Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Mesures FLIM-FRET des interactions protéines-protéines chez les bactéries vivantes.
Dans cet article
Résumé
Nous décrivons ici un protocole pour caractériser les interactions protéine-protéine entre deux protéines fortement exprimées différemment dans pseudomonas aeruginosa vivants utilisant des mesures de FLIM-FRET. Le protocole comprend les constructions de souches bactériennes, l’immobilisation des bactéries, l’imagerie et les routines d’analyse des données post-imagerie.
Résumé
Les interactions protéine-protéine (IPP) contrôlent divers processus clés dans les cellules. La microscopie d’imagerie à vie par fluorescence (FLIM) combinée au transfert d’énergie par résonance Förster (FRET) fournissent des informations précises sur les IPP dans les cellules vivantes. FLIM-FRET s’appuie sur la mesure de la désintégration à vie par fluorescence d’un donneur fret à chaque pixel de l’image FLIM, fournissant des informations quantitatives et précises sur les IPP et leurs organisations cellulaires spatiales. Nous proposons ici un protocole détaillé pour les mesures FLIM-FRET que nous avons appliqué pour surveiller les IPP en direct Pseudomonas aeruginosa dans le cas particulier de deux protéines en interaction exprimées avec des numéros de copie très différents pour démontrer la qualité et la robustesse de la technique à révéler les caractéristiques critiques des IPP. Ce protocole décrit en détail toutes les étapes nécessaires à la caractérisation ppi - à partir de constructions mutantes bactériennes jusqu’à l’analyse finale à l’aide d’outils récemment développés offrant des possibilités avancées de visualisation pour une interprétation simple des données complexes FLIM-FRET.
Introduction
Les interactions protéine-protéine (IPP) contrôlent divers processus clés dans les cellules1. Les rôles des IPP diffèrent en fonction de la composition protéique, des fonctions d’affinités et des emplacements dans lescellules 2. Les IPP peuvent être étudiés au moyen de différentes techniques3. Par exemple, la co-immunoprécipitation est un outil relativement simple, robuste et peu coûteux couramment utilisé pour identifier ou confirmer les IPP. Toutefois, l’étude des IPP peut s’avérer difficile lorsque les protéines en interaction ont de faibles niveaux d’expression ou lorsque les interactions sont transitoires ou pertinentes uniquement dans des environnements spécifiques. L’étude des IPP se produisant entre les différentes enzymes de la voie de pyoverdine dans P. aeruginosa exige que la répression du répresseur général fer-co-factorisé Fur soit soulagée pour permettre l’expression de toutes les protéines de la voie de pyoverdine pour être exprimée dans la cellule4,5,6. Cette régulation commune pour toutes les protéines de la voie entraîne des expressions opportunes dans la cellule qui devrait favoriser leurs interactions. La diversité en termes de taille, de nature, de niveaux d’expression et le nombre de protéines de cette voie métabolique rendent difficile l’étude dans les systèmes reconstitués6. Il est donc essentiel d’explorer les IPP dans leur environnement cellulaire pour mieux comprendre les fonctions biologiques des protéines dans leur contexte natal.
Seules quelques méthodes, y compris la fluorescence, permettent d’explorer les IPP dans les cellulesvivantes 7. Parmi les différents paramètres de fluorescence qui peuvent être mesurés, la durée de vie de fluorescence (c.-à-d. le temps moyen qu’un fluorophore reste dans son état excité avant d’émettre un photon) est probablement l’un des paramètres les plus intéressants à explorer dans les cellules vivantes. La durée de vie de fluorescence d’un fluorophore est très sensible à son environnement et FLIM peut donc fournir des informations chimiques ou physiques concernant l’environnement fluorophore8. Cela inclut la présence d’un transfert d’énergie par résonance Förster (FRET) qui peut se produire en présence d’un « accepteur » de fluorescence situé à une courte distance d’un « donneur » de fluorescence. Le transfert d’énergie entraîne un raccourcissement important de la durée de vie de fluorescence du donneur (figure 1A), faisant de la microscopie d’imagerie à vie de fluorescence (FLIM) une approche puissante pour explorer les interactions protéine-protéine directement dans les cellules vivantes. FLIM peut en outre fournir des informations spatiales sur l’endroit où les interactions ont lieu dansles cellules 7,8. Cette approche est extrêmement puissante pour étudier les IPP dans les situations où l’étiquetage avec les fluorophores des deux partenaires en interaction est possible.
Pour fret de se produire - conditions critiques sur la distance entre deux fluorophores sont nécessaires8,9. Les deux fluorophores ne doivent pas être éloignés l’un de l’autre de plus de 10 nm. Par conséquent, il faut faire preuve de prudence lors de la conception d’expériences FLIM-FRET pour s’assurer que le donneur et l’accepteur de fluorescence ont une chance d’être situés l’un près de l’autre dans le complexe en interaction. Bien que cela puisse sembler contraignant, il s’agit en fait d’un véritable avantage, car la dépendance à distance du FRET fait en sorte que deux protéines étiquetées subissant fret doivent interagir physiquement (Figure 1A). Les difficultés à obtenir des réponses claires sur l’IPP dans les expériences de colocalisation (deux protéines colocalisées peuvent ne pas nécessairement interagir) ne sont donc pas un problème à l’aide de FLIM-FRET.

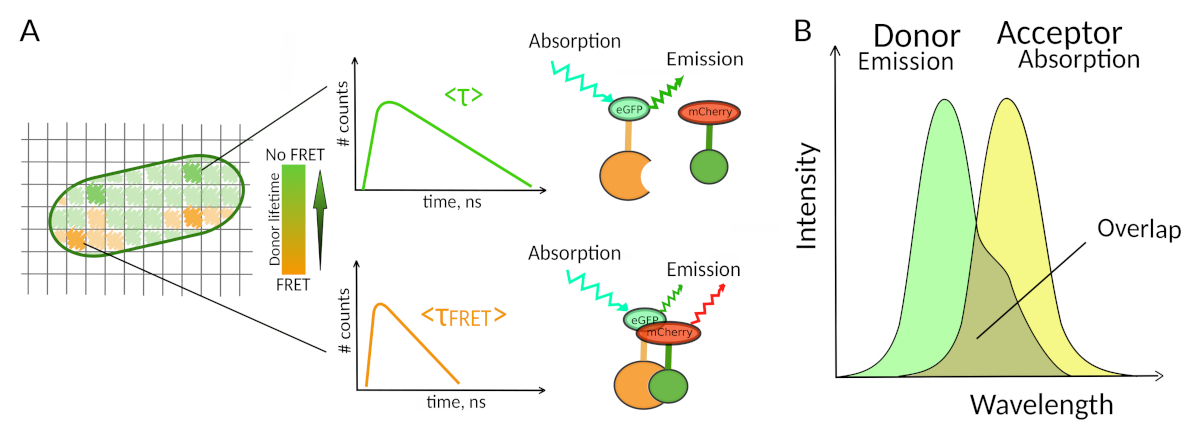
Figure 1 : Principe d’analyse FLIM-FRET. Chaque pixel de l’image multidimensionnelle FLIM-FRET contient des informations sur la désintégration de fluorescence enregistrée à cet endroit particulier (#counts = nombre de photons détectés dans le canal t). (R) La représentation classique de l’image FLIM est généralement une image 2D codée à vie en fausse couleur (à gauche). Une diminution de la durée de vie moyenne de fluorescence du donneur - comme en voit un changement dans l’échelle des couleurs - peut être observée en présence de FRET et est instructive sur la présence d’IPP dans cette zone spatiale. ( B )Unchevauchement entre le spectre des émissions des donneurs et le spectre d’absorption de l’accepteur est nécessaire pour que le FRET se produise. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
Une deuxième exigence du FRET est que le spectre d’émission du donneur et les spectres d’absorption de l’accepteur sechevauchent 8 (figure 1B). L’excitation de fluorescence du donneur doit être à des longueurs d’onde qui contribuent très peu à l’excitation directe de fluorescence de l’accepteur. Toutes les combinaisons de fluorophores ne sont pas possibles et nous recommandons en outre d’utiliser préférentiellement les donneurs ayant des désintégrations monoexponentielles de fluorescence pour faciliter les interprétations flim-FRET10. Plusieurs couples de protéines de fluorescence répondent à ces exigences, y compris le populaire couple eGFP-mCherry11 (pour un examen sur la palette de protéines fluorescentes disponibles fret pairesvoir 12,13).
FLIM-FRET permet de mesurer la désintégration à vie par fluorescence d’un donneur fret à chaque pixel d’une image FLIM (Figure 1A). Il existe deux techniques majeures pour déterminer la durée de vie de la fluorescence qui diffèrent dans l’acquisition et l’analyse : le domaine de fréquence (FD)14 et le domaine du temps (TD). TD FLIM est plus répandue et est effectuée à l’aide d’un éclairage pulsé combiné avec différentes configurations de détection possibles, y compris les méthodes de gating15,strie caméra 16 ou en corrélation avec le temps simple comptage des photons (TCSPC) techniques8. Tant pour les techniques de DF que pour les techniques de DT, la durée de vie de la fluorescence n’est pas mesurée directement, mais nécessite une analyse des données mesurées pour estimer la durée de vie ou la présence d’interactions. Pour les techniques tcspc, l’analyse la plus largement utilisée repose sur l’ajustement des désintégrations avec des fonctions exponentielles simples ou multiples en utilisant les reconvolutions itératives les moins carrées qui minimisent la somme pondérée des résidus.
Enfin, FLIM-FRET peut être effectué à la fois en utilisant un seul photon ou excitations multiphoton. Les derniers ont plusieurs avantages comme la réduction de l’autofluorescence et photodamage hors du plan focal. Les excitations multiphoton permettent également une plus longue profondeur d’excitation si l’on travaille dans des échantillons 3Dépais 8. Au contraire, l’excitation simple de photons est habituellement plus efficace car les sections transversales d’absorption de deux photons des protéines fluorescentes sontlimitées 17.
Ici, nous proposons un protocole pour les mesures FLIM-FRET des IPP en direct P. aeruginosa dans le cas particulier de deux protéines en interaction (PvdA et PvdL) exprimées avec un nombre très différent de copies pour démontrer la qualité et la robustesse de la technique à révéler les caractéristiques critiques des IPP. Les protéines PvdA et PvdL sont impliquées dans la biosynthèse de la pyoverdine. PvdA est un L-ornithine N5-oxygenase et synthétise le L-N5-formyl-N5-hydroxyornithine de L-ornithine par hydroxylation (PvdA) et formylation (PvdF)18. Le PvdL est une enzyme de synthèse peptidique non ribosomique (NRPS) composée de quatre modules. Le premier module catalyse l’acylation de l’acide myristique. Le deuxième module catalyse l’activation de L-Glu et sa condensation vers le myristic-coA. Ensuite, le troisième module condense un acide aminé L-Tyr qui est ensuite isomérisé en D-Tyr. Enfin, le quatrième module lie un acide aminé L-Dab (acide diaminobutyrique) pour former le tripepide acylé L-Glu/D-Tyr/L-Dab6. PvdL est donc responsable de la synthèse des trois premiers acides aminés du précurseur de la pyoverdine. L’interaction de la protéine PvdA avec le PvdL est surprenante car le PvdL, au contraire du PvdI et du PvdJ, ne porte pas de module spécifique à la L-N5-formyl-N5-hydroxyornithine. Cette interaction suggère que toutes les enzymes responsables de la biosynthèse précurseur de la pyoverdine sont disposées dans de grands complexes multi-enzymatiques transitoires etdynamiques 19,20.
Dans ce rapport, nous expliquons en détail comment construire les souches bactériennes exprimant nativement les deux protéines étiquetées eGFP et mCherry en interaction. Nous décrivons également la préparation de l’échantillon et les conditions d’imagerie cellulaire FLIM-FRET efficace. Enfin, nous proposons un didacticiel étape par étape pour l’analyse d’images, y compris un outil récemment développé offrant des possibilités avancées de visualisation pour une interprétation simple des données complexes FLIM-FRET. Avec ce rapport, nous tenons à convaincre non seulement les aventuriers, mais la plupart des biologistes que FRET-FLIM est une technique accessible et puissante capable de répondre à leurs questions sur les IPP directement dans l’environnement cellulaire natif.
Protocole
1. Construction plasmide
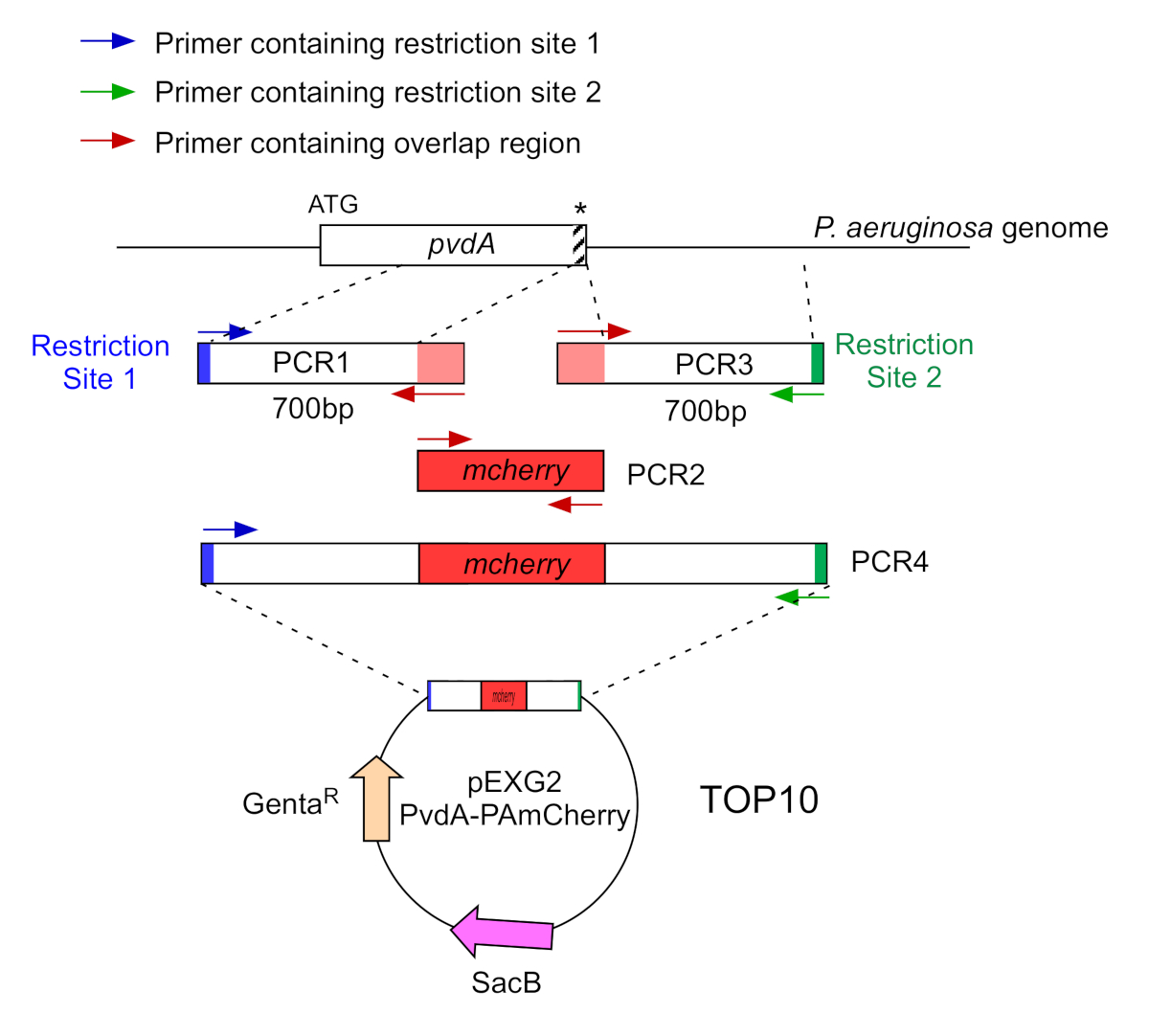
- Amplifier par deux PCR (PCR1 et 3) les séquences d’ADN (utiliser l’ADN génomique de P. aeruginosa PAO1) des 700 paires de base en amont et en aval des régions correspondant au site d’insertion dans le génome de P. aeruginosa avec polymése d’ADN haute fidélité. Ajouter des sites de restriction aux amorces en bleu et vert et ajouter une séquence qui se chevauche avec mCherry aux amorces en rouge( Figure 2).
- Pour le PvdA étiqueté au C-terminus avec eGFP, amplifiez la région de 700 bp en amont par rapport au codon d’arrêt par les amorces en bleu, et amplifiez la région aval de 700 bp contenant le codon d’arrêt avec les amorces en vert.
- Pour le PvdL étiqueté au N-terminus avec mCherry, amplifiez la région de 700 bp en amont du gène PvdL, y compris le codon de départ, par les amorces en bleu, et amplifiez la région aval de 700 bp avec les amorce en vert.

Figure 2 : Aperçu de la stratégie pcr et de la construction des plasmides utilisés pour la construction du PvdA-mCherry. Voir le texte pour plus de détails - pvdA code une enzyme impliquée dans la biosynthèse de la pyoverdine siderophore, un métabolite secondaire impliqué dans l’acquisition de fer. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
- Amplifiez l’ADN codant eGFP (sans le début et arrêtez les codons) avec des amorces en rouge avec polymése d’ADN haute fidélité.
- Purifier les produits PCR sur une colonne de nettoyage PCR (Table of Materials).
- Mélangez les produits PCR qui se chevauchent en rapport équimolaire et effectuez un deuxième PCR à l’aide d’amorces avec le site de restriction utilisé pour pcr 1 et 3 (vert et bleu dans la figure 2).
- Migrez le produit PCR dans le gel agarose-1x TAE (Tris-Base Acétate EDTA pH 8.0), coupez la bande correspondante et extrayez l’amplicon avec un kit de nettoyage PCR (Table of Materials).
REMARQUE : Le protocole peut être mis en pause ici. - Digérer l’amplicon PCR et le plasmide pEXG2 à l’aide des enzymes de restrictioncorrespondantes 21.
- Ligate plasmide et insérer avec ligase D’ADN T4 en utilisant 90 ng de plasmide et le rapport moléculaire 1:1 (plasmide:insert).
- Transformer la construction des plasmides en cellules chimiquement compétentes E. coli TOP10 cellules en mélangeant le produit de ligature et 100 μL de TOP10. Incuber le mélange bactéries/plasmides compétent sur la glace pendant 30 minutes avant de procéder à un choc thermique de 42 °C pendant 60 s. Ensuite, mettez le tube sur la glace pendant 10 min.
- Ajouter 1 mL de bouillon de lysogène (LB) aux bactéries et incuber à 37 °C pendant 1 h.
- Plaque 100 bactéries μL sur l’agar LB contenant 15 μg/mL de gentamicine.
- Incuber toute la nuit à 37 °C.
- Filtrer la présence de l’insert par la colonie PCR: à partir d’une colonie transformatrice isolée, ramasser une quantité infime de bactéries à ajouter à un mélange PCR contenant des amorces hybridant sur le plasmide de telle sorte que la présence de l’amplicon pourrait être détecté en exécutant le produit sur un gel d’agarose (polymérose de l’ADN). De la même colonie utilisée pour la PCR, transférer une petite quantité de bactéries sur une plaque fraîche contenant 15 μg/mL de gentamicine à isoler et à utiliser pour l’extraction des plasmides. Enfin, isoler et purifier le plasmide (Tableau des matériaux) et vérifier l’insert par séquençage.
- Conserver les bactéries TOP10 contenant le plasmide en LB avec 20 % de glycérol dans un microtube de 1,5 mL à -80 °C et le plasmide purifié à -20 °C dans un tube de 1,5 mL.
REMARQUE : Le protocole peut être mis en pause ici
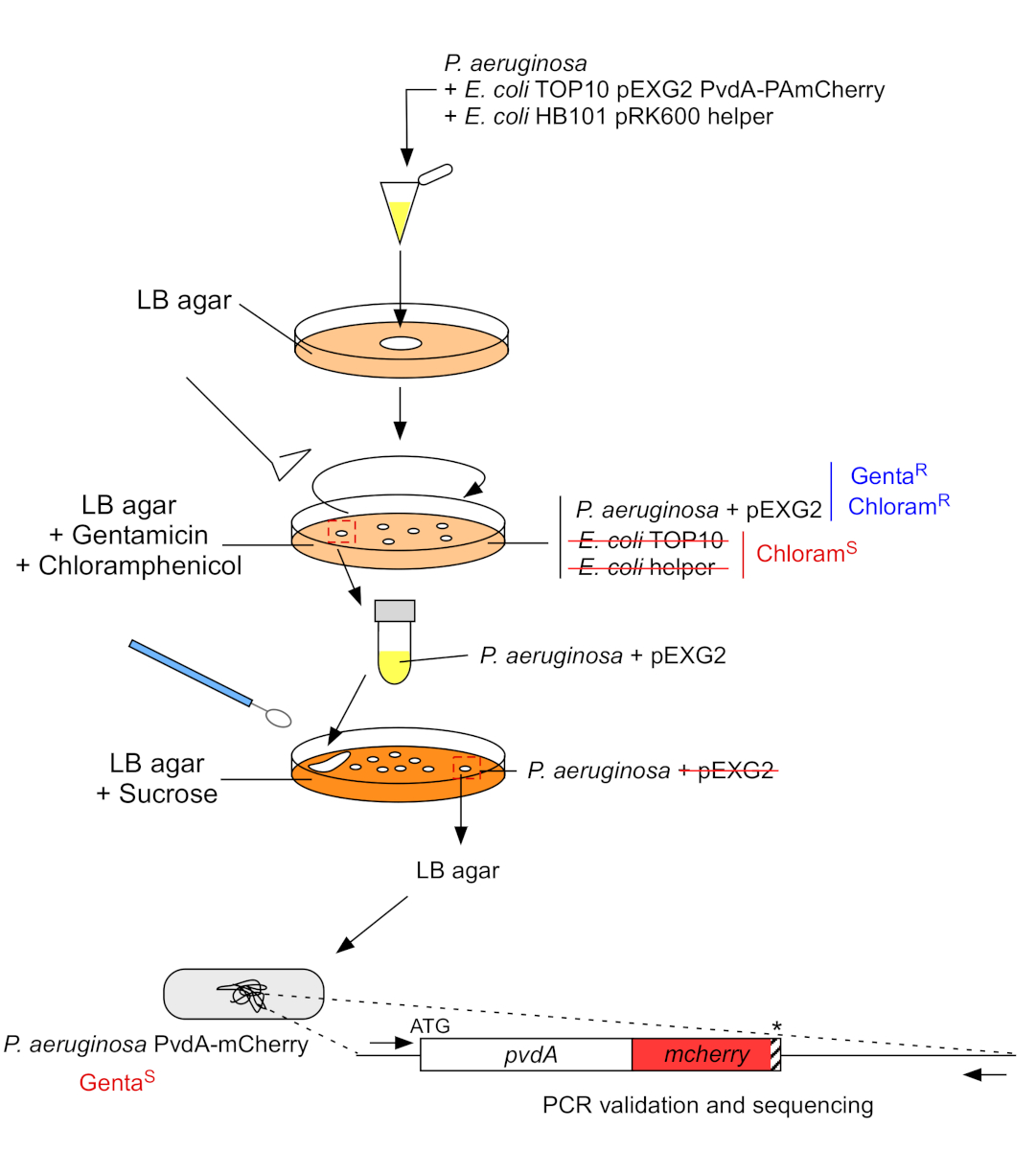
2. Insertion d’étiquettes fluorescentes dans le génome chromosomique de P. aeruginosa (Figure 3)
- Cultiver P. aeruginosa, TOP10 et E. coli bactéries d’aide, chacun dans 5 mL de LB sans antibiotique à 30 °C sous secousse orbitale pendantla nuit 22. Générer l’insertion d’étiquettes fluorescentes dans le génome de P. aeruginosa en transférant le plasmide de E. coli TOP10 dans la souche PAO1 et en intégrant le plasmide dans le génome par recombinaison homologue. Un deuxième événement de croisement excisant le vecteur génère le mutant correspondant.
- Mesurer la densité optique à 600 nm (OD600 nm)de la culture bactérienne et mélanger une quantité égale de P. aeruginosa (500 μL, OD600 nm = 1,0) avec E. coli TOP10 pEXG2 (500 μL, OD600 nm = 1,0) et E. coli HB101 pRK600 helper (500 μL, OD600 nm = 1,0) en microtube de 1,5 mL.
- Centrifugeuse 5 min à 9300 x g pour granuler les bactéries.
REMARQUE : Les outils en ligne peuvent être utilisés pour convertir la force g centrifuge en rotation par minute (régime) pour ajuster la vitesse de centrifugeuse. - Gardez la pastille bactérienne et jetez le surnatant.
- Resuspendez la pastille contenant des bactéries dans 50 μL de LB.
- Plaquer une tache (~50 μL) du mélange au milieu de l’agar LB (préchauffer à 37 °C) et incuber 5 h à 37 °C.
- Raclez l’endroit avec une boucle stérile d’inoculation et résuspendez dans 1 mL de LB.
- Plaque 100 μL de cette suspension bactérienne sur l’agar LB contenant 10 μg/mL de chloramphéniphéniol pour éliminer E. coli (E. coli TOP10 pEXG2 et E. coli HB101 pRK600 aide sont sensibles au chloramphéniphénol, mais P. aeruginosa est naturellement résistant) et 30 μg/mL gentamicine et incuber 2 jours à 37 °C.
- Resuspendez une colonie en 1 mL LB et incubez à 37 °C sous la secousse orbitale de 4h.
- Centrifugeuse 3 min à 9 300 x g et jeter 950 μL de supernatant. Resuspendez la pastille dans 50 μL de LB et isolez le mélange sur l’agar LB contenant du saccharose et sans NaCl.
- Incuber toute la nuit à 30 °C.
- Repérer les colonies isolées sur l’agar LB et l’agar LB contenant 15 mg/mL de gentamicine afin de vérifier la sensibilité à la gentamicine.
- Vérifiez l’insertion eGFP ou mCherry par colonies PCR (polyméseau d’ADN) et séquençage à l’aide d’amorces spécifiques.

Figure 3 : Protocole de construction des souches de P. aeruginosa par insertion d’étiquettes fluorescentes. Voir le texte pour plus de détails. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
3. Mesure de pyoverdine
- Cultiver des bactéries dans 5 mL de LB à 30 °C sous secousse orbitale pendant la nuit.
- Bactéries granulées par centrifugation, lavage et culture dans 5 mL de SM (Succinate Medium, composition: 6 g◊L−1 K2HPO4, 3 g◊L−1 KH2PO4, 1 g◊L−1 (NH4)2 SO4, 0,2 g −L−1 MgSO4, 7 H2O et 4 g◊L−1 succinate de sodium avec le pH ajusté à 7,0 en ajoutant NaOH) à 30 °C sous secousse orbitale pendant la nuit. SM est un milieu privé de fer - l’absence de fer activera l’expression des protéines de la voie de pyoverdine normalement réprimée en présence de fer.
- Mesurer l’OD600 nm et diluer à nouveau les bactéries dans le milieu SM frais à600 nm OD = 0,1 et les cultiver à 30 °C sous secousse orbitale pendant la nuit.
- Mesurez od600 nm pour déterminer la quantité de bactéries dans chaque échantillon.
- Préparer une cuvette à quartz contenant 100 μL de culture P. aeruginosa et compléter jusqu’à 1 mL de SM (900 μL). Préparer une cuvette à quartz contenant 1 mL de SM moyen (blanc).
- À l’aide d’un spectrophotomètre visible par les UV, mesurez l’absorption au maximum du pic d’absorption. Au pH 7.0, le maximum d’absorption de pyoverdine se produira à ~400 nm. Déterminer la concentration de pyoverdine (forme d’apo) dans l’échantillon à l’aide de la loi Beer-Lambert à l’aide d’un coefficient d’extinction molaire à 400 nm de e = 19 000 M-1 -1-1.
REMARQUE : La pyoverdine peut être quantifiée dans la gamme d’absorption de ~0.1 à ~1 (selon le spectrophotomètre UV-Visible) dans laquelle l’absorption augmente linéairement avec la concentration.
4. Culture des bactéries et conditions pour les cellules d’exprimer PvdA, PvdL et PvdJ
- Le jour 1, inoculer un tube avec 5 mL de LB à partir du stock approprié de glycérol de bactéries et de cultiver des bactéries pendant la nuit à 30 °C à 200 rpm dans un incubateur shaker orbital.
- Le jour 2, pelleter les cellules par centrifugation à 3000 x g pendant 3 min et jeter le supernatant.
- Resuspendez les cellules en 10 mL de SM.
- Répétez les étapes 4.2-4.3 une fois et cultivez des bactéries en SM pendant la nuit à 30 °C 200 rpm.
- Le jour 3, diluer 1/10 de la culture des bactéries dans sm frais.
- Cultivez à nouveau des bactéries diluées pendant la nuit dans les mêmes conditions.
REMARQUE : La présence de pyoverdine peut être détectée visuellement car elle colore en jaune-vert le média en croissance. Il montre que l’expression des protéines de la voie de pyoverdine ont été activées et que des enzymes d’intérêt sont exprimées dans les cellules.
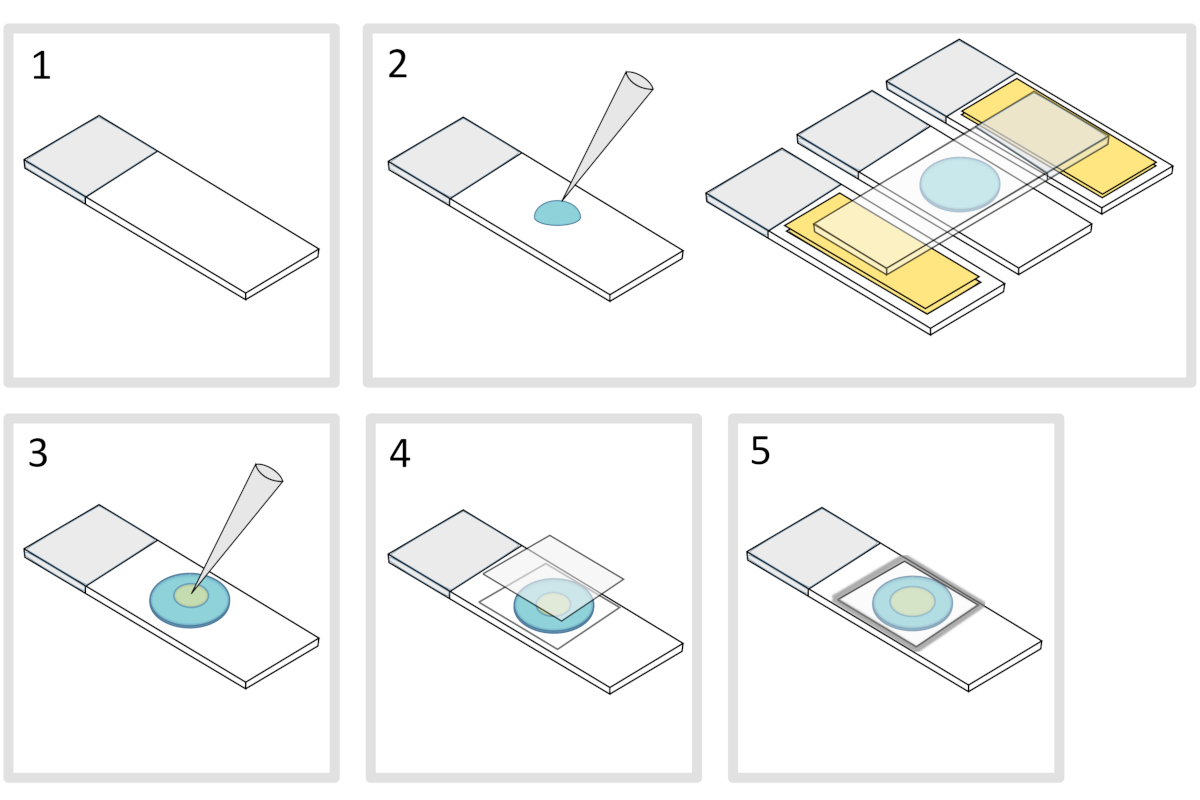
5. Préparation de la garniture d’agarose( figure 4)
- Placez un microscope en verre-glisser sur une surface horizontale plate. Disposer deux glissières en verre surmontées de deux couches de ruban adhésif de chaque côté de la glissière initiale.
REMARQUE : Gardez un espace de 1 à 2 mm entre les trois glissières alignées pour éviter que l’agarose maillé ne se propage éventuellement sur les glissières avec du ruban adhésif. - Pipette et verser une gouttelette de 70 μL de 1% d’agarose fondue sur la glissière de verre. Ajouter une quatrième diapositive sur le dessus pour aplatir la gouttelette d’agarose et appuyez doucement vers le bas. Attends une minute.
- Enrôlez la glissière supérieure et déposez avec une pipette environ 3 μL de bactéries en 3 à 4 endroits à différents endroits sur la garniture d’agarose.
- Couvrir d’un couvercle en verre microscopie (par exemple, un couvercle de 22x22 mm #1,5 d’épaisseur).
REMARQUE : La planéité et l’épaisseur des coverslips sont importantes pour travailler avec des excitations à deux photons. Les couvertures de précision avec flatness uniforme contrôlé et faible autofluorescence sont généralement un bon choix. - Fixer le coverslip avec de la paraffine fondue pour sceller le coverslip sur la lame de verre. Commencez par fixer les quatre coins du coverslip.

Figure 4 : Préparation des coussinets Agarose. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
6. Imagerie avec une microscopie à deux photons
REMARQUE : Nous utilisons un microscope inversé à balayage à balayage à deux photons fait maison avec un objectif d’immersion d’eau 60x 1.2NA fonctionnant en mode de collecte de fluorescence dé-scanné. La longueur d’onde excitation à deux photons est fixée à 930 nm. Il est fourni par un laser Ti:Sapphire (taux de répétition de 80 MHz, ≈ 70 fs de largeur d’impulsion) travaillant à 10-20 mW. Des photons de fluorescence ont été rassemblés par un filtre court de passage de 680 nm et un filtre de bande-passage de 525/50 nm avant d’être dirigés vers une photo-diode d’avalanche fibre-couplée reliée à un module de comptage simple de photons (TCSPC) corrélé dans le temps. Le microscope est également équipé d’une lampe de fluorescence de transmission. Plusieurs microscopes FLIM-FRET sont maintenant disponibles dans le commerce et de nombreuses installations d’imagerie sont équipées d’installations capables d’effectuer des mesures FLIM-FRET.
- Utilisez la lampe de fluorescence pour concentrer l’objectif sur le monocouche des bactéries dans l’échantillon et certaines régions d’intérêt.
- Vérifiez que l’obturateur laser excitation est fermé et que la lumière infrarouge provenant du laser est bloquée et n’entre pas dans le microscope.
Attention : Une attention particulière et une vigilance constante doivent être accordées en travaillant avec des lasers pulsés IR car la lumière laser ne peut pas être vue par les yeux, mais toute exposition directe et même transitoire ou réflexion laser peut être extrêmement nocive et créer des lésions oculaires irréversibles. Veuillez consulter les procédures et la formation locales en matière de sécurité laser avant d’utiliser des configurations de microscopie. - Placez la diapositive de microscopie sur la scène avec les coverslips face à l’objectif.
- Vérifiez que la lampe de fluorescence est allumée.
- Tournez la tourelle du cube filtrant pour sélectionner le cube eGFP et ouvrez l’obturateur de la lampe de fluorescence.
- Envoyez la lumière de fluorescence vers l’oculaire du microscope.
Attention: Assurez-vous que les filtres appropriés sont éliminés dans le chemin lumineux pour éliminer la lumière d’excitation directe provenant de la lampe de fluorescence qui peut endommager les yeux. - Concentrez l’objectif sur les bactéries à l’aide du bouton microscope.
- Sélectionnez une région d’intérêt pour l’échantillon en la traduisant à l’aide du joystick contrôlant l’étape motorisée
REMARQUE : La mise au point est plus facile avec un échantillon hautement fluorescent permettant de voir la fluorescence directement avec les yeux. - Changez l’excitation pour le laser 2PE pour les mesures FLIM-FRET.
- Renvoyez le chemin d’émission de fluorescence vers le détecteur.
- Tournez en arrière la tourelle cube filtre pour sélectionner pour le cube dichroïque pour le laser 930 nm.
- Réglez la puissance laser à 20 mW.
- Réglez la taille de la région d’intérêt à 30 μm. Cette opération ajuste la tension d’exploitation des miroirs galvo et définit la portée de leurs mouvements (Figure 5).
- Allumez le détecteur et commencez à scanner l’échantillon - les boutons de démarrage et d’arrêt contrôlant la numérisation contrôlent également l’ouverture et la fermeture de l’obturateur laser pour des raisons de sécurité et pour limiter le photobleaching de l’échantillon (Figure 5).

Figure 5 : Représentation schématique de l’interface du logiciel de contrôle au microscope. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
- Si nécessaire, ajustez la mise au point en déplaçant légèrement le bouton de mise au point fine du microscope.
- Choisissez le champ de vision pour l’imagerie en déplaçant finement la scène de l’interface de l’ordinateur. Cela peut être fait sur la configuration en déplaçant la croix sur l’image dans le logiciel de contrôle du microscope (Figure 5) qui définira le nouveau centre de l’image et en appuyant sur Move Stage. Un bon champ de vision pour l’acquisition correspond à une image avec 10-30 bactéries immobiles, toutes correctement ciblées (toutes les bactéries sont sur le même plan). Si vous êtes intéressé à extraire des données FLIM-FRET à cellules individuelles, assurez-vous que les bactéries sont bien individualisées (la segmentation de l’image sera beaucoup plus facile).
- Ouvrez le logiciel SPCM (logiciel commercial pour l’acquisition de données) et vérifiez que le taux de comptage des photons n’est pas trop élevé pour éviter l’effet de carambolage qui peut affecter les mesures à vie. Si nécessaire, abaissez l’intensité laser pour maintenir le taux de nombre de photons bas (environ 1% du taux de répétition laser).
REMARQUE : L’effet pile-up décrit les effets des photons perdus à des taux élevés de nombre de photons en raison de l’heure morte des dispositifs de comptage des photons simples corrélés dans le temps (TCSPC). Si le carambolage se produit, la durée de vie moyenne mesurée devient artificiellement plus courte avec peut-être un composant plus court supplémentaire qui peut apparaître dans la décomposition en raison de l’amortissement des photons émettant rapidement. - Ajuster les paramètres d’acquisition, y compris le temps de collecte des acquisitions (généralement de 60 à 180 s sont nécessaires pour recueillir suffisamment de photons).
- Appuyez sur le bouton Démarrer et attendez la fin de l’acquisition.
- Enregistrez les données.
- Arrêtez de scanner l’échantillon et éteignez le détecteur.
- Sélectionnez un autre champ de vision dans l’échantillon et répétez les étapes 6.14-6.22 ou imagez une nouvelle diapositive de microscopie en répétant les étapes 6.1-6.22.
REMARQUE : P. aeruginosa peut vivre et diviser jusqu’à 6-8 heures à température ambiante sur la garniture d’agarose (correspondant à la dernière ~4 temps de doublement à 20°C). Idéalement, n’attendez pas trop longtemps pour effectuer la mesure FLIM-FRET pour éviter d’observer un tampon complètement recouvert de bactéries.
7. Analyse des données

Figure 6 : Panneau principal de la fenêtre d’analyse des données du logiciel SPCImage. Image d’intensité (boîte bleue), image à vie (boîte violette), histogramme à vie (en haut à droite), courbe de décomposition à une position sélectionnée (boîte verte) et paramètres de décomposition à la position sélectionnée (boîte cyan) d’un représentant PvdA-eGFP pourriture enregistrée en direct P. aeruginosa à l’aide d’une carte d’acquisition bh SPC830 sur une configuration maison Two-Photon Excitation-FLIM-FRET. La courbe expérimentale de décomposition du pixel pointée dans l’image ci-dessus, son ajustement mono-exponentiel (courbe rouge) décontvoltant la décomposition de sa fonction de réponse instrumentale calculée (courbe verte) peuvent être vus dans le panneau vert. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
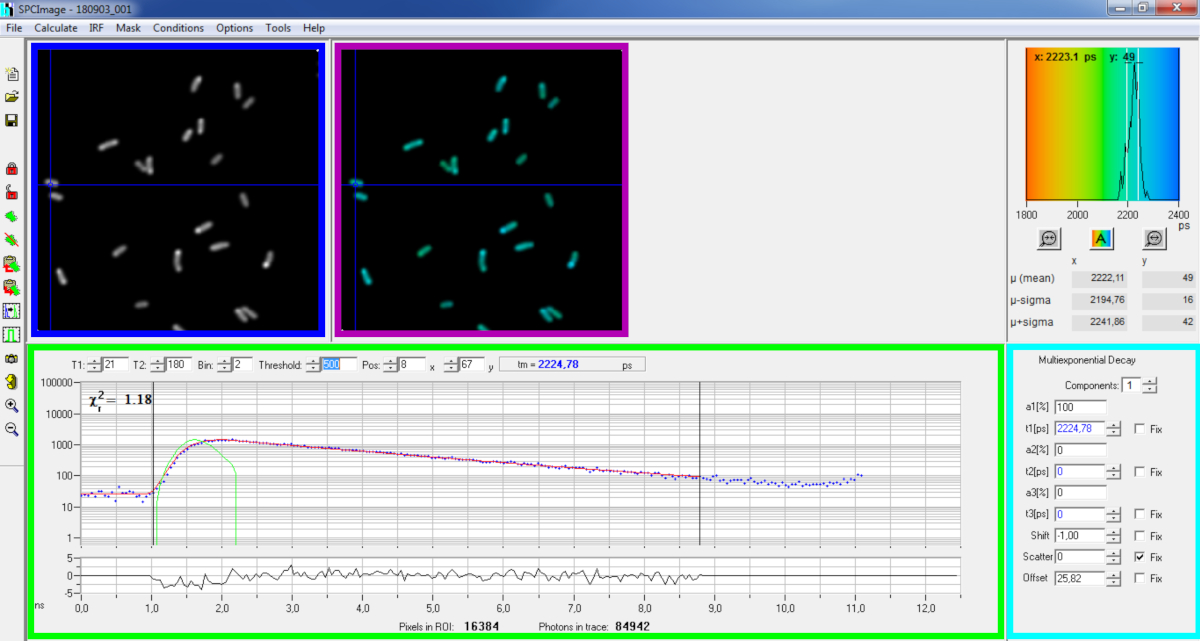{kind=link}
- Analyse de base
- Exécutez le logiciel SPCImage.
- Importez le fichier SPCM enregistré. L’image d’intensité est affichée sur le panneau supérieur gauche du logiciel(figure 6 boîte bleue).
- Examinez la fenêtre courbe de décomposition(figure 6 boîte verte) qui affiche les données de décomposition correspondant au pixel sélectionné dans l’image d’intensité(figure 6 boîte bleue). Les numéros de photons de chaque canal de temps sont montrés comme points bleus et l’ajustement de la décomposition est dessiné comme une ligne rouge. Notez qu’après le chargement des données, le logiciel affiche le pixel le plus lumineux de l’image. Déplacez la croix bleue à travers l’image pour examiner les pixels avec une intensité plus faible. La fenêtre de décomposition se actualisera automatiquement à chaque nouvelle position de pixel.
REMARQUE : Il est très difficile de mesurer la fonction de réponse instrumentale (IRF) d’un système de balayage laser. Un IRF calculé à partir du bord ascendant des courbes de désintégration de fluorescence dans les données flim peut être utilisé pour la déconvolution de la désintégration. C’est l’option faite par défaut dans SPCimage(courbe verte figure 6). - Ajustez la plage de montage en déplaçant les canaux de départ et de fin de la boîte de montage (T1 et T2 dans la boîte verte). T1 devrait commencer par les premiers canaux de la décomposition croissante et T2 définir le dernier canal à la fin de la décomposition et peut être choisi comme l’un des derniers canaux de la décomposition avec un certain nombre de photons au-dessus du nombre de photons compensés (c.-à-d., les niveaux de photons comptés avant la décomposition augmente).
- Choisissez le binning en changeant la valeur bin. La décomposition de la courbe intègre le comptage photon du pixel sélectionné ainsi qu’une zone de pixels i autour de la position du curseur définie par le paramètre du bac (l’augmentation du binning augmentera le nombre de photons dans la décomposition et peut être utile pour atteindre le nombre de photons requis pour les modèles multi-exponentiels).
- Ajustez la valeur seuil. Les pixels qui n’ont pas au moins un canal avec un certain nombre de photons supérieurs à la valeur seuil ne seront pas inclus dans la procédure de montage. Bien sûr, plus le nombre de pixels à adapter est élevé, plus l’analyse est longue.
REMARQUE : Les données FLIM peuvent contenir un grand nombre de pixels et de canaux de temps. Les dernières versions du logiciel permettent d’utiliser GPU (Graphics Processor Unit) pour traiter un grand nombre de pixels en parallèle, ce qui réduit considérablement les temps de traitement. Il peut être intéressant d’ajuster les paramètres de binning et de seuil à l’aide d’images correspondant aux constructions bactériennes présentant l’intensité de fluorescence la plus faible (p. ex., avec des souches bactériennes ayant les niveaux d’expression les plus bas). Cela permettra de s’assurer que les désintégrations pertinentes observées dans ces échantillons répondront aux critères de filtrage et seront incluses dans l’analyse. Ces paramètres peuvent ensuite être utilisés pour toutes les images. - Ajuster, si nécessaire, les paramètres de décomposition (boîte cyan). Que le décalage varie, la plupart du temps la dispersion et le décalage peuvent être fixés à zéro si un coup d’oeil aux fonctions de décomposition montrent que leur contribution est négligeable. Le décalage peut être estimé en regardant les premiers canaux de la décomposition - notez que l’imagerie pendant une longue période en raison de la faible fluorescence dans l’échantillon se traduit généralement par un décalage non-zéro. La dispersion se produit principalement dans des échantillons épais et peut être considérée comme négligeable autrement.
- Avant d’exécuter l’ajustement, sélectionnez l’algorithme de montage. Ouvrez la fenêtre paramètres de l’algorithme dans afficher/masquer les options de modèle. Sélectionnez l’algorithme d’estimation de probabilité maximale (MLE)( Figure 7A).
- Exécutez le montage de l’image en cliquant sur Calculer | de décomposition. Une fois terminée, l’image FLIM codée à vie apparaît dans le panneau d’image à vie(figure 6 boîte violette).
REMARQUE : Sur la fenêtre courbe de décomposition(figure 6 boîte verte), il est possible de voir la valeur à vie qui correspond à chaque pixel de l’image en déplaçant la croix bleue.
REMARQUE : pour traiter automatiquement un grand nombre de fichiers de données FLIM similaires, un mode de traitement par lots peut être utilisé. - Vérifiez la qualité de l’ajustement en regardant les résidus (idéalement répartis au hasard autour de 0) et une valeur carrée Chi près de 1.
- Les données adaptées peuvent être exportées dans différents formats. Pour exporter des fichiers dans des fichiers txt, rendez-vous sur File | Exportation. Dans la fenêtre Options d’exportation (Figure 7B), choisissez Sélectionnez Tous, puis cliquez sur Exportation.
- Enfin enregistrer le fichier d’analyse. Les fichiers d’analyse sont enregistrés sous forme de fichiers *.img et peuvent être rouverts directement dans SPCImage.
REMARQUE : Dans des cas particuliers de quantités déséquilibrées de donneurs/accepteurs, le FLIM-FRET peut révéler des sous-populations dans un mélange de complexes protéiques en interaction - en particulier lorsque les concentrations des deux partenaires sont très différentes, ce qui donne lieu à des mélanges d’espèces complexes et libres. Les espèces non interagissant (caractérisées par une décomposition très semblable à la désintégration du donneur seulement) peuvent être discriminées à partir d’espèces en interaction en supposant une invariance spatiale des composantes de la durée de vie des donneurs dans l’ensemble de données. De même, des complexes d’interaction non stoichiométriques avec plus de donneurs ou plus d’accepteur de fluorescence peuvent se former. Les désintégrations de fluorescence de ces complexes sont généralement difficiles à interpréter. Un diagramme FLIM peut être utilisé pour fournir des informations critiques sur la stoichiométrie et le mode de liaison desIPP 20,23. L’intrigue du diagramme FLIM est une représentation graphique de la composante la plus courte durée de vie en fonction de son amplitude. Il peut être utilisé pour visualiser des pixels avec des signatures de décomposition similaires. Pour dessiner de telles représentations, les désintégrations expérimentales de fluorescence doivent être équipées d’un modèle exponentiel deux. Les étapes suivantes peuvent être un guide à travers ce processus. - Commencez par analyser les données du donateur uniquement la construction. Il permettra de déterminer la valeur à vie du donneur. Idéalement, mesurez cette valeur par rapport à plusieurs images enregistrées dans les mêmes conditions que les constructions donneur/accepteur pour récupérer une valeur à vie robuste pour le donneur.
- Une fois déterminé, adaptez les désintégrations de fluorescence des constructions de donneurs/accepteurs à un modèle à deux exponentielles. Dans la boîte de paramètres de désintégration du cyan (figure 6), réglez le nombre de composants à 2. Fixez le paramètre t2(ps) à la valeur à vie robuste du donneur déterminé à l’étape 1 et cochez la case pour fixer ce paramètre.
REMARQUE : Il est important de fixer la durée de vie de longue durée τ2 afin de limiter le surajustage, d’améliorer la convergence des raccords et d’obtenir des paramètres d’ajustement deux exponentielsplus fiables 24,25,26. - Enregistrez le fichier *img et les données d’exportation sous forme de fichiers *.asc comme à l’étape 7.1.11.

Figure 7 : (A) Paramètres de l’algorithme pour équiper les désintégrations de modèles exponentiels. Sélection de MLE (algorithme de probabilité maximale ou estimation de probabilité maximale, MLE) comme modèle d’ajustement, et fenêtre d’options d’exportation (B). S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
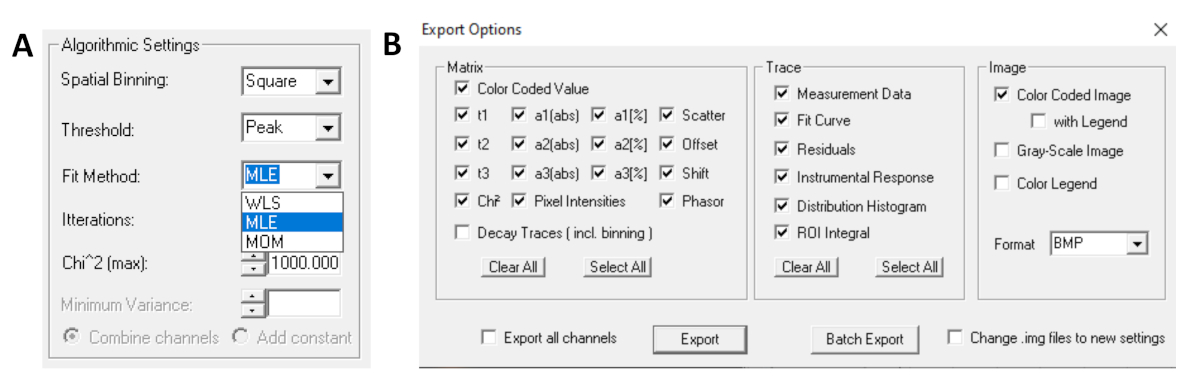{kind=link}
- Analyse avancée des images FLIM en R
- Installez R (https://cran.r-project.org) et RStudio (https://rstudio.com) si nécessaire.
- Ouvrez RStudio et créez un nouveau projet.
- Déplacez toute l’analyse *.asc fichier dans un dossier appelé « données » dans le dossier principal du projet.
- Ouvrez un nouveau fichier script (ou ouvrez le script supplémentaire FLIM_analysis. R).
- Installez le paquet flimDiagRam dédié pour l’analyse des données flim https://github.com/jgodet/flimDiagRam. Appelez le paquet dans l’espace de travail. (Voir l’avis HowTo_FlimDiagRam)
REMARQUE : L’installation des paquets ne doit être effectuée qu’une seule fois. Une fois installés, les paquets peuvent être appelés à partir de n’importe quelle nouvelle session R. Le téléchargement de paquets R à partir de github nécessite l’installation de paquets de « devtools ». L’installation de « devtools » peut prendre plusieurs minutes. Le paquet flimDiagRam peut être utilisé pour représenter les paramètres et les distributions des données FLIM, pour extraire des données FLIM au niveau des cellules individualisées individuelles, pour comparer les résultats de la FLIM entre les conditions ou les souches et pour explorer les données FLIM à l’aide d’outils de visualisation avancés comme le diagramme FLIM. - Utilisez le code commenté étape par étape et les données sont mises à disposition pour reproduire indépendamment tous les sous-chiffres présentés dans la section Résultats représentatifs ci-dessous. Ce tutoriel peut être trouvé dans l’avis HowTo_FlimDiagRam à https://github.com/jgodet/flimDiagRam/blob/master/HowTo.pdf. Le code peut être facilement transposé pour analyser les données.
Résultats
Les fonctions cumulatives empiriques de distribution (ecdf) des durées de vie de fluorescence mesurées pour les différentes souches bactériennes sont indiquées à la figure 8. Si fret se produit, les ecdfs sont déplacés vers les durées de vie plus courtes(figure 8A,8B). Notez que lorsque l’interaction des deux protéines entraîne une longue distance entre les deux fluorophores, aucun FRET ne peut se produire (Figu...
Discussion
FLIM-FRET offre certains avantages clés par rapport à l’imagerie FRET basée sur l’intensité. La durée de vie de fluorescence est un paramètre intrinsèque du fluorophore. Par conséquent, elle ne dépend pas des concentrations locales de fluorophores ni de l’intensité de l’excitation lumineuse. La durée de vie de fluorescence est en outre également mal affectée par le photo-blanchiment. Il est particulièrement intéressant de mettre en évidence les IPP dans les cellules où les concentrations de prot?...
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous reconnaissons le Dr Ludovic Richert pour son aide précieuse dans l’acquisition de données FLIM et pour la maintenance technique et le développement de la configuration FLIM. Ces travaux ont été financés par des subventions de la Fondation pour la Recherche en Chimie (https://icfrc.fr/). VN est financé par la Fondation pour la Recherche Médicale (FRM‐SPF201809006906). YM remercie l’Institut Universitaire de France (IUF) pour son soutien et son temps supplémentaire consacré à la recherche. L’IJS et JG reconnaissent l’Institut de livraison de médicaments de Strasbourg pour son soutien financier.
matériels
| Name | Company | Catalog Number | Comments |
| 525/50 nm band-pass filter | F37-516, AHF, Germany | ||
| 680 nm short pass filter | F75-680, AHF, Germany | ||
| Agarose | Sigma-Aldrich | A9539 | |
| Ammonium Sulfate (NH4)2SO4 | Sigma-Aldrich | A4418 | |
| DreamTaq DNA polymerase 5U/μL | ThermoFisher Scientific | EP0714 | |
| E. coli TOP10 | Invitrogen | C404010 | |
| Fiber-coupled avalanche photo-diode | SPCM-AQR-14- FC, Perkin Elmer | ||
| Glass coverslips (Thickness No. 1.5, 20×20mm | Knitel glass | MS0011 | |
| High-Fidelity DNA polymerase Phusion 2U/μL | ThermoFisher Scientific | F530S | |
| Lysogeny broth (LB) | Millipore | 1.10285 | |
| Magnesium Sulfate Heptahydrate (MgSO4 . 7H2O) | Sigma-Aldrich | 10034-99-8 | |
| Microscope slides (25×75mm) | Knitel glass | MS0057 | |
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | 740609.50 | |
| NucleoSpin Plasmid | Macherey-Nagel | 740588.10 | |
| Potassium Phosphate Dibasic (K2HPO4) | Sigma-Aldrich | RES20765 | |
| Potassium Phosphate Monobasic (KH2PO4) | Sigma-Aldrich | P5655 | |
| Sodium Succinate (Disodium) | Sigma-Aldrich | 14160 | |
| SPCImage, SPCM software | Becker & Hickl | ||
| Sterile inoculating loop | Nunc | 7648-1PAK | |
| T4 DNA ligase 1U/μL | ThermoFisher Scientific | 15224017 | |
| TCSPC module | SPC830, Becker & Hickl, Germany | ||
| Ti:Sapphire laser | Insight DeepSee, Spectra Physics | ||
| Tubes 50mL | Falcon | 352070 |
Références
- Braun, P., Gingras, A. C. History of protein-protein interactions: From egg-white to complex networks. Proteomics. 12, 1478-1498 (2012).
- Nooren, I. M. A., Thornton, J. M. Structural characterisation and functional significance of transient protein-protein interactions. Journal of Molecular Biology. 325, 991-1018 (2003).
- Hayes, S., Malacrida, B., Kiely, M., Kiely, P. A. Studying protein-protein interactions: Progress, pitfalls and solutions. Biochemical Society Transactions. 44, 994-1004 (2016).
- Guillon, L., Altenburger, S., Graumann, P. L., Schalk, I. J. Deciphering protein dynamics of the siderophore pyoverdine pathway in Pseudomonas aeruginosa. PLoS ONE. 8, 1-9 (2013).
- Ringel, M. T., Brüser, T. The biosynthesis of pyoverdines. Microbial Cell. 5, 424-437 (2018).
- Schalk, I. J., Rigouin, C., Godet, J. An overview of siderophore biosynthesis among fluorescent Pseudomonads and new insights into their complex cellular organization. Environmental Microbiology. 22, 1447-1466 (2020).
- Cui, Y., et al. Techniques for detecting protein-protein interactions in living cells: principles, limitations, and recent progress. Science China Life Sciences. , (2019).
- Day, R. N., Mazumder, N., Sun, Y., Christopher, K. G. FRET microscopy: Basics, issues and advantages of FLIM-FRET imaging. Springer Series in Chemical Physics. 111, 249-276 (2015).
- Bastiaens, P. I. H., Squire, A. Fluorescence lifetime imaging microscopy: Spatial resolution of biochemical processes in the cell. Trends in Cell Biology. 9, 48-52 (1999).
- Yasuda, R. Imaging spatiotemporal dynamics of neuronal signaling using fluorescence resonance energy transfer and fluorescence lifetime imaging microscopy. Current Opinion in Neurobiology. 16, 551-561 (2006).
- Tramier, M., Zahid, M., Mevel, J. -. C., Masse, M. -. J., Coppey-Moisan, M. Sensitivity of CFP/YFP and GFP/mCherry Pairs to Donor Photobleaching on FRET Determination by Fluorescence Lifetime Imaging Microscopy in Living Cells. Microscopy Research and Technique. 71, 146-157 (2008).
- Bajar, B. T., Wang, E. S., Zhang, S., Lin, M. Z., Chu, J. A guide to fluorescent protein FRET pairs. Sensors (Switzerland). 16, 1-24 (2016).
- Piston, D. W., Kremers, G. J. Fluorescent protein FRET: the good, the bad and the ugly. Trends in Biochemical Sciences. 32, 407-414 (2007).
- Leray, A., et al. Optimized protocol of a frequency domain fluorescence lifetime imaging microscope for fret measurements. Microscopy Research and Technique. 72, 371-379 (2009).
- Elson, D. S., et al. Real-time time-domain fluorescence lifetime imaging including single-shot acquisition with a segmented optical image intensifier. New Journal of Physics. 6, 1-13 (2004).
- Rajoria, S., Zhao, L., Intes, X., Barroso, M. FLIM-FRET for Cancer Applications. Current Molecular Imaging. 3, 144-161 (2014).
- Drobizhev, M., Makarov, N. S., Tillo, S. E., Hughes, T. E., Rebane, A. Two-photon absorption properties of fluorescent proteins. Nature Methods. 8, 393-399 (2011).
- Visca, P., Ciervo, A., Orsi, N. Cloning and nucleotide sequence of the pvdA gene encoding the pyoverdin biosynthetic enzyme L-ornithine N5-oxygenase in Pseudomonas aeruginosa. Journal of Bacteriology. 176, 1128-1140 (1994).
- Imperi, F., Visca, P. Subcellular localization of the pyoverdine biogenesis machinery of Pseudomonas aeruginosa: A membrane-associated 'siderosome'. FEBS Letters. 587, 3387-3391 (2013).
- Gasser, V., et al. In cellulo FRET-FLIM and single molecule tracking reveal the supra-molecular organization of the pyoverdine bio-synthetic enzymes in Pseudomonas aeruginosa. Quarterly Reviews of Biophysics. , 1-11 (2019).
- Rietsch, A., Mekalanos, J. J. Metabolic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Molecular Microbiology. 59, 807-820 (2006).
- Herrero, M., De Lorenzo, V., Timmis, K. N. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. Journal of Bacteriology. 172, 6557-6567 (1990).
- Godet, J., Mély, Y. Exploring protein-protein interactions with large differences in protein expression levels using FLIM-FRET. Methods and Applications in Fluorescence. 8, 014007 (2019).
- El Meshri, S. E., et al. Role of the nucleocapsid domain in HIV-1 gag oligomerization and trafficking to the plasma membrane: A fluorescence lifetime imaging microscopy investigation. Journal of Molecular Biology. 427, 1480-1494 (2015).
- Becker, W. The bh TCSPC Handbook. Scanning. , 1 (2010).
- Richert, L., Didier, P., de Rocquigny, H., Mély, Y. Monitoring HIV-1 protein oligomerization by FLIM FRET microscopy. Springer Series in Chemical Physics. , 111 (2015).
- Fereidouni, F., Blab, G. A., Gerritsen, H. C. Phasor based analysis of FRET images recorded using spectrally resolved lifetime imaging. Methods and Applications in Fluorescence. 2, (2014).
- Fereidouni, F., Gorpas, D., Ma, D., Fatakdawala, H., Marcu, L. Rapid fluorescence lifetime estimation with modified phasor approach and Laguerre deconvolution: a comparative study. Methods and Applications in Fluorescence. 5, 035003 (2017).
- Margineanu, A., et al. Screening for protein-protein interactions using Förster resonance energy transfer (FRET) and fluorescence lifetime imaging microscopy (FLIM). Scientific Reports. 6, (2016).
- Guzmán, C., Oetken-Lindholm, C., Abankwa, D. Automated High-Throughput Fluorescence Lifetime Imaging Microscopy to Detect Protein-Protein Interactions. Journal of Laboratory Automation. 21, 238-245 (2016).
- Liu, W., Cui, Y., Ren, W., Irudayaraj, J. Epigenetic biomarker screening by FLIM-FRET for combination therapy in ER+ breast cancer. Clinical Epigenetics. 11, 1-9 (2019).
- Liu, X., et al. Fast fluorescence lifetime imaging techniques: A review on challenge and development. Journal of Innovative Optical Health Sciences. 12, 1-27 (2019).
- Padilla-Parra, S., Auduge, N., Coppey-Moisan, M., Tramier, M. Non fitting based FRET-FLIM analysis approaches applied to quantify protein-protein interactions in live cells. Biophysical Reviews. 3, 63-70 (2011).
- Padilla-Parra, S., Audugé, N., Coppey-Moisan, M., Tramier, M. Quantitative FRET analysis by fast acquisition time domain FLIM at high spatial resolution in living cells. Biophysical Journal. 95, 2976-2988 (2008).
- Stringari, C., et al. Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proceedings of the National Academy of Sciences of the United States of America. 108, 13582-13587 (2011).
- Digman, M. A., Caiolfa, V. R., Zamai, M., Gratton, E. The phasor approach to fluorescence lifetime imaging analysis. Biophysical Journal. 94, 14-16 (2008).
- Liang, Z., Lou, J., Scipioni, L., Gratton, E., Hinde, E. Quantifying nuclear wide chromatin compaction by phasor analysis of histone Förster resonance energy transfer (FRET) in frequency domain fluorescence lifetime imaging microscopy (FLIM) data. Data in Brief. 30, 105401 (2020).
- Grimm, J. B., Heckman, L. M., Lavis, L. D. The chemistry of small-molecule fluorogenic probes. Progress in Molecular Biology and Translational Science. 113, (2013).
- Li, L., Sun, H. Next Generation of Small-Molecule Fluorogenic Probes for Bioimaging. Biochemistry. 59, 216-217 (2020).
- Yao, R., Ochoa, M., Yan, P., Intes, X. Net-FLICS: fast quantitative wide-field fluorescence lifetime imaging with compressed sensing - deep learning approach. Light: Science and Applications. 8, 1-7 (2019).
- Smith, J. T., et al. Fast fit-free analysis of fluorescence lifetime imaging via deep learning. Proceedings of the National Academy of Sciences of the United States of America. 116, 24019-24030 (2019).
- Yao, R., Ochoa, M., Intes, X., Yan, P. Deep compressive macroscopic fluorescence lifetime imaging. Proceedings - International Symposium on Biomedical Imaging. 2018, 908-911 (2018).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.