
Method Article
Quantitative Approaches for Scoring in vivo Neuronal Aggregate and Organelle Extrusion in Large Exopher Vesicles in C. elegans
* These authors contributed equally
In This Article
Summary
This protocol describes approaches for detection and quantitation of large aggregate and/or organelle extrusions (~4 µm) produced by C. elegans cells in the form of membrane-bound exophers. We describe strains, growth conditions, scoring criteria, timing, and microscopy considerations needed to facilitate dissection of this debris expulsion mechanism.
Abstract
Toxicity of misfolded proteins and mitochondrial dysfunction are pivotal factors that promote age-associated functional neuronal decline and neurodegenerative disease across species. Although these neurotoxic challenges have long been considered to be cell-intrinsic, considerable evidence now supports that misfolded human disease proteins originating in one neuron can appear in neighboring cells, a phenomenon proposed to promote pathology spread in human neurodegenerative disease.
C. elegans adult neurons that express aggregating proteins can extrude large (~4 µm) membrane-surrounded vesicles that can include the aggregated protein, mitochondria, and lysosomes. These large vesicles are called “exophers” and are distinct from exosomes (which are about 100x smaller and have different biogenesis). Throwing out cellular debris in exophers may occur by a conserved mechanism that constitutes a fundamental, but formerly unrecognized, branch of neuronal proteostasis and mitochondrial quality control, relevant to processes by which aggregates spread in human neurodegenerative diseases.
While exophers have been mostly studied in animals that express high copy transgenic mCherry within touch neurons, these protocols are equally useful in the study of exophergenesis using fluorescently tagged organelles or other proteins of interest in various classes of neurons.
Described here are the physical features of C. elegans exophers, strategies for their detection, identification criteria, optimal timing for quantitation, and animal growth protocols that control for stresses that can modulate exopher production levels. Together, details of protocols outlined here should serve to establish a standard for quantitative analysis of exophers across laboratories. This document seeks to serve as a resource in the field for laboratories seeking to elaborate molecular mechanisms by which exophers are produced and by which exophers are reacted to by neighboring and distant cells.
Introduction
The neurotoxic challenges of aggregates and dysfunctional mitochondria have long been considered to be cell-intrinsic, but more recently it has become clear that misfolded human disease proteins originating in one neuron can also spread to neighboring cells, promoting pathology1. Likewise, mammalian mitochondria can be sent out of the cell of their original production for transcellular degradation2 or for rescue of mitochondrial populations in challenged neighboring cells3. Vesicles of various sizes have generally been observed to transfer cellular materials to neighboring cells or to fluid surroundings4. Some extruded vesicles approach the size of the average neuronal soma (average touch neuron soma ~ 6 µm) and can accommodate large aggregates and organelles.
A striking example of large vesicle extrusion that can carry protein aggregates and organelles occurs in C. elegans touch receptor neurons that express a high copy number reporter construct encoding a noxious aggregation-prone, degradation-resistant mCherry5. Extrusions from the touch neurons, called exophers, are ~4 µm average diameter, selectively include mCherry or other aggregates, and are delivered directly into the neighboring hypodermis, which normally surrounds the touch receptor neurons. The hypodermis attempts lysosome-based degradation, but some non-digestible contents such as mCherry aggregates can be re-extruded by the hypodermis into the fluid-filled pseudocoelom of the animal, from which the mCherry can be taken up by remote scavenger cells called coelomocytes for long term storage (Figure 1, Figure 2)5.
The large extruded exopher vesicles leave the cell surrounded by touch receptor plasma membrane and can contain aggregated human disease proteins, mitochondria, and lysosomes. The process of exopher production appears to involve sorting of potentially toxic species (for example an aggregation-prone expressed mCherry is segregated from soluble, inoffensive proteins like GFP that remains mostly in the neuronal soma). In this way, directed expulsion of the threatening entities is accomplished by the neuron5. A proteostasis challenge, such as stress induced by autophagy knockdown, MG132-mediated proteasome inhibition, or transgenic expression of human disease proteins such as Huntington’s disease-associated expanded polyglutamine Q128 or Alzheimer’s disease-implicated fragment Aβ1-42, can increase the numbers of neurons that produce exophers5.
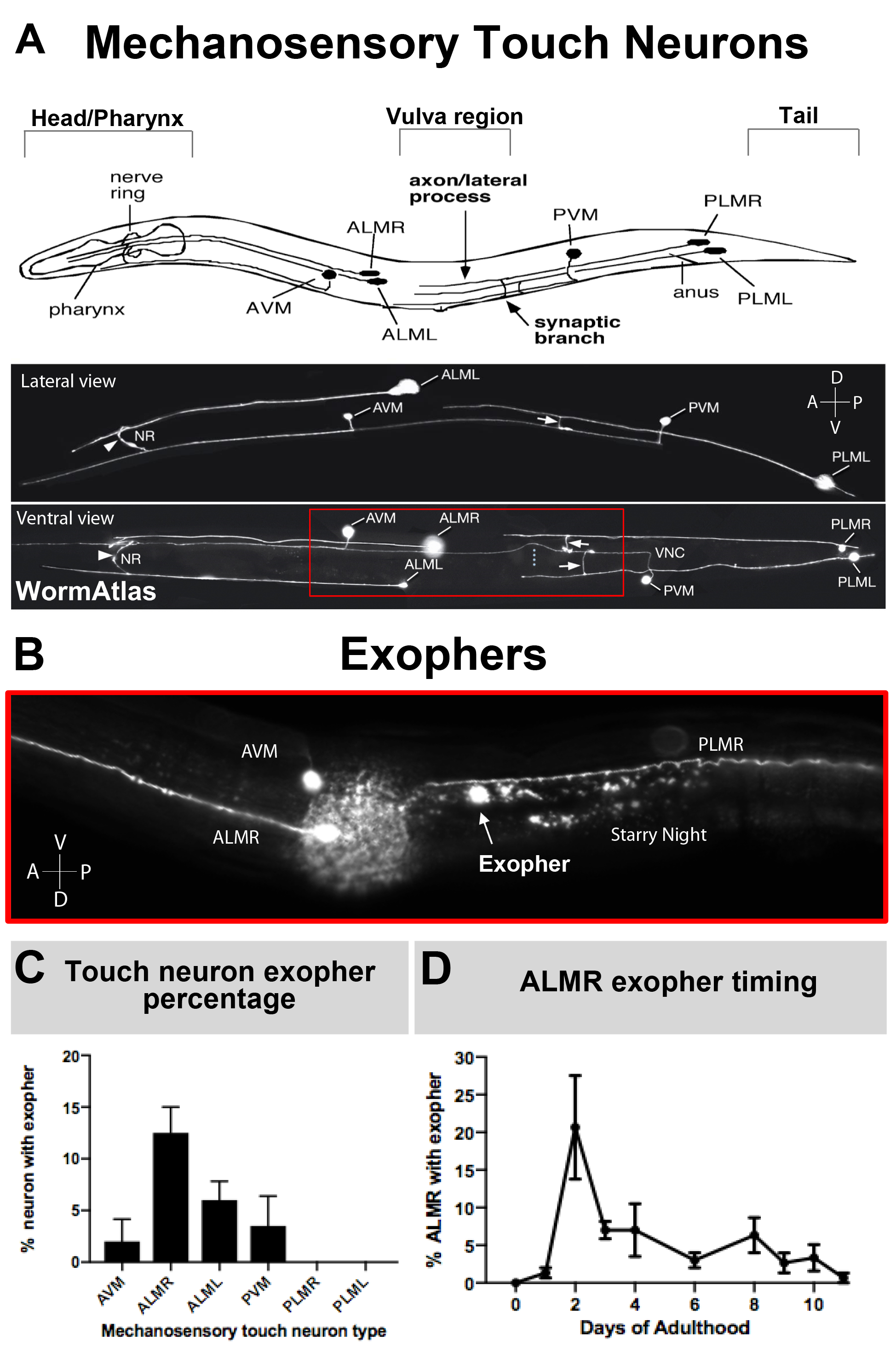
As exophers have only recently been documented, what is known of their biology merits description. Exophers were discovered in, and are the most well studied in, C. elegans touch receptor neurons. There are six C. elegans mechanosensory touch neurons that have cell bodies distributed around the body (Figure 3A) and are called microtubule cells because their ultrastructure features distinctive 15 protofilament microtubules. The touch receptor neurons are the anterior AVM (anterior ventral microtubule neuron), ALMR, and ALML (anterior lateral microtubule neurons right and left), the more central PVM (posterior ventral microtubule neuron), and the posterior PLMR and PLML (posterior lateral microtubule neurons right and left) in the tail. Interestingly, the six touch receptor neurons produce exophers at different rates, despite expressing the same offensive transgene (Figure 3C). Of the six mechanosensory touch receptor neurons, the ALMR neuron undergoes exophergenesis more often than the other touch neurons. Quantitation of exopher numbers from touch neurons is thus usually established by focusing upon the ALMR.
Exophergenesis is a dynamic process that typically begins with swelling of the neuronal cytoplasm (Figure 1A-B). Cellular contents, organelles, or protein aggregates are collected to one side of the neuronal soma, most commonly toward the posterior end of the ALMR neuron (away from the projecting neurite), forming a pre-exopher domain (PED) (Figure 1B). The early protrusion is observed as the PED begins to project outwards, forming a recognizable protruded bud. The late bud is defined when the widest diameter of the pre-exopher domain is approximately ⅓ larger than the diameter of the constriction of the soma-exopher neck (Figure 1C). Exophers can be ejected in nearly any direction from the soma, but most exophers exit posteriorly from the cell body and remain in approximately the same focal plane as the originating soma.
The exopher can move away from the originating soma as the neck of the bud narrows into a thin filament. Exophers can remain attached to the soma via this filament (Figure 1D, arrow) and can later become detached. Cellular contents such as calcium, aggregates, and mitochondria can be transferred via this filament into the attached exopher5, although the bulk of extruded material is put into the exopher compartment by the massive budding event. Exophers are considered mature when there is no visible connecting tube or thin filament and the exopher is fully separated from the sending soma (Figure 1E).
Exophers produced by C. elegans touch neurons immediately encounter the hypodermis, the tissue that surrounds the touch neuron. Most commonly, the exopher vesicle appears to travel within the hypodermis posteriorly towards the tail, and can be fairly distant from the soma before exopher contents appear targeted for degradation (for example, the distance can be ~100 µm away from the soma (Figure 1F)). The fluorescent exopher vesicle breaks up into many smaller vesicles within the hypodermis, taking on an appearance referred to as “starry night” (Figure 1G and Figure 2). In the “starry night” stage, punctate fluorescent material can be observed scattered across the hypodermal syncytium into many smaller points of fluorescence as compared to the original solitary exopher. Starry night can look punctate under low magnification and with higher magnification, can look punctate and/or networked within the hypodermis. The fluorescent signal of the starry night is typically dimmer than the exopher and the neuronally expressed fluorescence (Figure 2B-C). The dispersal of mCherry into many punctate vesicles is thought to involve phagosome maturation and fusion with the endosomal/lysosomal network of the hypodermal cell. Some exopher materials are likely degraded in the hypodermal lysosomal network, but residual species that are resistant to degradation (such as mCherry aggregates) are thrown out of the hypodermis into the pseudocoelom, a fluid compartment that can contain cellular debris. The fluorescent material is later taken in by remote scavenger cells called coelomocytes (Figure 2C), which can concentrate, store, and again attempt degradation of mCherry.
The phenomenon of aggregate extrusion and transfer appears conserved across phyla, having been reported in genetic models such as C. elegans5,6,7 and D. melanogaster8,9 as well as in multiple mammalian models. Exopher-like extrusions have been reported for mammalian cells10, an observation suggesting that conserved mechanisms might underlie aggregate and organelle expulsion. Exopher production may thus be a conserved mechanism of cellular debris management that constitutes a fundamental, but formerly unrecognized, branch of neuronal proteostasis and mitochondrial quality control, which, when imbalanced, might actively contribute to neurodegenerative disease. Identification of the molecules involved in debris discrimination and sorting, transport to a distinct subcellular locale, extrusion, formation/scission of the tubular connection linking the soma and late exopher, and recognition of the large extruded vesicle for remote degradation by a neighboring cell remain for future work. Studies in nematode and fly models will be critically important to defining mechanisms of aggregate and organelle collection and transfer, utilizing unbiased genetic approaches and powerful cell biological tools offered by these models to identify participating molecules in physiological context.
Critical first steps in deciphering mechanisms operative in exopher biology involve defining protocols for reproducible in vivo exopher quantification. The C. elegans model offers a particular advantage for such efforts since the body is transparent and exophers can be readily observed when they contain fluorescently tagged proteins or organelles. Exophers have been reported to be generated by C. elegans dopaminergic neurons PDE and CEP, ASE and ASER sensory neurons, and dye-filling amphid neurons5. Because exophers produced by touch receptor neurons are best characterized, the focus here is on the use of touch neurons for exopher analysis. However the basic approach can be applied to measure exopher production from any cell. Protocols to detect and quantitate exophers produced by C. elegans touch receptor neurons that transgenically express mCherry protein are outlined, with an emphasis on cargoes that can be monitored and temporal constraints in scoring. This article defines approaches toward in vivo exopher identification, and the quantitation of environmental and genetic conditions that modulate exopher production. Protocols emphasize critical attention to constant non-stress conditions for the determination of baseline exopher production and for comparisons across genotypes.
Protocol
1. Strains useful for exopher detection
- Select a strain that expresses fluorescent cargoes within the neurons of C. elegans to readily visualize exophers.
NOTE: Table 1 lists strains that have been used to visualize exophers produced in touch receptor neurons5,11,12. In principle, any cell or neuronal type can be tested for exopher production by using a cell or tissue specific promoter to drive expression of a fluorescent protein that aggregates or is otherwise selected for extrusion. - Alternatively, use a dye-filling assay to visualize exophers in the amphid head neurons, which are open to the environment and amenable to backfilling5,13.
2. Growth media
- Prepare standard nematode growth media (NGM) to culture strains according to standard methods14,15.
NOTE: Lack of food, or fluoro-deoxyuridine (FuDR), used commonly to block progeny production, and can dramatically affect exopher production. Keep the population continuously fed (avoid even short periods of bacterial food exhaustion) and maintain animals at a constant temperature.
3. Animal husbandry critical for consistent exopher production
- Raise animals on consistent media and with consistent bacterial food sources. Animals must not run out of bacterial food, even for short periods of time since food limitation can dramatically change exopher production levels.
- Keep media recipes and preparation uniform throughout a study.
NOTE: Changing media can affect basal levels of exopher production. Agar batches can influence baseline exopher levels, so when supply lots change, make a note of the date. Throw out stock plates after two weeks to ensure healthy bacterial food and to prevent dried agar, which causes changes in agar osmolarity that influence exopher levels. - For basal conditions, keep animals at a constant temperature of 20 °C. Rearing animals at variable temperatures (even temporary changes in temperature) can cause variations in the timing of maximal exopher production.
NOTE: Temperature variability is not limited to culture conditions. Temperatures variations during experiments or at the lab bench can be impactful. For example, temperatures within a microscope room should not differ dramatically from the culture incubator or lab bench. - Do not use pharmacological anti-fertility interventions because fertilized eggs are critical for early adult production of exophers.
NOTE: Use of Fluoro-deoxyuridine (FuDR)16 or C2217, must be avoided. When performing life-span or old-aged animal experiments, age-synchronized populations should be maintained by physically removing adults from their smaller progeny by picking them onto fresh plates spread with bacteria rather than using common pharmacological anti-fertility interventions. - Do not use contaminated cultures; reinitiate experiments in the event of biological compromise of the population or the plate. Bacterial or fungal contamination can induce stresses and metabolic changes in animals and must be absent from experimental populations.
- To maximize reproducible results, maintain cultures for at least two healthy, well-fed, contamination-free generations at 20 °C before experimentation to avoid potential environmentally-induced epigenetic changes.
4. Age synchronization for exopher scoring by bleaching, sucrose flotation, or L4 larvae picking
- Keep experimental populations the same biological age, as exopher detection patterns vary with adult age and comparison of animals of mixed age populations can confound results. Always ensure successful synchronization of experimental animal populations by checking for the “white crescent” vulva morphology at the L4 stage.
NOTE: Generally, peak exopher production for C. elegans mechanosensory ALMR neurons occurs on adult day 2-3 (Figure 3D), as measured from days post L4 stage. Adult day 1 is 24 hours after the L4 larval stage that is distinguished by “white crescent” vulva morphology (Figure 5E). - Prepare synchronized egg populations by bleaching gravid adults.
- Collect gravid adults filled with eggs by washing animals growing on an NGM plate. To wash, flood the plate with 1 mL of M9 buffer, pipet up and down to collect liquid with suspended animals and pipet into a 1.5 mL microcentrifuge tube. Pellet animals by gravitational settling or gentle centrifugation with a mini centrifuge and remove the supernatant.
- Add 150 µL of 5M NaOH and 150 µL of 6% sodium hypochlorite (bleach) in 1 mL in H2O and mix by inversion for approximately 5 minutes.
NOTE: Fresh bleaching solution ensures that the animal cuticle can be disrupted for egg-harvest. Progress in disrupting the cuticle can be monitored under a dissecting microscope; adults should break and release eggs at the point that bleaching should be stopped. - Gently centrifuge with a minicentrifuge tube for 20 s and remove the supernatant. Add 1 mL of M9 buffer and centrifuge again, leaving about 100 µL on top of the pellet.
- Repeat steps 4.2.3 twice to remove traces of bleach solution.
- Resuspend the eggs in the remaining volume and transfer to a fresh seeded NGM plate. Adults will be lysed, but many viable eggs should be in the preparation.
- Prepare synchronized populations by timed egg-lay.
- Pick 20 gravid adults to a seeded NGM plate using standard transfer protocols14.
- Allow animals to freely crawl and lay eggs for 1.5 h (mutant strains with low brood sizes may require the introduction of more adult animals).
- Remove all adult animals from the plate by picking, leaving the synchronized egg population behind. Check plates a few hours later to verify that no viable adults have been missed during adult removal.
- Prepare synchronized egg populations by sucrose flotation selection of eggs.
- Collect animals and eggs from five NGM plates upon which gravid animals have been laying eggs for at least 24 hours by flooding plates with M9 solution with 0.1% detergent (such as Tween 20 or Triton X-100) and collecting into a 15 mL tube. Pellet adults by gentle centrifugation at room temperature (2,000 x g for 30 s).
- Remove supernatant and wash animals in 15 mL of fresh M9 three times, discarding the supernatant after each wash, being sure to keep the pellet enriched in animals and eggs.
- Retain 2 mL of supernatant and resuspend the pellet. Add 2 mL of 60% weight by volume sucrose.
- Centrifuge at 2000 x g for 5 min. The solution will now display an upper phase highly enriched in eggs.
- Transfer about 2.5 mL of the upper phase to a new 15 mL tube and add 10 mL of M9.
- Mix by inversion for 1 min, and then centrifuge 2000 x g for 1 min.
- Remove the supernatant and wash the egg-enriched pellet in M9. 10-15 µL of the egg-pellet can be distributed to a fresh OP50-seeded NGM plate.
NOTE: This method prepares a great number of eggs; do not allow collected animals to run out of OP50 E. coli food.
- Prepare synchronized populations by picking animals at the L4 stage of development.
- Grow animals on seeded NGM plates as described above.
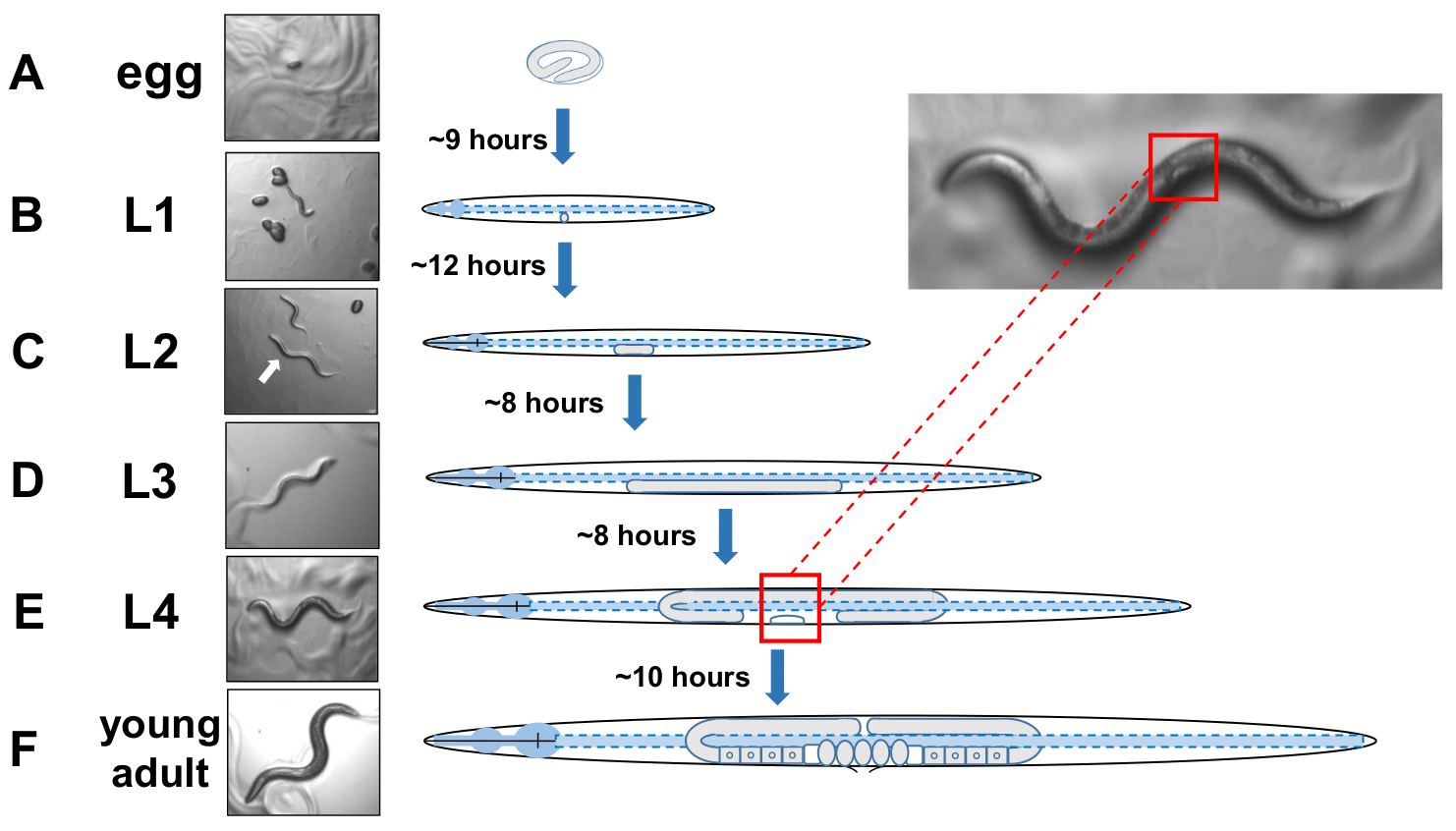
NOTE: C. elegans develop in four discrete stages. At 20 °C, a newly laid egg takes about 9 hours to hatch (Figure 5A). Post hatch, an animal passes through larval stage 1 (L1) to larval stage 4 (L4), with each stage lasting 8-12 hours between each molt (Figure 5A-F). Therefore, a plate prepared by inoculating with eggs should have many L4 animals to pick about 40 hours after eggs are introduced. - Identify L4-staged animals by locating the white half-moon crescent shape of the developing vulva (Figure 5E).
NOTE: Animals at the L4 stage are uniform in size and in body pigmentation. Pick animals with the white crescent to a fresh growth plate for examination of staged animals. The following day (~ 24 hours later) should be counted as adult day 1. - Score a population of animals daily on adult day 2.
NOTE: Exophers are typically scored at day 2 of adulthood, which is the peak exopher production under basal conditions. However, because the exophergenesis peak and timing may be shifted by environmental or genetic changes that are being studied, it is advised to score a population of adult animals daily over four days to generate the most comprehensive picture (Figure 3D).
- Grow animals on seeded NGM plates as described above.
5. Detection of exophers using a fluorescent microscope
- Observe exophers using a high-magnification pseudo-stereo dissecting microscope that is outfitted for fluorescence microscopy.
- Immobilize animals on NGM plates by pipetting 100-200 µL of 10-100 mM levamisole/tetramisole solution onto the NGM agar plate surface. After 2-4 minutes, the animals become paralyzed and can be observed directly on the agar plate.
NOTE: Immobilization treatments are not absolutely required, such that with a trained eye, neuronal identification and exopher presence can be scored by visually following crawling animals under the microscope on the plate when determining whether or not an exopher has been produced. - Observe fluorescent neurons using a total magnification of 100x to accomplish dissecting microscope detection of exophers.
NOTE: Scoring exopher events using dissection microscopy allows for the observation of large numbers of animals with relative ease directly on the agar plates on which they are reared. - Live imaging and mounting reporter strains for exopher studies using confocal microscopy
- Use a confocal microscope to live-image intracellular dynamics and characteristics of exophergenesis.
NOTE: Live imaging is an advantageous approach for observing subtle details of exopher production because exopher production is a dynamic process. - Restrict animal movement for high resolution live imaging using convenient methods, including utilization of levamisole or tetramisole at 10-100 mM or the application of hydrogel polystyrene microbeads (with diameters of 15 µm, 30 µm or 40 µm)18.
- Use a confocal microscope to live-image intracellular dynamics and characteristics of exophergenesis.
- Slide preparation for compound and confocal microscopy
- Mount 20-50 animals into an immobilizing agent on a microscope slide. Reusable ringed cytology slides painted with 13 mm diameter raised-rings are useful for mounting.
- Pick live animals into 5-20 µL of a paralytic such as 10-100 mM levamisole or tetramisole within the painted circle or on the agar pad.
- Wait 4 minutes for paralysis, and then cover the slide with a coverslip (recommend No. 1½ (0.16 – 0.19 mm) or No. 2 (0.17 - 0.25 mm).
- Mounting small numbers of animals
- Do not crush mounted animals; when observing only a few (less than 20) animals per slide, there is a risk of crushing some of the animals due to unequal pressure of the coverslip. This risk can be minimized by using a low percentage agarose pad for mounting.
- Make a 2-4% agarose pad slide, and then add 2-15 µL of paralytic solution to the pad. Keep in mind levamisole and tetramisole diffuse into the pad, decreasing their effective concentration.
- Mount by picking animals into a 2-15 µL drop of paralytic solution or microbeads resting on the agar pad. Place the coverslip on top and check that the animals are intact18.
- Agar pad preparation
- To prepare 2% agar pads, heat 2% agarose in M9 solution and microwave until the agarose is in a homogeneous and molten state.
- To achieve an agar pad of sufficient quality, alternate mixing and microwaving at low power for less than 20 seconds. Avoid the inclusion of air bubbles within the pad by placing boiling agar on a heating block and allowing the bubbles to rise to the surface.
- Use a Pasteur pipette to draw agar from deep within the molten solution below the risen bubbles.
- Prepare two taped slides and place on either side of a clean glass microscope slide upon a flat surface. To make the taped slides place two 5 cm strips of laboratory tape upon each slide (Figure 6A).
- Using a Pasteur pipet, place a single drop of agar upon the clean microscope slide sandwiched between the taped slides (Figure 6B).
- Carefully and quickly, cover the drop of molten agar with a fourth clean slide by placing in across the taped slides (Figure 6Cc).
NOTE: The slide should gently press the molten agar into a flattened circle about 0.4 mm thick (the thickness of the tape) (Figure 6D). The agar should quickly cool. - Remove the top slide by sliding it off (Figure 6E). Agar pads dry quickly and are best used within minutes. Once the top slide is removed, use the gel pad immediately for mounting animals. Avoid using pads with air bubbles.
- Store agar pads up to 30 minutes encased between the two glass slides. Dried agar causes animals to clump together and desiccate. Mount animals within 2-15 µL of paralytic solution or microbeads and cover with coverslip; screen the slide within 20 minutes of paralysis and mounting (Figure 6).
NOTE: Because stress conditions can alter exopher rates, avoid paralytics that can induce oxidative stress (ex: sodium azide) when screening for exophers.
- Detection of exophers using a spinning-disk confocal microscope
- Observe cell biological features such as organelles and other contents with 1.4 numerical aperture objectives at 63x and 100x.
- Use software capable of stage-control and image acquisition utilizing the multidimensional acquisition. Microscopes and image processing software should also be suitable for imaging and data collection as these steps involve standard imaging approaches.
6. Identifying touch neurons and scoring for exophers with mounted animals
- Mount paralyzed adult animal (Figure 6).
- Identify the desired Z-plane. Use low magnification bright-field (10-40x) to identify the suitable Z-plane of the animal, taking note of the positioning of the animal, head-tail orientation, and location of the vulva - which are landmarks for later neuronal and exopher identification (Figure 3A & Figure 5E).
- Focus on the fluorescence signal of the chosen reporter. Staying in the same Z-plane, switch to widefield fluorescence viewing at 10-40x for the chosen cytosolic reporter.
NOTE: In this example fluorescent expression is driven by the mec-4 mechanosensory touch neuron-specific promoter. High copy arrays, and different fluorophores have variability in expression and therefore variable fluorescent intensity. Adjust if needed. - Scroll within the Z axis to observe the depth of the animal and fluorescent expression in the focal plane. While doing so, confirm the head-tail orientation; the head/pharynx will have the fluorescent nerve ring and in this case, the tail will contain 1-2 visible PLM somas (Figure 3A).
- Identify touch neurons
- Identify whether the animal is mounted on the left or the right side (Figure 3A).
NOTE: Considering the 3-dimensionality of the animal, the best imaging resolution is accomplished on the side closest to the optics. - Identify the soma (ALM, ALMR, AVM) by observing - start at the head to identify the nerve ring and lateral neuronal processes.
- At 10-40x magnification, slowly scroll through the Z-axis to identify the attached process.
- Once the process is identified, follow it laterally in the posterior direction towards the vulva, where the soma will be apparent, marked by a round cell body at the end of the process. Once the most in-focus neuronal soma is found, it can be identified by using other neuronal landmarks as follows:
- Use the AVM, a nearby ventral neuron, to help assign animal orientation. If the AVM neuron is in the same plane as the ALM then the animal is resting upon its side and the neuron outside that plane is the ALMR . If the AVM neuron is not in the same plane as the ALM in question, the closest touch neuron to the focal plane is ALML.
- Identify the PVM neuron, another ventral touch neuron located near the tail, to indicate whether the anterior touch neuron is in the same plane. If so, the touch neuron observed is ALML.
- Get a sense of the position of other soma bodies, near the area of interest (fluorescent neurons located on either side of the soma), and in all Z-planes, even if it is not possible to get the deepest neuron set in clear focus.
NOTE: The identification of all touch neuron somas is important because out-of-focus soma can be mistaken for exophers.
- Identify whether the animal is mounted on the left or the right side (Figure 3A).
7. Identifying and scoring for exophers
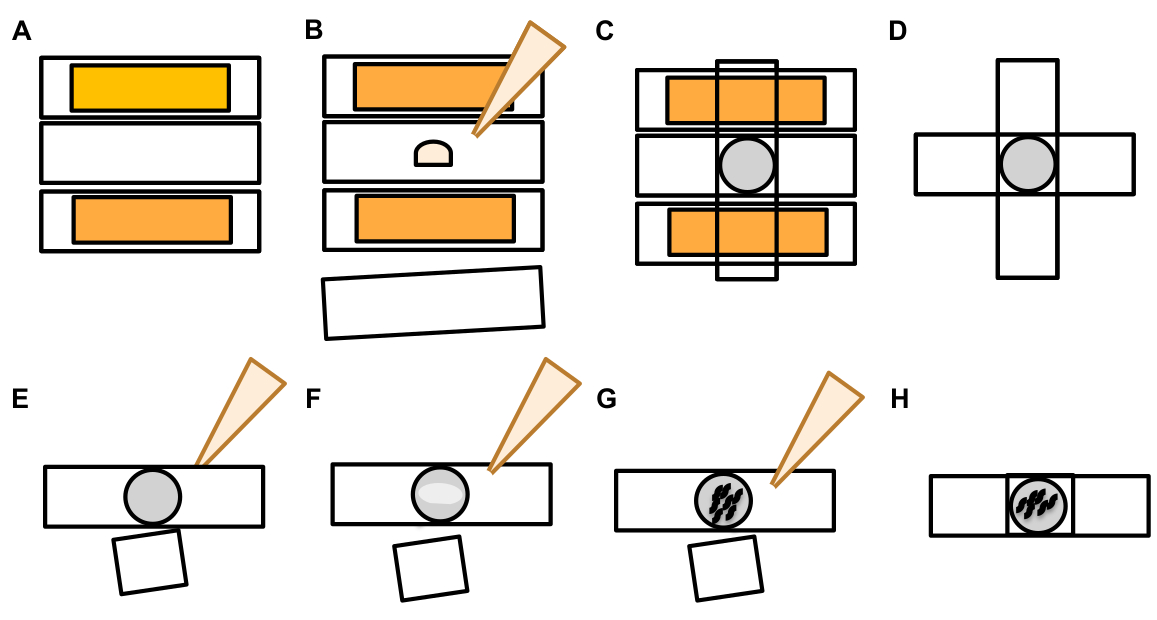
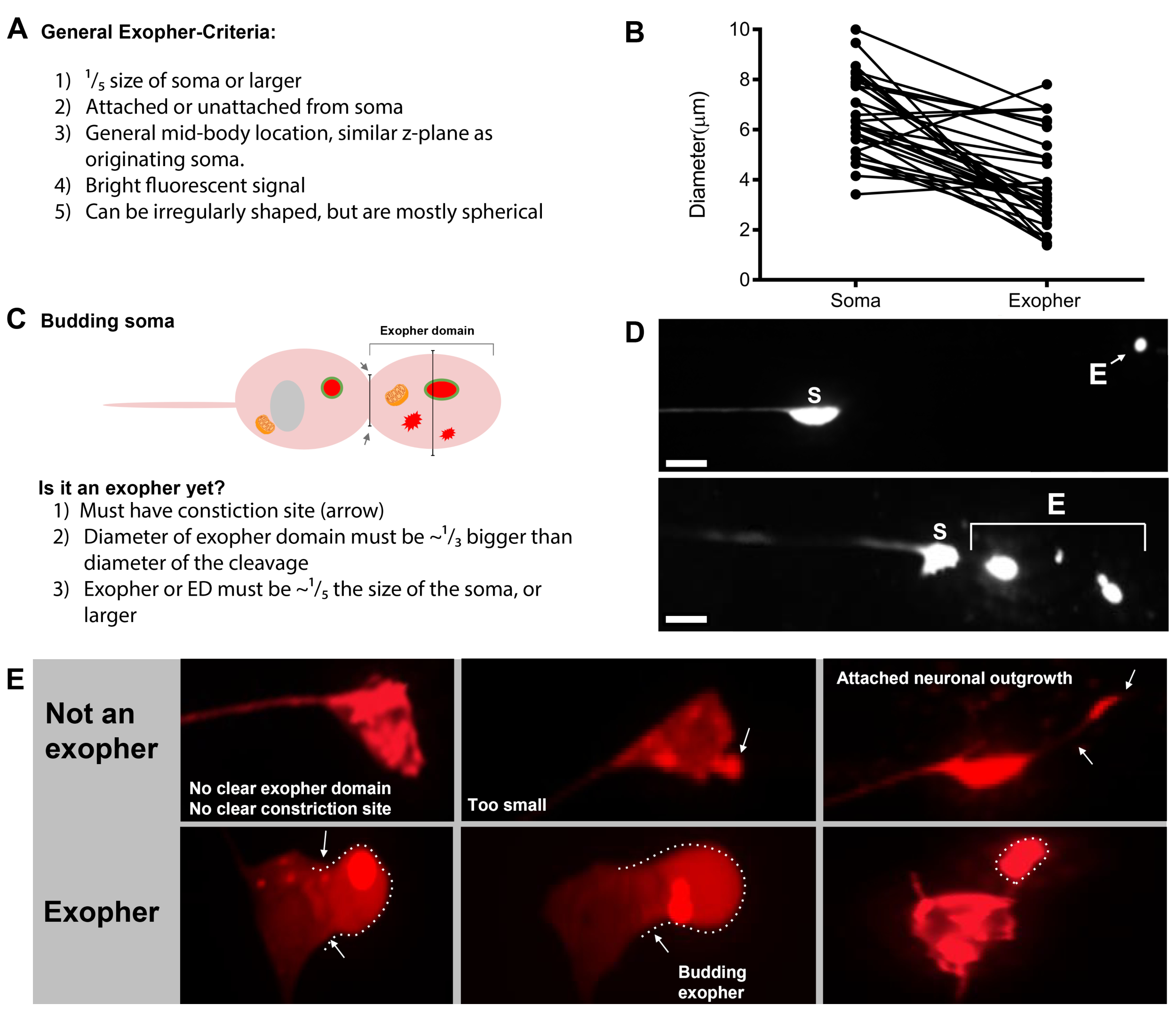
- Once a touch neuron is found, inspect it for large protrusions (exopher domains) large enough to be considered a bud exopher, (reaching at least 1/5th the size of the originating soma) (Figure 1C).
NOTE: The average exopher measures around 2-8 µm in diameter, while the average soma of a (ZB4065 bzIs166[Pmec-4::mCherry]) animal measures 6-10 µm in day 2 adults (Figure 7B). - If no bud or exopher domain is observed, inspect the neuronal soma for an attached thin filament emanating from the soma. Attached exophers tend to be located closer to the originating soma and in a similar Z-plane.
NOTE: Exophers do not always remain attached to the soma. The detection of an attached filament is a definitive indication that the object is an exopher. - To identify an unattached exopher, look for the contents of an exopher. Exophers can concentrate expelled fluorescent proteins and are therefore often brighter than the soma.
NOTE: The contents of exophers are heterogeneous and variable. Cellular organelles such as lysosomes and mitochondria can also be extruded within exophers (Figure 4C-E). - Look for unattached exophers in different focal planes than the plane in which the originating soma was found. Although exophers have been seen to protrude from the ALM soma in any direction, it is typical that exophers protrude away from the soma, in a posterior direction from the neuronal process.
- Check for large, spherical objects that are not positioned and identified as neuronal somas. Exophers can be irregularly shaped but are typically spherical structures. Exophers become degraded over time, so older exophers tend to have a more irregular shape.
NOTE: Mature or older exophers are distinguished from the dispersed "starry night" stage via the brighter fluorescence intensity of exophers and their spherical shape. - Investigate the “starry night” phenotype as evidence of earlier exophergenesis. Exophers progress into a “starry night” stage as the exopher breaks up into smaller vesicles and the surrounding hypodermis attempts to degrade the exopher contents (Figure 1G, 2B, 3B & 7A).
NOTE: The starry night stage is marked by fragmented and scattered (sometimes networked) fluorescent entities that have lost structural integrity and displays a dim fluorescence in comparison to the touch neurons and exopher structures. - Look for instances of “multiple exopher events”. Exophers are usually produced as a singular occurrence (1 exopher emanating from 1 soma) but under some circumstances more than one exopher can be released from a single soma (Figure 7D).
NOTE: Mature exophers can degrade into multiple vesicles as they are degraded in the hypodermis. Distinguishing whether each exopher was generated by an independent exophergenesis event or whether one original exopher split to create an additional vesicle can only be determined by time-lapse observation. - Keep in mind that not all morphological abnormalities mature into exophers.
- Do not score a distended soma as an exopher. An extended or pointed soma can be observed on occasion (especially with age or under stress), but an extension without a clear constriction site is not scored as an exopher.
- Reject small resolved buds that do not attain 1/5th the size of the soma in exopher event quantification.
- Do not count neurite outgrowths as exophers. Mature neurites can extend dramatically with age (usually in the opposite direction of the neuronal process) and fluorescent protein can migrate into the distal end of such structures19.
NOTE: These neurite outgrowths are not exophers as they have a distinct developmental pattern over days and weeks, do not form buds, and do not detach (Figure 7E).
- Identify fluorescent entities that are not exophers.
NOTE: It is important to get an idea of background fluorescence to ensure correct identification of the extruded fluorescent entity vs. autofluorescence.- Transgenic fluorescent expression vs autofluorescence. Do not mistake autofluorescence for transgenic expression. The true exopher signal will not be in the intestine or gut (DIC confirmation can be used to identify these tissues) and exopher signal will be significantly brighter than background autofluorescence.
NOTE: Autofluorescence is caused by gut granule intestinal fluorescent pigmentation and accumulates with age. It is heterogenous, especially as viewed with different wavelengths. - Signal from embryos. Do not mistake embryo signal for exophergenesis. Confirm suspicions of embryo signal by switching from fluorescence to brightfield illumination and checking for associations of signal with eggs in the uterus.
- Out of plane or nearby soma bodies. Avoid mistaking an out of plane soma for an exopher by identifying all nearby soma bodies, even out-of-focus somas at the start of the observation.
NOTE: If scoring for exophers from ALMR, identify and account for the location of AVM and ALMR somas. More details on soma body identification are described in Figure 3A.
- Transgenic fluorescent expression vs autofluorescence. Do not mistake autofluorescence for transgenic expression. The true exopher signal will not be in the intestine or gut (DIC confirmation can be used to identify these tissues) and exopher signal will be significantly brighter than background autofluorescence.
8. Scoring and statistics
- Score exophers as binary (yes, there is an exopher/no, there is not an exopher).
- Consider exopher detection as an "exopher-event" for a given neuron. An exopher event can constitute observation of a single exopher near a soma or multiple exophers.
NOTE: To quantify numbers of individual exophergenesis events use time-lapse observation. - Count exopher events per a particular identified cell because different cells do not produce exophers at the same rate (see for example Figure 3C). ALMR neurons produce the most baseline exophers in the strains described herein and thus often this is the cell selected for exopher quantification from touch receptor neurons.
- For statistics, in general, conduct at least 3 biological trials, of at least 30 animals scored per trial with the corresponding number of observations required for analysis of the disruption.
- For multiple trials involving one or two mutants/treatments compared to control, the Cochran-Mantel-Haenszel test is appropriate to determine p values.
- For trials involving more than two mutants of treatments compared to control, it is also appropriate to use a binary logistic regression analysis to evaluate significance for any number of categorical predictors.
Results
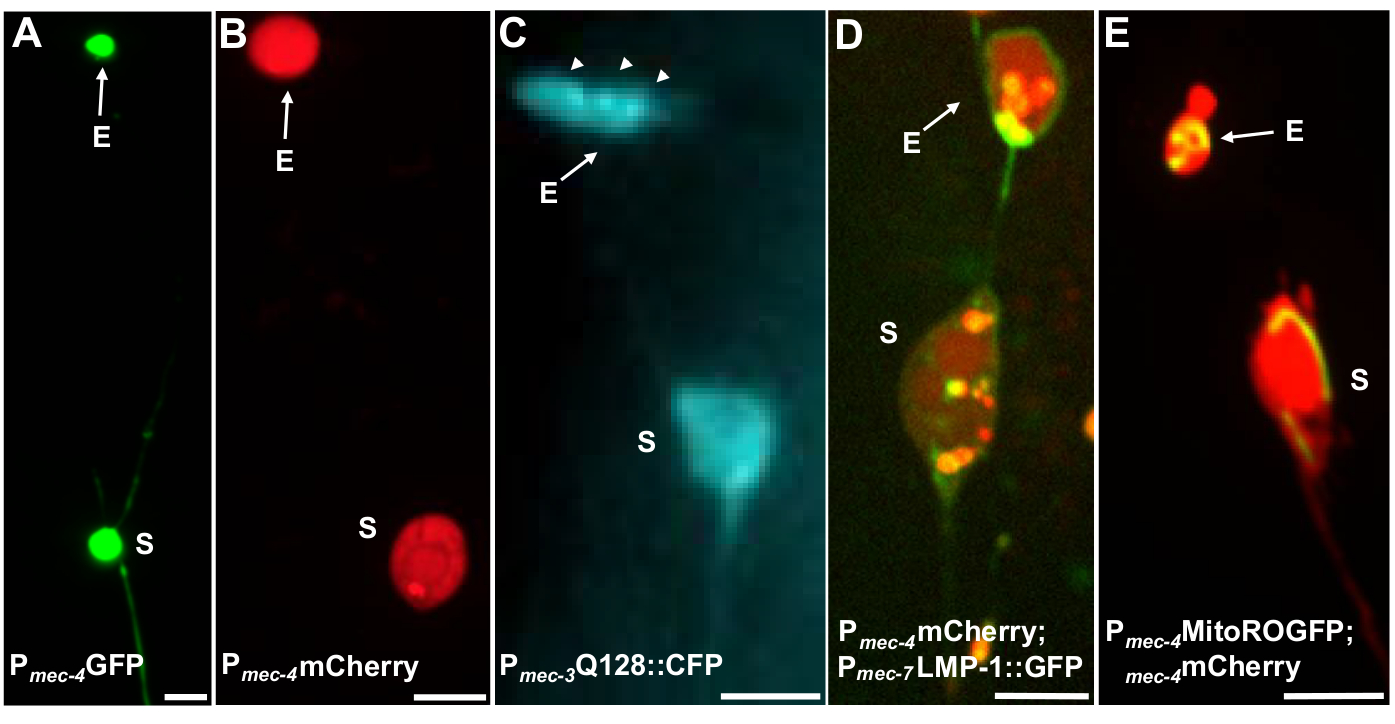
Multiple fluorescent reporters can be used to measure exophers. Touch neuron exophers are readily visualized in vivo via fluorescent tagging of proteins that may be selected for extrusion, by labelling of organelles that can be extruded, or by tagging cell membranes. Table 1 identifies touch neuron expressed fluorescent reporters that have been used to monitor exophers, with representative examples included in Figure 4. Cargoes that are known to be extruded in exophers include a fusion of the N-terminal domain of human huntingtin to expanded polyglutamine (Q128) (Figure 4B), lysosomes that are GFP-tagged with lysosomal associated membrane protein (LMP-1) (Figure 4C), and mitochondria tagged with matrix-localized GFP (Figure 4D). Cytoplasmic GFP is not strongly expelled and is preferentially retained in the soma5, although GFP can weakly visualize exophers (Figure 4A). When GFP is fused to proteins that are expelled, this tag can be used to visualize exophers. An important point is that by tagging different proteins, a large range of questions on the expulsion of specific cargoes and organelles, as well as on the proteins and membranes that make up exophers, can be addressed.
A pseudo-stereomicroscope setup is an effective tool for viewing exophers in animals upon agar plates. This setup is a hybrid of compound and stereoscopic technology that includes high numerical aperture optics on each magnification, pseudo-stereo technology (discrete objectives over a stereoscopic base), and a zoom operating switch for viewing at magnifications intermediate to installed objectives. A microscope such as this should be equipped with 10x eyepieces and objectives powerful enough for observing neuronal morphology and exopher production for high-throughput scoring (2x objective used for scanning/picking, 10x objective used for identifying and scoring).
While magnification capabilities of standard stereomicroscopes typically have high enough resolution to see the network of touch neurons expressing fluorescent proteins, standard dissecting microscopes are not sufficient for observing subcellular details of exophers like the tubular connections of soma to exopher. Such observations necessitate confocal microscopy (see the Table of Materials for equipment details).
Exopher quantitation studies require strict controls to eliminate experimental stresses. The attentive maintenance of consistent growth conditions is required for reproducible exopher production. More specifically, exopher production is stress-responsive such that consistent feeding, constant temperature, and contamination-free growth across generations are critical for reproducibility. Under basal growth conditions with high neuronal expression of mCherry, exopher production is relatively low (5-25% of ALMRs produce exophers) but some stresses, including osmotic and oxidative stress, can increase exopher rates. While mCherry expression can be thought of as stress, a corollary of the stress-sensitivity of exopher levels is that, if properly controlled, experimental stress introduction can be a strategy to more easily induce and observe exophergenesis.
Timing and anticipated exopher production levels. Exophers are virtually absent during larval development. The period of peak exopher production in young adult life appears to be highly restricted to during adult days 1-4, most commonly being evident at adult day 2 or 3. Because the peak can shift ahead or back a little, the most complete evaluation of an exopher production profile is to score multiple trials daily over adult days 1-4. In general, an ALMR produces one major exopher, with the vesicle persisting for at least 24 hours. The exopher can be produced fairly quickly (on the order of minutes at its fastest). Most commonly, only one major exopher is produced per neuron in early adult life, but production of multiple exophers is possible.
In general exopher production by ALMRs expressing mCherry under basal conditions ranges from 5-25% of ALMRs examined within the optimal timeframe of adult day 2-3 (Figure 3D). Proteostasis crises5, as well as exposure to other stresses can modulate exopher level. Stress or genetic perturbations can increase exopher production to detection rates as high as 90% of ALMR neurons producing exopher extrusions.
Feeding-based RNAi for testing roles of specific genes in exophergenesis. The nematode C. elegans is commonly subjected to RNAi knock down by feeding animals transformed E. coli strain HT115 that express a double stranded RNA (dsRNA) targeting a gene of interest20. HT115 bacteria can be used when scoring for exophers in feeding RNAi5. While transcripts in most tissues can be targeted by RNAi using this technique, neurons are more refractory. Sensitivity to RNAi can be calibrated using animals that express the transgenic dsRNA transporter SID-1 under a neuron-specific promoter. In this way neuronal tissue can be sensitized to RNAi21.
Tissue-specific knockdown of a gene of interest can be accomplished by expressing a component of endogenous RNAi metabolism within a mutant that is deficient in that component. For example: the Argonaute protein RDE-1 can be expressed specifically in the neurons of rde-1 mutant animals to achieve knockdown of a gene of interest only in neurons when animals are exposed to an RNAi intervention targeting that gene.
Using standard nematode RNAi protocols20,22, exposure of the parents at the L4 stage to the RNAi and allowing their progeny to develop consuming transformed HT115 bacteria until adulthood generates the strong genetic knock-down but be attentive to potential developmental delays induced by RNAi as experimental animals may grow differently than an empty vector control. It is important to always include the empty vector control for negative control comparison. HT115 bacteria can be used when scoring for exophers in feeding RNAi. However, note that some genes are effective at changing exophergenesis rates even during shorter periods of RNAi exposure5. If targeting of certain genes leads to developmental failure, avoid exposing animals to lifelong knockdown, animals can simply be picked at the L4 stage onto RNAi plates for exposure from L4 to adult D2 or D3.
| Strain name | Genotype | Description | Exopher percentage | Reference |
| SK4005 | zdIs5[Pmec-4GFP] | Cytosolic expression of GFP in touch neurons. | 1-8% ALM | Figure 4A, Melentijevic 2017 |
| ZB4065 | bzIs166[Pmec-4::mCherry] | Overexpression of mCherry (bzIs166) in touch neurons, produces both cytosolic signal and mCherry aggregates. bzIs166 is an exopher inducer. mCherry aggregates are predictors of exophergenesis and are preferentially extruded in exophers. | 3-20% ALM (normal conditions). 20-80% ALM (fasting conditions). | Figure 4B, Melentijevic 2017 |
| ZB4067 | bzIs167[Pmec-4mitogfp Pmec-4mCherry4]; igIs1[Pmec-7YFP Pmec-3htt57Q128::cfp lin-15+]; | YFP cytosolically labels mec-7 touch neurons. Co-expressed Q128::CFP aggregates and induces exophers. CFP preferentially silences. | ~25% | Figure 4C, Meletijevic 2017 |
| ZB4509 | bzIs166[Pmec-4mCherry]; bzIs168[Pmec-7LMP-1::GFP] | bzIs168 LMP-1::GFP labels plasma membranes and lysosomal membranes. bzIs168 can be used to identify neuronal membranes, exophers (as they are membrane bound), and lysosomal-membrane structures. | 3-20% ALM | Figure 4D, Melentijevic 2017 |
| ZB4528 | bzIs166[Pmec-4mCherry]; zhsEx17 [Pmec-4mitoLS::ROGFP] | Allele zhsEx17 is a mitochondrially localized reporter that changes its peak excitation wavelength from 405nm (oxidized) to 476nm (reduced) according to the local oxidative environment. It is expressed in the touch neurons and can be used on its own to identify mitochondria in touch neurons and in mito-exophers. | 3-20% ALM proteo-exopher. % ALM mito-exopher quantitation in progress. | Figure 4E, Melentijevic 2017, Cannon 2008, Ghose 2013 |
Table 1. Strains that have been used for visualization of touch neurons, touch neuron-exophers, and exopher contents.

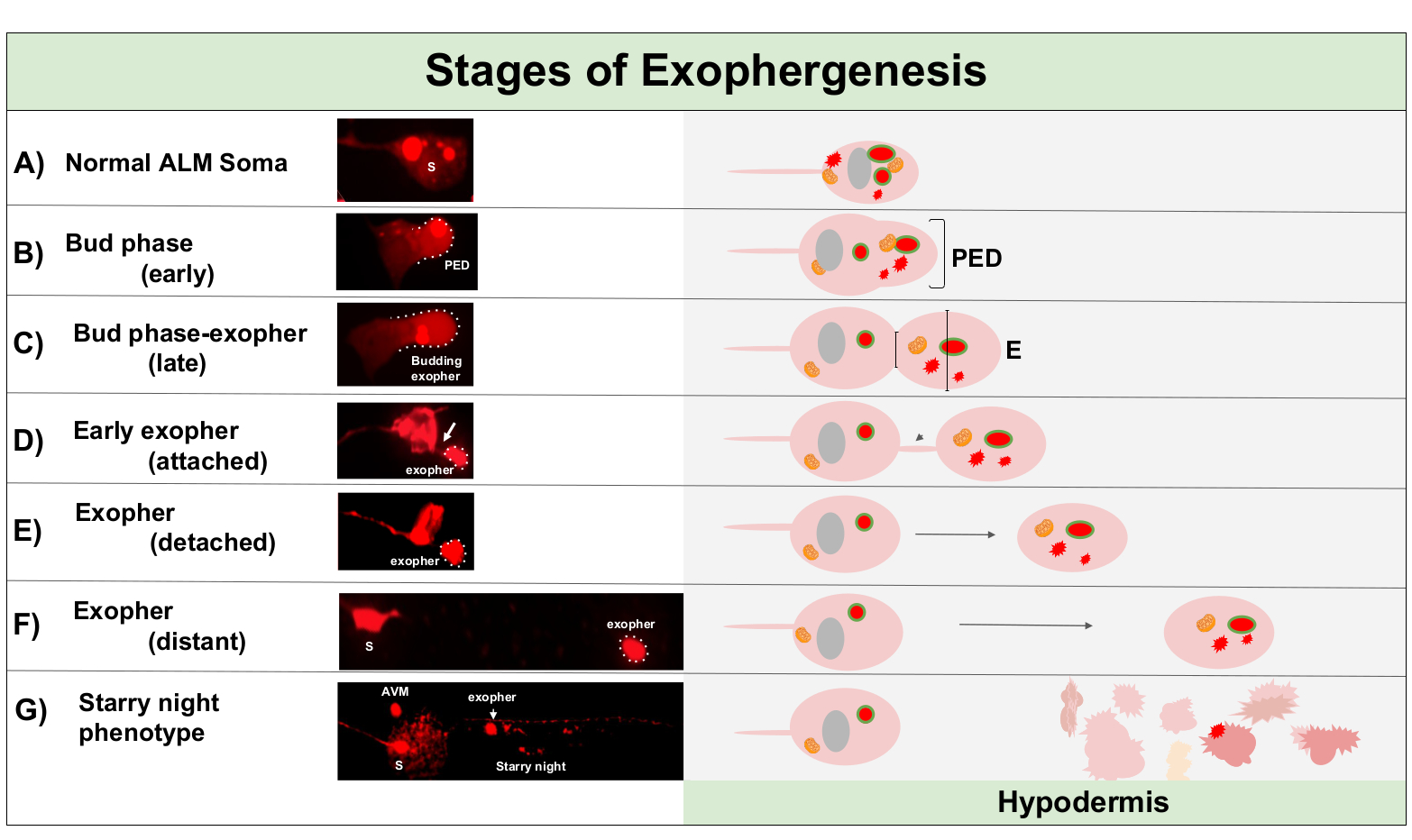
Figure 1: Stages of Exophergenesis. The process of making and ejecting an exopher is called ‘exopher-genesis’. The dynamic process of exopher formation can take several minutes to several hours. Depicted are examples of soma and exopher morphology at specific steps during the dynamic exophergenesis process in a high-exopher producing strain, ZB4065 bzIs166[Pmec-4mCherry]. All images are of day 2 adult ALM neurons taken with a 100x objective. (A) Normal soma. Adult mechanosensory touch neuron ALM transgenically expressing Pmec-4mCherry. The soma morphology depicted is typical of young adult neurons in this strain, with mCherry concentrations in the cytoplasm. (B) Early bud phase. The first observable step of exophergenesis involves polarization of selected cytoplasmic material to the edge of the soma membrane. This step is often accompanied by an expansion or swelling of the soma. In the case of the touch neurons, the pre-exopher domain (PED) extends into the surrounding hypodermis (not visible here). Note the greater concentration of mCherry material into the early bud domain. (C) Late bud phase. Upon further cellular polarization and an expansion of the pre-exopher domain, a constriction between the soma and exopher (arrow) becomes evident. This event signals the transition to the late bud phase. Although in the late bud stage the cell exhibits a clear constriction site and separate soma and exopher domains, it is not yet pinched off completely from the soma; the budding exopher may be attached by a thick stalk (arrow). The budding domain is considered an early exopher when the diameter of the exopher domain in question is roughly ⅓ larger than the diameter of the construction site/stalk. (D) Early-exopher phase. Early exophers can be attached by a stalk from the departing soma—the diameter of this connection can thin as the exopher moves away from the soma. Cytoplasmic material can be transferred from the soma to the exopher via this tube, although most material is loaded during the process of budding out. Exophers can detach from the soma as depicted in (E), separated exophers are considered mature exophers (F). The mature exopher can transit through the surrounding hypodermal tissue, moving away from the departing soma. (G) Breakdown of the mCherry-labelled exopher into smaller vesicles within the hypodermis results in a scattered punctate appearance of the mCherry material, most likely as it enters the hypodermal endolysosomal network. The dispersed punctate signal is called the “starry night” phase. Degradation of some exopher contents is likely accomplished by hypodermal lysosomes, but some material is not fully degraded and is often re-extruded by the hypodermis into the pseudocoelom. The post-exophergenesis mCherry transit is described in more detail in Figure 2. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: mCherry extruded from touch neurons in exophers engages the surrounding hypodermal lysosomal network but can later be extruded into the pseudocoelom where coelomocytes can store/degrade the mCherry. (A) Cartoon summary of how mCherry extruded in exophers transits the body after expulsion by neurons. During exophergenesis selected cellular contents such as mCherry become localized and bud off from the sending neuronal soma in an independent vesicle surrounded by the neuronal and hypodermal plasma membranes. Since the touch neurons are embedded in the hypodermal tissue, as the exopher domain buds outwards it moves further into the hypodermis. The exopher can transit the hypodermis, and after hours to days, exopher contents can fragment within the endolysosomal network of the hypodermis. The mCherry can appear as scattered puncta throughout the hypodermis, a stage called “starry night”. After a few days, some of the mCherry can pass out of the hypodermis into the surrounding pseudocoelom, where scavenger cells called coelomocytes can get access to, and take up, mCherry that can be stored. (B) Example of the appearance of the starry night mCherry vesicles. Image of an ALM soma tagged with mCherry with large exopher fragments and starry night vesicles. Strain is ZB4065 bzIs166[Pmec-4mCherry]. (C) Example of mCherry concentration in distant coelomocytes. Sideview of an adult animal day 10 of strain ZB4065 bzIs166[Pmec-4mCherry] showing mCherry concentrated in coelomocytes (arrows). Some starry night vesicles are also evident. In general coelomocyte concentration becomes evident after about adult day 5 of life. (B bottom) Cartoon reproduction of (B), with touch neurons and processes outlined in red, as are brightest exopher fragments; scattered small vesicles of different Z-depths are shown in lighter pink. (C bottom) Cartoon version of image of (C), showing neuronal process in red, starry night in pink and coelomocytes in green. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Mechanosensory touch neurons produce exophers at different levels with a precise temporal profile. (A) (Top) Cartoon depiction of mechanosensory touch neurons in spatial relation to key anatomical landmarks of C. elegans including the pumping pharynx and the neuron-dense nerve ring at the head of the animal, the vulva at the mid body, and the tapered tail. (Bottom) Fluorescently labeled touch neurons expressing GFP as viewed from the top and left side (images adapted from WormAtlas). The red box depicts the area where ALM exophers are typically located. (B) High magnification view of the mid body region at which ALM-derived exophers are produced in a strain expressing [Pmec-4mCherry]. AVM and ALMR neuron are depicted, and shown is an ALMR exopher along with mCherry starry night. ALMR neurons most readily produce exophers. (C) ALMR mechanosensory touch neurons more readily produce exophers compared to other touch neurons in hermaphrodites under basal conditions. Mechanosensory touch neuron exopher production on adult day 2, as scored for individual touch receptor neurons is indicated. Strain: ZB4065 bzIs166[Pmec-4mCherry], N>150, error bars are SEM. (D) ALMR touch neurons produce more exophers during days 2 and 3 of adulthood compared with the adolescent L4 stage or with animals in advanced age. Strain: ZB4065 bzIs166[Pmec-4mCherry], N>150, error bars are SEM. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Examples of some fluorescent reporters that tag exopher contents. A straightforward way to observe exophers is by creating transgenic animals that express fluorophores from neuronal promoters. The fluorophores allow for visualization of the exopher and transgenic expression induces aggregation and/or proteostress that increases exophergenesis. Exophers produced by amphid neurons can also be observed under native conditions, using dye filling for visualization. Shown are examples of common strains that can be used to observe exophers, (E) exopher, (S) soma. (A) Soma and exopher from an ALM of an adult of strain SK4005 zdIs5[Pmec-4GFP],100x objective used for photography, scale bar 3μm. In this strain, exophers that include soluble GFP are measured, but exopher production occurs infrequently. Fusing GFP to proteins that can be preferentially extruded in exophers in other studies confirms that GFP fusions can be detected in mature exophers. (B) ALM soma and exopher of an adult of strain ZB4065 bzIs166[Pmec-4mCherry], which expresses mCherry and induces touch neuron exopher production. 100x objective used for photography, scale bar 5 μm. (C) ALM soma and exopher of an adult of strain ZB4067 bzIs167[Pmec-4mitogfp Pmec-4mCherry4]; igIs1[Pmec-7YFP Pmec-3htt57Q128::cfp lin-15+]; selective blue channel used for image of htt57Q128::CFP. The exopher contains htt57Q128::CFP aggregates (arrows), that appear more concentrated in the exopher than in the soma. 40x objective used for photography, scale bar 5μm. (D-E) Exophers can contain organelles and organelle-specific tagging with fluorescent proteins enables monitoring of organelle extrusion. (D) Lysosomal membrane tag LMP-1::GFP outlines the soma and exopher membrane and tags plasma membranes weakly (plasma membrane localization is a trafficking step on the way to lysosomal targeting) and labels lysosomal organelles strongly. Shown is an adult ALM soma co-expressing Pmec-4mCherry and the Pmec-7LMP-1::GFP that localizes to membranes and lysosomes. The soma has an attached exopher with other smaller extrusions likely to be exopher fragments (arrows). GFP positive structures are included in the soma and are present in the large exopher, strain: ZB4509 bzIs166[Pmec-4mCherry]; bzIs168[Pmec-7LMP-1::GFP]. 100x objective used for photography, scale bar 5 μm. E) A mitochondrial GFP marker can be used to identify mitochondria in soma and exophers. Shown is an adult ALM soma expressing Pmec-4mCherry and mito::ROGFP, which localizes to the mitochondrial matrix. mito::ROGFP expressed alone, without the mCherry, can also readily be used to identify neurons and score for exophers that include mitochondria. Strain: ZB4528 bzIs166[Pmec-4mCherry]; zhsEx17 [Pmec-4mitoLS::ROGFP]. 100x objective used for photography; scale bar 5μm. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Developmental cycle of C. elegans and L4 identification. (A) At 20 °C an egg takes approximately 9 hours to hatch once laid by the mother. (B) A newly hatched animal is in larval stage 1 (L1) and molts into an L2 larva after 12 hours. (C) Animals remain in the L2 and the (D) L3 larval stages for about 8 hours each. (E) Adolescent animals are considered the fourth larval stage (L4) and are marked by a conspicuous developing vulva that appears as a white crescent near the mid body. The presence of this while crescent enables easy identification and picking of L4 staged animals to establish synchronized cultures that later facilitate scoring for exophers. Animals remain in the L4 stage for about 10 hours before their final molt into gravid adults, F) identified by developing eggs, visible spermatheca, and the initiation of egg-laying. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Preparation of microscope slide agar pad. (A) Prepare two slides with a single strip of laboratory tape placed lengthwise across the top. Place a non-taped microscope slide in between as pictured. B) Place a drop of molten agarose on top of the slide. (C) Place a clean slide gently on top of the drop, pressing the agarose into a deflated circle pad. (D) Remove the taped slides, which act to accomplish an even flattening of the agar that is needed to create an even pad. (E) Remove the top slide once the agarose pad has dried. (F) Pipette a paralytic solution (levamisole or tetramisole) on top of the agar pad. (G) Pick appropriately staged animals into the paralytic. (H) Gently cover the animals with a coverslip and ensure animals are alive. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Characters of exophers and exopher identification criteria. (A) General criteria that identify an exopher. (B) Diameter comparisons between the sending soma and the extruded exopher, measured in μm. Adult ALM somas, N=35, strain: ZB4065 bzIs166[Pmec-4mCherry] - 6.53 μm average size of soma and 3.83 μm average size of exopher. (C) Defining criteria for differentiating between an exopher domain and a budding exopher. (D) Most commonly, individual neurons make one large exopher, which later splits or fragments as the hypodermis attempts to degrade its contents. Still, multiple exophers may be observed next to one touch neuron that might derive from either multiple exopher events from one neuron or alternatively, exophers can also bud or fragment themselves. The origin of multiple exopher-like entities can only be determined using time lapse microscopy. Top depicts an ALMR touch neuron soma with a single distant exopher. Bottom depicts an ALMR touch neuron soma with multiple exopher-like extrusions. (E) Common morphological features in adult ALM touch neuron somas that may be mistaken for exopher events. Top left - A distended ALM soma, with no clear exopher domain or constriction site. Top middle - Neurons can have small extracellular protrusions that may be analogous to exophers, but do not meet size requirement criteria to be considered an exopher. Top right – With age, touch neurons can develop outgrowths along their minor neurite. Often mCherry material can be collected at the tip of the neurite outgrowth. This is not scored as an exopher if the collected mCherry does not meet exopher-to-soma size requirements. Bottom depicts adult ALM neurons that have defining criteria for an exopher domain or an exopher. Botom left - ALM soma that has a prominent exopher domain that selectively includes mCherry cytosol and mCherry tagged aggregates. The exopher domain constriction site is marked by arrows and meets the size criteria (at least 1/5th the size of the soma). The largest diameter of the exopher domain is almost ⅓ bigger than the diameter of the constriction site, meeting criteria for an exopher event. Bottom middle - ALM soma that has a prominent budding exopher that meets the size criteria. There is a clear constriction site. Bottom right - ALM soma that has an attached mCherry-filled exopher that meets exopher size requirements. The exopher is attached by a thin connecting filament. All images are from strain ZB4065 bzIs166[Pmec-4mCherry]. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The characterization of the in vivo molecular mechanisms of aggregate and organelle elimination in the form of large exophers is in its infancy. Questions as to the designation of cargoes for expulsion, the polarized collection of these cargoes within the cell, the regulation of the decision to generate exophers, the machinery that mediates extrusions, and the interaction of exophers with the degradative machinery in a neighboring cell all remain to be addressed. Furthermore, the in vivo visualization of tubular connections that can pass biological materials that include calcium, aggregates, and mitochondria is interesting and understudied biology in its own right. Questions of why certain cells are more prone to exopher production than others also are unresolved, but can begin to be genetically dissected with the approaches outlined in this protocol.
Described in detail in this protocol are the approaches to achieving reproducible scoring of exopher production, with attention to distinguishing exophers from nearby cell somas, timing of analyses to capture peak of exopher production, and strict control of growth conditions to eliminate unintended stresses that can modulate exopher levels. Both distinction of the large early exopher, or the “starry night’ dispersion in the surrounding hypodermis can be quantitated as evidence of exopher production. That being said, neurons expressing mCherry under basal conditions are most often associated with 5-25% of neurons of a specific type producing an exopher. Controlled introduction of stress conditions could be applied to increase exopher production to detection as high as 90% of neurons producing extrusions, particularly useful for genetic or pharmacological screens for modifiers.
In human neurodegenerative disease, large aggregates can transfer from diseased neurons into neighboring cells to promote pathology spread. The exopher mechanism might transpire via a conserved mechanism used for aggregate extrusion across phyla. Defining the in vivo molecules that either enhance the efficiency of this process (considered more effective proteostasis control) or block it might be harnessed to influence design of novel strategies for combating multiple neurodegenerative diseases. As such, the protocol described here could be used for classical genetic mutagenesis screens, genome-wide RNAi screens that systematically knock down genes to identify enhancers and suppressors, or for drug intervention studies that identify candidate pharmacological modifiers of this process. The approach is straightforward, although somewhat laborious. Exophers are so large they can be viewed with a high-magnification dissecting microscope. Still, C. elegans neurons are relatively small and looking at their organelles or their membranes require higher power confocal images and is a slow process. Options for higher throughput could involve high content imaging approaches in multi-well plate format.
The application of a standardized approach to exopher scoring should underlie a concerted genetic dissection of the process by which neurons can organize and eliminate cellular debris.
Disclosures
None
Acknowledgements
We acknowledge the following NIH grants: R01AG047101 and R37AG56510. Members of the Driscoll and Grant labs have contributed extensively to the development and fine tuning of protocols described, with rigorous experiments and strong communication.
Materials
| Name | Company | Catalog Number | Comments |
| 95B Scientific CMOS camera | Photometrics Prime | ||
| 1,000 μL low retention tips | Sarstedt | ||
| 10 mL serological pipette | Appleton Woods | CC214 | |
| 10 μL low retention tips | Sarstedt | 70.1130.105 | |
| 13% sodium hypochlorite | Acros Organics | AC219255000 | |
| 15 mL centrifuge tubes | Fisher Scientific | 05-539-12 | |
| 2 L erlenmeyer flasks | Scientific Laboratory Supplies | FLA4036 | |
| 25 mL serological pipette | Appleton Woods | CC216 | |
| 300 μL low retention tips | Sarstedt | 70.765.105 | |
| 50 mL serological pipette | Appleton Woods | CC117 | |
| 5-Fluoro-2'-deoxyuridine 98% | Alfa Aesar | L16497.ME | |
| 9 cm sterile Petri dishes | Fisher Scientific | 11309283 | |
| absolute ethanol | Vwr | 20821.33 | |
| Agar | Sigma Aldrich | A1296 | |
| C. elegans strain wild type | Supplied by CGC | N2 | C. elegans strain |
| calcium chloride dihydrate | Sigma Aldrich | C3881 | |
| cholesterol | Acros | 110190250 | |
| dibasic sodium phosphate | Sigma Aldrich | S3264 | |
| E. coli strain OP50 | Supplied by CGC | Op50 | E coli strain |
| FBS10 Standard microscope | Meyer Instruments | KSC 410-1-100-1 | FBS10 Standard with Plate Base, 100/100 Trinocular Head and Flip zoom |
| glass pipette 270 mm | Fisherbrand | FB50255 | |
| Heraeus Multifuge X3R | Thermofisher scientific | 75004515 | |
| Inoculating Spreaders | Fisher Scientific | 11821741 | |
| LB medium capsules | MP biomedicals | 3002-031 | |
| LDI – Laser Diode Illuminator | 89 North | ||
| levamisole | Sigma Aldrich | 16595-80-5 | |
| M4 multipette | Eppendorf | 4982000012 | |
| magnesium sulphate | Sigma Aldrich | M7506 | |
| monobasic potassium phosphate | Sigma Aldrich | P0662 | |
| Multitron Standard shaking incubator | Infors HT | INFO28573 | |
| Nalgene 1 L Centrifuge pots | Fisher Scientific | 3120-1000 | |
| P10 pipette | Eppendorf Research Plus | 3123000020 | |
| P1000 pipette | Eppendorf Research Plus | ||
| P200 pipette | Eppendorf Research Plus | 3123000055 | |
| pipeteboy 2 | VWR | 612-0927 | |
| Polystyrene microbeads | Sigma Aldrich | MFCD00131491 | |
| RC5C plus floor mounted centrifuge | Sorvall | 9900884 | |
| Reusable ringed cytology slides | ThermoFisher Scientific | 22037242 | |
| SK4005 zdIs5[Pmec-4GFP] | contract Driscoll lab | GFP expressed in touch neurons | |
| sodium chloride | Sigma Aldrich | 13422 | |
| Sodium hydroxide | Fisher Chemical | S/4880/53 | |
| Tactrol 2 Autoclave | Priorclave | ||
| Triton-X | Thermofisher scientific | 28313 | |
| Tween 20 | Sigma Aldrich | 9005-64-5 | |
| X-Light V2 Spinning Disk Confocal Unit | CrestOptics | ||
| ZB4065 bzIs166[Pmec-4mCherry] | contract Driscoll lab | mCherry expressed in touch neurons | |
| ZB4067 bzIs167[Pmec-4mitogfp Pmec-4mCherry4]; igIs1[Pmec-7YFP Pmec-3htt57Q128::cfp lin-15+] | contract Driscoll lab | Q128 expressed in touch neurons | |
| ZB4509 bzIs166[Pmec-4mCherry]; bzIs168[Pmec-7LMP-1::GFP] | contract Driscoll lab | mitoROGFP expressed in touch neurons | |
| ZB4528 bzIs166[Pmec-4mCherry]; zhsEx17 [Pmec-4mitoLS::ROGFP] | contract Driscoll lab | autophagy marker expressed in touch neurons | |
| ZEISS Axio Vert.A1 | Zeiss |
References
- Davis, A. A., Leyns, C. E. G., Holtzman, D. M. Intercellular Spread of Protein Aggregates in Neurodegenerative Disease. Annual Review of Cell and Developmental Biology. 34, 545-568 (2018).
- Davis, C. H., et al. Transcellular degradation of axonal mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 111 (26), 9633-9638 (2014).
- Torralba, D., Baixauli, F., Sanchez-Madrid, F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Frontiers in Cell and Developmental Biology. 4, 107 (2016).
- Stahl, P. D., Raposo, G. Extracellular Vesicles: Exosomes and Microvesicles Integrators of Homeostasis. Physiology (Bethesda, Md.). 34 (3), 169-177 (2019).
- Melentijevic, I., et al. C-elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature. 542 (7641), 367 (2017).
- Nussbaum-Krammer, C. I., Park, K. W., Li, L., Melki, R., Morimoto, R. I. Spreading of a prion domain from cell-to-cell by vesicular transport in Caenorhabditis elegans. PLoS Genetics. 9 (3), 1003351 (2013).
- Tyson, T., et al. Novel animal model defines genetic contributions for neuron-to-neuron transfer of alpha-synuclein. Scientific Reports. 7, (2017).
- Babcock, D. T., Ganetzky, B. Transcellular spreading of huntingtin aggregates in the Drosophila brain. Proceedings of the National Academy of Sciences of the United States of America. 112 (39), 5427-5433 (2015).
- Pearce, M. M. P., Spartz, E. J., Hong, W., Luo, L., Kopito, R. R. Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nature Communications. 6, 6768 (2015).
- Fu, H., Li, J., Du, P., Jin, W., Cui, D. Metabolic wastes are extracellularly disposed by excretosomes, nanotubes and exophers in mouse HT22 cells through an autophagic vesicle clustering mechanism. bioRxiv. 10 (1), (2019).
- Ghose, P., Park, E. C., Tabakin, A., Salazar-Vasquez, N., Rongo, C. Anoxia-reoxygenation regulates mitochondrial dynamics through the hypoxia response pathway, SKN-1/Nrf, and stomatin-like protein STL-1/SLP-2. PLoS Genetics. 9 (12), 1004063 (2013).
- Cannon, M. B., Remington, S. J. Redox-sensitive green fluorescent protein: probes for dynamic intracellular redox responses. A review. Methods in Molecular Biology. 476, 51-65 (2008).
- Perkins, L. A., Hedgecock, E. M., Thomson, J. N., Culotti, J. G. Mutant sensory cilia in the nematode Caenorhabditis elegans. Developmental Biology. 117 (2), 456-487 (1986).
- Stiernagle, T. Maintenance of C. elegans. WormBook: The Online Review of C. Elegans Biology. , 1-11 (2006).
- Sutphin, G. L., Kaeberlein, M. Measuring Caenorhabditis elegans life span on solid media. Journal of Visualized Experiments. (27), (2009).
- Mitchell, D. H., Stiles, J. W., Santelli, J., Sanadi, D. R. Synchronous growth and aging of Caenorhabditis elegans in the presence of fluorodeoxyuridine. Journal of Gerontology. 34 (1), 28-36 (1979).
- Weicksel, S. E., et al. A novel small molecule that disrupts a key event during the oocyte-to-embryo transition in C. elegans. Development. 143 (19), 3540-3548 (2016).
- Dong, L., et al. Reversible and long-term immobilization in a hydrogel-microbead matrix for high-resolution imaging of Caenorhabditis elegans and other small organisms. PloS One. 13 (3), 0193989 (2018).
- Toth, M. L., et al. Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. Journal of Neuroscience. 32 (26), 8778-8790 (2012).
- Conte, D., MacNeil, L. T., Walhout, A. J. M., Mello, C. C. RNA Interference in Caenorhabditis elegans. Current Protocols in Molecular Biology. 109, (2015).
- Calixto, A., Chelur, D., Topalidou, I., Chen, X., Chalfie, M. Enhanced neuronal RNAi in C. elegans using SID-1. Nature Methods. 7 (7), 554-559 (2010).
- Maher, K. N., Catanese, M., Chase, D. L. Large-scale gene knockdown in C. elegans using dsRNA feeding libraries to generate robust loss-of-function phenotypes. Journal of Visualized Experiments. (79), e50693 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved