorni dopo l'iniezione (dpi)) oppure i pesci vengono randomizzati e inseriti in gruppi sperimentali, trattati con farmaci e confrontati con il controllo del veicolo (screening farmacologico, imaging a 6 dpi). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Mescolare tutti i plasmidi insieme in 500 μL di terreno privo di siero, per consentire la miscelazione completa di tutti i plasmidi. Aggiungere 32 μl di reagente LipoD293 a 500 μl di DMEM senza siero e agitare per miscelare completamente. Mescola accuratamente entrambi i volumi. Lasciare che i plasmidi e il lipoD293 si aggreghino per 20 minuti.
- Aggiungere goccia a goccia a un pallone di coltura cellulare da 75cm2 contenente il 70% di cellule HEK293T confluenti contenenti 9 mL di terreno di coltura completo. Aggiungere la miscela di trasfezione direttamente allo strato cellulare utilizzando una pipetta sierologica (pallone con orientamento orizzontale).
- Sostituire il terreno con 20 mL di DMEM completo fresco 16 ore dopo la trasfezione. Raccogliere il surnatante dopo 72 ore dalla trasfezione. Aliquotare il surnatante virale in aliquote da 1 mL e conservare a -80 °C. Il surnatante lentivirale è stabile a -80 °C per almeno 1 anno.
4. Trasduzione lentivirale
- Prima della trasduzione lentivirale, stabilire una curva di uccisione quando si utilizza un costrutto lentivirale selezionabile.
- Per la curva di uccisione, piastra la linea cellulare da trasdurre in una piastra a 12 pozzetti (confluenza circa il 10-20%). Aggiungere una curva di dose del selettore (concentrazioni approssimative per le curve di uccisione: puromicina 0,5-10 μg/mL, blasticidina 1-20 μg/mL, geneticina (G418) 100-2000 μg/mL, igromicina 100-2000 μg/mL).
- Cambiare il fluido ogni tre giorni per garantire una concentrazione stabile del selettore scelto.
- Aggiungere 1 mL di surnatante lentivirale a 9 mL di terreno di coltura, contenente una concentrazione finale di 8 μg/mL di polibrene su cellule confluenti al 20-40%. I volumi possono essere ridotti, mantenendo questo rapporto tra surnatante/terreno.
- 16-24 ore dopo la trasduzione, sostituire il mezzo. Quando necessario, ripetere il passaggio precedente per migliorare la penetranza del fenotipo (controllare la fluorescenza per decidere se è necessaria un'altra trasduzione).
- 48 ore dopo la trasduzione, selezionare le cellule utilizzando l'antibiotico corrispondente al marcatore di resistenza incorporato nella cassetta lentivirale. La concentrazione da utilizzare per la selezione della popolazione cellulare trasdotta dovrebbe uccidere la popolazione wild-type entro 7 giorni dall'applicazione del selectante (cioè, consentendo alle cellule trasdotte di crescere più della popolazione wildtype).
- Applicare il surnatante virale in diverse molteplicità di infezioni (MOI) per garantire che la trasduzione e le lesioni genetiche subite dal genoma cellulare non influenzino negativamente la vitalità cellulare o la tumorigenicità.
5. Allevamento di pesci zebra
- Il giorno 0, 2 giorni prima dell'attecchimento delle cellule tumorali, accoppiare il pesce zebra adulto in modo "family cross" a temperatura ambiente (Figura 1).
- Rimuovere la vasca del pesce zebra dal sistema di stabulazione (mantenuto a 28,5 °C).
- Separare i pesci in piccoli gruppi di riproduzione con un rapporto 1:1 maschio: femmina, con 10 pesci per gruppo. Posizionare il pesce in piccole vasche di riproduzione, nell'acqua prelevata dal sistema di stabulazione, sopra una griglia inclinata (inclinata, per imitare le secche in cui il pesce zebra si riproduce naturalmente).
NOTA: Indotto dal calo della temperatura da 28,5°C a temperatura ambiente (25°C) e dall'ingresso nella successiva fase di luce del ciclo buio/luce, il pesce deporrà le uova. - Successivamente, rimuovere gli adulti e trasferirli nella loro vasca di alloggiamento.
- Raccogliete le uova e lavatele con acqua d'uovo usando un colino. Dividete le uova a circa 75-100 per piatto e mantenele a 28,5°C.
- Circa 6 ore dopo la raccolta, pulire le stoviglie dagli embrioni morti o malformati.
- La mattina dopo, scambia l'acqua dell'uovo e pulisci di nuovo i piatti dagli embrioni morti.
6. Raccolta delle cellule
NOTA: Una corretta preparazione delle cellule è fondamentale per la procedura di impianto, l'utilizzo di una quantità superflua di cellule consente una più facile elaborazione a valle. La terza fase di centrifugazione è fondamentale, in quanto questo ti lascerà con solo il pellet di cellule, il PBS rimanente bloccato sui lati della provetta da centrifuga supera di gran lunga il volume di risospensione finale.
- Prima dell'uso, preriscaldare tutti i terreni e le soluzioni utilizzati nelle colture cellulari in un bagno d'acqua a 37 °C.
- Aggiungere 2 mL di TryplE per pallone di coltura2 da 75 cm o 1 mL per pallone da 25 cm2 e incubare fino a quando tutte le cellule sono arrotondate. Per la maggior parte delle linee cellulari dovrebbero essere sufficienti 2-5 minuti. Per le cellule altamente epiteliali o fibroblastiche, 5-10 minuti dovrebbero consentire un corretto distacco (una tripsinizzazione insufficiente ostacolerà i processi a valle e faciliterà l'aggregazione cellulare durante l'impianto).
- Picchiettare delicatamente il lato del pallone per rimuovere le celle rimanenti.
- Sommare al volume di coltura originale del terreno completo. Pipettare su e giù delicatamente ma accuratamente con una pipetta sierologica per tagliare i grumi cellulari in una sospensione di singola cellula. Non generare schiuma durante questo processo poiché la schiuma è indicativa del taglio meccanico delle cellule.
- Trasferire in una provetta sterile da 15 mL e centrifugare per 5 minuti a 200 x g a temperatura ambiente. Aspirare il surnatante e aggiungere 1 mL di PBS sterile. Risospendere accuratamente e accuratamente le cellule utilizzando una pipetta sterile da 1000 μl.
- Rimuovere 20 μl di sospensione cellulare per il conteggio e trasferire la sospensione cellulare rimanente nella centrifuga. Centrifugare per 4 minuti a 200 x g a temperatura ambiente.
- PASSAGGIO CRITICO: Rimuovere tutto il PBS, centrifugare per 30 s a 200 x g a temperatura ambiente e rimuovere il PBS rimanente.
- Diluire le cellule a 250 cellule/nL in polivinilpirrolidon 40 al 2% (PVP40, 2% (p/v) in DPBS) come segue:

(ad esempio,
- Risospendere accuratamente le cellule, evitando la formazione di bolle d'aria (le cellule possono essere conservate per almeno 2 ore in PVP40 al 2% senza perdita di potenziale tumorigenico).
7. Modellazione degli xenotrapianti
Tutti gli esperimenti devono essere eseguiti in conformità con le normative locali sul benessere degli animali.
A seconda dell'applicazione, due principali variazioni nel disegno sperimentale sono classificate: una valutazione del fenotipo (7.1 la fase di pre-screening) e in secondo luogo 7.2, uno screening in cui le cellule sono state modificate prima dell'attecchimento o 7.3 in cui gli embrioni sono trattati con un inibitore chimico.
- Pre-screening e determinazione del potenziale tumorigenico
- Innestare larve di zebrafish di interesse (WT, linea transgenica o reporter) a 2 dpf con un numero variabile di cellule fluorescenti (ad esempio, 200, 400, 600 ±100).
- Esaminare le larve 16-24 ore dopo l'iniezione per rimuovere i valori anomali (numero di cellule estremamente alto o basso in circolazione per il modello ectopico, o cellule all'interno della testa per il modello ortotopico) e rimuovere i pesci erroneamente innestati. Indicare il numero di larve per gruppo sperimentale per l'analisi di gruppo rispetto all'analisi cinetica delle stesse larve.
- Monitorare le larve di zebrafish a intervalli regolari (1,2,4,6 giorni dopo l'iniezione (dpi)) e visualizzare 20 individui (come descritto nei passaggi 9 e 10), da un pool di ±50 larve.
- Monitorare il fenotipo generale e la progressione della malattia e successivamente quantificare con ImageJ (misurazione della densità integrata del segnale del fluoroforo nelle cellule tumorali).
- Tracciare i dati per visualizzare la cinetica di crescita delle cellule tumorali all'interno del pesce zebra (Figura 3).
- Modificare le cellule a priori (knock down o knock out di un gene di interesse) e innestarle nel pesce zebra.
- Innestate i pesci e rimuovete tutti i fenotipi indesiderati (per condizione).
- Immagine degli individui a 1 dpi (20 larve per gruppo). Gli individui possono essere ripresi a intervalli prestabiliti (1,2,4 e 6 dpi).
- A 6 dpi dopo l'imaging, sopprimere il pesce con un sovradosaggio di tricaina (dosaggio 10 volte superiore a 0,4 mg/mL) e gettarlo su carta assorbente che riveste un imbuto.
- Trattare i pesci con farmaci dopo l'attecchimento.
- Prima dell'applicazione del farmaco su zebrafish trapiantato, determinare la dose massima tollerata (MTD) su zebrafish (titolare da 10 μM a 0,150 nM, utilizzando il volume più alto di solvente come controllo negativo) abbiamo impostato l'MTD come concentrazione in cui il >80% degli individui sopravvive all'intero trattamento.
- Un giorno dopo l'iniezione, rimuovere i fenotipi indesiderati.
- Dividere casualmente i pesci in gruppi (36-48 individui/condizione) e conservare in una piastra da 24 pozzetti con 6 larve per pozzetto in 1 mL di acqua d'uovo.
- Applicare i farmaci 24 ore dopo l'attecchimento. Come controllo utilizzare la stessa quantità di solvente (DMSO, EtOH ecc.) al volume più alto applicato per un gruppo sperimentale.
- Iniziare il trattamento farmacologico alla dose massima tollerata. Cambia l'acqua dell'uovo contenente il farmaco a giorni alterni. Rimuovere l'acqua d'uovo e le larve morte il più completamente possibile durante ogni cambio.
8. Iniezione
NOTA: Utilizzare un controllore di impulsi pneumatico accoppiato a una linea di aria compressa, che fornisca una pressione in eccesso di 100 psi. Ciò consente di ottenere una pressione sufficiente sia per iniettare (≈20 psi) che per espellere eventuali aggregati di celle (≈100 psi). La pressione e il tempo di avviamento devono essere di circa 200 ms a 20 psi. Se una delle due deve essere diminuita di oltre il 50% all'inizio dell'iniezione, la sospensione cellulare è troppo fluida (la cella o la concentrazione di PVP40 sono troppo basse) o l'apertura dell'ago è troppo grande.
- Rimuovere con cautela un ago capillare dal suo contenitore. Rompere l'ago per formare un'apertura di ø20 μm, utilizzando una pinza da orologiaio di precisione.
- Risospendere accuratamente e con cura le cellule utilizzando un puntale per pipetta da 20 μl. Pipettare la sospensione cellulare nell'ago capillare di vetro aperto utilizzando una punta lunga (microloader). Caricare l'ago nel micromanipolatore.
- Posizionare ~20-40 larve anestetizzate in 0,04 mg/mL di tricaina su una piastra di agarosio utilizzando una pipetta di trasferimento. Rimuovere l'umidità in eccesso per immobilizzare le larve utilizzando una pipetta di trasferimento. Le larve saranno per lo più orientate in modo laterale a causa della presenza di un sacco vitellino ancora relativamente grande.
- Iniettare le larve con circa 200, 400 e 600 cellule attraverso il dotto di Cuvier (doC) per il modello ectopico.
- Allo stesso modo, iniettare le larve per via retro-orbitale (RO). Per ottenere il modello ortotopico (iniezione di 100 ±50 cellule), modificare la lunghezza dell'impulso pneumatico sulla picopompa (iniziare a ~20 psi, 200 ms e regolare di conseguenza). Durante l'iniezione assicurarsi che le larve non si secchino. Assicurarsi che tutte (o la maggior parte) delle larve vengano iniettate.
- Sciacquare le larve iniettate con acqua fresca d'uovo e trasferirle in una capsula di Petri pulita ed etichettata (raggruppando fino a 150 individui per piastra). Ripetere questo processo fino a quando non viene iniettato un numero sufficiente di larve.
- Dopo l'attecchimento, mantenere il pesce a 34 °C in un'incubatrice umidificata, dove 34 °C è la temperatura più alta facilmente tollerata dal pesce zebra e consente un efficiente attecchimento delle cellule tumorali dei mammiferi.
NOTA: In generale, con l'iniezione di singole linee cellulari sia in doC che in RO abbiamo osservato una morte approssimativa dovuta a danno meccanico del <5% (il danno meccanico uccide le larve tra 1-16 ore dopo l'iniezione).
9. Screening
- Utilizzando un microscopio a stereofluorescenza, esaminare i pesci per il fenotipo appropriato 1 ora dopo l'impianto quando si confrontano le cellule modificate a priori (o 1 giorno dopo l'impianto, quando si selezionano i farmaci, prima dell'assegnazione casuale nei gruppi di trattamento).
- Le larve impiantate attraverso la doC dovrebbero avere cellule nella coda tra 1 ora e 16 ore dopo l'impianto. Rimuovere tutti gli altri pesci, compresi i pesci che mostrano anomalie, dalla piscina iniettata.
NOTA: Le larve impiantate retro-orbitalmente dovrebbero avere cellule solo nell'interstizio dietro l'occhio, le larve che hanno cellule sparse in tutta la testa o nel corpo vengono rimosse dalla piscina. - Pulire le larve selezionate positivamente e assegnarle in modo casuale ai gruppi sperimentali.
- Dopo l'attecchimento, mantenere i pesci a 34 °C in un'incubatrice umidificata e monitorarli quotidianamente. La disseminazione ematogena delle cellule impiantate attraverso la doC è quasi istantanea, mentre la diffusione metastatica delle cellule impiantate nella cavità RO si diffonderà dopo 2-4 giorni.
10. Imaging epifluorescente di larve di pesce zebra
- Anestetizzare le larve di zebrafish con 0,2 mg/mL di tricaina, aggiungendo tricaina all'acqua del pesce o spostando una sottopopolazione di pesci dalla capsula di mantenimento a una capsula contenente 0,2 mg/mL di tricaina.
- Tenere il pesce zebra in un piatto con tricaina fino a quando non rimane fermo, fino a quando la stimolazione della linea laterale non induce un comportamento di volo.
- Trasferire il pesce in una capsula di Petri ricoperta di agarosio, circa 10 per piatto. Rimuovere la maggior parte dell'acqua sollevando delicatamente un'estremità della capsula (lasciando che l'acqua si accumuli delicatamente nell'estremità inferiore della capsula di Petri). Se fatto con attenzione, tutti i pesci si allineeranno, con la coda rivolta verso il basso.
- Immagina tutti i pesci dalla parte superiore del piatto verso il basso. Quindi lavare il pesce con acqua d'uovo in un piatto senza tricaina.
- Ripetere l'operazione fino a quando non viene ripreso un numero sufficiente di persone.
- Quindi trasferire le larve a 34 °C o abbatterle (a 6 dpi) attraverso un sovradosaggio di tricaina (cioè 0,5 mg/mL, incubando per 10 minuti, prima di scartarle su carta assorbente che riveste un imbuto).
11. Imaging confocale di larve di zebrafish (innestate)
- Anestetizzare il pesce zebra con 0,2 mg/mL di tricaina come descritto in precedenza.
- Posizionare un piatto confocale con fondo di vetro sotto uno stereomicroscopio e mettere a fuoco il fondo del piatto. Trasferire 5-10 larve in un piatto confocale con fondo di vetro. Rimuovere quanta più acqua possibile.
- Coprire le larve con agarosio a 42 °C, 1% a basso punto di fusione sciolto in acqua d'uovo. Assicurarsi che l'agarosio si sia raffreddato ad almeno 42 °C prima dell'uso; Temperature più elevate potrebbero danneggiare o uccidere le larve.
- Usando lo stereomicroscopio, orientare rapidamente ma delicatamente le larve spingendolo verso il basso, utilizzando una punta del micro caricatore ribassata. Se è richiesto un orientamento ventrale, tenere le larve in posizione con le pinze di una pinza da orologiaio (senza toccare l'embrione).
- Mentre i set di agarosio regolano finemente l'orientamento delle larve. Lasciare che le larve si solidifichino completamente prima di trasferirle al microscopio confocale.
12. Impostazione del microscopio confocale
- Attivare le linee laser di eccitazione verde (488 nm) e rossa (564 nm). Posizionare la capsula confocale nel supporto del microscopio confocale. Usando l'epifluorescenza, muovi il fascio di luce in modo che si unisca al primo pesce (impostazione x e y). Attraverso l'oculare impostare il fuoco in modo che coincida con il centro delle larve (impostazione z).
- Imposta il guadagno di 700 su entrambi i canali fluorescenti, 1-5% di potenza laser. Aumenta la potenza del laser e diminuisci l'offset per approssimare l'intera gamma dinamica. Non saturare eccessivamente il segnale, ma migliorare il segnale per mostrare solo alcuni pixel saturi.
- Quando si cattura un punto, impostare l'inizio e la fine delle larve lungo un asse ( x o y), se impostato lungo un asse un intero embrione può essere ripreso in 1 x 4 segmenti e può essere post-elaborato in un'unica immagine utilizzando ImageJ.
- Dopo l'imaging, rimuovere le larve dall'agarosio strappandolo delicatamente attorno alle larve incorporate usando una pinza da orologiaio. In caso contrario, sopprimere le larve con un sovradosaggio di tricina non diluita, coprendo l'agarosio con uno strato di tricina e incubando per 10 minuti.
13. Analisi dei dati
- Aprire separatamente i singoli set di dati in ImageJ/Fiji (ad esempio, controllo, farmaco A, farmaco B, farmaco A+B), a partire dal controllo del veicolo.
- Aprire la macro di analisi (script con annotazioni disponibile) (http://doi.org/10.5281/zenodo.4290225).
- In breve, l'analisi macro fa quanto segue: concatena tutte le immagini aperte (una condizione); divide le immagini nei canali separati che compongono l'immagine; chiude tutti i canali accessori (lasciando il canale delle cellule tumorali); esegue un algoritmo di thresholding, sull'intera sequenza concatenata; misura la densità integrata di ogni singola immagine; e salva le misure come foglio excel nella cartella principale.
- Esegui l'analisi macro in tutte le condizioni.
- Combina le misurazioni (in generale almeno n=2*20) e rimuovi i valori anomali (Q-test nel Graph pad Prism v8).
- Normalizzare le misurazioni al controllo del solvente o al giorno 1 (a seconda del tipo di esperimento, il primo per un esperimento di inibizione di farmaci e il secondo per un esperimento di cinetica di crescita). Esprimere le misurazioni come carico normalizzato delle cellule tumorali (asse y ) nel tempo o nella condizione (asse x ), come mostrato rispettivamente nella Figura 3 e nella Figura 4.
Risultati
Abbiamo fornito istruzioni passo dopo passo per un approccio rapido e semplice al passaggio da una nuova linea cellulare alla sua analisi. Iniziamo con la sovraespressione di un tracciante fluorescente utilizzando una cassetta di sovraespressione lentivirale (passaggi 3 e 4). Segue la preparazione delle cellule per garantire il minor volume morto possibile durante l'iniezione, consentendo di iniettare un numero elevato di cellule sia nella doC che nello spazio retro-orbitale (fasi 6 e 7). Successivamente, eseguiamo l'acquisizione di dati ad alto rendimento utilizzando la microscopia stereofluorescente e la microscopia confocale ad alto ingrandimento per l'analisi qualitativa della disseminazione delle cellule tumorali di tutto il corpo (Figura 2 e passaggi 10, 11 e 12). È necessario prestare attenzione durante l'acquisizione dei dati, poiché per garantire la riproducibilità sia per l'imaging al microscopio stereo che per quello confocale, vengono delineate le impostazioni generiche e la standardizzazione (passaggi 11 e 12). Viene discussa l'analisi dei dati (utilizzando imageJ/Fiji) 16, insieme alla standardizzazione utilizzando le macro imageJ (passaggio 13).
Nella fase 3 abbiamo menzionato la marcatura transitoria delle cellule (tumorali) per eseguire un rapido pre-screening per valutare il potenziale tumorigenico di una nuova linea cellulare tumorale. Un avvertimento importante è che, sebbene facile da usare e di lunga durata, la colorazione transitoria qui descritta ha la possibilità di formare artefatti (cioè, è necessario prestare attenzione per garantire che i frammenti cellulari possano essere distinti dalle cellule intere, come è stato ampiamente eseguito da Fior e colleghi 9). Nella nostra esperienza la formazione di questi artefatti è direttamente collegata all'estrema stabilità della macchia e alla luminosità (anche dopo la morte cellulare), dove i frammenti cellulari vengono dispersi e assorbiti dalle cellule immunitarie, che potrebbero successivamente essere erroneamente concluse derivare da metastasi attive.
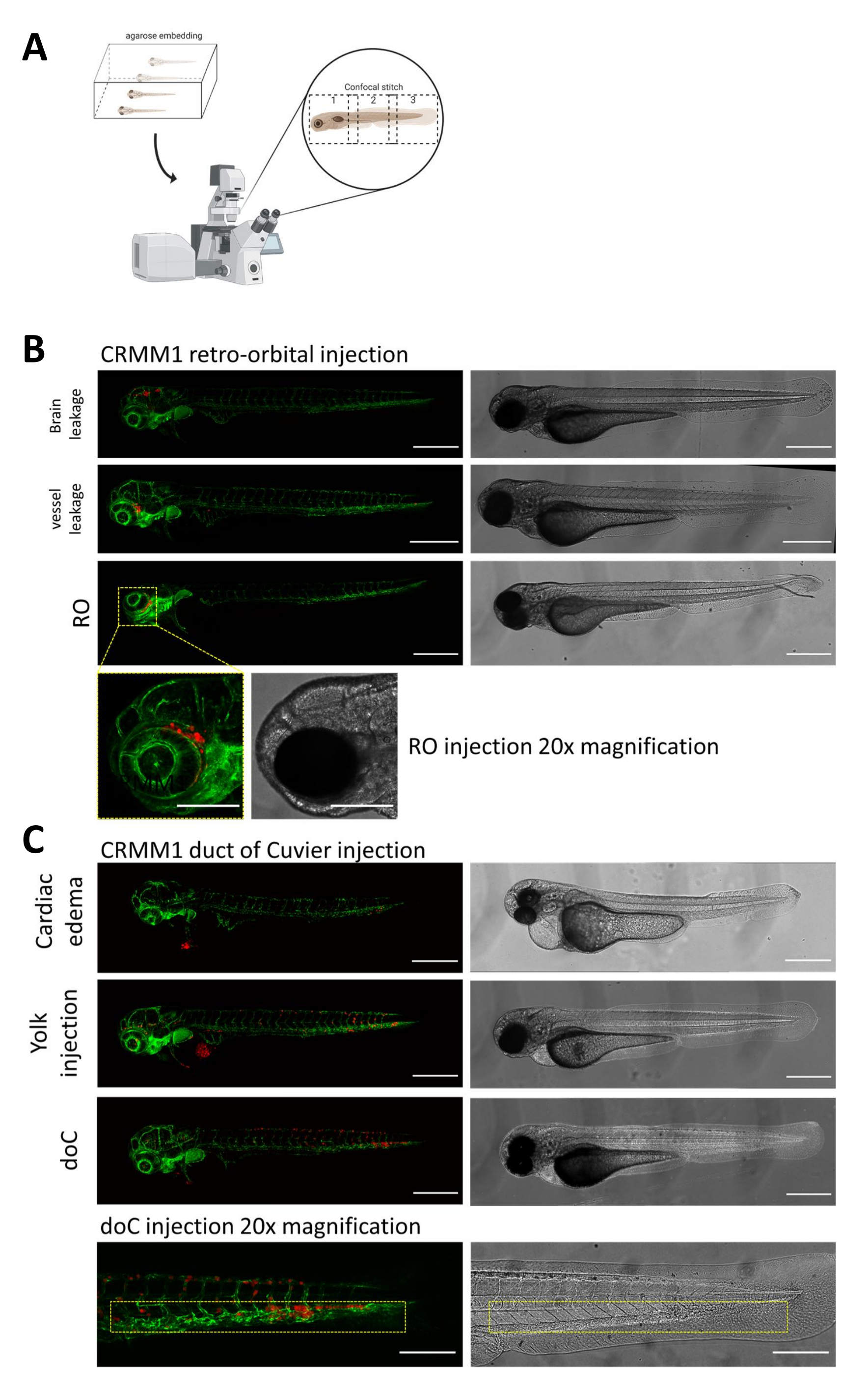
In entrambi i modelli descritti, l'attecchimento sistemico attraverso la doC e l'attecchimento localizzato nello spazio retro-orbitale, lo screening approfondito delle larve un giorno dopo l'iniezione è di fondamentale importanza. Come mostrato nella Figura 2B , tutte le larve che mostrano uno spostamento meccanico delle cellule innestate nell'area della testa (oltre il sito retro-orbitale) nel modello retro-orbitale e nelle cellule nel sacco vitellino, o che mostrano un edema nel pool iniettato con doC, devono essere rimosse. Tutti i fenotipi selezionati negativamente sono visualizzati come punti confocali ad alta risoluzione nella Figura 2, ma possono essere facilmente visti e rimossi attraverso l'osservazione stereomicroscopica.
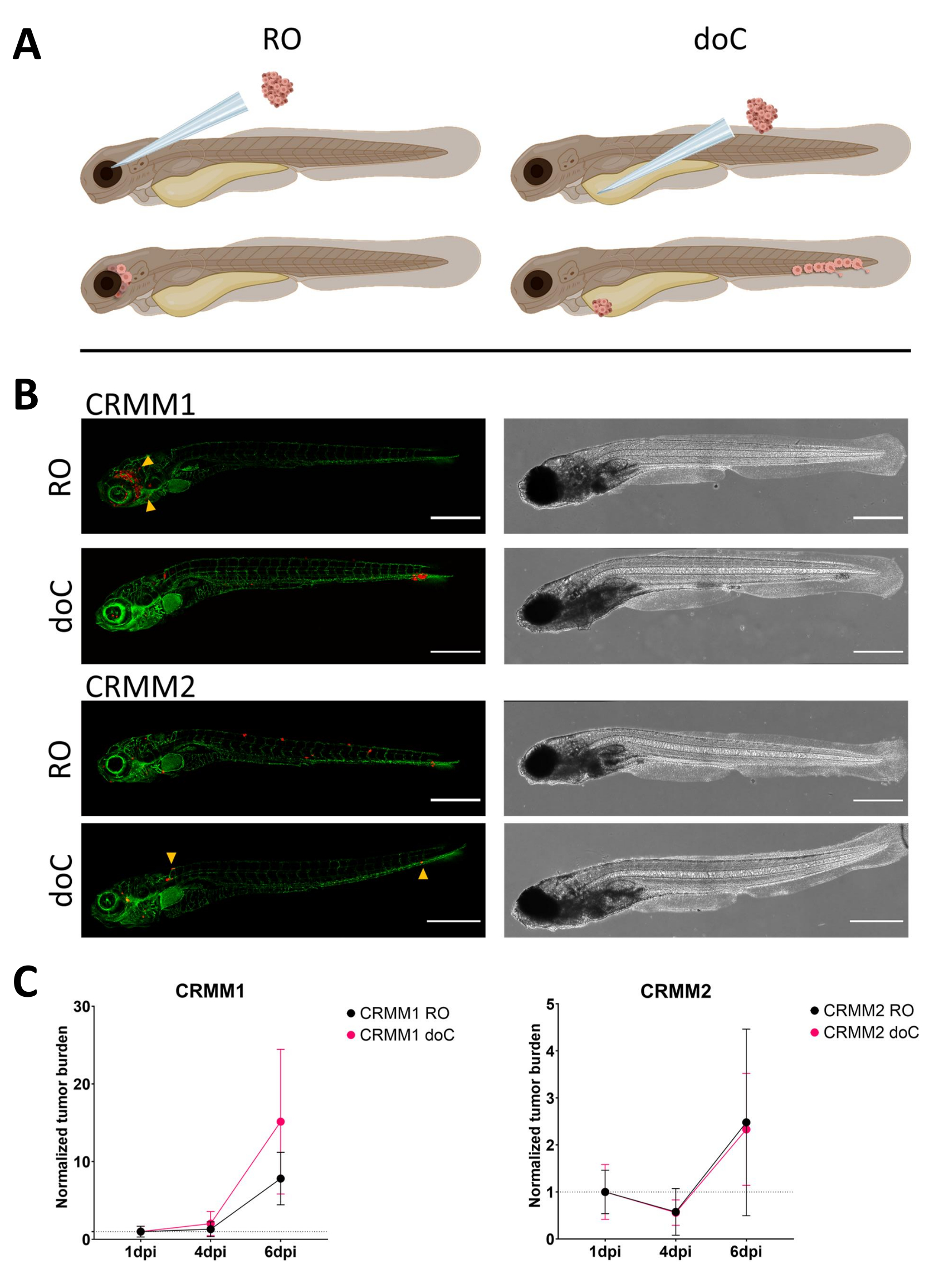
Nel corso del tempo, le cellule migreranno e prolifereranno. Per il modello retro-orbitale, abbiamo osservato l'infiltrazione nei tessuti vicini per CRMM1, ma abbiamo osservato una minore proliferazione per CRMM2. Abbiamo sorprendentemente osservato metastasi a distanza che si verificano tra 2-4 dpi in alcuni individui (20%), dove abbiamo misurato una differenza significativa a 6 dpi, come mostrato nella Figura 4. Per entrambe le linee cellulari, abbiamo testato il potenziale proliferativo quando iniettato in entrambi i siti. Per CRMM1 c'è stato un aumento significativo (p<0,0001) del numero di cellule tumorali per o nei siti di iniezione, quando visualizzato come carico di cellule tumorali normalizzato, normalizzandosi al primo giorno per ciascun modello (aumento di 7,8 volte, ±3,2 per il modello RO e un aumento di 15 volte ±8,8 per il modello doC). CRMM2 non ha mostrato una crescita significativa quando normalizzato al primo giorno per ogni singolo modello (aumento di 2,4 volte, aumento di ±1,9 e 2,3 volte, ±1,14 per RO e doC). È stato riscontrato che CRMM1 prolifera facilmente sia nel tessuto retro-orbitale che nel tessuto ematopoietico caudale dopo l'attecchimento. La linea cellulare CRMM2 era meno proliferativa in entrambi i modelli, ma è interessante notare che è stata trovata in grado di metastasi a distanza quando iniettata nello spazio retro-orbitale, come mostrato nella Figura3B, C.
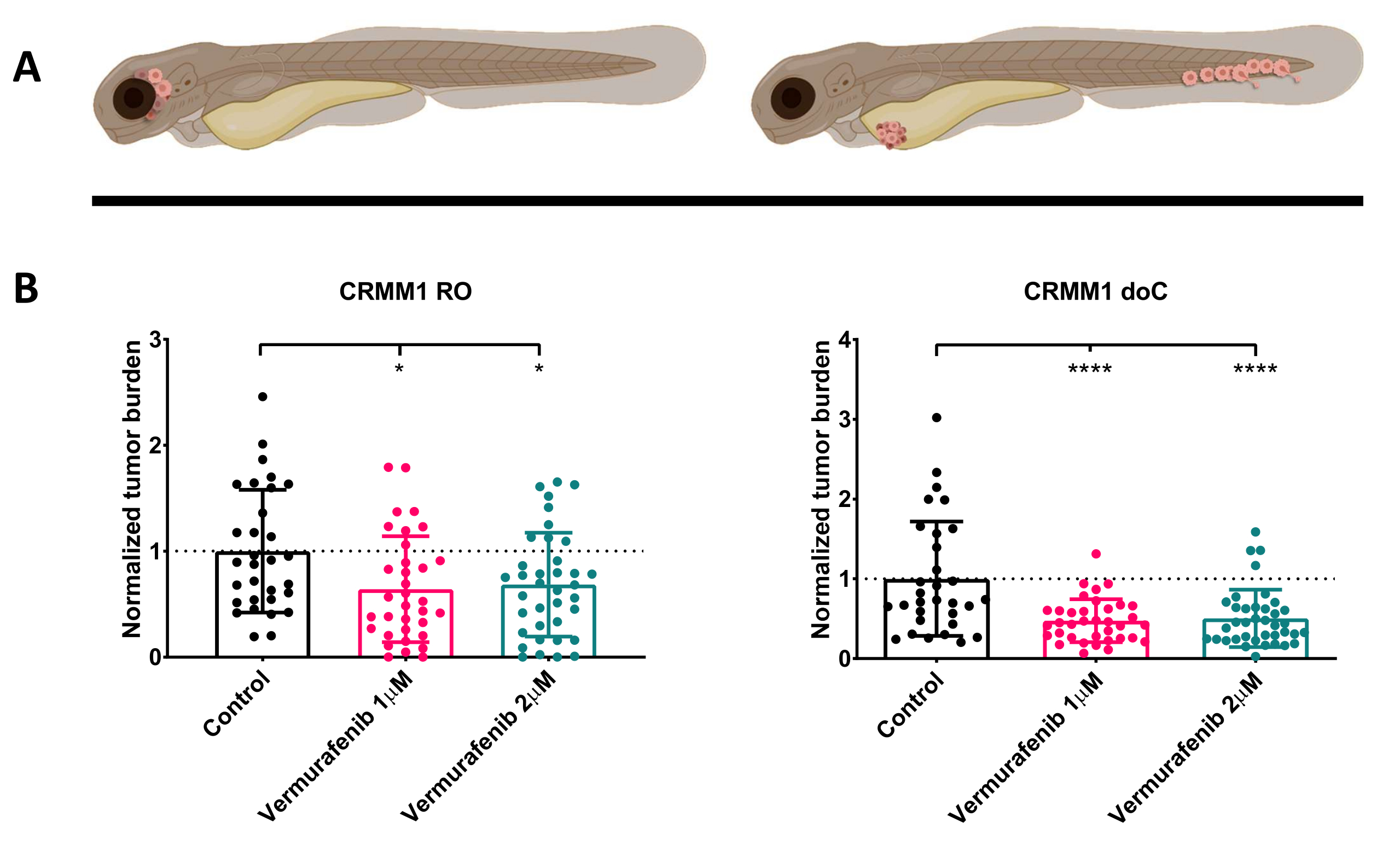
Dopo lo screening delle larve iniettate a 1 dpi e l'assegnazione casuale degli individui al gruppo di trattamento o di controllo, i pesci sono stati trattati per 6 giorni, cambiando l'acqua contenente Vemurafenib (questo inibitore può essere facilmente sostituito con qualsiasi altro composto antitumorale titolato). Abbiamo scelto di elaborare il modello di disseminazione del melanoma congiuntivale ematogeno precedentemente pubblicato che attiantò CRMM114, testando l'efficacia di Vemurafenib su CRMM1 impiantato ortotopicamente. CRMM1 ha mostrato una forte riduzione significativa del gruppo ectopicamente innestato trattato con Vemurafenib (P<0,0001) e una risposta stentata ma significativa per il modello ortotopicamente innestato (p<0,05), come mostrato nella Figura 4.

Figura 2. Valutazione fenotipica e screening dopo l'iniezione. A) Rappresentazione schematica della generazione di punti confocali xenotrapianti di zebrafish, che produce immagini senza soluzione di continuità e ad alta risoluzione dopo l'integrazione della successiva proiezione confocale. Qui gli xenotrapianti di zebrafish sono incorporati in agarosio a basso punto di fusione all'1% e montati su una capsula confocale con fondo di vetro (come descritto al punto 11.3). B) Tutti i possibili esiti dell'attecchimento retro-orbitale e del dotto di Cuvier sono visualizzati iniettati in zebrafish reporter di vasi sanguigni fluorescenti verdi (TG:fli:GFP), con cellule colorate attraverso l'espressione lentivirale di tdTomato). Denotiamo il corretto attecchimento a 1 dpi (pannello RO) e i fenotipi indesiderati (sia perdita cerebrale che perdita di vasi sanguigni). Le ultime due popolazioni devono essere rimosse per garantire che non confondano i risultati sperimentali a valle. C) I fenotipi indesiderati per l'attecchimento ematogeno attraverso il dotto di Cuvier (doC) sono contorni in cui le larve edematose cardiache (edema cardiaco) e le larve con cellule che fuoriescono nel sacco vitellino (iniezione di tuorlo) devono essere rimosse per evitare interferenze con le misurazioni a valle. Le larve correttamente iniettate vengono inserite in gruppi sperimentali come descritto al punto 7.1. (Tutte le immagini sono state acquisite a 1 dpi, utilizzando un microscopio confocale, barre della scala 200 μm. Le caselle gialle indicano i siti metastatici per gli attecchimenti RO e doC, rispettivamente per la regione della testa e il tessuto ematopoietico caudale). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3. L'analisi comparativa delle linee cellulari di melanoma congiuntivale CRMM1 e CRMM2 mostra una capacità metastatica e di crescita differenziale. A) Rappresentazione schematica dei modelli di iniezione, del modello retro-orbitale (RO) e del modello di attecchimento ematogeno (doC) i pesci utilizzati sono reporter di vasi sanguigni verdi TG(fli:GFP), con cellule che sovraesprimono tdTomato mostrate in rosso. B) Fenotipi rappresentativi dei pesci innestati con CRMM1 e CRMM2, CRMM1 mostra un attecchimento efficiente (sia RO che doC) e un'invasione su piccola scala nel tessuto circostante il sito di attecchimento RO (RO, punte di freccia gialle). CRMM2 mostra un'efficienza di attecchimento notevolmente inferiore per entrambi i modelli di attecchimento, ma mostra metastasi a distanza quando iniettato per via retro-orbitale (come mostrato in RO, indicato dalle punte di freccia). (Tutte le immagini sono state acquisite a 6 dpi, un microscopio confocale, barre della scala 200 μm. Le punte di freccia gialle indicano i siti metastatici sia per gli attecchimenti RO che per quelli doC, rispettivamente per la regione della testa e il tessuto ematopoietico caudale). C) Grafici di attecchimento cinetico sia per CRMM1 che per CRMM2, confrontando entrambi i modelli di attecchimento con il giorno 1 (normalizzando al giorno 1), si osserva un aumento significativo (p<0,0001) del carico tumorale normalizzato per la linea cellulare CRMM1 (tra 1 dpi e 6 dpi) dove si verifica una tendenza al rialzo (non significativa) per CRMM2. CRMM1 rivela una differenza significativa tra la crescita RO e doC, dove il modello doC mostra un tasso di espansione tumorale più elevato (circa 2 volte superiore per le larve trapiantate doC). I grafici mostrano la media e l'errore standard della media (SEM). Tutti i gruppi sono stati normalizzati a 1 dpi per ogni singola condizione. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4. L'inibitore di BRAF V600E Vemurafenib inibisce significativamente le larve di zebrafish innestate di melanoma congiuntivale RO e doC. A) Rappresentazione schematica dei fenotipi del pesce zebra, dei modelli RO e doC. B) Sia le larve innestate RO che doC, iniettate con la linea cellulare di melanoma congiuntivale CRMM1, mostrano una significativa riduzione del carico tumorale normalizzato (p<0,05 e P<0,001 rispettivamente). I modelli di zebrafish trapiantati con doC indicano una maggiore risposta al farmaco e una relazione dose-indipendente con l'inibizione del farmaco, indicando una possibile saturazione dell'inibizione). I grafici mostrano la media e l'errore standard della media (SEM), tutti i gruppi sono stati normalizzati per controllare ogni singola linea cellulare. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Reagente | Volume |
| psPAX2 | 1,71 pmol (12,14μg) |
| pMD2.G | 0,94 pmol (3,66μg) |
| Plasmide di trasferimento* | 1,64 pmol (Calcola il volume esatto) |
Tabella 1.
Discussione
Qui, abbiamo definito un approccio meticoloso per modellare il melanoma oculare primario e metastatico negli xenotrapianti di zebrafish. Combinando sia un'iniezione localizzata ortotopica che un modello di iniezione sistemica ed ectopica, abbiamo ricapitolato l'eziologia della cancerogenesi per un cancro in cui in precedenza non erano disponibili modelli animali. La trasparenza intrinseca della larva precoce di zebrafish consente il tracciamento di cellule tumorali marcate in fluorescenza a livello di intero animale, garantendo la facile visualizzazione di potenziali siti metastatici17. Inoltre, l'analisi microscopica confocale ad alto ingrandimento ci permette di tracciare le cellule ad una risoluzione subcellulare10.
Abbiamo fornito istruzioni passo dopo passo per un approccio rapido e semplice al passaggio da una nuova linea cellulare alla costituzione dello xenotrapianto e alla sua analisi. Iniziamo con la sovraespressione di un tracciante fluorescente utilizzando una cassetta di sovraespressione lentivirale (fase 3 e 4) seguita dalla preparazione cellulare per garantire il minor volume morto possibile durante l'iniezione. Ciò consente l'iniezione di un numero elevato di cellule sia nella doC che nello spazio retro-orbitale (fase 7 e 8). Quindi eseguiamo l'acquisizione di dati ad alto rendimento utilizzando la microscopia stereofluorescente e la microscopia confocale ad alto ingrandimento per l'analisi qualitativa della disseminazione delle cellule tumorali di tutto il corpo (Figura 2 e passaggi 9 e 10). È necessario prestare attenzione durante l'acquisizione dei dati, poiché per garantire la riproducibilità sia per l'imaging al microscopio stereo che per quello confocale, vengono delineate le impostazioni generiche e la standardizzazione (passaggi 11 e 12). Viene discussa l'analisi dei dati (utilizzando imageJ/Fiji) 16, insieme alla standardizzazione utilizzando le macro ImageJ (passaggio 13).
Nella fase 3 menzioniamo la marcatura transitoria delle cellule (tumorali) per eseguire un rapido pre-screening per valutare il potenziale tumorigenico di una nuova linea cellulare tumorale. Un avvertimento importante è che, sebbene facile da usare e di lunga durata, la colorazione transitoria qui descritta ha la possibilità di formare artefatti (ad esempio, è necessario prestare attenzione per garantire che i frammenti cellulari possano essere distinti dalle cellule intere, come è stato ampiamente eseguito da Fior e colleghi 9). Nella nostra esperienza la formazione di questi artefatti è direttamente collegata all'estrema stabilità della macchia e alla luminosità (anche dopo la morte cellulare), dove i frammenti cellulari vengono dispersi e assorbiti dalle cellule immunitarie, che potrebbero successivamente essere erroneamente concluse derivare da metastasi attive.
Utilizzando questi modelli, abbiamo simulato lo sviluppo del tumore primario confinando fisicamente le cellule trapiantate all'interno dell'interstizio retro-orbitale. Il successivo screening approfondito a 1 giorno dopo l'attecchimento assicura che le cellule trovate in un sito distante più avanti nell'esperimento abbiano metastatizzato attivamente (intravasato e disseminato, per poi stravasare nella nicchia metastatica). L'attecchimento attraverso la doC, la vena cardinale comune embrionale, consente un impianto facile e altamente riproducibile di grandi quantità di cellule (con un surplus di 600 cellule se adeguatamente concentrate), eludendo efficacemente le fasi primarie della cascata metastatica (intravasazione) e permettendoci di concentrarci sulle fasi successive della cascata metastatica (adesione, stravaso e crescita). Sebbene strumenti potenti se utilizzati correttamente, entrambi i modelli dovrebbero essere monitorati ampiamente durante il primo giorno dopo l'attecchimento per garantire che non vengano tratte conclusioni false positive durante le fasi successive dell'esperimento.
In linea con le pubblicazioni precedenti, abbiamo dimostrato che le linee di melanoma congiuntivale formano facilmente colonie metastatiche dopo la disseminazione in tutto il sistema di circolazione sanguigna del pesce zebra14. Qui riportiamo l'espansione del repertorio di attecchimento con l'iniezione retro-orbitale come modello ortotopico, e la successiva metastasi attiva al tessuto ematopoietico caudale della linea cellulare CRMM2. Successivamente riportiamo l'efficacia dell'inibitore specifico di BRAF V600E Vemurafenib anche sulla forma primaria di melanoma congiuntivale quando modellato in larve di zebrafish.
Utilizzando i metodi sopra menzionati, un ricercatore esperto è in grado di generare oltre centinaia di larve innestate al giorno (circa 200 all'ora) di entrambi i modelli proposti. In un arco di tempo di due settimane un farmaco può essere titolato per la dose massima tollerata e sottoposto a screening su un modello di xenotrapianto stabilito. Dall'inizio alla fine, utilizzando una linea cellulare non trasdotta, è possibile ottenere un profilo di sensibilità al farmaco nel modello di zebrafish entro un mese (dato che la linea cellulare iniettata è tumorigenica all'interno del modello di zebrafish). Nelle nostre mani solo 20 larve per esperimenti e due ripetizioni biologiche hanno prodotto in modo riproducibile una robusta inibizione del farmaco, quando due singoli esperimenti entrano in conflitto (o non producono un'inibizione della crescita statisticamente significativa) può essere condotta una terza ripetizione biologica.
Attraverso piccoli aggiustamenti, questi modelli ci hanno permesso di adattare rapidamente queste strategie di impianto per il glioblastoma (iniezione nella cavità cerebrale posteriore), il cancro al seno (iniezione di doC) e l'osteosarcoma (doC), tra gli altri 18,19,20,21. Questi modelli possono essere successivamente utilizzati sia per la ricerca di base che per lo screening preclinico sia di singoli farmaci che di strategie farmacologiche combinatoriali. Recentemente, abbiamo descritto diversi regimi di somministrazione di farmaci e la loro fotoattivazione utilizzando questi modelli 13.
Divulgazioni
Nessuno.
Riconoscimenti
Questo lavoro è stato sostenuto da finanziamenti del programma di ricerca e innovazione Horizon 2020 dell'Unione Europea nell'ambito dell'accordo di sovvenzione n. 667787 (progetto UM Cure 2020, www.umcure2020.org). Il Chinese Scholarship Council è gentilmente riconosciuto per una borsa di dottorato a J.Y.
Materiali
| Name | Company | Catalog Number | Comments |
| 2.5mm box filament | Science products | FB255B | for pulling micro injection needles using a Sutter P97 or P1000 |
| 3mL transfer pipettes | Merck | Z350796 | for transfer and selection of zebrafish embryos |
| Agarose | Milipore | 2120 | 1.5% (w/v) in eggwater, 1.5 g in 100 mL DPBS, microwave to dissolve, for injecting and stereofluorescence imaging of zebrafish larvae |
| Capillaries: borosilicate glass outer | World precision instruments | BF100-78-10 | Borosilicate glass capillaries used for needle preparation |
| DMSO | Sigma | D8418 | Often used as solvent in drug treatments, should be stored at 2-8°C the dark. |
| DPBS | Thermo Fischer Scientific | 14190144 | Dulbecco’s phosphate buffered saline, without Mg2+ and Ca2+ for washing the cells, lack of Ca2+ impairs cell-cell adhesion through cadherins and prevents cell aggregation during injection |
| Egg water | Instant ocean | SS15-10 | 0.6 mg/L final concentration sea salt in demineralized water |
| GFP encoding lentiviral transfer plasmid | Addgene | Plasmid #106172 | Generated in Snaar lab, available at Addgene |
| Hek293T | ATCC | CRL-3216 | Stable cell line for generating lentiviral particles, contains SV40-T antigen required for the generation of lentiviral particles |
| Leica sp8 confocal | Leica | Leica TCS SP8 | automated stage confocal microscope with 405/488/514/635nm lasers |
| LipodD293 | Signagen | SL100668 | Highly efficient HEK293t optimized transfection reagent |
| Low-melting agarose | Milipore | 2070 | 1% (w/v) in eggwater 1.5 g in 100 mL DPBS, microwave to dissolve, for embedding zebrafish larvae for confocal imaging |
| Micro loader tips | Fischer scientific | 10289651 | flexible microloader tips |
| Micro manipulator | World precision instruments | M3301R | x/y/z manual micro manipulator for microinjection |
| Needle puller: P-97 or P-1000 | Sutter | P-97 | needle puller used for generating standardized micro engraftment needles |
| Nr.5 watchmakers forceps | VWR | HAMMHSC818-11 | fine watchmakers forceps used for breaking back needles |
| Picopump | World precision instruments | SYS-PV820 | pulse controller supplying pressure for microinjection |
| pMD2.G | Addgene | plasmid #12259 | Gifted by Didier Trono, 2nd generation lentiviral virulence plasmid |
| psPAX2 | Addgene | plasmid #12260 | Gifted by Didier Trono, 2nd generation lentiviral packaging plasmid |
| PVP40 | Sigma-Aldrich | PVP40 | Polyvinylpyrrolidone average mol wt 40,000) PVP40 2% (w/v) in DPBS, 1 g PVP40 in 50 mL DPBS. Vortex and incubate at 37°C to facilitate dissolving. Store at room temperature. |
| tdTomato encoding lentiviral transfer plasmid | Addgene | Plasmid #106173 | Generated in Snaar lab, available at Addgene |
| transmitted light stereo microscope | Leica | leica M50 with (MDG33 base) | leica transmitted light microscope with mirror adjustable illumination. |
| Tricaine | Sigma-Aldrich | E10521 | Ethyl 3-aminobenzoate methanesulfonate or MS-222 |
| TryplE | Thermo Fischer Scientific | 12604-01 | Synthetic trypsine replacement, less damaging to the cells and allows for the gentle dispersion of strongly adherent cells. (Thermo- |
| willco dish | WillCo wells | GWST-5040 | 50mm glass bottom dishes, allow for the embedding of up to 20 zebrafish larvae, enabling the imaging of multiple conditions in one dish due to its large optical glass surfac |
Riferimenti
- Yang, J., Manson, D. K., Marr, B. P., Carvajal, R. D. Treatment of uveal melanoma: where are we now. Therapeutic Advances in Medical Oncology. 10, (2018).
- Wong, J. R., Nanji, A. A., Galor, A., Karp, C. L. Management of conjunctival malignant melanoma: A review and update. Expert Review of Ophthalmology. 9, 185-204 (2014).
- Nguyen, D. X., Bos, P. D., Massagué, J. Metastasis: from dissemination to organ-specific colonization. Nature Reviews Cancer. 9, 274-284 (2009).
- White, R. M., et al. Transparent Adult Zebrafish as a Tool for In Vivo Transplantation Analysis. Cell Stem Cell. 2, 183-189 (2008).
- Zon, L. I., Peterson, R. T. In vivo drug discovery in the zebrafish. Nature Reviews Drug Discovery. 4, 35-44 (2005).
- Howe, K., et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 496, 498-503 (2013).
- Palmblad, M., et al. Parallel deep transcriptome and proteome analysis of zebrafish larvae. BMC Research Notes. 6, 428 (2013).
- Yan, C., et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell. 177, 1903-1914 (2019).
- Fior, R., et al. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proceedings of the National Academy of Sciences of the United States of America. 114, 8234-8243 (2017).
- Campbell, P. D., Chao, J. A., Singer, R. H., Marlow, F. L. Dynamic visualization of transcription and RNA subcellular localization in zebrafish. Development. 142, 1368-1374 (2015).
- Campeau, E., et al. A Versatile Viral System for Expression and Depletion of Proteins in Mammalian Cells. PLoS One. 4, 6529 (2009).
- vander Helm, D., et al. Mesenchymal stromal cells prevent progression of liver fibrosis in a novel zebrafish embryo model. Scientific Reports. 8, 16005 (2018).
- Chen, Q., et al. TLD1433 photosensitizer inhibits conjunctival melanoma cells in zebrafish ectopic and orthotopic tumour models. Cancers. 12, (2020).
- Pontes, K. C. d. e. S., et al. Evaluation of ( fli:GFP ) Casper Zebrafish Embryos as a Model for Human Conjunctival Melanoma. Investigative Opthalmology & Visual Science. 58, 6065 (2017).
- Liverani, C., et al. Innovative approaches to establish and characterize primary cultures: an ex vivo 3D system and the zebrafish model. Biology Open. 6, 133-140 (2017).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9, 676-682 (2012).
- White, R. M., et al. Transparent Adult Zebrafish as a Tool for In Vivo Transplantation Analysis. Cell Stem Cell. 2, 183-189 (2008).
- Mercatali, L., et al. Development of a patient-derived xenograft (PDX) of breast cancer bone metastasis in a Zebrafish model. International Journal of Molecular Sciences. 17, (2016).
- Tulotta, C., et al. Imaging cancer angiogenesis and metastasis in a zebrafish embryo model. Advances in Experimental Medicine and Biology. 916, 239-263 (2016).
- Paauwe, M., et al. Endoglin expression on cancer-associated fibroblasts regulates invasion and stimulates colorectal cancer metastasis. Clinical Cancer Research. 24, 6331-6344 (2018).
- Cao, J., et al. Overexpression of EZH2 in conjunctival melanoma offers a new therapeutic target. Journal of Pathology. 245, (2018).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati