Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Mikrosatelliten-DNA-Genotypisierung und Durchflusszytometrie-Ploidy-Analysen von formalinfixierten Paraffin-eingebetteten Hydatidiform-Molargeweben
In diesem Artikel
Zusammenfassung
Hydatidiforme Maulwürfe sind abnorme menschliche Schwangerschaften mit heterogenen Ätiologien, die nach ihren morphologischen Merkmalen und dem elterlichen Beitrag zu den Molgenomen klassifiziert werden können. Hier werden Protokolle der Multiplex-Mikrosatelliten-DNA-Genotypisierung und Durchflusszytometrie von formalinfixiertem Paraffin-eingebettetem Molgewebe detailliert beschrieben, zusammen mit der Interpretation und Integration der Ergebnisse.
Zusammenfassung
Hydatidiform Maulwurf (HM) ist eine abnorme menschliche Schwangerschaft, die durch übermäßige trophoblastische Proliferation und abnorme embryonale Entwicklung gekennzeichnet ist. Es gibt zwei Arten von HM, die auf mikroskopischer morphologischer Auswertung basieren, vollständiges HM (CHM) und partielles HM (PHM). Diese können auf der Grundlage des Elternbeitrags zu den Molgenomen weiter unterteilt werden. Eine solche Charakterisierung von HM durch Morphologie und Genotypanalysen ist entscheidend für das Patientenmanagement und für das grundlegende Verständnis dieser faszinierenden Pathologie. Es ist gut dokumentiert, dass die morphologische Analyse von HM einer breiten Interobserver-Variabilität unterliegt und allein nicht ausreicht, um HM genau in CHM und PHM zu klassifizieren und von hydropischen nicht-molaren Abtreibungen zu unterscheiden. Die Genotypisierungsanalyse wird hauptsächlich an DNA und Geweben aus formalinfixierten Paraffin-eingebetteten (FFPE)-Produkten der Empfängnis durchgeführt, die eine weniger optimale Qualität aufweisen und folglich zu falschen Schlussfolgerungen führen können. In diesem Artikel werden detaillierte Protokolle für Multiplex-Genotypisierung und Durchflusszytometrieanalysen von FFPE-Molarengeweben sowie die Interpretation der Ergebnisse dieser Methoden, deren Fehlerbehebung und Integration in die morphologische Bewertung bereitgestellt. , p57KIP2-Immunhistochemie und Fluoreszenz-In-situ-Hybridisierung (FISH), um eine korrekte und robuste Diagnose zu erhalten. Hier teilen die Autoren die Methoden und Lehren aus der Analyse von rund 400 Empfängnisprodukten aus den letzten 10 Jahren.
Einleitung
Ein hydatidiformer Maulwurf (HM) ist eine abnorme menschliche Schwangerschaft, die durch abnorme embryonale Entwicklung, Hyperproliferation des Trophoblasten und hydropische Degeneration von Chorionzotten (CV) gekennzeichnet ist. Historisch gesehen wurde HM in zwei Typen unterteilt, vollständiges HM (CHM) und partielles HM (PHM) nur auf der Grundlage der morphologischen Bewertung1. Es hat sich jedoch gezeigt, dass die morphologische Bewertung allein nicht ausreicht, um HM in die beiden Subtypen (CHM und PHM) einzustufen und sie von nicht-molaren Fehlgeburten2,3,4zu unterscheiden.
Da CHM und PHM unterschiedliche Neigungen zu malignen Erkrankungen haben, ist es daher wichtig, den genotypischen Typ von HM genau zu bestimmen, um den Patienten eine angemessene Nachbeobachtung und Behandlung zu ermöglichen. Infolgedessen wurden in den letzten Jahrzehnten mehrere Methoden entwickelt und entwickelt, um den elterlichen Beitrag zu den Molgeweben zu identifizieren und eine korrekte Klassifizierung von HM zu erreichen. Dazu gehören Karyotypie-Analyse, chromosomaler Banding-Polymorphismus, humanes Leukozyten-Antigen (HLA) serologische Typisierung, Restriktionsfragmentlänge Polymorphismus, variable Anzahl von Tandem-Wiederholungen, Mikrosatelliten-Genotypisierung, Durchflusszytometrie und p57 KIP2-Immunhistochemie. Dies hat eine genaue Unterteilung von HM-Konzepten auf der Grundlage des elterlichen Beitrags zu ihren Genomen ermöglicht, wie folgt: CHM, die diploid undrogenetisch monospermisch oder diploid androgenetisch dispermisch sind, und PHM, die triploid sind, dispermisch in 99% und monospermin in 1% der Fälle5,6,7,8. Darüber hinaus gibt es eine andere genotypische Art von HM, die in den letzten zwei Jahrzehnten entstanden ist, die diploid biparental ist. Letzteres ist meist wiederkehrendeund und kann ein einzelnes Familienmitglied (einfache Fälle) oder mindestens zwei Familienmitglieder (familiäre Fälle) betreffen. Diese diploiden biparentalen Maulwürfe werden meist durch rezessive Mutationen in NLRP7 oder KHDC3L bei den Patienten9,10,11,12verursacht. Diploid biparental HM bei Patienten mit rezessiven Mutationen in NLRP7 kann als CHM oder PHM durch morphologische Analyse diagnostiziert werden und dies scheint mit der Schwere der Mutationen bei den Patienten13,14verbunden zu sein. Neben der Klassifizierung von HM nach ihren Genotypen ermöglichte die Einführung und Verwendung mehrerer Genotypisierungsmethoden die Unterscheidung der verschiedenen Molentitäten von nicht-molaren Fehlgeburten, wie z. B. aneuploiden diploiden biparentalen andere Arten von Konzepten5,15. Solche Vorstellungen können einige Trophoblast-Proliferation und abnorme villous Morphologie haben, die bis zu einem gewissen Grad einige morphologische Merkmale von HM imitieren.
Der Zweck dieses Artikels besteht darin, detaillierte Protokolle für Multiplex-Genotypisierung und Durchflusszytometrie von formalinfixierten Paraffin-eingebetteten Geweben (FFPE) sowie umfassende Analysen der Ergebnisse dieser Methoden und deren Integration mit anderen Methoden für korrekte und schlüssige Diagnose von Molgeweben.
Protokoll
Diese Studie wurde vom McGill Institutional Review Board genehmigt. Alle Patienten gaben ihre schriftliche Zustimmung zur Teilnahme an der Studie und zur Abrufung ihrer FFPE-Empfängnisprodukte (POCs) aus verschiedenen Pathologieabteilungen.
HINWEIS: Zwar gibt es mehrere Methoden zur Genotypisierung und Ploidie-Bestimmung durch Durchflusszytometrie, aber die hier bereitgestellten Protokolle beschreiben eine Analysemethode mit jeweils einer Plattform.
1. Genotypisierung
-
Auswahl des besten FFPE-Blocks
- Für jedes FFPE-Empfängnisprodukt (POC) 4 m dicke Hämatoxylin- und Eosin-Gefärbte Abschnitte wie in den Abschnitten 1.2 und 1.3 beschrieben, einen für jeden verfügbaren Block, für die morphologische Auswertung mittels Mikroskopie vorbereiten.
- Wählen Sie mit den H&E-Dias und einem Lichtmikroskop den FFPE-Block aus, der die größte Menge an Chorionzotten (CV) enthält, und wenn möglich den Block, der den Lebenslauf hat, der von mütterlichem Gewebe getrennt ist und nicht mit ihm vermischt wird.
-
Schneiden
- Legen Sie den gewählten Block für 15 min auf Eis, um den Schnitt zu erleichtern.
- Passen Sie das Mikrotome an Schnittabschnitte an, die für die mikroskopische morphologische Auswertung 4 m dick und für die DNA-Extraktion 10 m dick sind.
- Legen Sie den Kaltblock in das Mikrotome und schneiden Sie einen Abschnitt aus jedem Block für die H&E-Färbung und 10 bis 30 Abschnitte aus dem gewählten Block, abhängig von der Menge des CV im Block, für die DNA-Extraktion.
HINWEIS: Für Blöcke, die voller CV sind, reichen 10 Abschnitte für die DNA-Extraktion aus. Wenn nur etwa 10% des Blocks CV enthält, während der Rest mütterliches Gewebe sind, dann werden 20 bis 30 Abschnitte benötigt, um ausreichende Mengen an DNA zu gewährleisten. - Mit Zangen jeden Abschnitt in ein 45 °C-Wasserbad übertragen. Nehmen Sie den Abschnitt aus dem Wasserbad mit einer positiv geladenen Folie (Materialtabelle) auf, die zuvor mit der Proben-Identifikationsnummer mit einem Bleistift beschriftet ist.
- Legen Sie die Dias mit den Abschnitten in einen Ofen bei 65 °C, damit die Abschnitte an den Dias haften können. Halten Sie die Dias für H&E 25 min im Ofen. Bewahren Sie die Dias für die DNA-Extraktion 20 min im Ofen auf.
HINWEIS: Die kürzere Inkubationszeit macht das Gewebe etwas weniger an den Dias haftend und erleichtert somit die Entfernung des mütterlichen Gewebes.
-
H&E Färbung
- Lassen Sie die Dias auf Raumtemperatur abkühlen (10 min).
- Reagenz-Präparation
- Vorbereiten der Arbeitslösung Eosin Y (0,25%) gemäß Tabelle 1. Gut mischen und bei Raumtemperatur aufbewahren.
- Bereiten Sie die Arbeitshämatoxylinlösung vor, indem Sie die Vorratslösung von Hämatoxylin 5x in Wasser verdünnen (d. h. 80 ml Wasser mit 20 ml Hämatoxylin mischen).
HINWEIS: Wrap Lagerlösung von Hämatoxylin in Folie für die Lagerung.
- Bereiten Sie Färbegläser mit den richtigen Reagenzien unter einer Dunstabzugshaube gemäß Tabelle 2vor.
- Führen Sie die H&E-Färbung durch Untertauchen der Dias in die entsprechenden Färbegläser für den richtigen Zeitraum gemäß Tabelle 2durch.
- Montieren Sie die 4'm-Abschnitte für die morphologische Analyse mit Montagemedium und Deckslip mit Glasabdeckungen (Materialtabelle).
HINWEIS: Die 10 m Abschnitte für die Genotypisierung sollten nicht abgedeckt werden. - Lassen Sie die 10 m Abschnitte unter der Dunstabzugshaube für mindestens 3 h, damit sich die giftigen Xylolgerüche auflösen.
ACHTUNG: Alle Färbeschritte müssen unter einer Dunstabzugshaube durchgeführt werden. Xylol-Produkte müssen jederzeit unter der Haube aufbewahrt werden, da Xylolgerüche giftig sind. Darüber hinaus müssen Xylol und Hämatoxylin in speziellen Behältern entsorgt werden. Sobald diese Behälter voll sind, müssen sie wie von der Sicherheitsorganisation des Labors empfohlen entsorgt werden.
| Reagenz | menge |
| Eosin Y Aktienlösung (1%) | 250 ml |
| 80% Ethanol | 750 ml |
| Eishaltige Essigsäure (konzentriert) | 5 mL |
Tabelle 1: Arbeitslösung Eosin Y (0,25%) vorbereitung.
| Verwendete Reagenz (100 ml pro Behälter) | dauer |
| 1) Xylol | 5 Min. |
| 2) Xylol | 5 Min. |
| 3) 100% Ethanol | 2 Min. |
| 4) 95% Ethanol | 2 Min. |
| 5) 70% Ethanol | 2 Min. |
| 6) 50% Ethanol | 2 Min. |
| 7) Destilliertes Wasser | 5 Min. |
| 8) Hämatoxylin | 4 Min. |
| 9) Destilliertes Wasser | 5 Min. |
| 10) Eosin | 1 Min. |
| 11) 95% Ethanol | 5 Min. |
| 12) 100% Ethanol | 5 Min. |
| 13) Xylol | 5 Min. |
| 14) Xylol | 5 Min. |
Tabelle 2: Reagenzien und Dauern für das H&E-Färbeprotokoll.
-
Isolierung des Lebenslaufs
- Verwenden Sie unter einem leichten Stereomikroskop Zangen und kleine Stücke von wasserbefeuchteten Papiertüchern (Materialtabelle), um unerwünschte smütterliche Gewebe aus H&E-gefärbten 10 m dicken Abschnitten abzukratzen.
HINWEIS: Das Endziel ist es, nichts als CV oder fetale Membranen (wenn vorhanden) auf den Dias zu halten und damit alle anderen Gewebe zu entfernen. Dieser Schritt kann viel Zeit und Geduld erfordern, je nach Block, da er sorgfältige Aufmerksamkeit auf Details erfordert. - Lassen Sie eine zweite Person die Dias nach der Reinigung überprüfen, um sicherzustellen, dass sie frei von mütterlichem Gewebe sind.
- Nehmen Sie Fotos von den gereinigten Dias auf oder dokumentieren Sie Folgendes, um bei der Dateninterpretation zu helfen: 1) ob das Gewebe schwer zu reinigen, hämorrhagisch oder sehr sauber war, 2) die Anzahl der verwendeten Abschnitte und 3) die ungefähre Menge an gereinigten Geweben.
HINWEIS: Abbildung 1 zeigt ein Beispiel für eine Folie, die leicht zu reinigen ist. Für einen Block, der ungefähr diese Menge an CV enthält, reichen 10 Abschnitte für die DNA-Extraktion aus. Die Folie in Abbildung 2 hat nur sehr wenige Lebensläufe, die mit mütterlichem Gewebe vermischt sind, was die Reinigung sehr schwierig und zeitaufwändig macht. Für einen Block, der ungefähr diese Menge an CV enthält, werden 30 Abschnitte für die DNA-Extraktion benötigt. - Sammeln Sie den Lebenslauf mit kleinen befeuchteten Papiertüchern. Mit der Zange ein winziges Stück aus den befeuchteten Papiertüchern reißen und mit ihm den Lebenslauf sammeln.
- Legen Sie die Papiertücher mit ihrem beigefügten Lebenslauf in ein beschriftetes 1,5 ml Rohr.
- Minimieren Sie die Menge an Papiertüchern, die in diesem Schritt verwendet werden, da zu viel die DNA-Extraktionsspalte verstopfen und folglich die endgültige Menge der gesammelten DNA reduzieren kann. Im Durchschnitt sollen weniger als sieben kleine Papiertücher pro Probe verwendet werden. Wenn dies aufgrund des Vorhandenseins großer Mengen von CV nicht möglich ist, teilen Sie die Probe auf zwei Rohre auf, um die Extraktion zu erleichtern.
- Verwenden Sie unter einem leichten Stereomikroskop Zangen und kleine Stücke von wasserbefeuchteten Papiertüchern (Materialtabelle), um unerwünschte smütterliche Gewebe aus H&E-gefärbten 10 m dicken Abschnitten abzukratzen.

Abbildung 1: Repräsentative Folie für Genotypisierung. Oben: Eine Rutsche, die "gereinigt" werden muss, um frei von mütterlichem Gewebe zu werden. Unten: Das gleiche Dia, das nach der Reinigung gezeigt wurde und nun nichts als CV für die DNA-Extraktion enthält. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Repräsentative Folie für Genotypisierung. Oben: Eine Rutsche, die "gereinigt" werden muss, um frei von mütterlichem Gewebe zu werden. Unten: Das gleiche Dia, das nach der Reinigung gezeigt wurde und nun nichts als CV für die DNA-Extraktion enthält. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Folgen Sie dem Protokoll der DNA-Extraktion aus FFPE-Kit (Tabelle der Materialien) dna-Extraktion durchzuführen.
HINWEIS: Einige Kits empfehlen die Verwendung von 15 x 20 l Elutionspuffer für die endgültige Elution. Erfahrungsgemäß eignet sich die Elution mit 15 L Elutionspuffer für die meisten Proben gut. Verdünnungen können bei Bedarf aus der Bestands-DNA hergestellt werden.
-
DNA-Quantifizierung
- Mit Hilfe eines Laborspektrophotometers laden Sie 1 L DNA und messen die Absorption bei 260 nm für die Quantifizierung.
- Laden Sie 1 l DNA auf ein 2% Agarose-Gel und führen Sie die Gelelektrophorese bei einer Spannung von 80 bis 100 V zur qualitativen Auswertung aus.
- Wählen Sie auf der Grundlage der Ergebnisse der Schritte 1.6.1 und 1.6.2 das Volumen der DNA aus, das bei der Multiplex-Kurz-Tandem-Repeat-Amplifikation (STR) Polymerase-Kettenreaktion (PCR) verwendet werden soll. Ziel ist es, mindestens 1000 ng DNA in der folgenden PCR-Verstärkung zu verwenden.
ANMERKUNG: Abbildung 3 zeigt repräsentative Beispiele für Gele zusammen mit den Konzentrationen der DNA (basierend auf den Spektralphotometer-Ergebnissen) und dem Volumen der DNA-Lösung, die für die nachfolgende Multiplex-STR-PCR empfohlen wird.

Abbildung 3: Repräsentatives Gel für die DNA-Quantifizierung. Enthalten sind die Konzentrationen jeder DNA, gemessen mit einem Spektralphotometer, und die für die Multiplex-PCR verwendeten Mengen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
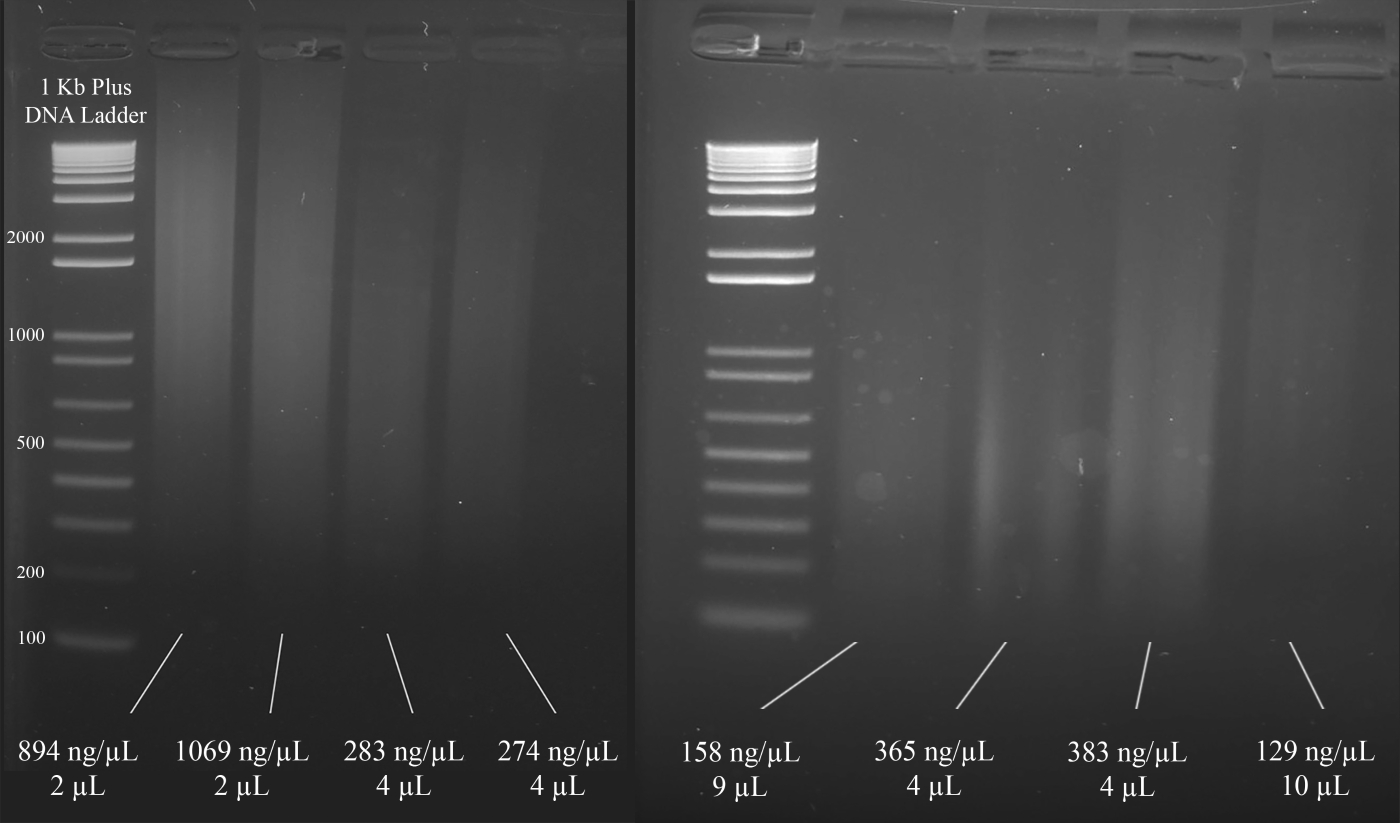{kind=link}
-
PCR-Verstärkung
- Durchführen einer fluoreszierenden Mikrosatellitengenotypisierung mit einem Multiplex-STR-System (Materialtabelle).
- Verwenden Sie die in Abbildung 4 dargestellten PCR-Bedingungen für die PCR-Verstärkung mit dem Multiplex-STR-System (Materialtabelle).
HINWEIS: Die folgenden Primer werden in diesem Multiplex-STR-System verwendet: D18S51, D21S11, TH01, D3S1358, Penta E, FGA, TPOX, D8S1179, vWA, Amelogenin, CSF1PO, D16S539, D7S820, D13S317, D5S818 und Penta D.

Abbildung 4: PCR-Zyklusbedingungen für das Multiplex-STR-System. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
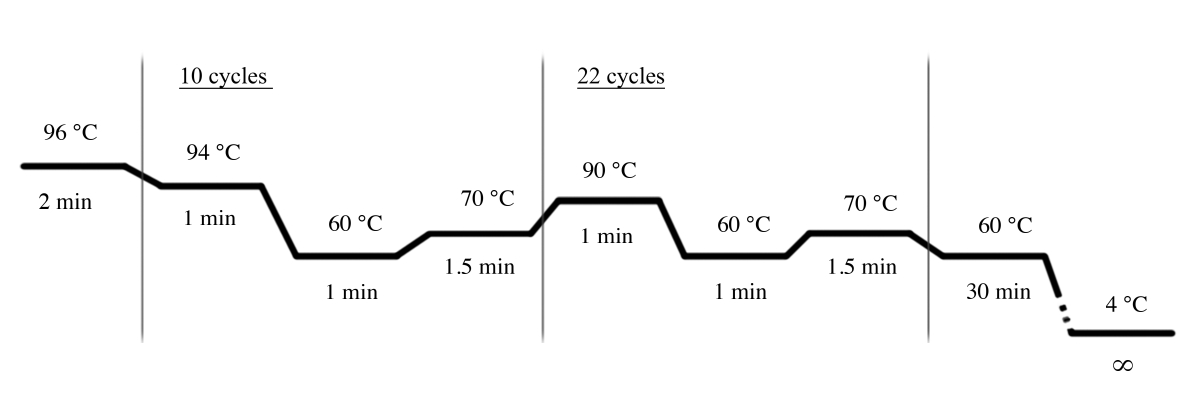{kind=link}
-
Lösen Sie die PCR-Produkte durch Kapillarelektrophorese.
- Suspendieren Sie 1 l jeder verstärkten Probe in 0,5 l der internen Standardspur des Multiplexsystems und 9,5 l hochdeionisiertem Formamid (Materialtabelle).
- Führen Sie Proben durch ein Kapillarelektrophorese-Instrument (Tabelle der Materialien) mit einer geeigneten Trennmatrix (Materialtabelle) für das Instrument und den Farbstoffsatz des Multiplexsystems.
- Datenanalyse
- Analysieren Sie die Daten mit einer DNA-Fragmentanalyse-Software und vergleichen Sie die POC-Allele mit den Elternallelen, um deren Herkunft zu bestimmen.
- Richten Sie einen Größenstandard ein.
HINWEIS: Dadurch kann die Software die Leiter erkennen, die im Multiplex-STR-System verwendet wird, und den Amplicons basierend auf der Leiter Basepairs zuweisen. Die folgenden Schritte beziehen sich auf eine bestimmte Software(Tabelle der Materialien), können jedoch auch beim Einrichten anderer Softwaretypen hilfreich sein.- Öffnen Sie die Software. Klicken Sie auf Neues Projekt starten und dann auf New Size Standard.
- Geben Sie dem Größenstandard einen Namen (z. B. ABI_600).
- Geben Sie im Feld Enter New Size Standard Definition folgendes ein:60, 80, 100, 120, 140, 160, 180, 200, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475, 500, 550, 600. Klicken Sie dann auf Größe hinzufügen(en).
HINWEIS: Die eingegebenen Zahlen werden unter dem Feld auf der rechten Seite angezeigt, das den Namen Aktuelle Größenstandarddefinition trägt (siehe Abbildung 5). - Klicken Sie auf Speichern.
- Um eine Datei zu importieren und zu analysieren, klicken Sie auf Dateien hinzufügen, und wählen Sie die zu analysierende fsa-Datei aus. Klicken Sie auf Ausgewählte Dateien hinzufügen und dann auf OK. Führen Sie dann die folgenden Schritte aus:
- Suchen Sie die Spalte Größenstandard, und wählen Sie ABI_600 (oder den Namen, der dem Größenstandard gegeben wurde).
- Klicken Sie unter Analysemethodeauf Sizing Default - NPP und dann auf die grüne Schaltfläche Analysieren.
- Die Datei kann nun angezeigt werden. Passen Sie die Anzeigeoptionen an, um die Daten wie gewünscht anzuzeigen.
- Troubleshooting - Analysemethode
HINWEIS: Die Software kann manchmal nicht spitzen identifizieren und sie richtig ausrichten. Dies geschieht, wenn die Spitzen entweder zu niedrig oder zu hoch sind. Die folgenden beiden Analysemethoden können dies korrigieren und sollten versucht werden, bevor eine Probe erneut getestet wird.- Analysemethode 1 für hohe Spitzen:
- Klicken Sie auf Neue Analysemethode und nennen Sie sie High Peaks (oder einen anderen Namen gemäß persönlichen Vorlieben).
- Klicken Sie auf Bereich und dann auf Teilbereich für die Analyse und Größenänderung. Geben Sie dann 100 für den Startpunkt und die Startgröße ein.
- Geben Sie für den Stopppunkt10.000 ein. Geben Sie für die Stop-Größe1000 ein.
- Klicken Sie dann auf Minimale Spitzenhöhen und ändern Sie die Zahlen so, dass die Spitzenschwelle für die Farben wie folgt lautet: Blau: 50; Grün: 50; Gelb: 20; Rot: 100; Orange: 5000.
- Speichern Sie die neue Analysemethode.
- Analysemethode 2 für niedrige Spitzen:
- Klicken Sie auf Neue Analysemethode und nennen Sie sie Low Peaks (oder einen anderen Namen gemäß persönlichen Vorlieben).
- Klicken Sie auf Bereich und dann auf Teilbereich für die Analyse und Größenänderung. Geben Sie dann 100 für den Startpunkt und die Startgröße ein.
- Geben Sie für den Stopppunkt10.000 ein. Geben Sie für die Stop-Größe1000 ein.
- Klicken Sie dann auf Qualitätsflaggen und ändern Sie den Passbereich so, dass er von 0,5 auf 1lautet. Ändern Sie den Bereich "Niedrige Qualität", sodass er von 0,0 auf 0,0lautet. Ändern Sie die Linearität in Folgendes: von (bp) 100.0 bis (bp) 800.0.
- Speichern Sie die neue Analysemethode.
HINWEIS: Es ist nun möglich, eine Datei neu zu analysieren, indem Sie Low Peaks oder High Peaks unter Analysemethode auswählen und dann auf die grüne Schaltfläche Analysieren klicken.
- Analysemethode 1 für hohe Spitzen:

Abbildung 5: Screenshot mit dem Größenstandard-Editor. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
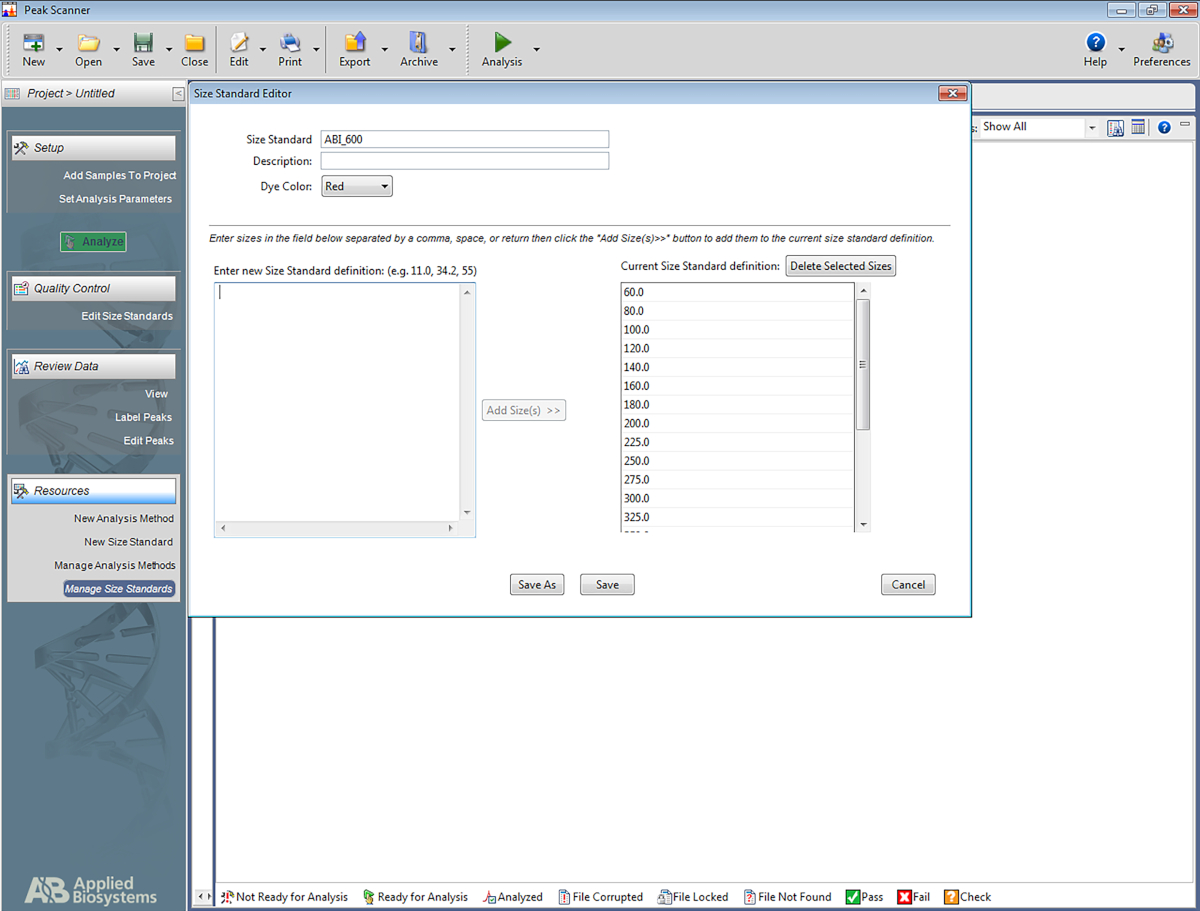{kind=link}
2. Durchflusszytometrie
-
Die Wahl des idealen FFPE-Blocks
- Wählen Sie mit H&E-Dias und einem Lichtmikroskop einen FFPE-Block aus, der etwa 50 bis 70 % seines Gewebes aus CV enthält.
ANMERKUNG: Abbildung 6 ist ein repräsentatives Beispiel für einen geeigneten Block für die Durchflusszytometrieanalyse, da er aus etwa 50 % CV (rechte Hälfte des Abschnitts) und 50 % mütterlichem Gewebe (linke Hälfte) besteht. Das Vorhandensein von mütterlichem Gewebe ist wichtig, weil sie als interne Kontrolle für den diploiden Gipfel dienen. - Für Blöcke, die nicht die ideale Menge an Lebenslauf haben, bereichern Sie für CV, da der Schnitt durchgeführt wird. Identifizieren Sie dazu, welche Seite der frisch geschnittenen Abschnitte mehr Lebenslauf gemäß der entsprechenden H&E-Folie enthält. Verwenden Sie auf dieser Grundlage eine Klinge, um die andere Hälfte abzuschneiden, die verworfen werden muss, um sich für den Lebenslauf zu bereichern.
ANMERKUNG: Abbildung 7 zeigt einen Block, der nicht über einen ausreichenden Cv für die Durchflusszytometrieanalyse verfügt. Bei Blöcken wie diesem müssen die Abschnitte so geschnitten werden, dass die Hälfte, die weniger Lebenslauf enthält, verworfen wird, um die Mengen an Lebenslauf in Bezug auf mütterliche Gewebe zu erhöhen, wie in der Abbildung dargestellt. Achten Sie darauf, mehr Abschnitte zu schneiden, um zu kompensieren, was verworfen wird.
- Wählen Sie mit H&E-Dias und einem Lichtmikroskop einen FFPE-Block aus, der etwa 50 bis 70 % seines Gewebes aus CV enthält.

Abbildung 6: H&E-Abschnitt, der einen POC-Block darstellt, der sich ideal für die Durchflusszytometrie eignet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 7: H&E-Abschnitt, der einen schwierigeren Block für die Durchflusszytometrie darstellt. Dieser repräsentative H&E-Abschnitt zeigt, dass nur die untere Hälfte dieses Abschnitts für die Analyse der Durchflusszytometrie verwendet werden sollte, mit dem Ziel, den Lebenslauf zu bereichern. Der umrissene Bereich mit der Bezeichnung "CV" besteht größtenteils aus Lebenslauf. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Schneiden
- Lassen Sie die Blöcke für 15 min auf Eis, um den Schnitt zu erleichtern.
- Schneiden Sie mit dem bestmöglichen FFPE-Block vier Abschnitte, die 50 m dick sind (oder zwei 100 m dicke Abschnitte), mit einem Mikrotome.
HINWEIS: Bei der Durchflusszytometrie ist es vorzuziehen, dickere Abschnitte zu haben. - Für den Fall, dass kein idealer FFPE-Block verfügbar ist, soll das Verhältnis von Lebenslauf zu mütterlichem Gewebe dennoch erhalten bleiben. Wenn z. B. nur 30 % des Blocks aus Lebenslauf bestehen, während der Rest mütterliche Gewebe hat, entfernen Sie mindestens die Hälfte des Abschnitts, der das mütterliche Gewebe enthält, und verwenden Sie weitere Abschnitte zum Ausgleich (siehe Abbildung 7).
- Legen Sie die Abschnitte in beschriftete 15 ml Rohre.
HINWEIS: Achten Sie darauf, die Etiketten zu kleben, da die im nächsten Schritt verwendeten organischen Reagenzien Tinte auflösen und entfernen können.
- Flow-Zytometrie-Protokoll aus FFPE-Geweben
- Deparaffinierung und Rehydratation
- Führen Sie die folgenden Wärpunkte (Tabelle 3) unter einer Dunstabzugshaube durch.
- Füllen Sie das 15 ml-Rohr nach der in Tabelle 3 beschriebenen Reihenfolge mit 6 ml des entsprechenden Reagenzes, lassen Sie die Abschnitte in den Reagenzien für die jeweilige Dauer und entfernen Sie dann das Reagenz mit Vakuumsaugung und einer Glaspasteurpipette.
- Zwischen jedem Schritt tauchen Sie die Pasteur-Pipette zuerst in 70% Ethanol, dann in destilliertes Wasser, und fahren Sie dann mit dem nächsten Schritt fort.
- Achten Sie sehr darauf, keine Gewebestücke zusammen mit dem Reagenz zu entfernen. Neigen Sie das 15 ml-Rohr in einen 60-Grad-Winkel, um das Absaugen des flüssigen Reagenzes ohne Gewebezuziehen zu erleichtern.
ACHTUNG: Die ausrangierten Flüssigkeiten enthalten Xylol und sollten in Xylol-Abfallbehältern entsorgt werden.
- Lösungsvorbereitung
- Citratlösung vorbereiten, indem Man 2 g Zitronensäure in 1 L doppelt destilliertes Wasser auflösen. Den pH-Wert auf 6 bringen. Bei 4 °C lagern.
- Bereiten Sie die Pepsinlösung vor, indem Sie 0,01 g Pepsin in 2 ml 9 Teilen pro tausend NaCl, pH 1,64 auflösen. Dies gilt für eine Stichprobe.
ACHTUNG: Pepsin ist giftig und kann sich leicht zerstreuen und in die Luft gelangen. Tragen Sie eine Maske, wenn Sie Pepsin in seiner Pulverform behandeln und wischen Sie den gesamten Arbeitsbereich nach der Verwendung ab. - Propidium Iodid (PI)-Ribonuclease Eine Lösungsvorbereitung für eine Probe.
- Mischen Sie 50 l PI mit 450 l PBS (zu verdünnen 10x).
- Fügen Sie dem Gemisch 50 l Ribonuklease A (1 mg/ml) hinzu. Immer in Folie eingewickelt halten.
- Verdauung und Färbung
- 4 °C Citratlösung in die 15 ml-Rohre geben und dann 2 h in ein 80 °C-Wasserbad geben.
- Lassen Sie die Lösung auf Raumtemperatur abkühlen (15 min). Entfernen Sie die Citratlösung.
- Fügen Sie 6 ml 1x PBS, Wirbel, und warten Sie 1 x 2 min, damit sich das Gewebe nach unten absetzen kann. Entfernen Sie die 1x PBS mit Vakuumsaugung und einer Glaspasteurpipette.
- 1 ml Pepsinlösung (auf 37 °C vorgewärmt) hinzufügen und 30 min. Vortex alle 10 min in ein 37 °C Trockenbad geben. Bereiten Sie die PI-Ribonuklease-A-Lösung in den letzten 10 min dieser Inkubation vor.
- Fügen Sie 6 ml 1x PBS, Wirbel, und warten Sie 1 x 2 min, damit sich das Gewebe nach unten absetzen kann. Entfernen Sie die 1x PBS mit Vakuumsaugung und einer Glaspasteurpipette.
- Fügen Sie 550 l der PI-Ribonuklease-A-Lösung hinzu und legen Sie die Proben 30 min in ein 37 °C-Trockenbad.
HINWEIS: An dieser Stelle können die Proben in Folie eingewickelt und über Nacht bei 4 °C bis zum nächsten Morgen gelassen werden. - Filtern Sie die Lösung durch ein 48-m-Filtrationsnetz. Sammeln Sie das Filtrat in Polystyrol-Rundbodenrohren, die mit dem Durchflusszytometer verwendet werden können. Verwenden Sie Zangen, um ein 5 cm x 5 cm großes Stück Filternetz in den oberen Teil des Rohres zu legen, so dass die Flüssigkeit durch das Netz und in das Rohr geleitet werden kann.
HINWEIS: Die Proben können nun mit dem Durchflusszytometer ausgeführt werden. Bewahren Sie sie in Folie eingewickelt auf, bis sie zum Laufen bereit sind.
- Führen Sie Proben mit einem Durchflusszytometer mit Hilfe des Technikers der Durchflusszytometrieplattform der Organisation aus.
HINWEIS: Der PE-Kanal wird verwendet, um die PI-befleckte DNA zu erkennen, und die Durchflussrate sollte während der Erfassung auf Langsam eingestellt werden. Stellen Sie sicher, dass die Spannung so gewählt ist, dass die diploide Spitze ungefähr 200 entlang der PE-A x-Achse beträgt, um die Analyse und Interpretation zu erleichtern. Ziel ist es, mindestens 20.000 Ereignisse pro Stichprobe aufzuzeichnen.
- Deparaffinierung und Rehydratation
| Verwendetes Reagenz (je 6 ml) | dauer |
| 1) Xylol | 2 x 10 min |
| 2) 100% Ethanol | 2 x 10 min |
| 3) 95% Ethanol | 10 Min. |
| 4) 70% Ethanol | 10 Min. |
| 5) 50% Ethanol | 10 Min. |
| 6) Destilliertes Wasser | 2 x 10 min |
Tabelle 3: Reagenzien und Dauern für die Deparaffinierung und Rehydratation.
-
Flusszytometrie-Datenanalyse
- Analysieren sie Daten mit einer Durchflusszytometrieanalysesoftware (Tabelle der Materialien).
HINWEIS: Die folgenden Schritte beziehen sich auf eine bestimmte Software(Tabelle der Materialien), können jedoch auch beim Einrichten anderer Softwaretypen hilfreich sein.- Nachdem Sie die Samples auf einem Durchflusszytometer ausgeführt haben, laden Sie FCS 2.0-Dateien zur Analyse herunter.
- Öffnen Sie die Flow-Zytometrie-Analyse-Software, klicken Sie auf Datei | Neues Dokument.
- Klicken Sie auf das
 Histogrammsymbol ( ), und ziehen Sie dann den Mauszeiger, um ein Rechteck zu erstellen.
Histogrammsymbol ( ), und ziehen Sie dann den Mauszeiger, um ein Rechteck zu erstellen. - Suchen Sie nach der FCS-Datei und klicken Sie dann auf Öffnen. Klicken Sie entlang der x-Achse auf FCS-A und wählen Sie dann PE-Aaus.
- Klicken Sie auf das
 Punktdiagrammsymbol ( ) und ziehen Sie dann den Mauszeiger, um ein weiteres Rechteck unter dem Histogrammdiagramm zu erstellen. Suchen Sie dann nach derselben FCS-Datei, die für das Histogramm ausgewählt wurde.
Punktdiagrammsymbol ( ) und ziehen Sie dann den Mauszeiger, um ein weiteres Rechteck unter dem Histogrammdiagramm zu erstellen. Suchen Sie dann nach derselben FCS-Datei, die für das Histogramm ausgewählt wurde. - Ändern Sie die x-Achse des Punktdiagramms in PE-A und die y-Achse in PE-W.
HINWEIS: Abbildung 8A zeigt das Erscheinungsbild der Diagramme an dieser Stelle. - Klicken Sie auf
 das Symbol Region ( ) und zeichnen Sie ein Feld auf dem Punktdiagramm, das vor dem diploiden Peak beginnt (etwa 100 auf der x-Achse in Abbildung 8B) und das um 700 auf der x-Achse endet, wie im Punktdiagramm in Abbildung 8 dargestellt. B.
das Symbol Region ( ) und zeichnen Sie ein Feld auf dem Punktdiagramm, das vor dem diploiden Peak beginnt (etwa 100 auf der x-Achse in Abbildung 8B) und das um 700 auf der x-Achse endet, wie im Punktdiagramm in Abbildung 8 dargestellt. B.
HINWEIS: Der diploide Spitzenwert in Abbildung 8 liegt ungefähr bei 200 auf der x-Achse. Dies wird willkürlich gewählt, da die Proben durch das Durchflusszytometer aufgezeichnet werden, einfach um die Analyse und Interpretation der Ergebnisse zu erleichtern. - Klicken Sie auf Plot | Bearbeiten Sie Regionen/Gates, und geben Sie dann R0 in die Zelle ein, die sich neben der G0-Zelle unter Strategiebefindet. Klicken Sie dann auf Schließen.
- Klicken Sie auf eine beliebige Stelle auf das Histogramm, dann auf Plot | Format Plot/Overlay. Wählen Sie unter Gate G0 = R0 aus und klicken Sie dann auf OK.
HINWEIS: Dies ist der Gating-Schritt, der es einem ermöglicht, die Ploidy-Spitzen besser zu visualisieren. Das Histogramm sollte nun wie das Histogramm in Abbildung 8Baussehen. Es ist möglich, mit dem erstellten Tor herumzuspielen (durch Verschieben der in Schritt 2.4.1.7 gezeichneten Box), um sich auf bestimmte Bereiche des Punktdiagramms zu konzentrieren. - Um die Plots zu beschriften,
 klicken Sie auf das Symbol Textbereich ( ), ziehen Sie dann den Mauszeiger, um ein Feld am oberen Rand des Dokuments zu erstellen, und geben Sie dann die folgenden Informationen ein: Patienten-ID, POC-ID und der verwendete Block (da es mehrere Blöcke für ein POC geben kann) , Prozent CV auf dem Block vorhanden, Spannung verwendet, um die Probe auszuführen, und das Datum.
klicken Sie auf das Symbol Textbereich ( ), ziehen Sie dann den Mauszeiger, um ein Feld am oberen Rand des Dokuments zu erstellen, und geben Sie dann die folgenden Informationen ein: Patienten-ID, POC-ID und der verwendete Block (da es mehrere Blöcke für ein POC geben kann) , Prozent CV auf dem Block vorhanden, Spannung verwendet, um die Probe auszuführen, und das Datum.
- Analysieren sie Daten mit einer Durchflusszytometrieanalysesoftware (Tabelle der Materialien).

Abbildung 8: Screenshot mit einem Histogramm und einem Punktdiagramm einer repräsentativen Stichprobe, die nicht (A) und gated (B) ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
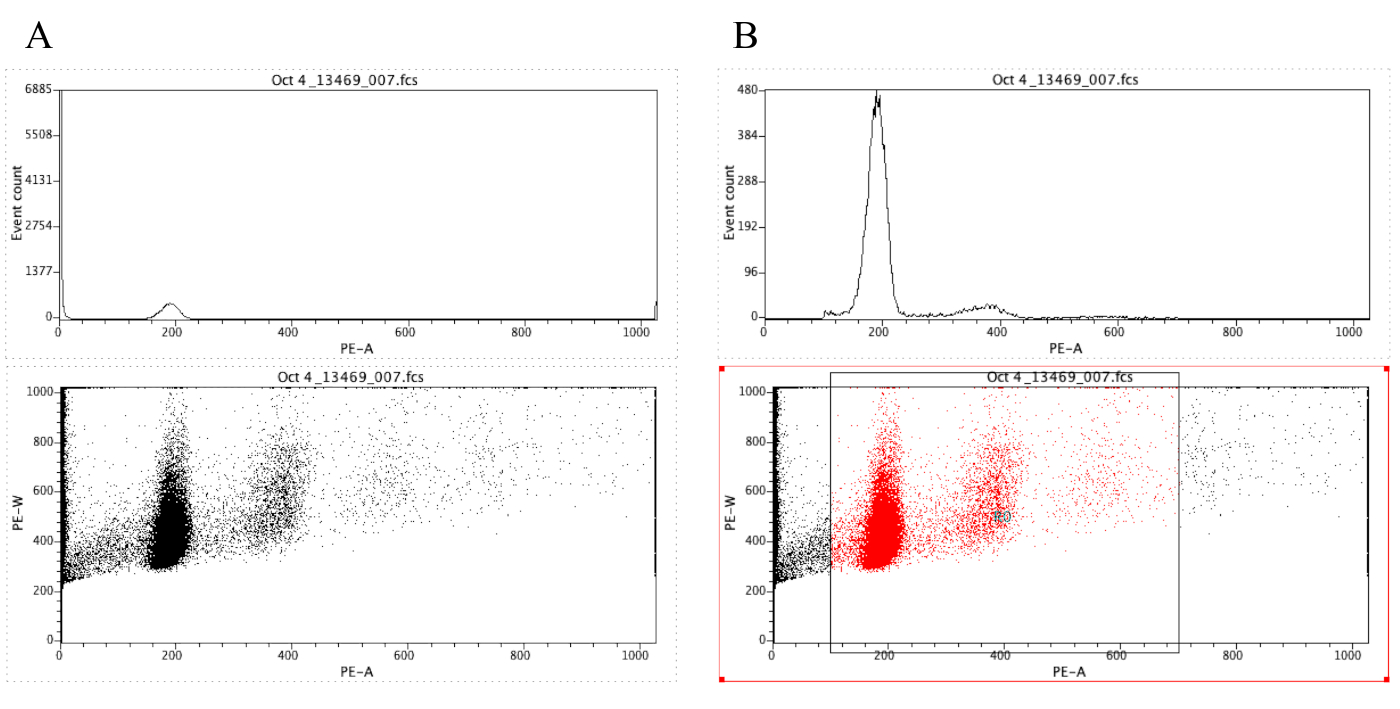{kind=link}
Ergebnisse
Die Komplexität von Molgeweben und die Tatsache, dass sie verschiedene Genotypen haben können, erfordern eine strenge Analyse und die Verwendung mehrerer Methoden wie morphologische Bewertung, p57-Immunhistochemie, Mikrosatellitengenotypisierung, Durchflusszytometrie und FISH. Zum Beispiel wurde ein Patient (1790) mit zwei PHM überwiesen, die nur durch Mikroarray-Analyse der POCs als triploid erwiesen wurden. Der Patient wurde daher mit wiederkehrendem PHM diagnostiziert. Die Mikrosate...
Diskussion
HM sind abnorme menschliche Schwangerschaften mit heterogenen Ätiologien und haben verschiedene histologische und genotypische Typen, was ihre genaue Klassifizierung und Diagnose schwierig macht. Die histopathologische morphologische Bewertung erwies sich oft als ungenau und ist daher allein unzuverlässig, um HM in CHM und PHM zu klassifizieren und von nicht-molaren Fehlgeburten zu unterscheiden. Daher erfordert eine genaue Diagnose von HM den Einsatz anderer Methoden wie Multiplex-Mikrosatelliten-DNA-Genotypisierung, ...
Offenlegungen
Die Autoren haben nichts zu verraten.
Danksagungen
Die Autoren danken Sophie Patrier und Marianne Parésy für die gemeinsame Nutzung des ursprünglichen Flow-Zytometrie-Protokolls und Promega und Qiagen für die Bereitstellung von Vorräten und Reagenzien. Diese Arbeit wurde von der Réseau Québécois en Reproduction und dem Canadian Institute for Health Research (MOP-130364) an R.S. unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| BD FACS Canto II | BD BioSciences | 338960 | |
| Capillary electrophoresis instrument: Genomes Applied Biosystems 3730xl DNA Analyzer | Applied biosystems | 313001R | Service offered by the Centre for Applied Genomics (http://www.tcag.ca) |
| Citric acid | Sigma | 251275 | |
| Cytoseal 60, histopathology mounting medium | Fisher | 23244257 | |
| Eosin Y stock solution (1%) | Fisher | SE23-500D | |
| FCSalyzer - flow cytometry analysis software | SourceForge | - | https://sourceforge.net/projects/fcsalyzer/ |
| FFPE Qiagen kit | Qiagen | 80234 | |
| Forceps | Fine Science Tools | 11295-51 | For sectioning and for the cleaning process |
| Glacial Acetic Acid (Concentrated) | Sigma | A6283-500mL | |
| Glass coverslips: Cover Glass | Fisher | 12-541a | |
| Hematoxylin | Fisher | CS401-1D | |
| Highly deionized formamide: Hi-Di Formamide | Thermofisher | 4311320 | |
| IHC platform: Benchmark Ultra | Roche | - | |
| Kimwipes | Ultident | 30-34120 | |
| Microtome | Leica | RM2135 | |
| Microtome blades | Fisher | 12-634-1C | |
| Nitex filtering mesh, 48 microns | Filmar | 74011 | http://www.filmar.qc.ca/index.php?filet=produits&id=51&lang=en ; any other filter is suitable, but this is an inexpensive and effective option from a non-research company |
| p57 antibody | Cell Marque | 457M | |
| Pasteur pipette | VWR | 53499-632 | |
| PCR machine | Perkin Elmer, Applied Biosystems | GeneAmp PCR System 9700 | |
| PeakScanner 1.0 | Applied Biosystems | 4381867 | Software for genotyping analysis. |
| Pepsin from porcine gastric mucosa | Sigma | P7012 | |
| Polystyrene round-bottom tubes | BD Falcon | 352058 | |
| Positively charged slides: Superfrost Plus 25x75mm | Fisher | 1255015 | |
| PowerPlex 16 HS System | Promega Corporation | DC2102 | |
| Propidium Iodide | Sigma | P4864 | |
| Ribonuclease A from bovine pancreas | Sigma | R4875 | |
| Separation matrix: POP-7 Polymer | Thermofisher | 4352759 | |
| UltraPure Agarose | Fisher | 16500-500 | |
| Xylene | Fisher | X3P1GAL |
Referenzen
- Szulman, A. E., Surti, U. The syndromes of hydatidiform mole. II. Morphologic evolution of the complete and partial mole. American Journal of Obstetrics & Gynecology. 132 (1), 20-27 (1978).
- Fukunaga, M., et al. Interobserver and intraobserver variability in the diagnosis of hydatidiform mole. The American Journal of Surgical Pathology. 29 (7), 942-947 (2005).
- Gupta, M., et al. Diagnostic reproducibility of hydatidiform moles: ancillary techniques (p57 immunohistochemistry and molecular genotyping) improve morphologic diagnosis for both recently trained and experienced gynecologic pathologists. The American Journal of Surgical Pathology. 36 (12), 1747-1760 (2012).
- Howat, A. J., et al. Can histopathologists reliably diagnose molar pregnancy?. Journal of Clinical Pathology. 46 (7), 599-602 (1993).
- Banet, N., et al. Characteristics of hydatidiform moles: analysis of a prospective series with p57 immunohistochemistry and molecular genotyping. Modern Pathology. 27 (2), 238-254 (2014).
- Lipata, F., et al. Precise DNA genotyping diagnosis of hydatidiform mole. Obstetrics & Gynecology. 115 (4), 784-794 (2010).
- Buza, N., Hui, P. Partial hydatidiform mole: histologic parameters in correlation with DNA genotyping. International Journal of Gynecologic Pathology. 32 (3), 307-315 (2013).
- Fisher, R. A., et al. Frequency of heterozygous complete hydatidiform moles, estimated by locus-specific minisatellite and Y chromosome-specific probes. Human Genetics. 82 (3), 259-263 (1989).
- Murdoch, S., et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nature Genetics. 38 (3), 300-302 (2006).
- Parry, D. A., et al. Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. American Journal of Human Genetics. 89 (3), 451-458 (2011).
- Nguyen, N. M., Slim, R. Genetics and Epigenetics of Recurrent Hydatidiform Moles: Basic Science and Genetic Counselling. Current Obstetrics and Gynecology Reports. 3, 55-64 (2014).
- Sebire, N. J., Savage, P. M., Seckl, M. J., Fisher, R. A. Histopathological features of biparental complete hydatidiform moles in women with NLRP7 mutations. Placenta. 34 (1), 50-56 (2013).
- Nguyen, N. M., et al. Comprehensive genotype-phenotype correlations between NLRP7 mutations and the balance between embryonic tissue differentiation and trophoblastic proliferation. Journal of Medical Genetics. 51 (9), 623-634 (2014).
- Brown, L., et al. Recurrent pregnancy loss in a woman with NLRP7 mutation: not all molar pregnancies can be easily classified as either "partial" or "complete" hydatidiform moles. International Journal of Gynecologic Pathology. 32 (4), 399-405 (2013).
- Colgan, T. J., Chang, M. C., Nanji, S., Kolomietz, E. A Reappraisal of the Incidence of Placental Hydatidiform Mole Using Selective Molecular Genotyping. The International Journal of Gynecological Cancer. 26 (7), 1345-1350 (2016).
- Murphy, K. M., McConnell, T. G., Hafez, M. J., Vang, R., Ronnett, B. M. Molecular genotyping of hydatidiform moles: analytic validation of a multiplex short tandem repeat assay. The Journal of Molecular Diagnostics. 11 (6), 598-605 (2009).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten