
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Citometria a flusso Protocolli per superficie e intracellulare Antigen analisi di tipi di cellule neurali
In questo articolo
Riepilogo
We provide a detailed description of a protocol for flow cytometric analysis of surface antigens and/or intracellular antigens in neural cell types. Critical aspects of experimental planning, step-by-step methodological procedures, and fundamental principles of flow cytometry are explained in order to enable neurobiologists to exploit this powerful technology.
Abstract
Flow cytometry has been extensively used to define cell populations in immunology, hematology and oncology. Here, we provide a detailed description of protocols for flow cytometric analysis of the cluster of differentiation (CD) surface antigens and intracellular antigens in neural cell types. Our step-by-step description of the methodological procedures include: the harvesting of neural in vitro cultures, an optional carboxyfluorescein succinimidyl ester (CFSE)-labeling step, followed by surface antigen staining with conjugated CD antibodies (e.g., CD24, CD54), and subsequent intracellar antigen detection via primary/secondary antibodies or fluorescently labeled Fab fragments (Zenon labeling). The video demonstrates the most critical steps. Moreover, principles of experimental planning, the inclusion of critical controls, and fundamentals of flow cytometric analysis (identification of target population and exclusion of debris; gating strategy; compensation for spectral overlap) are briefly explained in order to enable neurobiologists with limited prior knowledge or specific training in flow cytometry to assess its utility and to better exploit this powerful methodology.
Introduzione
Citometria a flusso è stato ampiamente sfruttato in immunologia, ematologia e oncologia per definire popolazioni di cellule con proprietà intrinseche scatter, espressione dell'antigene di superficie cellulare, e altri parametri di fluorescenza 1-3. Le nostre intuizioni sviluppo linea di sangue e malattie sono il risultato di un notevole grado di raffinatezza continua di questa metodologia, dopo la sua iniziale 4,5 attuazione. Una maggiore consapevolezza delle potenzialità analitica quantitativa e globale della citometria a flusso ha recentemente incoraggiato il suo uso più diffuso nel campo della ricerca sulle cellule staminali e può consentire allo stesso modo profondo il progresso in un arco di tempo più breve 6. Tuttavia, l'applicazione di citometria a flusso per analizzare specificamente e isolare popolazioni neuronali è stata a lungo percepita come stimolante. Contrariamente alle cellule ematopoietiche che esistono naturalmente in sospensione, tipi di cellule neurali sono tipicamente raccolte da fonti eccessivamente complesse che possono includere glia e vari other cellule circostanti, nonché una intricata rete di neuroni processo fruttiferi. Di conseguenza, neurobiologia ha ancora attuare la versatilità di citometria a flusso per il suo potenziale completo in routine di ricerca quotidiana. Tuttavia, fintanto che una sospensione valida singola cellula può essere generato (e protocolli è stato ideato e ottimizzata a tale scopo 7), citofluorimetria e classificare fluorescenza delle cellule attivate (FACS) può essere considerato un prezioso elemento del repertorio analitico neurobiologia 8-11.

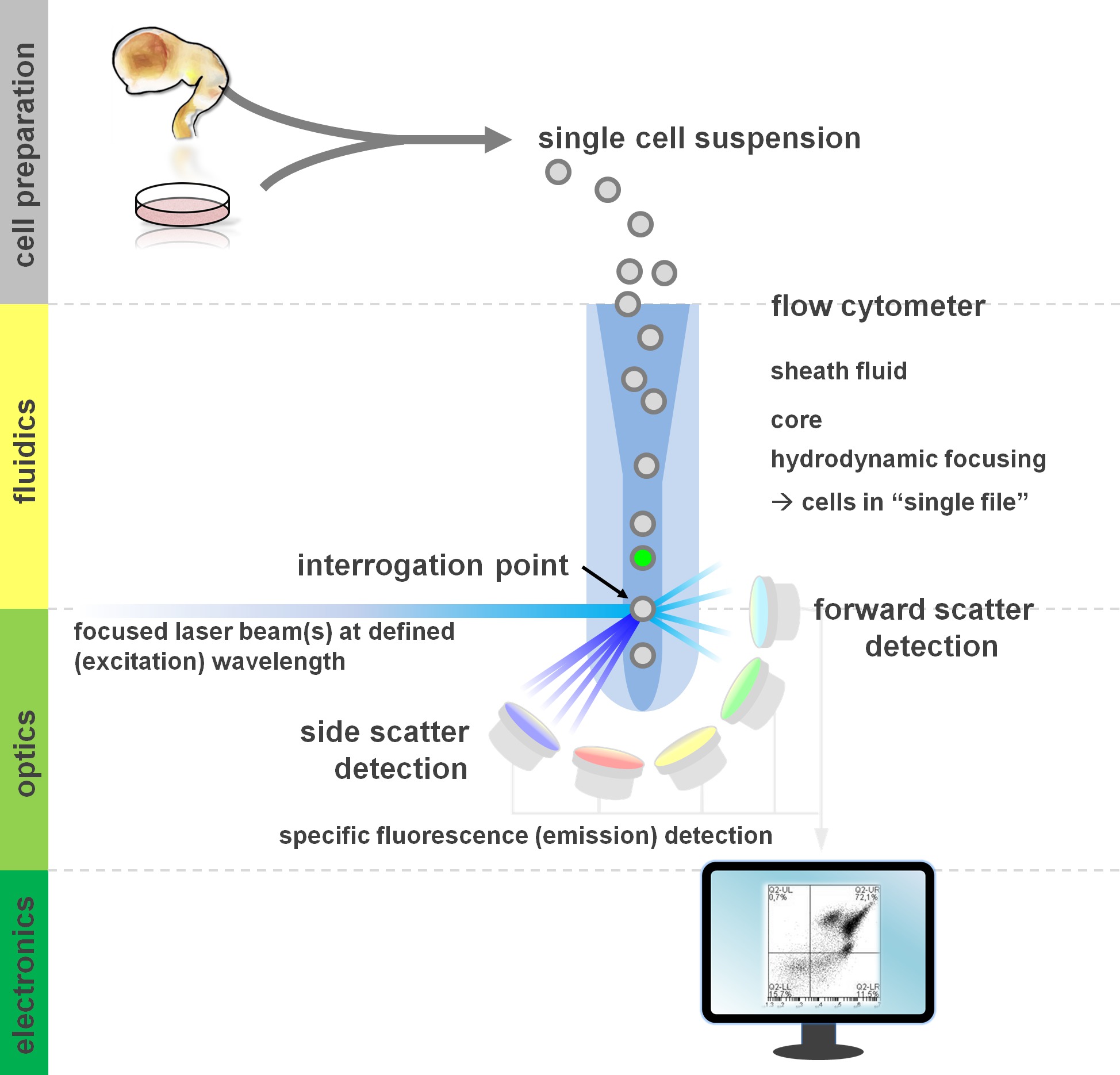
. Figura 1. Principio di citometria di flusso e componenti di un citofluorimetro a flusso citometri comprendono tre sistemi principali: fluidici, ottica ed elettronica. Un flusso semplificato di cellule in sospensione (preparata da tessuto primario o coltura in vitro) è compiuta dal flui guainad via focalizzazione idrodinamica, limitando il campione al suo nucleo centrale. Le parti ottiche sono composti di laser che illuminano il flusso di celle e filtri ottici che indirizzano il segnale ai rivelatori appropriati. I segnali luminosi rilevati sono convertiti in segnali elettronici, successivamente elaborati da un computer e visualizzati su un monitor per l'analisi dei dati e di gating. Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}
Gli utenti di flusso di metodi di citometria a profitto di almeno una conoscenza di base dei fondamenti di base, tra cui blocchi di costruzione di un citometro (per la revisione cfr 12,13; vedi anche figura 1). Un fascio laser interseca con un flusso fluidico idrodinamico concentrato che contiene le cellule in sospensione, che a loro volta passano attraverso il fascio laser in 'singolo file' uno dopo l'altro. Il interceptisu di una cella (o qualsiasi altra particella, per questo) con i risultati laser nella diffusione della luce da questo punto di interrogazione. Luce diffusa può essere rilevato in continuazione della direzione laser (forward scatter, associato con la dimensione della particella), nonché perpendicolare alla sua direzione (lato scatter, riflettendo la granulosità della particella / cella). Queste proprietà scatter cui sopra non richiedono etichettatura specifica, che è il motivo per cui un campione senza etichetta (o anche detriti cellulari, bolle d'aria, ecc) genera un segnale (evento) sul forward scatter bivariato contro lato scatter plot comunemente usato per gating iniziale. Utilizzando i laser e filtri specifici per il corrispondente spettri di eccitazione ed emissione appropriati, una cella può essere analizzata per la sua positività, livello di intensità, o assenza di marcatori fluorescenti. La maggior parte delle applicazioni citometria di flusso sono concentrati sulla caratterizzazione via antigeni di superficie cellulare. A differenza del lineag ematopoietichee, il lignaggio neurale è rimasta meno ampiamente definita in base alle superfici modelli di espressione epitopi 5. Un vantaggio di sfruttare antigeni di superficie è che le cellule vive possono essere sottoposti a separazione delle cellule paradigmi come il FACS. Al contrario, l'antigene intracellulare colorazione richiede fissazione e permeabilizzazione passi per mediare l'interazione epitopo-anticorpo, precludendo applicazioni a valle che richiedono cellule vitali. Da segnalare, questi approcci permettono ancora di numerose analisi quantitative 14 nonché analisi a valle per RNA e proteine espressione 15. Ematologia, immunologia e oncologia hanno spesso usato più di una dozzina di marcatori in collaborazione per definire particolari sottopopolazioni 16. Inoltre, citometria di massa o CyTOF possono ora essere utilizzati per analizzare fino a 30 parametri simultaneamente 17,18.
Per le applicazioni di cellule staminali neurali e colture primarie 14,19,20 l'eterogeneità delle cellulevitro è un fenomeno comune 21-23. Le cellule non rappresentano la popolazione bersaglio di interesse incarnano un potenziale fattore confondente per la lettura sperimentale 24,25. Convenientemente, i differenti sottoinsiemi cellulari presenti all'interno di una sospensione cellulare eterogenea recano profili di espressione dell'antigene distinte (noti o ancora da decifrare), che possono essere utilizzati per definire queste diverse popolazioni. Citometria a flusso può quindi svolgere un ruolo cruciale nel risolvere eterogeneità cellulare e, di conseguenza, facilitare applicazioni biomediche (test in vitro, terapia cellulare) e ottimizzare la lettura quantitativa concentrandosi sul sottoinsieme più rilevante 24,26. Varie combinazioni di antigene di superficie sono stati identificati negli ultimi anni per consentire la quantificazione e l'isolamento di particolari tipi di cellule neuronali. Questo include CD133 per l'arricchimento delle cellule staminali neurali 27, una combinazione delle CD15 / CD24 / antigeni di superficie CD29 per l'isolamento delle NSC, differentiated neurone e cellule della cresta neurale 28 o CD15 / CD24 / CD44 / CD184 / CD271 di isolare neurali e gliali sottoinsiemi 25, tra le altre firme 29,30. Al di là di neuroni, marcatori gliali includono A2B5 31, CD44 25, NG2 32 e GLAST 33. Una recente pubblicazione ha sfruttato il mesencefalo marcatore precursore fondello CORIN 34,35 per arricchire di precursori dopaminergici nel trapianto di cellule Parkinson paradigmi 36. Molecole di CD non sono solo i marcatori, ma funzionalmente rilevanti mediatori delle interazioni cellula-cellula e di capacità delle cellule di rispondere ai segnali di molecole della matrice extracellulare e fattori di crescita 37. Una strategia di potenziare ulteriormente l'arsenale di antigeni CD combinatorie a caratterizzare lo sviluppo lignaggio neurale è quella di utilizzare noti markers intracellulari per lo screening e definire combinazioni di antigene CD per un particolare tipo di cellule di interesse. Abbiamo recentemente sfruttato tale approccio e identificato CD49F - / CD200 alti modelli di espressione combinatorie come un nuovo approccio per arricchire sottoinsiemi neuronali da neurally differenziati pluripotenti indotte sistemi di coltura delle cellule staminali 38. Qui, includiamo e discutere il secondo protocollo (e varianti opzionali loro), in cui la colorazione superficiale e la colorazione intracellulare possono essere utilizzati contemporaneamente per definire sottopopolazioni di cellule neurali in citometria a flusso.

Figura 2. Schema di flusso di opzioni protocollo sperimentale. La figura illustra una rappresentazione schematica dei principali fasi del protocollo. Passaggi facoltativi (CFSE dye o etichettatura antigene intracellulare) sono indicati da caselle di colore grigio chiaro. Dopo la raccolta, è fondamentale per valutare il numero di vitalità e di cellule di sospensioni cellulari neuronali prima della colorazione della superficie cellulare. Positivo comecosì come controlli negativi devono essere inclusi in aggiunta ai campioni di interesse. I campioni possono essere analizzati mediante citometria a flusso e / o utilizzati in separazione delle cellule paradigmi. Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}
Mentre abbiamo precedentemente utilizzato anticorpo primario in combinazione con l'anticorpo secondario per la colorazione intracellulare 38, introduciamo ora etichettatura non covalente dell'anticorpo primario mediante frammenti Fab fluorescenti (etichettatura Zenon) come una lieve variazione, riducendo così le fasi di manipolazione cellulare 39. Inoltre, come ulteriore esempio della versatilità del protocollo, ci avvaliamo di una etichettatura facoltativa di un sottoinsieme sperimentale carbossifluoresceina estere succinimidyl (CFSE) prima superficie antigene colorazione. Tale CFSE pre-etichettatura consente confronto diretto immediato delle due linee cellulari o condizioni sperimentali (CFSE marcato vs. senza etichetta) all'interno di un unico tubo di esempio, la riduzione della varianza o sottili differenze di tempo di incubazione e di risparmio di anticorpi. CFSE è un colorante fluorescente stabilito che viene comunemente utilizzato per il monitoraggio delle cellule 40, in proliferazione 41,42 e codici a barre esperimenti 43,44. Infine, mentre passaggi effettivi (FACS, separazione immunomagnetica o immunopanning) non fanno parte di questo protocollo, in linea di principio, le modalità di raccolta e di etichettatura qui descritte fanno i campioni di rendimento che possono essere sottoposti a superficie antigene o intracellulari applicazioni di smistamento basate etichettatura- 15 25,28.
Con questo articolo, ci proponiamo di: sintesi di una superficie valida protocollo antigene colorazione 25,28, sintesi di un protocollo per la rivelazione di bersagli intracellulari e superficie combinata e intracellulare analisi dell'antigene 38, presenterà un intracellulare CFSE etichettatura tintura passo 41,45 come opzione sperimentale per comcomparata analisi di popolazioni di cellule neuronali, e riassumere approcci per l'analisi di citometria di flusso (controlli appropriati 13,46, gating strategia e dati presentazione 47).
Protocollo
1. Neural raccolta cellulare
- Valutazione al microscopio:
- Prima di avviare un esperimento, verificare lo stato della cultura in campo chiaro o contrasto di fase.
NOTA: Mentre tessuto neurale primaria ottenuti da dissezioni è, in linea di principio, ugualmente suscettibili di analisi citofluorimetrica 14,28, si ricorda che l'attenzione del protocollo è sulle cellule ottenute da sistemi cellulari neuronali in vitro.
- Prima di avviare un esperimento, verificare lo stato della cultura in campo chiaro o contrasto di fase.
- Cellule di vendemmia 7:
- Lavare delicatamente il piatto / boccetta di cellule aderenti con Mg 2+ / Ca 2+ tampone fosfato salino gratuito (PBS) a temperatura ambiente (per esempio, 5 ml di pallone T75, 3 ml per pozzetto per 6 pozzetti, o 10 ml 10 centimetri piatto).
NOTA: Il PBS utilizzato in tutto il protocollo è Mg 2+ / Ca 2+ gratuito.- Per l'esempio video, utilizzare un pallone T75 di cellule di neuroblastoma SH-SY5Y al 80% di confluenza. Applicare fasi di lavaggio supplementari nei casi in cuiconsiderevole detriti è presente nel piatto.
- Considerare lavaggi con siero-albumina contenenti PBS 48, Percoll, o Ficoll 49 sfumature di centrifugazione e / o perline disponibili in commercio 50,51. Rimozione di mielina e altri lipidi o altri contaminanti è fondamentale, soprattutto quando si utilizzano fonti primarie tessuti adulti.
- Aggiungere pre-riscaldato (37 ° C) la sostituzione tripsina in un volume adeguato che copre l'intera superficie del recipiente di coltura tissutale.
NOTA: In alternativa, prendere in considerazione altre opzioni digestione enzimatica Accutase o. Questo passaggio critico può influenzare negativamente l'espressione epitopi di superficie (vedi 7). - Incubare il piatto / matraccio a 37 ° C per 2 - 5 minuti (a seconda del tipo di cellula) per permettere alle cellule di staccare. Battere delicatamente il recipiente di coltura tissutale o filo con una pipetta sierologica per rimuovere le cellule. Evitare di digestione (come questo può causare la perdita di cellule e coagulazione a passi successivi).
- Quench la sostituzione tripsina aggiungendo due volte il volume di tampone di flusso (2% FBS in PBS) e raccogliere le cellule in una provetta conica da 15 ml.
- Triturare delicatamente la sospensione cellulare utilizzando una pipetta microlitro (100 - 1.000 ml) o una pipetta 5 ml sierologica per preparare una sospensione di cellule singole.
- Centrifugare le cellule a 220 xg per 5 min a 25 ° C. Con attenzione aspirare il surnatante lasciando il pellet alle spalle.
- Risospendere il pellet in un volume adeguato di tampone di flusso, a seconda delle dimensioni del pellet (ad esempio, per una beuta T75 confluenti di cellule SH-SY5Y la resa tipica è almeno 10 x 10 6 cellule, nel qual caso le cellule sono risospese in 5 ml di tampone di flusso).
NOTA: Se si osservano pezzi più grandi o di coagulazione, filtrare attraverso un 30 - 100 micron di maglia.
- Lavare delicatamente il piatto / boccetta di cellule aderenti con Mg 2+ / Ca 2+ tampone fosfato salino gratuito (PBS) a temperatura ambiente (per esempio, 5 ml di pallone T75, 3 ml per pozzetto per 6 pozzetti, o 10 ml 10 centimetri piatto).
- Conteggio delle cellule 52:
- Trasferire una piccola aliquota della sospensione di cellule in una provetta e diluire in un rapporto definito in un volume di trypan blu o una viabilità alternativa tintura prima del trasferimento ad un emocitometro o sistema di conteggio delle cellule automatizzato.
- Diluire la sospensione cellulare ad una concentrazione di 1 x 10 6 cellule vitali / ml aggiungendo il volume adeguato di tampone di flusso o PBS con 0,1% BSA (se procedere con etichettatura CFSE).
NOTA: propidio ioduro, 7-aminoactinomycin D, annessina V e disponibili in commercio kit di analisi fattibilità risolvibili rappresentano opzioni alternative al fine di valutare la vitalità delle cellule. Inoltre, utilizzando saggi apoptosi caspasi-3 fluorescenza come descritto in precedenza 53 possono essere utilizzati. Canali fluorescenti saranno "occupate" da questi reagenti che possono limitare le opzioni per passi successivi, se inclusi in tutti i campioni.
2. intracellulare Dye Labeling Utilizzando CFSE (Figura 3)
- Diluire il CFSE ad una concentrazione archivio desiderato che può essere usato facilmente.
NOTA: Per questi esperimenti una concentrazione di magazzino0.01 mM è stato utilizzato. Determinare la concentrazione ottimale di lavoro del CFSE empiricamente. - Aggiungere 10 ml di soluzione 0,01 mM CFSE per ml di cellule (Sezione 1.3.2, concentrazione di 1 x 10 6 cellule / ml in PBS + 0,1% BSA) per una concentrazione finale di 0,1 mM. Brevemente vortex per miscelare bene.
NOTA: concentrazioni CFSE usati qui sono circa dieci volte inferiore rispetto a quelli comunemente applicati in saggi di proliferazione, quindi cella tossicità del colorante è minima. Noi non osserviamo effetti negativi sulla vitalità cellulare. - Incubare per 5 minuti a RT, con agitazione costante (200 rpm). Proteggere dalla luce.
- Quench il colorante aggiungendo 5 volumi di tampone di flusso per i tubi. Centrifugare a 94 xg per 5 minuti a temperatura ambiente.
- Eliminare il surnatante lasciando il pellet alle spalle. Risospendere le cellule con 5 volumi di tampone di flusso.
- Centrifugare a 94 xg per 5 minuti a temperatura ambiente. Eliminare il supernatante e risospendere le cellule in tampone di flusso ad una concentrazione di 1 x 10 6 cellule / ml.
- Aggiungere un numero uguale di cellule non marcate di interesse alla sospensione cellulare macchiato. Procedere alla superficie protocollo di colorazione antigene (sezione 3).

Figura 3. Rilevamento di differenziale espressione dell'antigene di superficie CD tra due linee di cellule tramite cellule SH-SY5Y neuroblastoma CFSE tintura etichettatura. Sono pre-etichettati con CFSE per la successiva identificazione rispetto a fibroblasti BJ senza etichetta. Co-colorazione del campione misto (pannelli di destra) con marcatori di superficie CD24 o CD54 (sia coniugato con APC) dimostra che le linee cellulari sono facilmente distinguibili grazie alla colorazione CFSE (frecce = SH-SY5Y, punte di freccia = BJ fibroblasti). La maggior parte delle cellule SH-SY5Y esprimono CD24, ma non CD54 (ICAM-1). Al contrario, i fibroblasti BJ (CFSE-negativo) sono positivi per CD54, ma in gran parte negative per CD24.Target "https://www.jove.com/files/ftp_upload/52241/52241fig3highres.jpg" = "_ blank"> Clicca qui per vedere una versione più grande di questa figura.
3. Cella Surface colorazione
- Etichettare le provette compresi i campioni ed i controlli critiche (vedi Tabella 1).
| Tubo no. | Nome del campione | Antigen-fluoroforo | Diluizione |
| 1 | Cellule senza macchia | - | |
| 2 | Macchiato singolo | CD24-APC | 01:50 |
| 3 | Macchiato singolo | TUJ1-Alexa 488 nm Fluor | 1: 2.000 |
| 4 | Doppia macchiato | CD24-APC | 01:50 |
| TUJ1-Alexa 488 nm Fluor | 1: 2.000 | ||
| 5 | Macchiato singolo | Secondary solo: Alexa Fluor 488 nm | 1: 2.000 |
Tabella 1. L ist di tubi da includere in un tipico flusso citometria esperimento. La tabella mostra un insieme minimo di provette per campioni necessari per un esperimento di co-colorazione descritto in questo articolo video. Un esperimento ideale deve includere tutti i controlli necessari (positivi, così come controlli negativi) per compensazione accurata interpretazione dei risultati ottenuti.
- Aggiungere 100 ml di sospensione cellulare (dalla Sezione 2.7 o 1.3.2) per ogni provetta da 1,5 ml microcentrifuga.
NOTA: Assicurarsi che un minimo di 0,1 x 10 6 cellule sono presenti per 100 ml di sospensione cellulare. - Aggiungere fluoroforo anticorpo coniugato con il campione a una diluizione appropriata.
NOTA: Determinare diluizione di lavoro per ogni anticorpo prima dell'esperimento. Vedere la Tabella 2 per un elenco of antigeni di superficie neurali.
| Antigene | Tipo di cella | Riferimento |
| CD15 | Le cellule staminali neurali | [28, 67] |
| CD24 | Cellule neuronali | [28, 68] |
| CD29 | Le cellule staminali neurali | [28, 69, 70] |
| CD44 | Le cellule gliali | [25] |
| CD49f | Le cellule staminali neurali | [38] |
| CD56 (NCAM) | Cellule neuronali | [71] |
| CD133 | Le cellule staminali neurali | [27] |
| CD184 | Le cellule staminali neurali e cellule gliali | [25] |
| CD200 | Cellule neuronali | [38] |
| CD271 | Le cellule staminali della cresta neurale | [25] |
| A2B5 | Le cellule gliali | [31] |
| CORIN | Precursori dopaminergici | [35, 36] |
| FORSE1 | Le cellule staminali neurali (NSC) | [72] |
| GLAST | Le cellule gliali | [33] |
| NG2 | Le cellule gliali | [32] |
Tabella 2. Selezione di antigeni di superficie neurali. Questa tabella fornisce un elenco di epitopi di superficie trovati ad essere espressa da vari tipi di cellule neurali per esemplificare la crescente gruppo di antigeni di superficie utilizzate per caratterizzare il lignaggio neurale. Si noti che questa selezione è lungi dall'essere completo e che la maggior parte di questi marcatori sono espressi anche da una serie di altre cellule neuronali e non neuronali. Di conseguenza, le combinazioni di diversi marcatori saranno tenuti a definire meglio e isolare i sottoinsiemi neurali indicati.
- Incubare per 30 minuti su un agitatore orbitale (200 rpm) al buio.
- Flusso tampone di lavaggio
- Aggiungere 1 ml di tampone di flusso per i tubi. Centrifugare a 380 xg per 4 minuti a 4 ° C.
- Eliminare il surnatante lasciando il pellet alle spalle.
- Ripetere la fase di lavaggio.
- Dopo il secondo lavaggio, decantare il supernatante e risospendere le cellule in tampone portata ad un volume finale di 100 microlitri.
- Utilizzare campione per l'analisi di citometria di flusso. In alternativa, procedere alla sezione 4 e 5.
NOTA: Se le cellule devono essere ordinati e rimesso in cultura post-FACS (ie, sospensione di cellule vitali senza fissazione o permeabilizzazione), applicare tecniche asettiche durante la vendemmia, colorazione e fasi analitiche.
4. Fissazione e permeabilizzazione 38
- Fissazione con paraformaldeide (PFA):
- Preparare tampone di fissaggio contenente 2% PFA in PBS.
NOTA: PFA è nocivo per l'uomo e per l'ambiente. Utilizzare dispositivi di protezione adeguati e gettare rifiuti in conformità alla normativa vigente. - Aggiungere 500 pl di tampone di fissaggio a 100 ml di sospensione cellulare.
- Incubare le provette a RT per 15 minuti su un agitatore orbitale (100 rpm) al buio.
- Preparare tampone di fissaggio contenente 2% PFA in PBS.
- PBS lavaggio:
- Aggiungere 1 ml di PBS al tubo. Centrifugare a 380 xg per 3 min a 4 ° C.
- Scartare / decantare il surnatante lasciando circa 100 microlitri nel tubo.
- Permeabilizzazione con Tween-20:
- Preparare tampone permeabilizzazione contenente 0,7% Tween-20 in PBS.
- Aggiungere 500 pl di tampone di permeabilizzazione a 100 ml di sospensione cellulare.
- Incubare le provette a RT per 15 minuti su un agitatore orbitale (100 rpm) al buio.
- Lavare le cellule una volta con PBS (come descritto nel paragrafo 4.2) e rimuovere il surnatante dal tubi completely, lasciando solo il pellet dietro.
5. intracellulare Antigen Staining 38 (Figura 4)
- Preparazione di soluzioni di anticorpi primari:
- Diluire gli anticorpi primari in tampone di diluizione contenente 1% di albumina di siero bovino, siero 10% (per esempio, o di capra normale asino siero) e 0,5% Tween-20 in PBS.
NOTA: Scegliere il siero da utilizzare a seconda delle specie anticorpi secondari sono stati allevati in. - In alternativa, utilizzare Zenon etichettatura fluoresceina dell'anticorpo primario secondo le istruzioni del produttore.
- Preparare 1 mg di anticorpo primario in PBS ad una diluizione adeguata (volume totale ≤ 20 ml).
- Aggiungere 5 ml di Zenon fluoresceina IgG reagente etichettatura (Componente A) alla soluzione di anticorpi.
- Incubare la miscela per 5 minuti a temperatura ambiente.
- Aggiungere 5 ml di Zenon reagente bloccante (Componente B) alla miscela di reazione.
- Covarela miscela per 5 minuti a temperatura ambiente. Applicare l'anticorpo per il campione in 30 min.
- Diluire gli anticorpi primari in tampone di diluizione contenente 1% di albumina di siero bovino, siero 10% (per esempio, o di capra normale asino siero) e 0,5% Tween-20 in PBS.
- Primaria colorazione anticorpi:
- Aggiungere 100 ml di soluzione di anticorpo primario per il pellet e triturare delicatamente per miscelare.
- In alternativa, aggiungere Zenon fluoresceina anticorpo marcato per la sospensione cellulare alla diluizione appropriata.
- Incubare le provette a RT per 30 minuti su un agitatore orbitale (200 rpm), al riparo dalla luce. Lavare le cellule una volta con PBS (come descrive nella sezione 4.2) e rimuovere il surnatante dai tubi completamente, lasciando solo il pellet dietro.
- Colorazione anticorpo secondario (non richiesto per gli anticorpi Zenon fluoresceina marcati):
- Diluire gli anticorpi secondari in PBS ad una concentrazione adeguata.
- Aggiungere 100 ml di soluzione di anticorpo secondario al pellet e triturare delicatamente per miscelare. Incubare le provette a RT per 30 minuti su un agitatore (200 rpm) al buio.
- Lavare i campioni due volte con PBS (come descritto nella Sezione 4.2).
- Lavare una volta con tampone di flusso (vedi Sezione 3.5).
- Risospendere le cellule in circa 150 ml di tampone di flusso e analizzare il citometro a flusso.

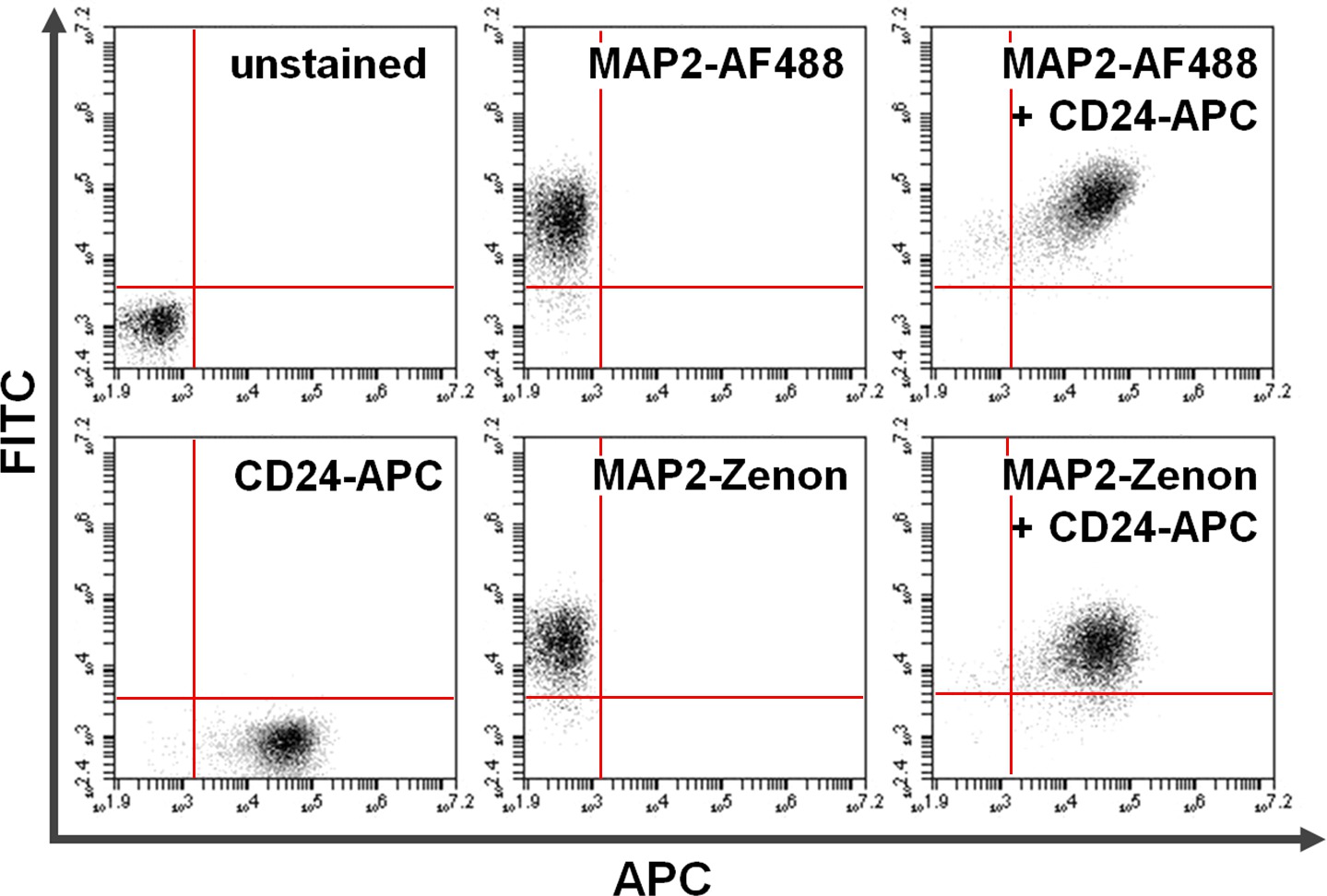
Figura 4. Co-colorazione di superficie e di proteine intracellulari. Citometria a flusso di dati esemplifica un confronto tra primaria + secondaria anticorpo-basata contro colorazione intracellulare a base di fluoresceina Zenon in combinazione con colorazione della superficie. Positività esclusiva on y (quadrante in alto a sinistra) e l'asse x (quadrante inferiore destro) mostra cellule colorate per MAP2 e CD24, rispettivamente. Dopo co-colorazione, MAP2 condiviso e di espressione CD24 può essere visto nel quadrante in alto a destra (pannelli di destra). Confronto di utilizzoAlexa Fluor 488. (Top; AF488) rispetto Zenon fluoresceina (in basso) per i rendimenti MAP2-etichettatura risultati simili Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}
6. analisi citofluorimetrica
- Condurre immediatamente mediante citometria a flusso, dopo il completamento del protocollo di colorazione utilizzando un citofluorimetro con filtri appropriati per il rilevamento del segnale. Utilizzare 488 nm laser rosso blu e 640 nm con FL-1 (533/30), FL-2 (585/40), e filtri FL-4 (675/25) passa-banda.
- Impostare porte principali sulla base del scatter in avanti e laterale esclusi detriti e cellule morte.
- Impostare cancelli fluorescenza per superficie e l'antigene intracellulare al ≤0.5% sulla base dei campioni senza macchia e la compensazione per sovrapposizione spettrale che utilizzano controlli colorati singoli.
Risultati
Il protocollo presentato qui permette approcci versatile sperimentali (Figura 2). Nella sua versione più breve (passi 1, 3 e 6), può essere considerato una guida per la semplice colorazione di antigeni di superficie. Nella sua forma più complessa, una serie di paradigmi co-etichettatura con una gamma di antigeni intracellulari può essere perseguito (fasi opzionali 2 e / o 4 a 5). Inoltre, la fase di...
Discussione
Il protocollo presentato qui è ben definito per colture cellulari neuronali derivate da cellule staminali umane, ma può essere ugualmente applicata ad altre fonti di cellule neurali compreso il tessuto primario o linee cellulari neurali. Oltre alle fonti embrionali, le cellule staminali o progenitrici neurali possono essere estratti dalle regioni neurogenici del cervello adulto 27. Inoltre, citometria a flusso e FACS possono essere sfruttati per quantificare, analizzare e isolare diverse popolazioni di cell...
Divulgazioni
The authors declare no potential conflicts of interest.
Riconoscimenti
Our research program is funded through the Emmy Noether-Program of the German Research Foundation (DFG), grant PR1132/3-1. Further support by the Müller-Fahnenberg Foundation of the University of Freiburg is gratefully acknowledged. This study was supported in part by the Excellence Initiative of the German Research Foundation (GSC-4, Spemann Graduate School).
Materiali
| Name | Company | Catalog Number | Comments |
| DMEM/F12 (1:1) (Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12) |  Life Technologies Life Technologies | 11330057 | |
| DPBS without Ca2+ Mg2+ | Life Technologies | 14190169 | |
| Fetal bovine serum, qualified, E.U.-approved, South America origin (FBS) | Life Technologies | 10270-106 | |
| MEM Non-essential amino acids (100x) | Life Technologies | 11140035 | |
| TrypLE Express | Life Technologies | 12604013 | |
| Trypan blue solution, 0.4% | Life Technologies | 15250061 | |
| Paraformaldehyde | Carl Roth | 335.3 | |
| Bovine serum albumin (BSA) Fraction V | PAA Laboratories, Coelbe | K41-001 | |
| Tween-20 Detergent | Calbiochem | 655205 | |
| Carboxyfluorescein succinimidyl ester (CFSE) | eBioscience | 65-0850-84 | |
| DMSO | AppliChem | A1584 | |
| Bottle top filters express plus 0.22 µm, 250 ml | Millipore | SCGPU02RE | |
| Cell culture treated flasks (T 25) | NUNC | 156367 | |
| Cell culture treated flasks (T 75) | NUNC | 156499 | |
| Conical tubes (15 ml) | Greiner Bio-One | 188271 | |
| Conical tubes (50 ml) | Greiner Bio-One | 227261 | |
| Pasteur pipet, glass (150 mm) | STEIN Labortechnik, Remchingen | S03710150 | |
| Pipet tips (0.1-10 µl) | Corning | 4125 | |
| Pipet tips (1-200 µl) | Corning | 4126 | |
| Pipet tips (100-1000 µl) | Corning | 4129 | |
| Serological pipets, 5 ml | Corning | 4051 | |
| Serological pipets, 10 ml | Corning | 4101 | |
| Serological pipets, 25 ml | Corning | 4251 | |
| Microcentrifuge tubes (0.5 ml) | Sarstedt | 72,699 | |
| Microcentrifuge tubes (1.5 ml) | Greiner Bio-One | 616201 | |
| Microcentrifuge tubes (2.0 ml) | Sarstedt | 72,695,500 | |
| Anti-Human CD24 APC monoclonal antibody | eBioscience | 17-0247-42 | Working dilution 1:50 |
| Anti-Human CD54 PE monoclonal antibody | eBioscience | 12-0549-42 | Working dilution 1:50 |
| Neuronal Class III β-Tubulin (Tuj1) polyclonal antibody | Covance | PRB-435P | Working dilution 1:2,000 |
| Alexa Fluor 488 Donkey anti Rabbit | Life Technologies | A21206 | Working dilution 1:2,000 |
| Zenon® Fluorescein Rabbit IgG Labeling Kit | Life Technologies | Z-25342 | |
| Neubauer-Improved counting chamber | Marienfeld | 0640010 | |
| Vortex | Scientific Industries | G560E | |
| Thermomixer comfort | Eppendorf | 5355 000.001 | |
| Accuri C6 flow cytometer | Becton Dickinson (BD) | 653118 | |
| Microcentrifuge refrigerated, PerfectSpin 24 R | Peqlab | 91-PSPIN-24R | |
| Orbital shaker, Unimax 1010 | Heidolph | 543-12310-00 | |
| Centrifuge refrigerated, Rotanta 96 RC | Hettich | 4480-50 | |
| Class II Biological safety cabinet Safe 2020 | Thermo Scientific | 51026640 | |
| CO2 Incubator, Heracell 240i | Thermo Scientific | 51026331 | |
| Vacuum system, Vacusafe comfort | Integra Biosciences | 158320 | |
| Microscope, Axiovert 40 CFL | Zeiss | 451212 | |
| Pipet controller, accu-jet pro | Brand | 26303 | |
| Micropipet, Pipetman neo P20N (2-20 µl) | Gilson | F144563 | |
| Micropipet, Pipetman neo P200N (20-200 µl) | Gilson | F144565 | |
| Micropipet, Pipetman neo P1000N (100-1000 µl) | Gilson | F144566 |
Riferimenti
- Herzenberg, L. A., et al. The History and Future of the Fluorescence Activated Cell Sorter and Flow Cytometry: A View from. 48 (10), 1819-1827 (2002).
- Chattopadhyay, P. K., Roederer, M. Cytometry: today’s technology and tomorrow's horizons. Methods (San Diego, Calif). 57 (3), 251-258 (2012).
- Jaye, D. L., Bray, R. A., Gebel, H. M., Harris, W. A. C., Waller, E. K. Translational applications of flow cytometry in clinical practice). J. Immunol. 188 (10), 4715-4719 (2012).
- Henel, G., Schmitz, J. Basic Theory and Clinical Applications of Flow Cytometry. Lab Med. 38 (7), 428-436 (2007).
- Seita, J., Weissman, I. L. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2 (6), 640-653 (2010).
- Ulrich, H., Bocsi, J. Phenotypes of stem cells from diverse origin. Cytometry. A. 77 (1), 6-10 (2010).
- Panchision, D. M., et al. Optimized flow cytometric analysis of central nervous system tissue reveals novel functional relationships among cells expressing CD133, CD15, and CD24. Stem cells. 25 (6), 1560-1570 (2007).
- Meyer, R. A., Zaruba, M. E., McKhann, G. M. Flow cytometry of isolated cells from the brain. Anal. Quant. Cytol. 2 (1), 66-74 (1980).
- Junger, H., Junger, W. G. CNTF and GDNF, but not NT-4, support corticospinal motor neuron growth via direct mechanisms. Neuroreport. 9 (16), 3749-3754 (1998).
- McLaren, F. H., Svendsen, C. N., Vander Meide, P., Joly, E. Analysis of neural stem cells by flow cytometry: cellular differentiation modifies patterns of MHC expression. J. Neuroimmunol. 112 (1-2), 35-46 (2001).
- Wang, S., Roy, N. S., Benraiss, A., Goldman, S. A. Promoter-based isolation and fluorescence-activated sorting of mitotic neuronal progenitor cells from the adult mammalian ependymal/subependymal. 22 (1-2), 167-176 (2000).
- Tanke, H. J., vander Keur, M. Selection of defined cell types by flow-cytometric cell sorting. Trends Biotechnol. 11 (2), 55-62 (1993).
- Baumgarth, N., Roederer, M. A practical approach to multicolor flow cytometry for immunophenotyping. J. Immunol. Methods. 243 (1-2), 77-97 (2000).
- Sergent-Tanguy, S., Chagneau, C., Neveu, I., Naveilhan, P. Fluorescent activated cell sorting (FACS): a rapid and reliable method to estimate the number of neurons in a mixed population. J. Neurosci. Methods. 129 (1), 73-79 (2003).
- Ernst, A., et al. Neurogenesis in the striatum of the adult human brain. Cell. 156 (5), 1072-1083 (2014).
- Perfetto, S. P., Chattopadhyay, P. K., Roederer, M. Seventeen-colour flow cytometry: unravelling the immune system. Nat. Rev. Immunol. 4 (8), 648-655 (2004).
- Bandura, D. R., et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 81 (16), 6813-6822 (2009).
- Bendall, S. C., et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 332 (6030), 687-696 (2011).
- Neveu, I., Rémy, S., Naveilhan, P. The neuropeptide Y receptors, Y1 and Y2, are transiently and differentially expressed in the developing cerebellum. Neuroscience. 113 (4), 767-777 (2002).
- Pruszak, J., Just, L., Isacson, O., Nikkhah, G. Isolation and culture of ventral mesencephalic precursor cells and dopaminergic neurons from rodent brains. Curr. Protoc. Stem Cell Biol. 2 (Unit 2D.5), (2009).
- Suslov, O. N., Kukekov, V. G., Ignatova, T. N., Steindler, D. A. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc. Natl. Acad. Sci. U.S.A. 99 (22), 14506-14511 (2002).
- Bez, A., et al. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 993 (1-2), 18-29 (2003).
- Pruszak, J., Isacson, O. Molecular and cellular determinants for generating ES-cell derived dopamine neurons for cell therapy. Adv. Exp. Med. Biol. 651, 112-123 (2009).
- Carson, C. T., Aigner, S., Gage, F. H. Stem cells: the good, bad and barely in control. Nat. Med. 12 (11), 1237-1238 (2006).
- Yuan, S. H., et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PloS One. 6 (3), e17540 (2011).
- Roy, N. S., Cleren, C., Singh, S. K., Yang, L., Beal, M. F., Goldman, S. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat. Med. 12 (11), 1259-1268 (2006).
- Uchida, N., et al. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. U. S. A. 97 (26), 14720-14725 (2000).
- Pruszak, J., Ludwig, W., Blak, A., Alavian, K., Isacson, O. CD15, CD24, and CD29 define a surface biomarker code for neural lineage differentiation of stem cells. Stem Cells. 27 (12), 2928-2940 (2009).
- Peh, G. S. -. L., Lang, R. J., Pera, M. F., Hawes, S. M. CD133 expression by neural progenitors derived from human embryonic stem cells and its use for their prospective isolation. Stem Cells Dev. 18 (2), 269-282 (2009).
- Golebiewska, A., Atkinson, S. P., Lako, M., Armstrong, L. Epigenetic landscaping during hESC differentiation to neural cells. Stem Cells. 27 (6), 1298-1308 (2009).
- Dietrich, J., Noble, M., Mayer-Proschel, M. Characterization of A2B5+ glial precursor cells from cryopreserved human fetal brain progenitor cells. Glia. 40 (1), 65-77 (2002).
- Nishiyama, A. NG2 cells in the brain: a novel glial cell population. Hum. Cell. 14 (1), 77-82 (2001).
- Jungblut, M., et al. Isolation and characterization of living primary astroglial cells using the new GLAST-specific monoclonal antibody ACSA-1. Glia. 60 (6), 894-907 (2012).
- Ono, Y., et al. Differences in neurogenic potential in floor plate cells along an anteroposterior location: midbrain dopaminergic neurons originate from mesencephalic floor plate cells. Development. 134 (17), 3213-3225 (2007).
- Chung, S., et al. ES cell-derived renewable and functional midbrain dopaminergic progenitors. Proc. Natl. Acad. Sci. U.S.A. 108 (23), 9703-9708 (2011).
- Doi, D., et al. Isolation of Human Induced Pluripotent Stem Cell-Derived Dopaminergic Progenitors by Cell Sorting for Successful Transplantation. Stem Cell Reports. 2 (3), 337-350 (2014).
- Solozobova, V., Wyvekens, N., Pruszak, J. Lessons from the embryonic neural stem cell niche for neural lineage differentiation of pluripotent stem cells. Stem Cell Rev. 8 (3), (2012).
- Turaç, G., et al. Combined flow cytometric analysis of surface and intracellular antigens reveals surface molecule markers of human neuropoiesis. PloS One. 8 (6), e68519 (2013).
- Buchwalow, I. B., Böcker, W. Chapter 2. Immunohistochemistry: Basics and Methods. , 9-17 (2010).
- Tario, J. D., et al. Optimized staining and proliferation modeling methods for cell division monitoring using cell tracking dyes. J. Vis. Exp. (70), e4287 (2012).
- Lyons, A. B., Parish, C. R. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 171 (1), 131-137 (1994).
- Hawkins, E. D., et al. Measuring lymphocyte proliferation, survival and differentiation using CFSE time-series data. Nat. Protoc. 2 (9), 2057-2067 (2007).
- Quah, B. J. C., Parish, C. R. The use of carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor lymphocyte proliferation. J. Vis. Exp. 44, (2010).
- Sukhdeo, K., et al. Multiplex flow cytometry barcoding and antibody arrays identify surface antigen profiles of primary and metastatic colon cancer cell lines. PloS One. 8 (1), e53015 (2013).
- Jiang, L., et al. Daucosterol promotes the proliferation of neural stem cells. The J. Steroid Biochem. Mol. Biol. 140, 90-99 (2014).
- Hulspas, R., et al. Considerations for the control of background fluorescence in clinical flow cytometry. Cytometry. B. 76 (6), 355-364 (2009).
- Moloney, M., Shreffler, W. G. Basic science for the practicing physician: flow cytometry and cell sorting. Annals of Allergy, Asthm., & Immunology: Official Publication of the American College of Allergy, Asthma., & Immunology. 101 (5), 544-549 (2008).
- Siebzehnrubl, F. A., et al. Isolation and characterization of adult neural stem cells. Methods Mol. Biol. 750, 61-77 (2011).
- Guez-Barber, D., et al. FACS purification of immunolabeled cell types from adult rat brain). J. Neurosci. Methods. 203 (1), 10-18 (2012).
- Tham, C. -. S., Lin, F. -. F., Rao, T. S., Yu, N., Webb, M. Microglial activation state and lysophospholipid acid receptor expression. Int. J. Dev. Neurosci. 21 (8), 431-443 (2003).
- Nguyen, H. X., Beck, K. D., Anderson, A. J. Quantitative assessment of immune cells in the injured spinal cord tissue by flow cytometry: a novel use for a cell purification method. J. Vis. Exp. (50), e2698 (2011).
- Marchenko, S., Flanagan, L. Counting human neural stem cells. J. Vis. Exp. (7), 262 (2007).
- Brunlid, G., Pruszak, J., Holmes, B., Isacson, O., Sonntag, K. C. Immature and neurally differentiated mouse embryonic stem cells do not express a functional Fas/Fas ligand system. Stem Cells. 25 (10), 2551-2558 (2007).
- Brewer, G. J. Isolation and culture of adult rat hippocampal neurons. J. Neurosci. Methods. 71 (2), 143-155 (1997).
- Cardona, A. E., Huang, D., Sasse, M. E., Ransohoff, R. M. Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat. Protoc. 1 (4), 1947-1951 (2006).
- Nielsen, J. A., Maric, D., Lau, P., Barker, J. L., Hudson, L. D. Identification of a novel oligodendrocyte cell adhesion protein using gene expression profiling. J. Neurosci. 26 (39), 9881-9891 (2006).
- Daneman, R., et al. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PloS One. 5 (10), e13741 (2010).
- Gräbner, R., Till, U., Heller, R. Flow cytometric determination of E-selectin, vascular cell adhesion molecule-1, and intercellular cell adhesion molecule-1 in formaldehyde-fixed endothelial cell monolayers. Cytometry. 40 (3), 238-244 (2000).
- Quah, B. J. C., Parish, C. R. New and improved methods for measuring lymphocyte proliferation in vitro and in vivo using CFSE-like fluorescent dyes. J. Immunol. Methods. 379 (1-2), 1-14 (2012).
- Lathia, J. D., et al. High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep. 6 (1), 117-129 (2014).
- Ganat, Y. M., et al. Identification of embryonic stem cell-derived midbrain dopaminergic neurons for engraftment. J. Clin. Invest. 122 (8), 2928-2939 (2012).
- Hedlund, E., et al. Embryonic stem cell-derived Pitx3-enhanced green fluorescent protein midbrain dopamine neurons survive enrichment by fluorescence-activated cell sorting and function in an animal model of Parkinson’s disease. Stem Cells. 26 (6), 1526-1536 (2008).
- Maroof, A. M., et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 12 (5), 559-572 (2013).
- Chivet, M., Hemming, F., Pernet-Gallay, K., Fraboulet, S., Sadoul, R. Emerging role of neuronal exosomes in the central nervous system. Front. Physiol. 3, 145 (2012).
- Graner, M. W., et al. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 23 (5), 1541-1557 (2009).
- Eldh, M., Lötvall, J. Isolation and characterization of RNA-containing exosomes. J. Vis. Exp. (59), e3037 (2012).
- Capela, A., Temple, S. LeX is expressed by principle progenitor cells in the embryonic nervous system, is secreted into their environment and binds Wnt-1. Dev. Biol. 291 (2), 300-313 (2006).
- Nieoullon, V., Belvindrah, R., Rougon, G., Chazal, G. mCD24 regulates proliferation of neuronal committed precursors in the subventricular zone. Mol. Cell. Neurosci. 28 (3), 462-474 (2005).
- Nagato, M., et al. Prospective characterization of neural stem cells by flow cytometry analysis using a combination of surface markers. J. Neurosci. Res. 80 (4), 456-466 (2005).
- Hall, P. E., Lathia, J. D., Miller, N. G. A., Caldwell, M. A., French-Constant, C. Integrins are markers of human neural stem cells. Stem Cells. 24 (9), 2078-2084 (2006).
- Hargus, G., et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. U.S.A. 107 (36), 15921-15926 (2010).
- Elkabetz, Y., et al. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 22 (2), 152-165 (2008).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati