
Method Article
Xeno-enxerto de peixe-zebra orto e ectópico de melanoma ocular para recapitular o tumor primário e o desenvolvimento de metástases experimentais
Neste Artigo
Resumo
Aqui, apresentamos um protocolo para estabelecer modelos versáteis de xenoenxerto ortotópico e ectópico de peixe-zebra para melanoma ocular para avaliar a cinética de crescimento do tumor primário, disseminação, extravasamento e formação de metástases perivasculares à distância e o efeito da inibição química sobre o mesmo.
Resumo
Atualmente, não existem modelos animais para melanoma ocular metastático. A falta de modelos de doença metastática tem dificultado muito a pesquisa e o desenvolvimento de novas estratégias para o tratamento do melanoma ocular metastático. Neste protocolo, delineamos uma maneira rápida e eficiente de gerar modelos embrionários de peixe-zebra para o estágio primário e disseminado do melanoma ocular, usando enxerto de células ectópicas ortotópicas retro-orbitais e intravasculares, respectivamente. Combinando essas duas diferentes estratégias de enxerto, podemos recapitular a etiologia do câncer em sua totalidade, progredindo do crescimento do tumor primário e localizado sob o olho para a formação de uma metástase perivascular na cauda. Esses modelos nos permitem modificar de forma rápida e fácil as células cancerígenas antes da implantação com marcação específica, interferência genética ou química; e tratar os hospedeiros enxertados com inibidores (pequenos moleculares) para atenuar o desenvolvimento do tumor.
Aqui, descrevemos a geração e quantificação de enxerto ortotópico e ectópico de melanomas oculares (melanoma conjuntival e uveal) usando linhagens celulares estáveis marcadas com fluorescência. Este protocolo também é aplicável para enxerto de células primárias derivadas de biópsia de paciente e material derivado de paciente/PDX (manuscrito em preparação). Poucas horas após o enxerto, a migração e proliferação celular podem ser visualizadas e quantificadas. Ambos os focos tumorais estão prontamente disponíveis para imagens com microscopia de epifluorescência e microscopia confocal. Usando esses modelos, podemos confirmar ou refutar a atividade de estratégias de inibição química ou genética em menos de 8 dias após o início do experimento, permitindo não apenas uma triagem altamente eficiente em linhagens celulares estáveis, mas também permite a triagem direcionada ao paciente para abordagens de medicina de precisão.
Introdução
A disseminação metastática é considerada a principal causa de morte do melanoma ocular; atualmente não existe um esquema de tratamento viável para o melanoma ocular disseminado 1,2. Além disso, não há modelos animais disponíveis para melanoma ocular que reflitam a doença metastática. Para preencher essa lacuna, geramos dois modelos distintos de peixe-zebra que recapitulam a formação de tumores primários ou os estágios iniciais da disseminação metastática, permitindo assim prontamente o estudo desses processos normalmente difíceis de estudar 3. Os modelos de micrometástase permitem a análise das últimas fases da disseminação metastática, incluindo homing, colonização e extravasamento. Intervenções genéticas ou químicas nesta fase e além podem fornecer um apoio poderoso no tratamento do melanoma ocular metastático.
O uso das larvas de peixe-zebra como receptoras de xeno- e aloenxertos é apoiado pelas forças intrínsecas desta espécie, como sua transparência óptica nos estágios iniciais de desenvolvimento (ou todo o seu ciclo de vida para mutantes casper 4), alta fecundidade e fertilização ex utero 5. A alta homologia transcricional em vertebrados garante a retenção dos principais mecanismos de sinalização entre o peixe-zebra e os humanos e, portanto, alto potencial de traduzibilidade dos resultados 6, embora as abordagens genéticas às vezes sejam prejudicadas ou complicadas devido à duplicação do genoma do teleósteo 7. Desenvolvimentos recentes ressaltaram a importância dos modelos de xenoenxerto de peixe-zebra como "avatares" pré-clínicos de doenças humanas8, produzindo efetivamente uma infinidade de modelos personalizados de terapia contra o câncer para a avaliação pré-clínica de estratégias de tratamento de um único experimento de peixe-zebra 9.
Considerando a falta de modelos animais e a falta concordante de opções de tratamento para melanoma ocular metastático, nossos modelos fornecem uma plataforma translacional rápida e fácil para rastrear alterações genéticas (intrínsecas de células cancerígenas) ou desenvolver estratégias de intervenção química em um ambiente pré-clínico. Dentro do mesmo modelo, podemos visualizar e medir a cinética de crescimento de células cancerígenas, taxa de enxerto / potencial metastático e homing celular em um nível de animal inteiro usando ampliação de baixo nível em um microscópio fluorescente estéreo e fazer medições semelhantes usando análise microscópica confocal de média ou alta ampliação para dissecar diferentes etapas da progressão do melanoma ocular em resolução subcelular 10.
Aqui, descrevemos protocolos abrangentes e detalhados para: a geração de células cancerígenas marcadas com fluorescência usando transdução lentiviral altamente otimizada11; enxertos intravenosos e retroorbitais (RO) subsequentes dessas células em larvas de peixe-zebra 2 dias após a fertilização (dpf) para gerar modelos ectópicos e ortotópicos, respectivamente; seguida de aquisição e análise de dados. Esses métodos, embora abrangentes para as aplicações aqui descritas, podem ser modificados para enxertar células na cavidade cerebral posterior, fígado e espaço perivitelina quando necessário (apenas alterando o local da injeção ou o momento da injeção)12,13.
Como prova de conceito, elaboramos as descobertas de Pontes et al. 2018, onde mostramos uma resposta específica de dose e mutação intrínseca celular de linhagens celulares de melanoma conjuntival no modelo de peixe-zebra 14. Elaboramos esses achados mostrando a eficácia do inibidor específico da mutação BRAF V600E vemurafenibe em modelos de melanoma conjuntival metastático e primário.
Protocolo
Todos os experimentos com animais foram aprovados pelo Comitê de Experimentos com Animais (Dier Experimenten Commissie, D.E.C.) sob licença AVD1060020172410. Todos os animais foram mantidos de acordo com as diretrizes locais usando protocolos padrão (www.ZFIN.org).
1. Preparação
- Reagentes
- Prepare água com ovo: 0,6 mg/L de sal marinho na concentração final.
- Prepare 5 mg / mL de Tricaína 25x estoque: Misture 5 g de Tricaína (etil 3-aminobenzoato de metanossulfonato ou MS-222) em pó, 900 mL de água desmineralizada e 21 mL de 1 M Tris (pH 9). Ajuste para pH 7 e encha até 1 L. A tricaína pode ser armazenada a 4 °C por um curto período (até seis meses) ou pode ser armazenada em temperatura ambiente por um mês em temperatura ambiente quando protegida da luz solar.
- Prepare agarose a 1,5% (p/v) em água com ovo: 1,5 g em 100 mL de DPBS. Microondas para dissolver.
- Prepare 1% (p / v) de agarose de baixo ponto de fusão em água de ovo: 1,5 g em 100 mL de DPBS. Microondas para dissolver.
- Prepare 2% (p / v) de estoque de PVP40 em DPBS: 1 g de PVP40 em 50 mL de DPBS. Vórtice e incubar a 37 °C para facilitar a dissolução. Armazene em temperatura ambiente.
- Use DMSO. É frequentemente usado como solvente em tratamentos medicamentosos e deve ser armazenado a 2-8 °C no escuro.
- Use TrypLE, um substituto sintético de tripsina que é menos prejudicial às células e permite a dispersão suave de células fortemente aderentes.
- Prepare a solução salina tamponada com fosfato de Dulbecco (DPBS) sem Mg2+ e Ca2+ para lavar as células. A falta de Ca2+ prejudica a adesão célula-célula através das caderinas.
- Prepare plasmídeos lentivirais: psPAX2 (plasmídeo # 12260) e pMD2.G (plasmídeo # 12259) doados por Didier Trono e um plasmídeo de transferência de codificação GFP (Plasmídeo # 106172) ou tdTomato (Plasmídeo # 106173) (Addgene).
- Use LipodD293: Reagente de transfecção altamente eficiente HEK293T otimizado.
- Prato de agarose
NOTA: Ao usar pratos que foram armazenados por muito tempo, certifique-se de adicionar um pequeno volume de água de ovo aos pratos antes de iniciar a injeção (isso evitará que o peixe seque muito rápido).- Prepare pratos revestidos de agarose a 1,5% (p / v) (agarose dissolvida em água com ovo).
- Use imediatamente ou armazene a 4 °C na posição invertida.
2. Agulhas
NOTA: Certifique-se de que os capilares foram calibrados no filamento utilizado. Ao trocar o filamento ou o capilar, determine o valor de rampa dos capilares no filamento usado (consulte o manual do extrator de agulhas).
- Um capilar de vidro produzirá duas agulhas de microinjeção. Antes de fazer agulhas, verifique a integridade estrutural do filamento (filamento de caixa de 2,5 mm) do extrator de agulhas.
- Certifique-se de que o filamento e o capilar estejam calibrados para obter o valor de rampa correspondente. Quando a integridade estrutural dos filamentos estiver comprometida (ou seja, irregular, furos, fundidos, etc.), troque o filamento.
- Use o seguinte programa (Agulha # 99, Calor = rampa + 15, puxar = 95, velocidade = 60, tempo = 90). Armazene as agulhas em uma placa de Petri designada (contendo argila ou fita adesiva para colar as agulhas)
3. Geração de partículas lentivirais
NOTA: Para evitar perda de tempo e recursos, uma rápida verificação da tumorigenicidade pode ser realizada antes da transdução lentiviral. Isso é feito para garantir que a linhagem celular a ser utilizada seja suficientemente tumorigênica no modelo de peixe-zebra, para isso as células podem ser coradas com um CMdiI (ou marcador análogo) conforme descrito em Liverani et al. 2017 15.
- Plaquear células HEK 293t um dia antes da transfecção para atingir uma confluência de aproximadamente 70% (feito rotineiramente dividindo um frasco cheio no mesmo frasco de cultura de volume em uma diluição 1:3 um dia antes).
- No dia da transfecção, co-transfeccione os plasmídeos de embalagem necessários psPAX2 e pMD2.G expressando o plasmídeo que expressa o envelope viral junto com um GFP (Plasmídeo # 106172) ou tdTomato (Plasmídeo # 106173) codificando o plasmídeo de transferência. A quantidade exata de plasmídeo usada é especificada na Tabela 1.
NOTA: Tanto o psPAX2 quanto o pMD2.G foram doados por Didier Trono (plasmídeo Addgene # 12260 e # 12259, respectivamente).

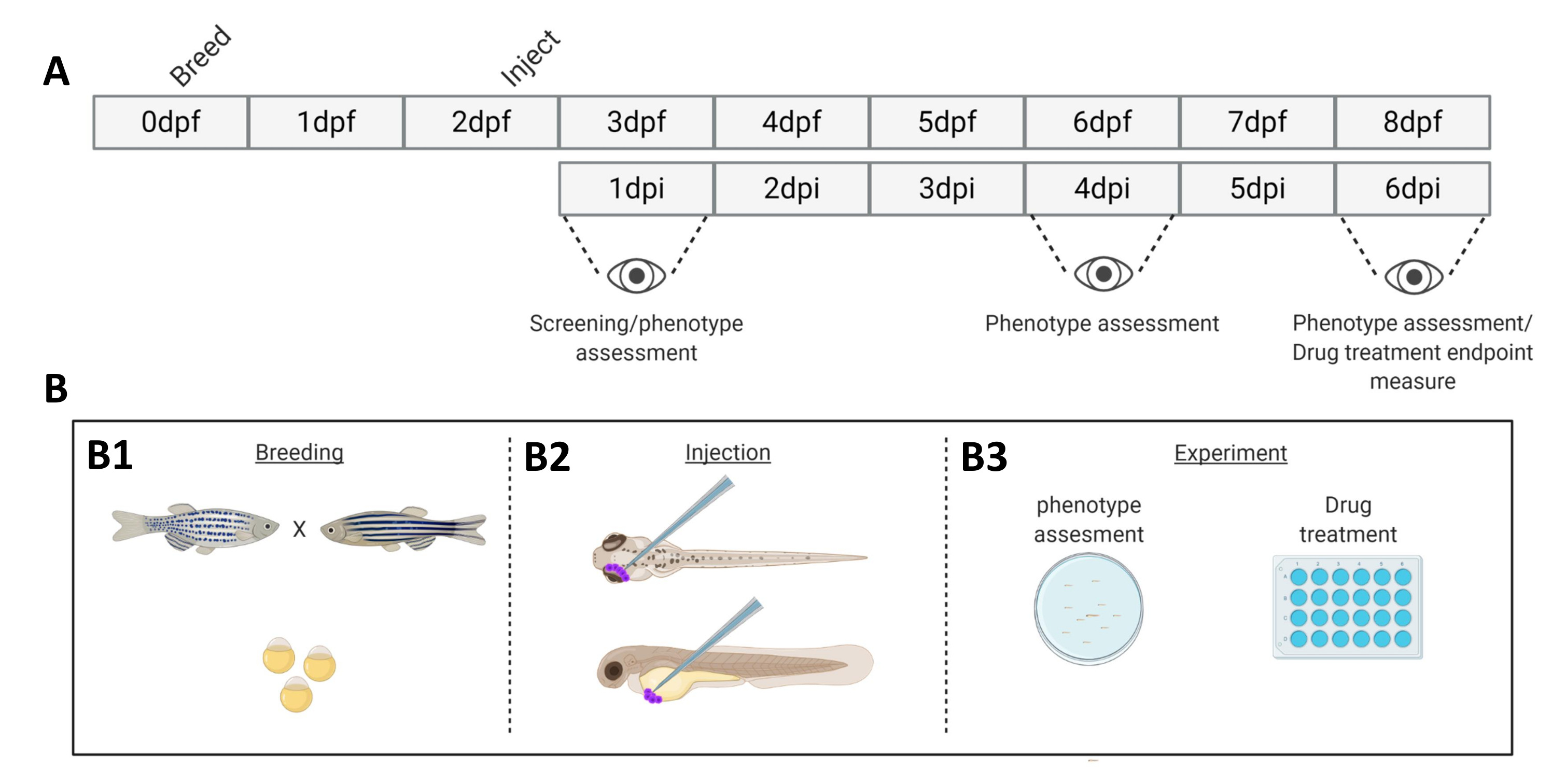
Figura 1. Representação esquemática do sistema de enxerto de peixe-zebra descrito. A) A linha do tempo da abordagem, com a reprodução do peixe-zebra no dia 0 (B1). Os peixes são colhidos pela manhã após o cruzamento do peixe (dia 1). Após 48-54 horas, os peixes eclodiram em grande parte (perdendo seu córion) e os peixes são injetados (retroorbitalmente ou sistemicamente, B2) após a limpeza da água dos detritos do córion (dia 2). As larvas são posteriormente rastreadas usando um microscópio fluorescente estéreo e todas as larvas que exibem fenótipos indesejados são descartadas (dia 3). Dependendo do objetivo do experimento, as larvas são visualizadas ao longo do tempo (B3, cinética de enxerto, imagens em 1, 4 e 6 dias após a injeção (dpi)) ou os peixes são randomizados e inseridos em grupos experimentais, tratados com drogas e comparados ao controle do veículo (triagem de drogas, imagem a 6 dpi). Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Misturar todos os plasmídeos em 500 μL de meio livre de soro, para permitir a mistura completa de todos os plasmídeos. Adicione 32 μL de reagente LipoD293 a 500 μL de DMEM sem soro e vortex para misturar completamente. Misture bem os dois volumes. Deixar os plasmídeos e o lipoD293 complexarem durante 20 minutos.
- Adicionar gota a gota a um balão de cultura de2 células de 75 cm contendo 70% de células HEK293T confluentes contendo 9 ml de meio de cultura completo. Adicionar a mistura de transfecção directamente à camada celular utilizando uma pipeta serológica (balão na orientação horizontal).
- Substitua o meio por 20 mL de DMEM completo fresco 16 horas após a transfecção. Colha o sobrenadante após 72 horas após a transfecção. Alíquota viral sobrenadante em alíquotas de 1 mL e armazenar a -80 °C. O sobrenadante lentiviral é estável a -80 °C por pelo menos 1 ano.
4. Transdução lentiviral
- Antes da transdução lentiviral, estabeleça uma curva de morte ao usar uma construção lentiviral selecionável.
- Para a curva de eliminação, coloque a linha celular a ser transduzida em uma placa de 12 poços (confluência de aproximadamente 10-20%). Adicione uma curva de dose do seletor (concentrações aproximadas para curvas de eliminação: puromicina 0,5-10 μg / mL, blasticidina 1-20 μg / mL, geneticina (G418) 100-2000 μg / mL, higromicina 100-2000 μg / mL).
- Mudar o meio de três em três dias para assegurar uma concentração estável do selector escolhido.
- Adicione 1 mL de sobrenadante lentiviral a 9 mL de meio de cultura, contendo uma concentração final de 8 μg / mL de polibreno em 20-40% de células confluentes. Os volumes podem ser reduzidos, mantendo essa proporção de sobrenadante/meio.
- 16-24 horas após a transdução, troque o meio. Quando necessário, repita a etapa anterior para aumentar a penetrância do fenótipo (verifique a fluorescência para decidir se outra transdução é necessária).
- 48 horas após a transdução, selecionar as células usando o antibiótico correspondente ao marcador de resistência incorporado ao lentiviral. A concentração a ser usada para a seleção da população de células transduzidas deve matar a população do tipo selvagem dentro de 7 dias após a aplicação do seletor (ou seja, permitindo que as células transduzidas superem a população do tipo selvagem).
- Aplicar sobrenadante viral em diferentes multiplicidades de infecção (MOI's) para garantir que a transdução e as lesões genéticas incorridas pelo genoma celular não afetem negativamente a viabilidade celular ou a tumorigenicidade.
5. Criação de peixe-zebra
- No dia 0, 2 dias antes do enxerto de células cancerígenas, acasalar o peixe-zebra adulto em forma de "cruzamento familiar" à temperatura ambiente (Figura 1).
- Remova o tanque de peixe-zebra do sistema de alojamento (mantido a 28.5 °C).
- Separe os peixes em pequenos grupos de reprodução na proporção de 1:1 macho: fêmea, com 10 peixes por grupo. Coloque os peixes em pequenos tanques de reprodução, em água retirada do sistema de alojamento, acima de uma grade inclinada (inclinada, para imitar as águas rasas em que o peixe-zebra desovaria naturalmente).
NOTA: Induzido pelo declínio da temperatura de 28,5 ° C para a temperatura ambiente (25 ° C) e a entrada na próxima fase de luz do ciclo escuro / claro, os peixes irão desovar. - Em seguida, remova os adultos e transfira para o tanque de alojamento.
- Recolha os ovos e lave com água de ovo usando uma peneira. Divida os ovos em aproximadamente 75-100 por prato e mantenha a 28,5 ° C.
- Aproximadamente 6 horas após a coleta, limpe a louça de embriões mortos ou malformados.
- Na manhã seguinte, troque a água do ovo e limpe novamente os pratos de embriões mortos.
6. Colheita de células
NOTA: A preparação adequada das células é a chave para o procedimento de implantação, o uso de uma quantidade supérflua de células permite um processamento mais fácil a jusante. A terceira etapa de centrifugação é crítica, pois isso o deixará apenas com o pellet da célula, o PBS restante preso nas laterais do tubo da centrífuga excede em muito o volume final de ressuspensão.
- Pré-aqueça todos os meios e soluções utilizados na cultura de células em banho-maria a 37 °C antes da utilização.
- Adicionar 2 ml de TryplE por balão de cultura de 75 cm2 ou 1 ml por balão de 25 cm2 e incubar até que todas as células estejam arredondadas. Para a maioria das linhagens celulares, 2-5 minutos devem ser suficientes. Para células altamente epiteliais ou células fibroblásticas, 5 a 10 minutos devem permitir o descolamento adequado (a tripsinização insuficiente dificultará os processos a jusante e facilitará a agregação celular durante a implantação).
- Bata suavemente na lateral do frasco para desalojar as células restantes.
- Adicione ao volume de cultura original do meio completo. Pipetar para cima e para baixo suavemente, mas completamente, com uma pipeta serológica para cortar aglomerados de células em suspensão unicelular. Não gere espuma durante este processo, pois a espuma é indicativa de cisalhamento mecânico das células.
- Transfira para um tubo estéril de 15 mL e centrifugue por 5 minutos a 200 x g em temperatura ambiente. Aspire o sobrenadante e adicione 1 mL de PBS estéril. Ressuspenda as células com cuidado e completamente usando uma pipeta estéril de 1000 μL.
- Remova a suspensão de células de 20 μL para contagem e transfira a suspensão de células restante para a centrífuga. Centrifugue por 4 minutos a 200 x g em temperatura ambiente.
- ETAPA CRÍTICA: Remova todo o PBS, centrifugue por 30 s a 200 x g à temperatura ambiente e remova o PBS restante.
- Diluir as células para 250 células/nL em polivinilpirrolidona 40 a 2% (PVP40, 2% (p/v) em DPBS) da seguinte forma:

(por exemplo,
- Ressuspenda completamente as células, evitando a formação de bolhas de ar (as células podem ser mantidas por pelo menos 2 horas em PVP40 a 2% sem perda do potencial tumorigênico).
7. Modelagem de xenoenxerto
Todos os experimentos devem ser realizados em conformidade com os regulamentos locais de bem-estar animal.
Dependendo da aplicação, duas variações principais no desenho experimental são classificadas como uma avaliação do fenótipo (7.1 o estágio de pré-triagem) e, em segundo lugar, 7.2 uma triagem onde as células foram modificadas antes do enxerto ou 7.3 onde os embriões são tratados com um inibidor químico.
- Pré-triagem e determinação do potencial tumorigênico
- Enxertar larvas de peixe-zebra de interesse (WT, transgênica ou linha repórter) a 2 dpf com um número variável de células fluorescentes (ou seja, 200, 400, 600 ±100).
- Rastreie as larvas 16-24 horas após a injeção para remover outliers (números de células extremamente altos ou baixos em circulação para o modelo ectópico, ou células dentro da cabeça para o modelo ortotópico) e remover peixes enxertados incorretamente. Indicar o número de larvas por grupo experimental para análise de grupo versus análise cinética das mesmas larvas.
- Monitore as larvas do peixe-zebra em intervalos regulares (1,2,4,6 dias após a injeção (dpi)) e obtenha imagens de 20 indivíduos (conforme descrito nas etapas 9 e 10), de um pool de ±50 larvas.
- Monitore o fenótipo geral e as progressões da doença e, posteriormente, quantifique com ImageJ (medindo a densidade integrada do sinal do fluoróforo nas células cancerígenas).
- Plote os dados para visualizar a cinética de crescimento das células cancerígenas dentro do peixe-zebra (Figura 3).
- Modifique as células a priori (derrube ou elimine um gene de interesse) e enxerte no peixe-zebra.
- Enxertar peixes e remover todos os fenótipos indesejados (por condição).
- Imagem dos indivíduos a 1 dpi (20 larvas por grupo). Os indivíduos podem ser fotografados em intervalos definidos (1,2,4 e 6 dpi).
- A 6 dpi após a imagem, eutanasiar o peixe por overdose de tricaína (10 vezes mais do que a dose de 0,4 mg / mL) e descartar em papel absorvente que forra um funil.
- Trate os peixes com medicamentos após o enxerto.
- Antes da aplicação do medicamento em peixe-zebra enxertado, determine a dose máxima tolerada (MTD) no peixe-zebra (titule de 10 μM a 0,150 nM, usando o maior volume de solvente como controle negativo), definimos o MTD como a concentração em que >80% dos indivíduos sobrevivem a todo o tratamento.
- Um dia após a injeção, remova os fenótipos indesejados.
- Divida aleatoriamente os peixes em grupos (36-48 indivíduos/condição) e mantenha em uma placa de 24 poços com 6 larvas por poço em 1 mL de água de ovo.
- Aplicar medicamentos 24 horas após o enxerto. Como controle, use a mesma quantidade de solvente (DMSO, EtOH etc.) no volume mais alto aplicado para um grupo experimental.
- Inicie o tratamento medicamentoso na dose máxima tolerada. Troque a água do ovo contendo o medicamento a cada dois dias. Remova a água do ovo e as larvas mortas o mais completamente possível durante cada troca.
8. Injeção
NOTA: Use um controlador de pulso pneumático acoplado a uma linha de ar comprimido, fornecendo pressão em excesso de 100 psi. Isso permite pressão suficiente para injetar (≈20 psi) e ejetar possíveis agregados celulares (≈100 psi). A pressão e o tempo de partida devem ser de aproximadamente 200 ms a 20 psi. Se um deles tiver que ser diminuído em mais de 50% no início da injeção, a suspensão celular é muito fluida (concentração de células ou PVP40 muito baixa) ou a abertura da agulha é muito grande.
- Remova cuidadosamente uma agulha capilar de seu recipiente. Quebre a agulha para formar uma abertura de ø20 μm, usando uma pinça de relojoeiro fino.
- Ressuspenda as células com cuidado e completamente usando uma ponta de pipeta de 20 μL. Pipetar a suspensão de células na agulha capilar de vidro aberta usando uma ponta longa (microcarregador). Carregue a agulha no micro manipulador.
- Coloque ~ 20-40 larvas anestesiadas em 0,04 mg / mL de tricaína em uma placa de agarose usando uma pipeta de transferência. Remova o excesso de umidade para imobilizar as larvas usando uma pipeta de transferência. As larvas serão orientadas principalmente de forma lateral devido à presença de um saco vitelino ainda relativamente grande.
- Injetar as larvas com aproximadamente 200, 400 e 600 células através do Ducto de Cuvier (doC) para o modelo ectópico.
- Da mesma forma, injete larvas retroorbitalmente (RO). Para produzir o modelo ortotópico (injetando 100 ±50 células), modifique o comprimento do pulso pneumático na picobomba (comece em ~ 20 psi, 200 ms e ajuste de acordo). Durante a injeção, certifique-se de que as larvas não sequem. Certifique-se de que todas (ou a maioria) das larvas sejam injetadas.
- Lave as larvas injetadas com água fresca de ovo e transfira para uma placa de Petri limpa rotulada (agrupando até 150 indivíduos por prato). Repita este processo até que larvas suficientes sejam injetadas.
- Após o enxerto, mantenha os peixes a 34 ° C em uma incubadora umidificada, onde 34 ° C é a temperatura mais alta prontamente tolerada pelo peixe-zebra e permite o enxerto eficiente de células cancerígenas de mamíferos.
NOTA: Em geral, com a injeção de linhagens celulares únicas em doC e RO, observamos uma morte aproximada devido a danos mecânicos de <5% (danos mecânicos matam as larvas entre 1-16 horas após a injeção).
9. Triagem
- Usando um microscópio de estereofluorescência, rastreie os peixes para o fenótipo apropriado 1 hora após a implantação ao comparar células modificadas a priori (ou 1 dia após a implantação, ao rastrear drogas, antes da atribuição aleatória em grupos de tratamento).
- As larvas implantadas através do doC devem ter células na cauda entre 1 hora e 16 horas após a implantação. Remova todos os outros peixes, incluindo peixes que apresentem anormalidade, da piscina injetada.
NOTA: As larvas implantadas retroorbitalmente devem ter células apenas no interstício atrás do olho, as larvas que têm células espalhadas por toda a cabeça ou corpo são removidas da piscina. - Limpe as larvas selecionadas positivamente e atribua aleatoriamente a grupos experimentais.
- Após o enxerto, manter os peixes a 34 °C em uma incubadora umidificada e monitorar diariamente. A disseminação hematogênica de células implantadas através do doC é quase instantânea, enquanto a disseminação metastática de células implantadas na cavidade RO se espalhará após 2-4 dias.
10. Imagem epifluorescente de larvas de peixe-zebra
- Anestesiar larvas de peixe-zebra com 0,2 mg/mL de tricaína, adicionando tricaína à água do peixe ou movendo uma subpopulação de peixes do prato de manutenção para um prato contendo 0,2 mg/mL de tricaína.
- Mantenha o peixe-zebra em um prato com tricaína até que permaneça estacionário, até que a estimulação da linha lateral não induza o comportamento de vôo.
- Transfira o peixe para uma placa de Petri coberta com agarose, aproximadamente 10 por prato. Remova a maior parte da água, levantando suavemente uma extremidade do prato (permitindo que a água se acumule suavemente na extremidade inferior da placa de Petri). Se feito com cuidado, todos os peixes se alinharão, com as caudas voltadas para baixo.
- Imagine todos os peixes da parte superior do prato até a parte inferior. Em seguida, lave o peixe com água de ovo em um prato sem tricaína.
- Repita até que indivíduos suficientes sejam fotografados.
- Em seguida, transfira as larvas de volta para 34 ° C ou descarte (a 6 dpi) por meio de overdose com tricaína (ou seja, 0,5 mg / mL, incubando por 10 min, antes de descartar em papel absorvente que reveste um funil).
11. Imagem confocal de larvas de peixe-zebra (enxertadas)
- Anestesiar o peixe-zebra com 0,2 mg/mL de tricaína conforme descrito anteriormente.
- Coloque um prato confocal com fundo de vidro sob um microscópio estéreo e concentre-se no fundo do prato. Transfira 5-10 larvas para um prato confocal com fundo de vidro. Remova o máximo de água possível.
- Cubra as larvas com agarose de baixo ponto de fusão a 42 °C, 1% dissolvida em água de ovo. Certifique-se de que a agarose esfriou até pelo menos 42 °C antes de usar; Temperaturas mais altas podem prejudicar ou matar as larvas.
- Usando o estereomicroscópio, oriente rápida mas suavemente as larvas empurrando-as para baixo, usando uma ponta de micro carregador aparada. Se for necessária uma orientação ventral, segure as larvas no lugar com as pinças de uma pinça de relojoeiro (sem tocar no embrião).
- Enquanto os conjuntos de agarose fazem ajustes finos na orientação das larvas. Deixe as larvas endurecerem completamente antes de transferi-las para o microscópio confocal.
12. Configurando o microscópio confocal
- Ligue as linhas de laser de excitação verde (488 nm) e vermelha (564 nm). Coloque a antena confocal no suporte do microscópio confocal. Usando a epifluorescência, mova o feixe de luz para coalescer com o primeiro peixe (configuração x e y). Através da ocular defina o foco para coincidir com o centro das larvas (configuração z).
- Defina 700 de ganho em ambos os canais fluorescentes, 1-5% de potência do laser. Aumente a potência do laser e diminua o deslocamento para aproximar a faixa dinâmica total. Não sature demais o sinal, mas aumente o sinal para mostrar apenas alguns pixels saturados.
- Ao capturar um ponto, defina o início e o fim das larvas ao longo de um eixo ( x ou y), se definido ao longo de um eixo, um embrião inteiro pode ser visualizado em 1 x 4 segmentos e pode ser pós-processado em uma imagem usando ImageJ.
- Após a imagem, remova as larvas da agarose rasgando-a suavemente ao redor das larvas embutidas usando uma pinça de relojoeiro. Caso contrário, eutanasiar as larvas por overdose de tricaína não diluída, cobrindo a agarose com uma camada de tricaína e incubando por 10 minutos.
13. Análise de dados
- Abra os conjuntos de dados individuais no ImageJ/Fiji (ou seja, controle, medicamento A, medicamento B, medicamento A+B) separadamente, começando com o controle do veículo.
- Abra a macro de análise (script anotado disponível) (http://doi.org/10.5281/zenodo.4290225).
- Em resumo, a análise macro faz o seguinte: concatena todas as imagens abertas (uma condição); divide as imagens nos canais separados que compõem a imagem; fecha todos os canais acessórios (saindo do canal da célula cancerígena); executa um algoritmo de limiar, em toda a sequência concatenada; mede a densidade integrada de cada imagem individual; e salva as medidas como uma planilha do Excel na pasta raiz.
- Execute a análise de macro em todas as condições.
- Combine medições (em geral, pelo menos n = 2 * 20) e remova outliers (teste Q no Graph pad Prism v8).
- Normalize as medições para o controle de solvente ou para o dia 1 (dependendo do tipo de experimento, o primeiro para um experimento de inibição de drogas e o último para um experimento de cinética de crescimento). Expresse as medições como carga de células cancerígenas normalizadas (eixo y ) ao longo do tempo ou condição (eixo x ), conforme mostrado na Figura 3 e na Figura 4, respectivamente.
Resultados
Fornecemos instruções passo a passo para uma abordagem rápida e fácil para progredir de uma nova linhagem celular para sua análise. Começamos com a superexpressão de um marcador fluorescente usando um de superexpressão lentiviral (etapas 3 e 4). Isso é seguido pela preparação da célula para garantir o menor volume morto possível durante a injeção, permitindo injetar um alto número de células no doC e no espaço retroorbital (etapas 6 e 7). Posteriormente, realizamos a aquisição de dados de rendimento semi-alto usando microscopia estereofluorescente e microscopia confocal de maior ampliação para análise qualitativa da disseminação de células cancerígenas de corpo inteiro (Figura 2 e etapas 10, 11 e 12). Deve-se ter cuidado ao adquirir dados, pois para garantir a reprodutibilidade para imagens microscópicas estéreo e confocais, as configurações genéricas e a padronização são delineadas (etapas 11 e 12). A análise dos dados é discutida (usando imageJ/Fiji) 16, juntamente com a padronização usando macros imageJ (etapa 13).
Na etapa 3, mencionamos a marcação transitória de células (cancerígenas) para realizar uma pré-triagem rápida para avaliar o potencial tumorigênico de uma nova linhagem de células cancerígenas. Uma ressalva importante é que, embora fácil de usar e de longa duração, a coloração transitória aqui descrita tem a possibilidade de formar artefatos (ou seja, deve-se tomar cuidado para garantir que os fragmentos celulares possam ser distinguidos de células inteiras, como foi realizado extensivamente por Fior e colegas 9). Em nossa experiência, a formação desses artefatos está diretamente ligada à extrema estabilidade da mancha e ao brilho (mesmo após a morte celular), onde fragmentos celulares são dispersos e absorvidos por células imunes, que podem posteriormente ser falsamente concluídas como derivadas de metástases ativas.
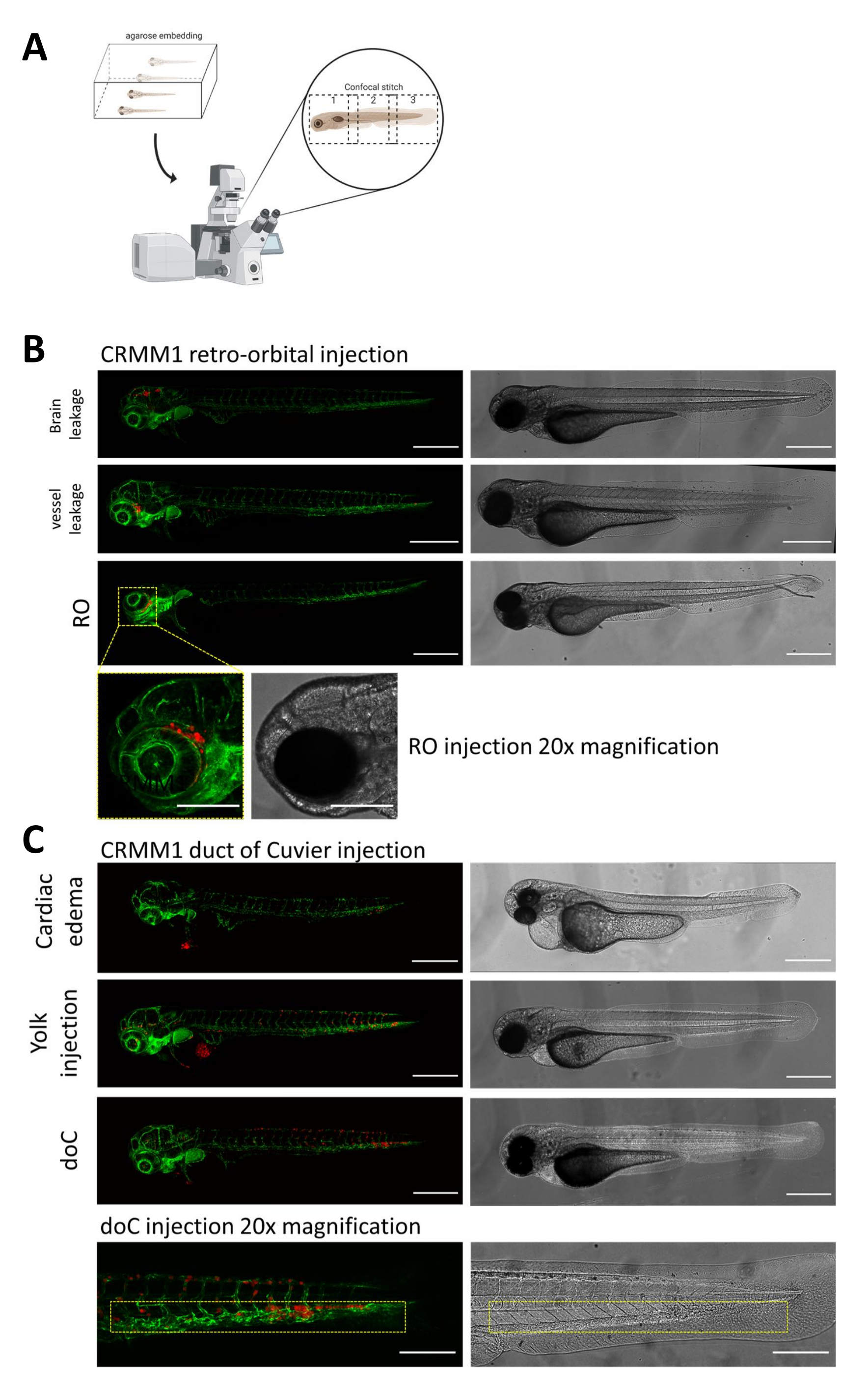
Em ambos os modelos descritos, o enxerto sistêmico através do doC e o enxerto localizado no espaço retro-orbital, a triagem completa das larvas um dia após a injeção é de suma importância. Conforme mostrado na Figura 2B , todas as larvas que exibem deslocamento mecânico das células enxertadas na área da cabeça (além do local retro-orbital) no modelo retro-orbital e células no saco vitelino, ou exibindo um edema no pool injetado de doC, devem ser removidas. Todos os fenótipos selecionados negativamente são exibidos como pontos confocais de alta resolução na Figura 2, mas podem ser facilmente vistos e removidos por meio de observação microscópica estereoscópica.
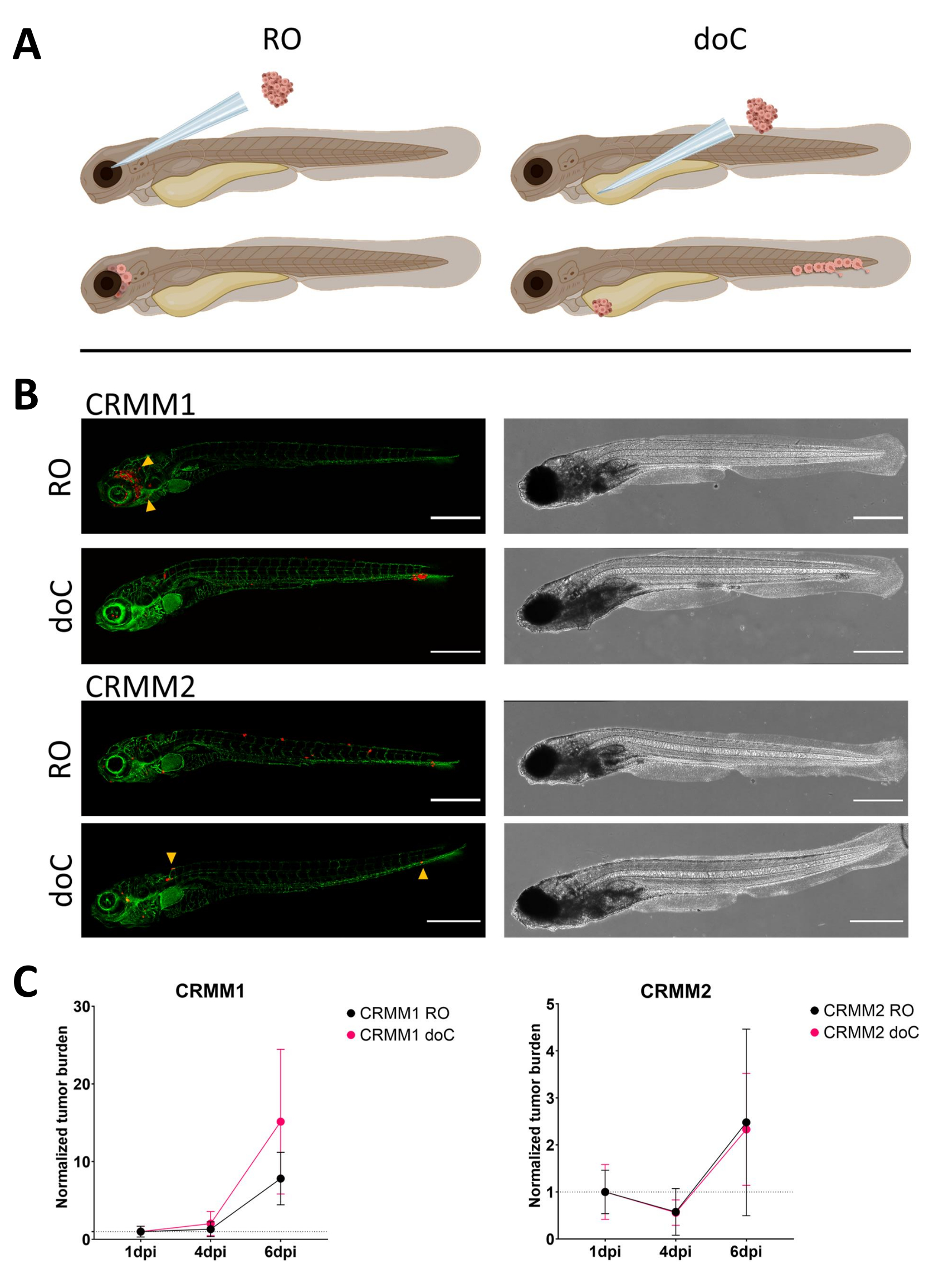
Com o tempo, as células migrarão e proliferarão. Para o modelo retro-orbital, observamos infiltração em tecidos vizinhos para CRMM1, mas observamos menor proliferação para CRMM2. Observamos metástases à distância entre 2-4 dpi em alguns indivíduos (20%), onde medimos uma diferença significativa em 6 dpi, conforme mostrado na Figura 4. Para ambas as linhagens celulares, testamos o potencial proliferativo quando injetado em ambos os locais. Para CRMM1, houve um aumento significativo (p<0,0001) no número de células cancerígenas para ou nos locais de injeção, quando exibido como carga de células tumorais normalizada, normalizando para o primeiro dia para cada modelo (aumento de 7,8 vezes, ±3,2 para o modelo RO e um aumento de 15 vezes ±8,8 para o modelo doC). O CRMM2 não apresentou crescimento significativo quando normalizado para o primeiro dia para cada modelo individual (aumento de 2,4 vezes, aumento de ± 1,9 e 2,3 vezes, ± 1,14 para RO e doC). Descobriu-se que o CRMM1 prolifera prontamente no tecido retro-orbital e no tecido hematopoiético caudal após o enxerto. A linhagem celular CRMM2 foi menos proliferativa em ambos os modelos, mas curiosamente foi considerada capaz de metástase à distância quando injetada no espaço retro-orbital, como mostrado na Figura3B, C.
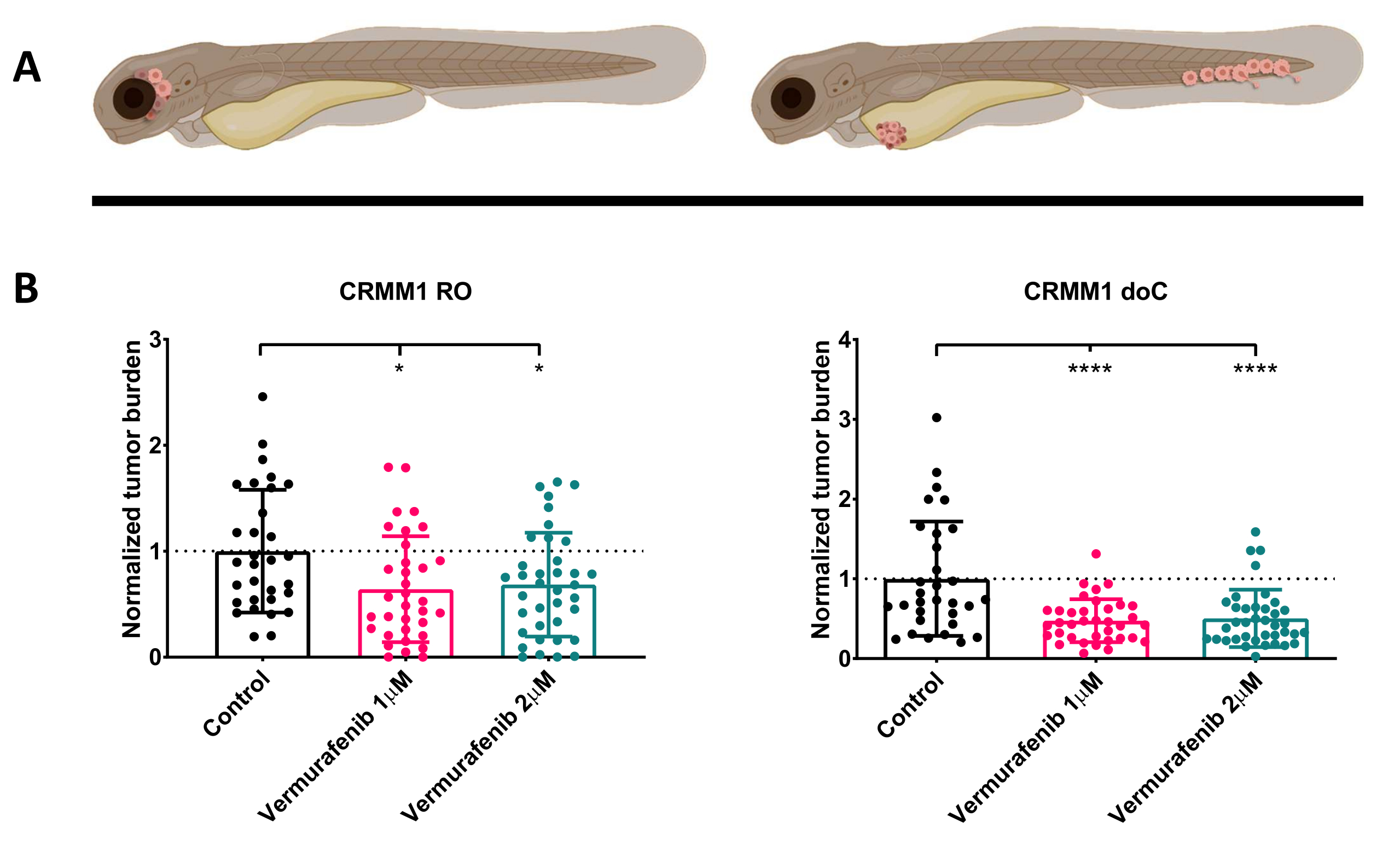
Após a triagem das larvas injetadas a 1 dpi e a atribuição aleatória dos indivíduos aos grupos de tratamento ou controle, os peixes foram tratados por 6 dias, trocando a água contendo Vemurafenibe (este inibidor pode ser facilmente trocado por qualquer outro composto antitumoral titulado). Optamos por elaborar o modelo de disseminação de melanoma conjuntival hematogeno publicado anteriormente enxertando CRMM114, testando a eficácia do Vemurafenibe em CRMM1 enxertado ortotopicamente. O CRMM1 mostrou uma forte redução significativa do grupo enxertado ectopicamente tratado com Vemurafenibe (P<0,0001) e uma resposta atrofiada, mas significativa, para o modelo enxertado ortotopicamente (p<0,05), conforme mostrado na Figura 4.

Figura 2. Avaliação fenotípica e triagem após a injeção. A) Representação esquemática da geração de ponto confocal de xenoenxerto de peixe-zebra, produzindo imagens perfeitas e de alta resolução após a integração da projeção confocal subsequente. Aqui, os xenoenxertos de peixe-zebra são incorporados em agarose de baixo ponto de fusão a 1% e montados em uma placa confocal com fundo de vidro (conforme descrito na etapa 11.3). B) Todos os resultados possíveis do enxerto retro-orbital e do ducto de Cuvier são exibidos injetados em peixe-zebra repórter de vaso sanguíneo fluorescente verde (TG: fli: GFP), com células coradas através de lentivírus sobre a expressão de tdTomato). Denotamos o enxerto correto a 1 dpi (painel RO) e os fenótipos indesejados (vazamento cerebral e vazamento de vasos sanguíneos). As duas últimas populações devem ser removidas para garantir que não confundam os resultados experimentais a jusante. C) Os fenótipos indesejados para o enxerto hematogênico através do ducto de Cuvier (doC) são contornos onde larvas edematosas cardíacas (edema cardíaco) e larvas com células vazando para o saco vitelino (injeção de gema) devem ser removidas para evitar interferência nas medições a jusante. As larvas corretamente injetadas são inseridas em grupos experimentais conforme descrito na etapa 7.1. (Todas as imagens adquiridas a 1 dpi, usando um microscópio confocal, barras de escala de 200 μm. As caixas amarelas indicam locais metastáticos para enxertos RO e doC, região da cabeça e tecido hematopoiético caudal, respectivamente). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3. A análise comparativa das linhagens celulares de melanoma conjuntival CRMM1 e CRMM2 mostra capacidade metastática e de crescimento diferencial. A) Representação esquemática de modelos de injeção, modelo retro-orbital (RO) e modelo de enxerto hematogênico (doC) os peixes utilizados são repórteres de vasos sanguíneos verdes TG (fli: GFP), com células superexpressando tdTomato mostradas em vermelho. B) Fenótipos representativos de peixes enxertados com CRMM1 e CRMM2, CRMM1 exibe enxerto eficiente (RO e doC) e invasão em pequena escala no tecido ao redor do local do enxerto de RO (RO, pontas de setas amarelas). CRMM2 exibe uma eficiência de enxerto notavelmente menor para ambos os modelos de enxerto, mas mostra metástase distante quando injetado retroorbitalmente (como mostrado em RO, denotado pelas pontas de seta). (Todas as imagens adquiridas a 6 dpi, um microscópio confocal, barras de escala de 200 μm. As pontas das setas amarelas indicam locais metastáticos para enxertos RO e doC, região da cabeça e tecido hematopoiético caudal, respectivamente). C) Gráficos de enxerto cinético para CRMM1 e CRMM2, comparando ambos os modelos de enxerto para o dia 1 (normalizando para o dia 1), há um aumento significativo (p<0,0001) na carga tumoral normalizada para a linha celular CRMM1 (entre 1 dpi e 6 dpi) onde há uma tendência ascendente (não significativa) para CRMM2. CRMM1 revela uma diferença significativa entre o crescimento de RO e doC, onde o modelo doC mostra uma taxa de expansão tumoral mais alta (aproximadamente 2 vezes maior para as larvas enxertadas de doC). Os gráficos exibem a média e o erro padrão da média (SEM). Todos os grupos foram normalizados para 1 dpi para cada condição individual. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4. O inibidor de BRAF V600E Vemurafenibe inibe significativamente as larvas de peixe-zebra enxertadas com melanoma conjuntival RO e doC. A) Representação esquemática dos fenótipos do peixe-zebra, modelos RO e doC. B) As larvas enxertadas com RO e doC, injetadas com a linhagem celular de melanoma conjuntival CRMM1, apresentam uma redução significativa da carga tumoral normalizada (p<0,05 e P<0,001, respectivamente). Os modelos de peixe-zebra enxertados com doC indicam uma resposta aumentada ao medicamento e uma relação independente da dose com a inibição do medicamento, indicando uma possível saturação da inibição). Os gráficos mostram a média e o erro padrão da média (SEM), Todos os grupos foram normalizados para controlar cada linha celular individual. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Reagente | Volume |
| psPAX2 | 1,71 pmol (12,14 μg) |
| pMD2.G | 0,94 pmol (3,66 μg) |
| Plasmídeo de transferência* | 1,64 pmol (calcular o volume exato) |
Tabela 1.
Discussão
Aqui, definimos uma abordagem meticulosa para modelar o melanoma ocular primário e metastático em xenoenxertos de peixe-zebra. Ao combinar uma injeção ortotópica localizada e uma injeção ectópica sistêmica, recapitulamos a etiologia da carcinogênese para um câncer onde nenhum modelo animal estava disponível anteriormente. A transparência inerente da larva inicial do peixe-zebra permite o rastreamento de células cancerígenas marcadas com fluorescência em um nível de animal inteiro, garantindo a fácil visualização de potenciais locais metastáticos17. Além disso, a análise microscópica confocal de alta ampliação nos permite rastrear células em uma resolução subcelular10.
Fornecemos instruções passo a passo para uma abordagem rápida e fácil para progredir de uma nova linhagem celular para o estabelecimento do xenoenxerto e sua análise. Começamos com a superexpressão de um marcador fluorescente usando um de superexpressão lentiviral (etapas 3 e 4) seguido de preparação celular para garantir o menor volume morto possível durante a injeção. Isso permite a injeção de um alto número de células no espaço doC e retroorbital (etapas 7 e 8). Em seguida, realizamos a aquisição de dados de rendimento semi-alto usando microscopia estereofluorescente e microscopia confocal de maior ampliação para análise qualitativa da disseminação de células cancerígenas de corpo inteiro (Figura 2 e etapas 9 e 10). Deve-se ter cuidado ao adquirir dados, pois para garantir a reprodutibilidade para imagens microscópicas estéreo e confocais, as configurações genéricas e a padronização são delineadas (etapas 11 e 12). A análise de dados é discutida (usando imageJ/Fiji) 16, juntamente com a padronização usando macros ImageJ (etapa 13).
Na etapa 3, mencionamos a marcação transitória de células (cancerígenas) para realizar uma pré-triagem rápida para avaliar o potencial tumorigênico de uma nova linhagem de células cancerígenas. Uma ressalva importante é que, embora fácil de usar e de longa duração, a coloração transitória aqui descrita tem a possibilidade de formar artefatos (por exemplo, deve-se tomar cuidado para garantir que os fragmentos celulares possam ser distinguidos das células inteiras, como foi realizado extensivamente por Fior e colegas 9). Em nossa experiência, a formação desses artefatos está diretamente ligada à extrema estabilidade da mancha e ao brilho (mesmo após a morte celular), onde fragmentos celulares são dispersos e absorvidos por células imunes, que podem posteriormente ser falsamente concluídas como derivadas de metástases ativas.
Usando esses modelos, simulamos o desenvolvimento do tumor primário confinando fisicamente as células enxertadas dentro do interstício retro-orbital. A triagem completa subsequente 1 dia após o enxerto garante que as células encontradas em locais distantes posteriormente no experimento tenham metástase ativamente (intravasadas e disseminadas, em última análise, para extravasar no nicho metastático). O enxerto através do doC, a veia cardinal comum embrionária, permite a implantação fácil e altamente reprodutível de grandes quantidades para as células (com um excedente de 600 células quando devidamente concentradas), contornando efetivamente os estágios primários da cascata metastática (intravasamento) e permitindo que nos concentremos nos estágios posteriores da cascata metastática (adesão, extravasamento e crescimento). Embora sejam ferramentas poderosas quando usadas corretamente, ambos os modelos devem ser monitorados extensivamente durante o primeiro dia após o enxerto para garantir que nenhuma conclusão falsa positiva seja tirada durante os estágios posteriores do experimento.
Em consonância com publicações anteriores, mostramos que as linhas de melanoma conjuntival formam prontamente colônias metastáticas após a disseminação por todo o sistema de circulação sanguínea do peixe-zebra14. Aqui relatamos a expansão do repertório de enxerto com a injeção retro-orbital como modelo ortotópico, e a subsequente metástase ativa para o tecido hematopoiético caudal da linhagem celular CRMM2. Posteriormente, relatamos a eficácia do inibidor específico de BRAF V600E Vemurafenibe também na forma primária de melanoma conjuntival quando modelado em larvas de peixe-zebra.
Usando os métodos acima mencionados, um pesquisador qualificado é capaz de gerar mais de centenas de larvas enxertadas por dia (aproximadamente 200 por hora) de qualquer um dos modelos propostos. Em um prazo de duas semanas, um medicamento pode ser titulado para a dose máxima tolerada e rastreado no modelo de xenoenxerto estabelecido. Do início ao fim, o uso de uma linhagem celular não transduzida para ter um perfil de sensibilidade a medicamentos no modelo de peixe-zebra pode ser alcançado em um mês (dado que a linha celular injetada é tumorigênica dentro do modelo de peixe-zebra). Em nossas mãos, apenas 20 larvas por experimentos e duas repetições biológicas produziram reprodutivelmente uma inibição robusta da droga, quando dois experimentos individuais entram em conflito (ou não produzem inibição de crescimento estatisticamente significativa), uma terceira repetição biológica pode ser conduzida.
Por meio de pequenos ajustes, esses modelos nos permitiram adaptar rapidamente essas estratégias de implantação para glioblastoma (injeção na cavidade cerebral posterior), câncer de mama (injeção de doC) e osteossarcoma (doC), entre outros 18,19,20,21. Esses modelos podem ser posteriormente utilizados tanto para pesquisa básica quanto para triagem pré-clínica de medicamentos únicos e estratégias combinatórias de medicamentos. Recentemente, descrevemos diferentes regimes de administração de medicamentos e sua fotoativação usando esses modelos 13.
Divulgações
Nenhum.
Agradecimentos
Este trabalho foi apoiado por financiamento do programa de investigação e inovação Horizonte 2020 da União Europeia ao abrigo do acordo de subvenção n.º 667787 (projeto UM Cure 2020, www.umcure2020.org). O Conselho Chinês de Bolsas de Estudo é gentilmente reconhecido por uma bolsa de doutorado para JY
Materiais
| Name | Company | Catalog Number | Comments |
| 2.5mm box filament | Science products | FB255B | for pulling micro injection needles using a Sutter P97 or P1000 |
| 3mL transfer pipettes | Merck | Z350796 | for transfer and selection of zebrafish embryos |
| Agarose | Milipore | 2120 | 1.5% (w/v) in eggwater, 1.5 g in 100 mL DPBS, microwave to dissolve, for injecting and stereofluorescence imaging of zebrafish larvae |
| Capillaries: borosilicate glass outer | World precision instruments | BF100-78-10 | Borosilicate glass capillaries used for needle preparation |
| DMSO | Sigma | D8418 | Often used as solvent in drug treatments, should be stored at 2-8°C the dark. |
| DPBS | Thermo Fischer Scientific | 14190144 | Dulbecco’s phosphate buffered saline, without Mg2+ and Ca2+ for washing the cells, lack of Ca2+ impairs cell-cell adhesion through cadherins and prevents cell aggregation during injection |
| Egg water | Instant ocean | SS15-10 | 0.6 mg/L final concentration sea salt in demineralized water |
| GFP encoding lentiviral transfer plasmid | Addgene | Plasmid #106172 | Generated in Snaar lab, available at Addgene |
| Hek293T | ATCC | CRL-3216 | Stable cell line for generating lentiviral particles, contains SV40-T antigen required for the generation of lentiviral particles |
| Leica sp8 confocal | Leica | Leica TCS SP8 | automated stage confocal microscope with 405/488/514/635nm lasers |
| LipodD293 | Signagen | SL100668 | Highly efficient HEK293t optimized transfection reagent |
| Low-melting agarose | Milipore | 2070 | 1% (w/v) in eggwater 1.5 g in 100 mL DPBS, microwave to dissolve, for embedding zebrafish larvae for confocal imaging |
| Micro loader tips | Fischer scientific | 10289651 | flexible microloader tips |
| Micro manipulator | World precision instruments | M3301R | x/y/z manual micro manipulator for microinjection |
| Needle puller: P-97 or P-1000 | Sutter | P-97 | needle puller used for generating standardized micro engraftment needles |
| Nr.5 watchmakers forceps | VWR | HAMMHSC818-11 | fine watchmakers forceps used for breaking back needles |
| Picopump | World precision instruments | SYS-PV820 | pulse controller supplying pressure for microinjection |
| pMD2.G | Addgene | plasmid #12259 | Gifted by Didier Trono, 2nd generation lentiviral virulence plasmid |
| psPAX2 | Addgene | plasmid #12260 | Gifted by Didier Trono, 2nd generation lentiviral packaging plasmid |
| PVP40 | Sigma-Aldrich | PVP40 | Polyvinylpyrrolidone average mol wt 40,000) PVP40 2% (w/v) in DPBS, 1 g PVP40 in 50 mL DPBS. Vortex and incubate at 37°C to facilitate dissolving. Store at room temperature. |
| tdTomato encoding lentiviral transfer plasmid | Addgene | Plasmid #106173 | Generated in Snaar lab, available at Addgene |
| transmitted light stereo microscope | Leica | leica M50 with (MDG33 base) | leica transmitted light microscope with mirror adjustable illumination. |
| Tricaine | Sigma-Aldrich | E10521 | Ethyl 3-aminobenzoate methanesulfonate or MS-222 |
| TryplE | Thermo Fischer Scientific | 12604-01 | Synthetic trypsine replacement, less damaging to the cells and allows for the gentle dispersion of strongly adherent cells. (Thermo- |
| willco dish | WillCo wells | GWST-5040 | 50mm glass bottom dishes, allow for the embedding of up to 20 zebrafish larvae, enabling the imaging of multiple conditions in one dish due to its large optical glass surfac |
Referências
- Yang, J., Manson, D. K., Marr, B. P., Carvajal, R. D. Treatment of uveal melanoma: where are we now. Therapeutic Advances in Medical Oncology. 10, (2018).
- Wong, J. R., Nanji, A. A., Galor, A., Karp, C. L. Management of conjunctival malignant melanoma: A review and update. Expert Review of Ophthalmology. 9, 185-204 (2014).
- Nguyen, D. X., Bos, P. D., Massagué, J. Metastasis: from dissemination to organ-specific colonization. Nature Reviews Cancer. 9, 274-284 (2009).
- White, R. M., et al. Transparent Adult Zebrafish as a Tool for In Vivo Transplantation Analysis. Cell Stem Cell. 2, 183-189 (2008).
- Zon, L. I., Peterson, R. T. In vivo drug discovery in the zebrafish. Nature Reviews Drug Discovery. 4, 35-44 (2005).
- Howe, K., et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 496, 498-503 (2013).
- Palmblad, M., et al. Parallel deep transcriptome and proteome analysis of zebrafish larvae. BMC Research Notes. 6, 428 (2013).
- Yan, C., et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell. 177, 1903-1914 (2019).
- Fior, R., et al. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proceedings of the National Academy of Sciences of the United States of America. 114, 8234-8243 (2017).
- Campbell, P. D., Chao, J. A., Singer, R. H., Marlow, F. L. Dynamic visualization of transcription and RNA subcellular localization in zebrafish. Development. 142, 1368-1374 (2015).
- Campeau, E., et al. A Versatile Viral System for Expression and Depletion of Proteins in Mammalian Cells. PLoS One. 4, 6529 (2009).
- vander Helm, D., et al. Mesenchymal stromal cells prevent progression of liver fibrosis in a novel zebrafish embryo model. Scientific Reports. 8, 16005 (2018).
- Chen, Q., et al. TLD1433 photosensitizer inhibits conjunctival melanoma cells in zebrafish ectopic and orthotopic tumour models. Cancers. 12, (2020).
- Pontes, K. C. d. e. S., et al. Evaluation of ( fli:GFP ) Casper Zebrafish Embryos as a Model for Human Conjunctival Melanoma. Investigative Opthalmology & Visual Science. 58, 6065 (2017).
- Liverani, C., et al. Innovative approaches to establish and characterize primary cultures: an ex vivo 3D system and the zebrafish model. Biology Open. 6, 133-140 (2017).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9, 676-682 (2012).
- White, R. M., et al. Transparent Adult Zebrafish as a Tool for In Vivo Transplantation Analysis. Cell Stem Cell. 2, 183-189 (2008).
- Mercatali, L., et al. Development of a patient-derived xenograft (PDX) of breast cancer bone metastasis in a Zebrafish model. International Journal of Molecular Sciences. 17, (2016).
- Tulotta, C., et al. Imaging cancer angiogenesis and metastasis in a zebrafish embryo model. Advances in Experimental Medicine and Biology. 916, 239-263 (2016).
- Paauwe, M., et al. Endoglin expression on cancer-associated fibroblasts regulates invasion and stimulates colorectal cancer metastasis. Clinical Cancer Research. 24, 6331-6344 (2018).
- Cao, J., et al. Overexpression of EZH2 in conjunctival melanoma offers a new therapeutic target. Journal of Pathology. 245, (2018).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados