Method Article
Hibridación fluorescente in situ en preparaciones de halo de ADN para revelar cromosomas completos, telómeros y loci de genes
En este artículo
Resumen
La combinación de preparaciones de halo de ADN con hibridación fluorescente in situ permite el análisis de alta resolución de las interacciones genómicas con el nucleoesqueleto. El genoma unido conduce a señales fluorescentes hibridadas ubicadas dentro de los núcleos extraídos residuales, mientras que el genoma no unido está en el halo de ADN que rodea los núcleos residuales.
Resumen
El genoma está asociado con varias estructuras dentro de los núcleos celulares, con el fin de regular su actividad y anclarla en lugares específicos. Estas estructuras se conocen colectivamente como el nucleoesqueleto e incluyen la lámina nuclear, los nucléolos y los cuerpos nucleares. Aunque existen muchas variantes de hibridación fluorescente in situ (FISH) para estudiar el genoma y su organización, a menudo están limitadas por la resolución y proporcionan información insuficiente sobre la asociación del genoma con las estructuras nucleares. El método del halo de ADN utiliza altas concentraciones de sal y detergentes no iónicos para generar bucles de ADN que permanecen anclados a estructuras dentro de los núcleos a través de regiones de unión dentro del genoma. Aquí, se extraen proteínas nucleares solubles, como histonas, lípidos y ADN que no están estrechamente unidos a la matriz nuclear. Esto conduce a la formación de un halo de ADN no unido que rodea un núcleo residual que a su vez contiene ADN estrechamente asociado con estructuras nucleares internas y proteínas resistentes a la extracción. Estas cadenas de ADN extendidas permiten una mayor resolución y pueden facilitar el mapeo físico. En combinación con FISH, este método tiene la ventaja añadida de estudiar las interacciones genómicas con todas las estructuras por las que está anclado el genoma. Esta técnica, denominada HALO-FISH, es muy versátil mediante la cual los halos de ADN se pueden acoplar con sondas de ácidos nucleicos para revelar loci de genes, cromosomas completos, satélite alfa, telómeros e incluso ARN. Esta técnica proporciona una visión de la organización y función nuclear en células normales y en la progresión de la enfermedad, como con el cáncer.
Introducción
La "matriz nuclear" fue descrita por primera vez por Berezney y Coffey en 19741. Después de realizar extracciones con molaridades de alta sal y tratamiento con nucleasas en núcleos hepáticos de rata, identificaron un marco estructural proteico. El procedimiento de halo de ADN se adaptó posteriormente de este método e implica la eliminación de proteínas solubles para que solo persistan la matriz nuclear (NM) y las proteínas y cromosomas asociados a NM. Las regiones de unión al ADN se encuentran en la base de los bucles de ADN y se denominan regiones unidas a la matriz (MAR) o regiones de unión a andamios (SAR), que son resistentes a la extracción con altas concentraciones de sal y detergente iónico litio-3,5-diyodosalicilato (LIS) respectivamente. En los halos de ADN, el ADN asociado con MARs/SARs está unido dentro del núcleo residual, mientras que los bucles de ADN se extienden lejos de estos sitios y forman el halo de ADN. Ahora sabemos que el genoma está anclado a través de dominios asociados a láminas (LADs) a la lámina nuclear y a través de regiones nucleolares asociadas (NADs) y posiblemente a través de otras estructuras nucleares como cuerpos nucleares específicos.
El método de halo de ADN se puede utilizar para el mapeo físico de ADN, genes y regiones cromosómicas, ya que el ADN extendido y la cromatina proporcionan una mayor resolución porque la cromatina se despoja de histonas y el ADN se estira 2,3,4,5,6. Sin embargo, existen algunas limitaciones al usar halos de ADN para esta aplicación. Por ejemplo, el ADN estrechamente asociado con núcleos residuales de halos de ADN puede ser inaccesible para las sondas, lo que lo excluye del análisis y el mapeo físico6. Otras técnicas como la fibra-FISH 2,4,5,7 y el peinado molecular8 también permiten el mapeo físico y tienen la ventaja de ser relativamente rápidos y fáciles de realizar. Ambos se utilizan preferentemente para el mapeo de ADN de genes sobre halos de ADN. Estos métodos extraen fibras de cromatina mediante el uso de extracciones de solventes o sales del núcleo, sin embargo, el peinado molecular tiende a tener mejor reproducibilidad 8,9.
Existe una creciente evidencia de que el nucleoesqueleto tiene un papel en el apoyo a procesos nucleares clave, como los sitios de unión para el ADN, la remodelación de la cromatina, la transcripción del ADN, la reparación del ADN y la replicación del ADN11,12. Como tal, la técnica del halo de ADN se desarrolló para investigar las interacciones entre el nucleoesqueleto y el genoma durante estas actividades celulares y se ha utilizado e informado rutinariamente en la investigación. Esta técnica también ha sido utilizada para investigar las interacciones entre el genoma y el nucleoesqueleto en relación con la progresión de la enfermedad con cambios asociados a la malignidad en la estructura nuclear identificados11.
La técnica del halo de ADN también ha sido utilizada para investigar la relación entre el genoma y el nucleoesqueleto durante el desarrollo y la diferenciación12. Varios estudios han utilizado una variación de la técnica de halo de ADN conocida como halosperma13 o SpermHalo-FISH si se combina con FISH14. La cromatina de los espermatozoides está estrechamente unida a proteínas conocidas como protaminas y esta técnica se desarrolló para mejorar el acceso al ADN de los espermatozoides. La halosperma se ha utilizado para investigar la integridad del ADN de los espermatozoides y determinar si hay daño en el ADN. Los espermatozoides con menos daño en el ADN se correlacionan con un tamaño de halo de ADN más grande, mientras que los espermatozoides con niveles aumentados de ADN fragmentado y dañado tenían halos pequeños o ninguno en absoluto. Por lo tanto, las halospermas se pueden utilizar como un marcador pronóstico potencial de la calidad embrionaria y el embarazo exitoso con FIV13. Este ejemplo enfatiza las posibles aplicaciones clínicas de esta técnica. En nuestro trabajo hemos utilizado HALO-FISH para evaluar los cambios en el comportamiento del genoma y el efecto de tratamientos farmacológicos específicos en la enfermedad de envejecimiento prematuro Síndrome de Progeria de Hutchinson-Gilford (HGPS)15.
Juntos, estos y otros estudios resaltan la amplitud de procesos / aplicaciones que la técnica de halo de ADN se puede utilizar para estudiar y la utilidad de la técnica.
Protocolo
1. Preparación de portaobjetos, esterilización y cultivo celular
- Prepare 500 ml de HCl al 10% (v/v) y vierta en un vaso grande.
- Deje caer el microscopio se desliza individualmente en el ácido e incubar durante 1 h a temperatura ambiente en un agitador ajustado a 2 x g.
PRECAUCIÓN: HCl es corrosivo e irritante. Puede causar quemaduras graves en la piel y daño ocular e irritación de la piel, los ojos y el sistema respiratorio. Asegúrese de que se use la protección personal adecuada, incluidos guantes de nitrilo, protección para los ojos y una bata de laboratorio. - Decantar el ácido del vaso de precipitados y lavar los toboganes diez veces en agua del grifo y luego otras diez veces en agua desionizada.
- Enjuague las diapositivas en metanol dos veces y manténgalas en metanol hasta la esterilización por llamas.
PRECAUCIÓN: El metanol es un líquido altamente inflamable y tóxico si se ingiere, en contacto con la piel o si se inhala. Además, el metanol puede causar daño a los órganos, es corrosivo y un irritante. Adhiérase a los límites de exposición en el lugar de trabajo y asegúrese de que se use la protección personal adecuada, incluidos guantes de goma de butilo, protección para los ojos y una bata de laboratorio. Siempre que sea posible, mango en un armario de humos de ventilación por extracción local (LEV). - Usando pinzas metálicas o pinzas largas, retire un portaobjetos de microscopio del vaso de precipitados que contiene metanol. Encienda sobre un quemador Bunsen para esterilizar y transfiéralo a un recipiente de cultivo celular rectangular que contenga cuatro compartimentos para portaobjetos, ubicado cerca del quemador Bunsen.
PRECAUCIÓN: La llama permite la esterilización inmediata de portaobjetos de microscopio antes de su uso; Sin embargo, este método tiene peligros asociados. Como el metanol es altamente inflamable, es importante que el vaso de precipitados que contiene los portaobjetos se coloque lejos del quemador Bunsen. Se deben usar pinzas largas o pinzas que sujeten firmemente los portaobjetos. El nivel de metanol en el vaso de precipitados solo debe cubrir los portaobjetos, tanto para minimizar la cantidad de metanol utilizado como para que solo los extremos de las pinzas / pinzas estén en contacto con el metanol. Asegúrese siempre de que el metanol se haya evaporado de las pinzas o pinzas después de su uso y que estos se hayan enfriado antes de volver a colocarlos en el vaso de precipitados que contiene los portaobjetos y el metanol. El vaso de precipitados debe estar cubierto por un trozo de papel de aluminio para privar de oxígeno en caso de que el metanol se incendie. Nunca se deslice la llama dentro de una campana de flujo laminar de Clase II donde circula el aire. - Alternativamente, realice los pasos 1.1 a 1.4, pero en lugar de quemar los portaobjetos después de incubar con metanol, coloque los portaobjetos sobre un tejido sin pelusa para que se sequen al aire. Una vez seco, envuélvalo en papel de aluminio y colócalo en un horno esterilizador o autoclave.
- Cultivar células en el medio apropiado con suero durante al menos 48 h a 37 °C enCO2 al 5% hasta alcanzar una confluencia del 60-70%. Este protocolo se realizó en un paso temprano de fibroblastos dérmicos humanos (HDF) y en fibroblastos clásicos del síndrome de progeria de Hutchinson-Gilford (HGPS) (AG06297) y fibroblastos HGPS tipo 2 atípicos (AG08466). Recolecte cada tipo de célula y cuente usando un hemocitómetro para determinar la densidad celular. Semilla 1 x 105 células en un medio de 10 ml por portaobjetos.
NOTA: La densidad celular es importante ya que los bucles de ADN de diferentes núcleos pueden converger si las células se vuelven demasiado confluentes. Las densidades de siembra pueden necesitar ser optimizadas dependiendo del tipo de célula utilizada, ya que las células transformadas pueden proliferar más rápidamente, mientras que los cultivos celulares de paso posterior pueden tardar más tiempo en alcanzar la confluencia deseada. - Si las células necesitan ser detenidas en G0 para quedar inactivas, entonces siembre 1 x 105 células (en un medio de 10 ml) por portaobjetos y deje crecer durante 24 h. Lave las células dos veces con medio libre de suero e incube en un medio estándar que contenga una concentración más baja de suero al 0,5% (suero de ternera recién nacido, NCS; o suero bovino fetal, FBS) durante 7 días.
- Si se requiere el estado proliferativo de las células para el ensayo de Halo de ADN, determine las células que han pasado por la fase S mediante la incorporación de 5-bromo-2'-desoxi-uridina (BrdU) en el ADN durante la replicación.
- Semilla de células como de costumbre y crecen durante 24 h. Retirar el medio de cultivo y sustituirlo por un medio que contenga BrdU y 5-fluoro-2'-desoxiuridina (3 μg/μL). Después de otras 24 h, retire el medio, lave las células una vez con medio (10% NCS) y luego vuelva a alimentar con medio fresco (10% NCS). Incubar durante 24 h adicionales y luego preparar los portaobjetos para el ensayo de halo de ADN.
2. Preparación de la sonda
- Sondas de pintura de cromosomas enteros y brazos
- Realizar sondas cromosómicas a partir de la amplificación de cromosomas clasificados por flujo o microdisecados mediante reacción en cadena de la polimerasa cebada con oligonucleótidos degenerados (DOP-PCR) utilizando el método de Telenius et al.16. Utilice DOP-PCR para marcar sondas cromosómicas con biotina-16-dUTP o digoxigenina-11-dUTP como se muestra en la Tabla 1. Consulte las instrucciones del fabricante para el perfil de amplificación, sin embargo, las condiciones utilizadas para este experimento se muestran en la Tabla 2.
- Preparar el brazo o la sonda cromosómica completa sumando 8 μL de producto de PCR marcado, 7 μL de ADN Cot-1, 3 μL de esperma de arenque, 1/20volumen de acetato de sodio 3 M (pH 5.4) y 2 volúmenes de etanol al 100%. Incubar la solución de la sonda durante un mínimo de 30 minutos a -80 °C.
NOTA: Este método se puede utilizar para crear sondas de un solo cromosoma o múltiples sondas cromosómicas si se utilizan diferentes etiquetas (es decir, biotina-16-UTP y digoxigenina-11-dUTP) para cada cromosoma de interés.
PRECAUCIÓN: El etanol es un líquido y vapor altamente inflamable y puede causar irritación ocular grave. Mantener alejado del calor, superficies calientes y fuentes de ignición. Adhiérase a los límites de exposición en el lugar de trabajo y asegúrese de que se use la protección personal adecuada, incluidos guantes de goma de butilo, protección para los ojos y una bata de laboratorio. Siempre que sea posible, mango en un armario de humos LEV. - Solución de sonda de centrífuga a 13.700 g durante 15 min a 4 °C y luego lavar con etanol al 70%. Repita el procedimiento de centrifugación y deseche el sobrenadante, teniendo cuidado de no alterar o perder el pellet de ADN. Deje que el pellet de ADN se seque.
- Añadir 12 μL de tampón de hibridación (50% formamida, 10% sulfato de dextrano, 10% 20x citrato de sodio salino (SSC; 3 M NaCl, 0,3 M citrato trisódico; pH 7,0), 1% (v/v) monolaurato sorbinal de polioxietileno (Tween-20)) al pellet de ADN. Dejar a 37 °C durante al menos 2 h para que el pellet de ADN se disuelva en el tampón de hibridación.
PRECAUCIÓN: La formamida es cancerígena y teratogénica, por lo que puede causar daños graves al feto. Si una mujer está embarazada o sospecha que está embarazada, debe evitar trabajar con formamida. La formamida debe utilizarse en una campana extractora LEV. Adhiérase a los límites de exposición en el lugar de trabajo y asegúrese de que se use la protección personal adecuada, incluidos guantes de goma de butilo, protección para los ojos y una bata de laboratorio.
- Aislamiento de ADN de cromosomas artificiales bacterianos (BAC)
- Rayar una pequeña porción de la reserva de glicerol del clon BAC en una placa de agar Luria-Bertani (LB) (1% (W/V) NaCl; 1% (p/v) triptona, 0,5% (p/v) extracto de levadura, 1,5% (p/v) Agar Technical, 12,5 μg/ml (p/v) cloranfenicol). Incubar durante la noche a 37 °C.
- Seleccione una sola colonia de la placa e inocule 10 ml de caldo LB (1% (p/v) NaCl, 1% (p/v) bactotriptona, 0,5% (p/v) extracto de levadura, 12,5 μg/ml (p/v) cloranfenicol). Dejar la solución en una incubadora agitadora durante la noche a 37 °C.
PRECAUCIÓN: Se sospecha que el cloranfenicol causa cáncer. Manejar con cuidado y reducir la exposición. - Cultivo por centrifugación a 1.700 x g durante 10 min a temperatura ambiente.
- Desechar el sobrenadante y añadir 300 μL de solución P1 (15 mM Tris (pH 8), 10 mM EDTA, 100 μg/ml RNasa A) al pellet. Vórtice vigorosamente y transfiera células a un tubo de microcentrífuga de 2 ml.
- Añadir 300 μL de solución de P2 (0,2 M NaOH, 1% (p/v) dodecil sulfato de sodio (SDS) gota a gota a las células. Invierta el tubo de microcentrífuga cerrado 5 veces y déjelo a temperatura ambiente durante un máximo de 5 minutos.
PRECAUCIÓN: El hidróxido de sodio es corrosivo y puede causar quemaduras graves en la piel y daño ocular. Puede ser corrosivo para los metales. Manejar con cuidado y reducir la exposición. Adhiérase a los límites de exposición en el lugar de trabajo y asegúrese de que se use la protección personal adecuada, incluidos guantes de goma de nitrilo, protección para los ojos y una bata de laboratorio. Siempre que sea posible, mango en un armario de humos LEV.
PRECAUCIÓN: El dodecil sulfato de sodio es un sólido inflamable, dañino si se ingiere y puede causar irritación cutánea y respiratoria. También puede causar daño ocular grave. Asegúrese de que se use la protección personal adecuada, incluidos guantes de goma de nitrilo, protección para los ojos y una bata de laboratorio. Siempre que sea posible, mango en un armario de humos LEV. - Añadir 300 μL de P3 (acetato de potasio 3 M) lentamente a las células y mezclar suavemente. Coloque el tubo de microcentrífuga sobre hielo durante 10 minutos.
- Centrifugar a 8.100 x g durante 10 min a 4 °C y transferir el sobrenadante a un tubo que contenga 800 μL de isopropanol helado. Invertir el tubo varias veces e incubar a -20 °C durante la noche.
PRECAUCIÓN: El isopropanol es un líquido y vapor altamente inflamable y puede causar irritación ocular grave, somnolencia o mareos. Mantener alejado del calor, superficies calientes y fuentes de ignición. Cumpla con los límites de exposición en el lugar de trabajo y asegúrese de que se use la protección personal adecuada, incluidos guantes de goma de nitrilo, protección para los ojos y una bata de laboratorio. Siempre que sea posible, mango en un armario de humos LEV). - Centrifugadora a 8.100 x g durante 15 min a 4 °C. Retire el sobrenadante y transfiéralo a otro tubo. Agregue 500 μL de etanol helado al 70%. Invertir el tubo varias veces y centrifugar a 8.100 x g durante 5 min a 4 °C.
- Retire el sobrenadante y seque al aire el pellet a temperatura ambiente. Una vez que el pellet esté seco, vuelva a suspender en 40 μL de agua tratada con pirocarbonato de dietilo (tratada con DEPC) y déjelo a 4 °C durante la noche. Una vez completamente resuspendido, retire 5 μL de solución y cargue un gel de agarosa al 1% para verificar la presencia de ADN.
- Preparación de una sonda de un solo gen de BAC mediante traducción de nick
- Utilice kits de etiquetado de traducción de níquel disponibles comercialmente. Como alternativa, utilice el siguiente protocolo. Véase el cuadro 3 para los componentes y volúmenes.
- Agregue los componentes de la Tabla 3 juntos en un tubo de microcentrífuga agregando la ADN polimerasa I al final, mezcle suavemente y centrifugue brevemente durante unos segundos. Incubar la solución a 15 °C durante 2 h.
- Para verificar el tamaño de los fragmentos, cargue 5 μL de la solución en un gel de agarosa al 2%. El rango de tamaño del fragmento de ADN debe estar entre 200-600 pb. Si los tamaños de los fragmentos de ADN son más grandes, continúe incubando la solución durante otros 15 minutos a 15 ° C y ejecute los productos en gel de agarosa al 2%.
- Detenga la reacción de traslación de nick agregando 10 mM EDTA, 0.1% SDS (2.5 μL de 0.5 M EDTA, pH 8.0 en 100 μL y 1 μL de 10% SDS en 100 μL). Calentar la solución a 65 °C durante 5 min.
- Para eliminar nucleótidos no incorporados, aplique la sonda BAC a una columna de espín. Las columnas de giro comerciales se pueden comprar, o se pueden crear usando una jeringa de la siguiente manera:
- Añadir 30 g de Sephadex G-50 a 500 ml de tampón de columna (10 mM Tris-HCl (pH8), 1 mM EDTA, 0,1% SDS). Autoclave la mezcla. Además, haga 500 ml de tampón de columna (sin Sephadex G-50) y autoclave.
- Haga columnas de centrifugado agregando lana de vidrio esterilizada en autoclave al fondo de una jeringa de 1 ml. Llene la jeringa de 1 ml con Sephadex G-50 en tampón de columna. Coloque la jeringa de 1 ml en un tubo de centrífuga de 15 ml que tenga un tubo de microcentrífuga sin tapa en la parte inferior. Centrifugar a 1.600 x g durante 5 min.
- Retire la jeringa y deseche el tubo de microcentrífuga en la parte inferior. Vuelva a añadir un tubo de microcentrífuga nuevo en el tubo de centrífuga de 15 ml. Agregue el tampón de columna (sin Sephadex G-50) a la jeringa de 1 ml y vuelva a insertarlo en el tubo de centrífuga de 15 ml. Centrifugar a 1.600 x g durante 5 min. Repita este paso de nuevo dos veces.
- Retire la jeringa e insértela en un tubo de centrífuga de 15 ml que contenga un nuevo tubo de microcentrífuga limpio. Aplique la sonda a la jeringa y recoja la sonda en el tubo de microcentrífuga.
- Para precipitar la sonda de ADN, agregue 5 μL de ADN de espermatozoide de arenque (10 mg / ml), 10 μL de acetato de sodio y 2,25 volúmenes de etanol al 100% a la solución de ADN. Mezclar suavemente la solución e incubar a -80 °C durante un mínimo de 1 h. Centrifugar a 13.700 x g durante 15 min a 4 °C.
- Desechar el sobrenadante y lavar el pellet con 200 μL de etanol helado al 70 % durante 15 minutos a 4 °C. Retire el sobrenadante y seque al aire. Una vez seco, vuelva a suspender el pellet en 20 μL de agua tratada con DEPC a temperatura ambiente durante varias horas o durante la noche a 4 °C. La sonda ya está lista para ser utilizada o puede almacenarse a -20 °C.
- Para cada portaobjetos, mezcle 5 μL de ADN de la sonda con 5 μL de ADN Cot-1 y séquelo con un concentrador de vacío Speed Vac. Una vez que el pellet se haya secado vuelva a suspender en 12 μL de mezcla de hibridación.
3. Preparación del ADN Halo
- Retire la placa de cultivo cuadrada que contiene los portaobjetos y las células adheridas de la incubadora. Deseche el medio, etiquete las diapositivas con un lápiz y colóquelas en un frasco de Coplin que contenga 50 ml de tampón del citoesqueleto helado (CSK): 100 mM NaCl, 3 mM MgCl2, 0.3 M sacarosa, 10 mM 1, 4-piperazinadiethanesulfónico ácido (PIPES; pH 7.8), 0.5% (v / v) Triton X-100 hecho en agua desionizada. Incubar durante 15 min sobre hielo o a 4 °C.
PRECAUCIÓN: Triton X-100 puede causar irritación de la piel y daño ocular grave. Maneje con el equipo de protección personal adecuado, incluidos guantes de nitrilo, gafas y bata de laboratorio. - Deseche el tampón CSK y enjuague rápidamente los portaobjetos en 50 ml de 1x tampón de halo de ADN (DHB; 140 mM NaCl, 27 mM KCl, 110 mM NaHPO 4, 15 mM KH2PO 4; pH7.4) tres veces, es decir, sumerja el portaobjetos en el frasco de Coplin que contiene DHB y retírelo.
- Transfiera los portaobjetos a un frasco de Coplin que contenga 50 ml de tampón de extracción: 2 M NaCl, 10 mM PIPES (pH 6.8), ácido etilendiaminotetraacético (EDTA) de 10 mM, 0.1% (p/v) de digitonina, 0.05 mM (v/v) de espermina, 0.125 mM (v/v) de espermidina. Incubar durante 4 min a temperatura ambiente.
PRECAUCIÓN: La digitonina es tóxica si se ingiere o entra en contacto con la piel y es mortal si se inhala. Asegúrese de que la digitonina se manipule en un armario de humos LEV y use una bata de laboratorio, guantes de nitrilo (doble canal), gafas de seguridad y máscara. Tanto la espermina como la espermidina pueden causar quemaduras graves en la piel y daño ocular, mientras que el EDTA causa irritación ocular grave, así que maneje cada producto químico con cuidado.
NOTA: Prepare la digitonina por separado disolviendo el polvo en agua a una temperatura de 60-70 °C. Agregue digitonina disuelta al tampón de extracción una vez enfriado. Agregue espermina, espermidina y digitonina al tampón de extracción para preservar la actividad biológica. - Incubar portaobjetos consecutivamente en 50 mL de 10x DHB (1.4 M NaCl, 270 mM KCl, 1.1 M NaHPO 4, 150 mM KH2PO4; PH7.4), 5x, 2x y 1x DHB durante 1 min cada uno.
- Sumerge las diapositivas (directas y directas) a través de una serie secuencial de etanol de 50 ml de etanol al 10%, 30%, 70% y 95% (v/v).
- Secar al aire los portaobjetos y almacenar a -80 °C hasta que se realice la hibridación bidimensional fluorescente in situ (2D FISH).
4. Hibridación bidimensional fluorescente in situ
- Hacer 20x SSC: 3 M NaCl, 0.3 M citrato trisódico, pH 7.0. Este tampón puede ser esterilizado en autoclave, almacenado a temperatura ambiente y diluido según sea necesario.
- Hacer formamida al 70% (v/v), 2x SSC pH 7.0 y calentar a 70 °C en un baño maría.
- Incubar portaobjetos, durante 5 min cada uno, a través de una serie secuencial de etanol de 50 ml de etanol de 70, 90 y 100%.
- Secar al aire en una placa de calentamiento y hornear en un horno a 70 °C durante 5 min.
- Desnaturalizar los portaobjetos introduciendo en la solución de formamida al 70%, 2x SSC durante 2 min a 70 °C.
NOTA: La temperatura y el tiempo son críticos para el paso 4.5. Si la temperatura es el ADN no se desnaturalizará y las sondas no se hibridarán, y no se obtendrá ninguna señal del halo de ADN FISH. - Coloque el portaobjetos desnaturalizado en 50 ml de etanol helado al 70% durante 5 minutos y tómelo a través de una serie de etanol de 90%, 95% y 100% a temperatura ambiente durante 5 minutos cada uno.
- Secar al aire en una placa de calentamiento
- Maneje directamente las sondas de cromosomas humanos totales etiquetadas de acuerdo con las instrucciones del fabricante. Para estos experimentos utilizamos pinturas de cromosomas enteros humanos 1, 13, 15, 17 y 18. Además, en este experimento, se utilizaron sondas de genes CCND1 y CTNNA1 .
NOTA: Tanto las sondas cromosómicas completas como las sondas específicas del gen BAC se marcaron con biotina-11-dUTP y se detectaron mediante estreptavidina conjugada con cianina 3 (Cy3). Para las sondas de pintura cromosómica hechas por (DOP-PCR) y BAC DNA marcadas por traducción nick, estas se denominarán sondas de ADN a partir de este punto en el protocolo y se tratarán de la siguiente manera. - Desnaturalizar la sonda de ADN (pintura cromosómica completa o sonda específica de genes) a 75 °C durante 10 minutos en un bloque caliente o baño de agua.
- Calentar las sondas de ADN a 37 °C durante 30 minutos en un bloque caliente o baño de agua antes de pipetear 10 μL en el portaobjetos apropiado.
NOTA: Este paso es importante para bloquear secuencias cromosómicas repetitivas. Si no se realizan se pueden producir señales no específicas en el ADN Halo FISH. - Sonda de recubrimiento con un cubreobjetos de 21 mm x 21 mm y sello con cemento de goma.
- Incubar portaobjetos durante un mínimo de 18 h a 37 °C en una cámara de hibridación humidificada.
NOTA: Las cámaras de hibridación humidificadas se pueden hacer a partir de cajas sándwich que contienen varias capas de tejido humedecido y una plataforma elevada construida a partir de pipetas de plástico cortadas de 10 ml para descansar los portaobjetos. Esto está cubierto con papel de aluminio para minimizar la exposición a la luz. - Retire el cemento de goma con cuidado con pinzas.
- Incubar portaobjetos en 50 ml de formamida al 50% (v/v), 2x SSC, solución de pH 7.0 que ha sido precalentada a 45 °C durante tres incubaciones de 5 minutos.
NOTA: Deje que el cubreobjetos se desprenda del portaobjetos en la primera incubación en formamida al 50% (v/v), 2x SSC, solución de pH 7.0. Esto evita daños a la preparación de ADN Halo que podrían ser causados por "arrastrar" el cubreobjetos. Los portaobjetos se pueden agitar en el amortiguador mediante el agarre con pinzas para ayudar a separar el cubreobjetos. - A continuación, coloque los portaobjetos en 50 ml de solución 0.1x SSC, pH 7.0 que se haya precalentado a 60 ° C pero se coloque en un baño de agua a 45 ° C. Incubar durante 5 min y reemplazar el tampón dos veces más con incubaciones de 5 min.
- Coloque los portaobjetos en un frasco de Coplin que contenga 50 ml de solución 4x SSC, pH 7.0 a temperatura ambiente e incube durante 15 minutos con tres cambios de tampón.
- Aplique 100 μL de solución de BSA al 4%, 4x SSC a cada portaobjetos y superponga con un trozo de película de parafina. Incubar a temperatura ambiente durante 10 min. Esto evita la unión no específica del anticuerpo.
- Para detectar la sonda marcada (biotina-16-dUTP), incubar con 100 μL de una estreptavidina-Cy3 1:200 (hecha en BSA al 1%, 4x SSC) durante 1 h a temperatura ambiente.
NOTA: Siga las instrucciones del fabricante con diluciones de anticuerpos y pruebe la dilución antes del experimento para asegurarse de que se produce una buena señal - Coloque los portaobjetos en un frasco de Coplin que contenga 50 ml de 4x SSC (0,5% Tween-20) solución de pH 7.0 a temperatura ambiente e incubar durante 15 minutos con tres cambios de tampón. Los portaobjetos se pueden montar en esta etapa como se muestra en el paso 4.21 si no se requiere inmunofluorescencia.
- Si se requiere el estado proliferativo de las células convertidas en halos de ADN, tiñe con anticuerpos anti-pKi67 después de los pasos de FISH, antes del montaje o tinción para BrdU incorporado.
- Lave los portaobjetos 3 veces durante 5 minutos cada uno en 50 ml de 1x solución salina tamponada con fosfato (PBS), seguido de bloqueo con NCS al 4% en PBS durante 1 h a temperatura ambiente.
- Aplicar 200 μL del anticuerpo primario necesario (pKi67 antihumano de conejo; anti-BrdU de ratón) al portaobjetos, superponer con una tira de película de parafina e incubar a temperatura ambiente durante 1 h.
- Lave los portaobjetos 3 veces durante 5 min en 1x PBS e incube a temperatura ambiente durante 1 h en 200 μL de anticuerpo secundario conjugado con fluorocromo (pKi67: anticonejo porcino TRITC; BrdU: burro anti-ratón Cy3). Realice 3 lavados más de 5 minutos con PBS. Todas las diluciones deben realizarse utilizando NCS al 1% (v/v) en PBS en el rango de diluciones sugeridas por el fabricante.
- Montar guías en 20 μL de montante que contiene DAPI y superposición con un cubreobjetos de 22 mm x 50 mm.
5. Telómero PNA FISH
- Para detectar telómeros, use el kit PINA FISH DE TELÓMEROS - FITC; Realice el procedimiento con las instrucciones del fabricante. El procedimiento debe ejecutarse a temperatura ambiente, a menos que se indique lo contrario.
- Sumerja los portaobjetos en solución salina tamponada con tris (TBS, pH 7.5) durante 2 min y luego colóquelos en formaldehído al 3.7% (en TBS; v / v) durante exactamente 2 min.
PRECAUCIÓN: La solución de TBS contiene trometamol al 10-30% y clorhidrato de propano-1,3-diol al 10-30% de 2-amino-2-(hidroximetil)propano-1,3-diol. Esto puede causar irritación grave de los ojos y la piel, así que use guantes protectores y gafas / protección facial. Usar en un área bien ventilada. - Lave los toboganes en un frasco de Coplin dos veces con TBS durante 5 minutos cada una.
- Sumerja las diapositivas en una solución de pretratamiento durante 10 minutos y luego lave dos veces con TBS durante 5 minutos por lavado.
- A continuación, tome las diapositivas a través de una serie de etanol helado que comprende 50 ml de etanol al 70%, 85% y 95% (v / v) durante 2 minutos por concentración. Después deje que los toboganes se sequen al aire.
- Aplique 10 μL de Telomere PNA Probe/FITC (o Cy3) dependiendo de la elección de la coloración de la etiqueta fluorescente, a cada capa de diapositiva y cubierta con un cubreobjetos. Incubar en un horno precalentado a 80 °C durante 5 min y luego colocar en la oscuridad durante aproximadamente 1 h.
PRECAUCIÓN: Telomere PNA Probe / FITC contiene 6-100% de formamida, que causa irritación ocular grave y es teratogénica, por lo que puede causar daños graves a un feto. Si una mujer está embarazada o sospecha que está embarazada, debe evitar trabajar con formamida. La formamida debe usarse en una campana extractora LEV y se debe usar protección adecuada para los ojos o la cara. - Para retirar los cubreobjetos, sumerja los portaobjetos en 'Solución de enjuague' durante 1 minuto y luego colóquelos en la 'Solución de lavado' durante 5 minutos a 65 °C.
PRECAUCIÓN: La solución de lavado contiene 1-5% de polioxietileno octilfenil éter y 1-5% de cloruro de sodio. Esto es corrosivo y puede causar daños oculares graves. Asegúrese de usar gafas o protección facial al manipular la solución de lavado. - Incubar los portaobjetos a través de una serie de etanol helado de 50 ml (70%, 85% y 95% (v/v)) durante 2 minutos por concentración y luego secar al aire. Una vez en seco, diapositiva de montaje con un montante que contenga DAPI y se superponga con un cubreobjetos.
6. Captura y análisis de imágenes
- Para visualizar los halos de ADN y los territorios cromosómicos, use un microscopio de epifluorescencia (por ejemplo, microscopio Leica DM4000) que capture imágenes utilizando un objetivo de aceite HC PL FLUOTAR 100X / 1.30 y una cámara DFC365FX.
- Capture imágenes en escala de grises y defina el color de cada canal capturado para permitir la pseudocoloración de las imágenes. En este experimento se utilizó un software comercial (por ejemplo, el software LAS AF versión 4.5.0). Los canales de color individuales se exportaron como TIFF.
- Analice imágenes utilizando el programa de procesamiento de imágenes Java Fiji ImageJ. Cargue la imagen presionando Archivo y Abrir.
- Cargue canales de imagen separados o divida una imagen compuesta en canales de escala de grises separados haciendo clic en Imagen | Color | Canales divididos. Seleccione un canal de imagen y haga clic en Imagen | Ajuste y luego seleccione Brillo y contraste. Modifique en consecuencia y repita con otros canales.
- Cree una máscara del núcleo residual seleccionando el canal teñido con DAPI que representa el núcleo. Haga clic en Imagen | Ajuste y, a continuación, seleccione Umbral. Aparecerá un cuadro de diálogo donde se puede alterar el umbral, marque la casilla Fondo oscuro . Modifique hasta que el núcleo residual esté despejado y pulse Aplicar y cierre el cuadro de diálogo.
NOTA: Esto crea una máscara binaria basada en la intensidad de píxeles, con píxeles blancos que muestran regiones de interés y píxeles negros que muestran fondo. Repita el mismo procedimiento en el canal de sonda. - Utilice la selección a mano alzada para delinear la periferia del núcleo residual y, a continuación, haga clic en Editar y Borrar exterior. Superponga el canal de la sonda en el núcleo residual. Esto se puede hacer presionando Imagen| Color | Combinar canales.
- Para establecer la escala de medición en ImageJ, dibuje una línea en la barra de escala o entre los puntos de dos distancias conocidas. Vaya a Analizar y presione Establecer escala. En el cuadro de diálogo, agregue la longitud de la distancia y haga clic en Aceptar. Para medir distancias, dibuje una línea entre los puntos que se están midiendo y haga clic en Analizar | Medir. Esto transferirá los valores de distancia a una ventana de datos.
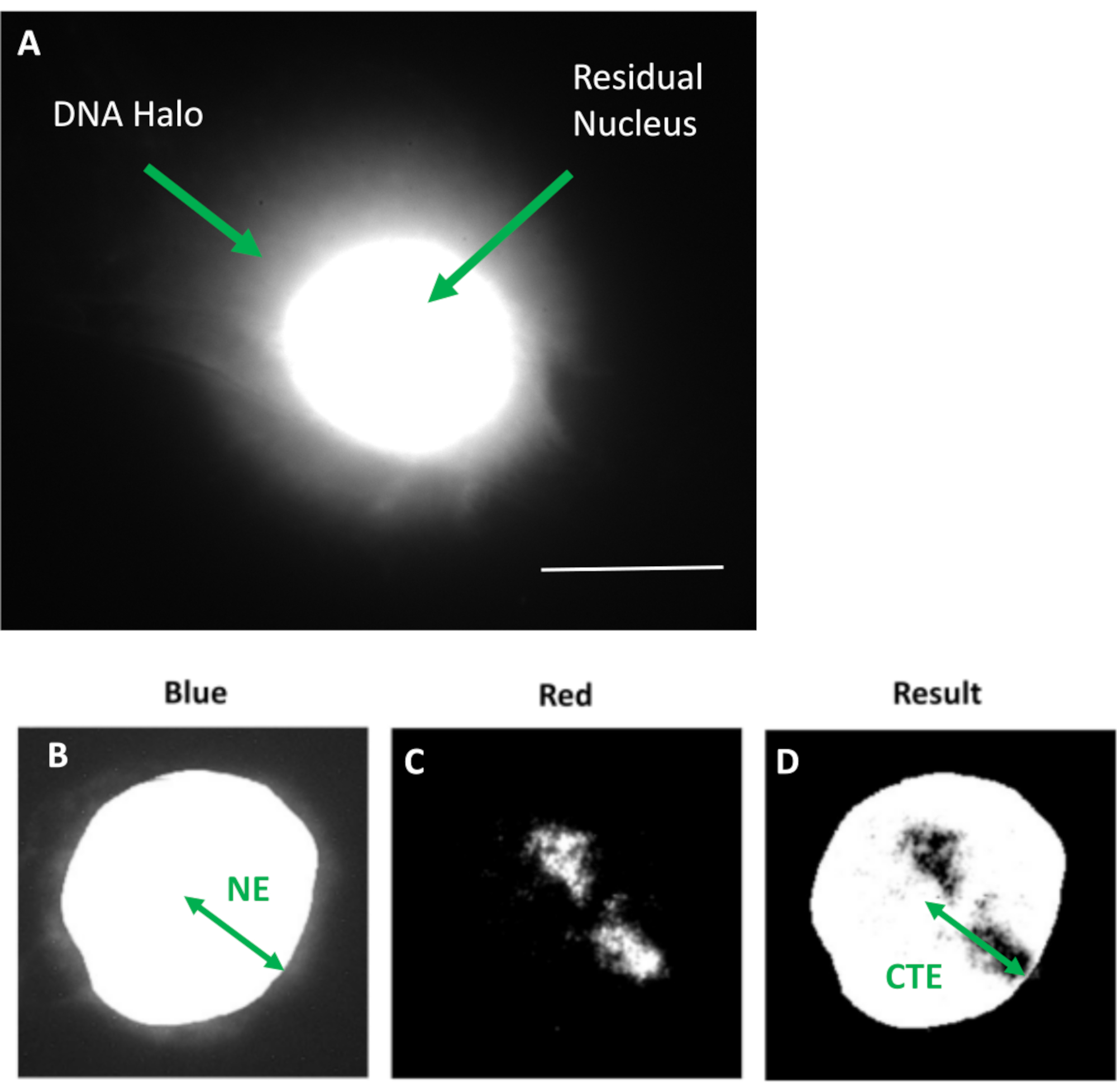
- Mida la intensidad DAPI más brillante, ya que coincide con el centro del núcleo. A partir de esto mide la distancia desde el centro nuclear hasta el borde del territorio cromosómico más lejano (CTE). Mida la distancia del centro nuclear al borde nuclear (NE).
- Asegúrese de que los resultados se representen como una relación CTE/NE. Aquí la distancia desde el centro nuclear a cada borde del territorio cromosómico más lejano (CTE) se divide por la distancia desde el centro nuclear a cada borde nuclear respectivo (NE). Esto debe realizarse en un mínimo de 50 núcleos. Esto se puede representar como un gráfico de barras o caja.
- Para el análisis de los telómeros, analice un mínimo de 30 núcleos por conjunto de datos. Las imágenes se pueden analizar utilizando Fiji ImageJ o manualmente para contar el número de telómeros dentro del núcleo residual y dentro del halo de ADN. BrdU o pKi67 permitieron la diferenciación de núcleos proliferantes (BrdU/piK67+) y senescentes/quiescentes (BrdU/pKi67-). Los datos pueden representarse en gráficos de barras con barras de error correspondientes al error estándar de la media (SEM).
- Utilice la prueba t de Student (no pareada) para comparar estadísticamente los resultados con p> 0,05 considerados significativos.
Resultados
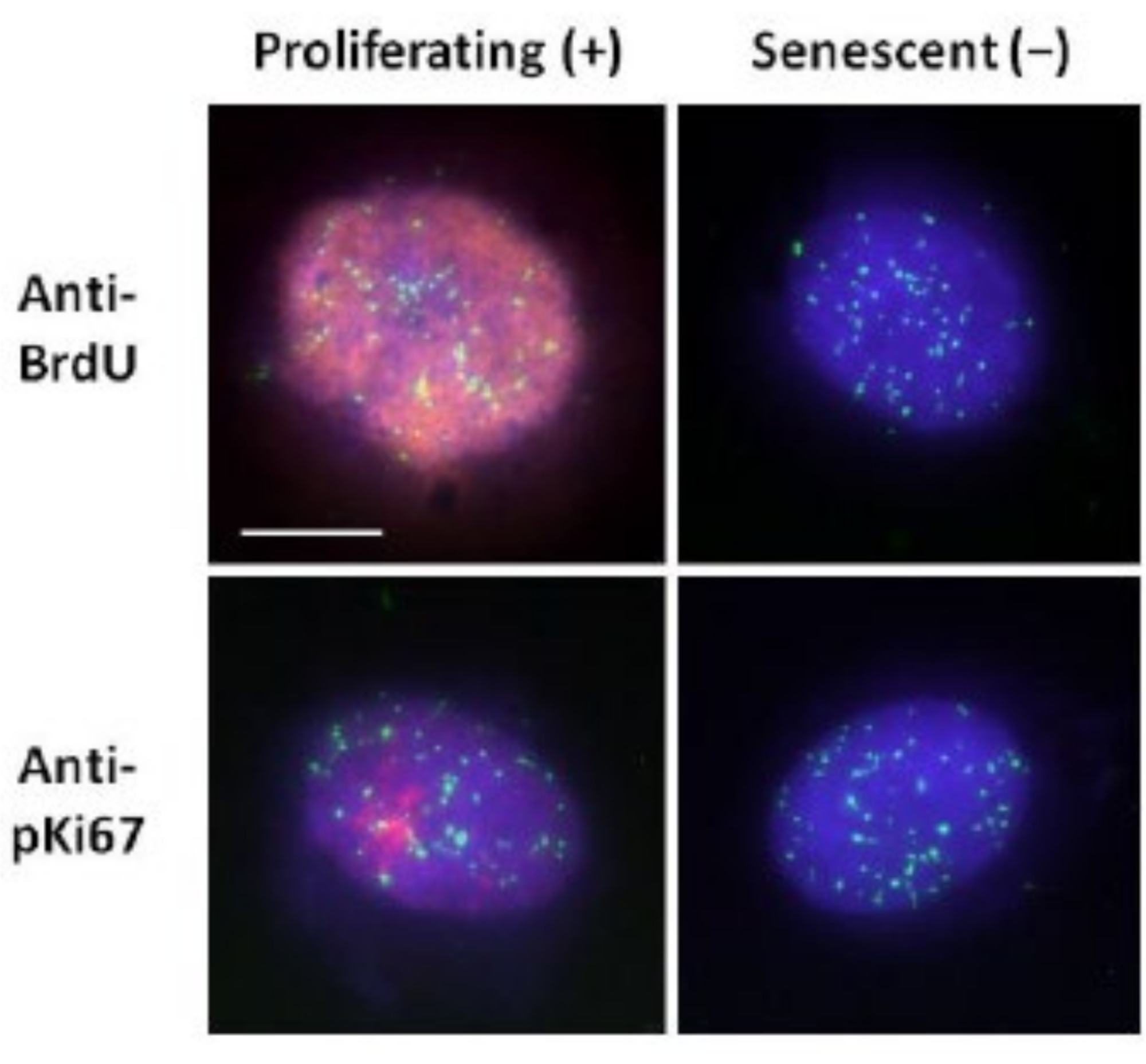
Este método de preparación del halo de ADN nos ha ayudado en nuestros esfuerzos por determinar las diferencias en el comportamiento del genoma dentro de las células jóvenes y viejas, pero también en las células derivadas de enfermedades de envejecimiento prematuro con proteínas nucleosqueléticas aberrantes15. La Figura 1 muestra ejemplos de halos de ADN donde es posible ver el borde de un núcleo residual, el ADN que queda dentro del núcleo residual y el ADN no unido que se ha enrollado en el área circundante creando un halo de ADN. También representa el análisis que muestra cómo se obtiene el núcleo residual y las mediciones NE y CTE. Es posible diferenciar entre células proliferantes y no proliferantes incorporando un nucleótido marcado como BrdU cuando las células están en fase S o empleando el marcador de proliferación diagnóstica anti-pKi67, que revela nucleolos y regiones de heterocromatina en células G117,18. Se supone que las células primarias cultivadas en suero alto sin lograr confluencia, que son negativas para los marcadores de proliferación, son senescentes. Las células primarias cultivadas en suero bajo o que se han vuelto confluentes, es decir, inhibidas por contacto que son negativas para los marcadores de proliferación, se consideran inactivas y podrían volver a entrar en el ciclo celular proliferativo dados los nutrientes y la situación correctos. Ser capaces de diferenciar entre células Ki67 positivas y negativas nos ha permitido determinar las diferencias entre los fibroblastos dérmicos humanos proliferantes, quiescentes y senescentes. La Figura 2 muestra halos de ADN de fibroblastos dérmicos humanos proliferantes creados a partir de células donde BrdU se incorporó a ellas durante la replicación del ADN, un mecanismo que no ocurre en células no proliferantes, y posteriormente teñido con anticuerpos anti-BrdU. La tinción con el marcador proliferativo anticuerpo anti-pKi67 también es visible en la Figura 2. Este es un antígeno robusto y sobrevive al protocolo FISH y, por lo tanto, puede teñirse para post-FISH y pre-montaje. Por lo tanto, las células proliferantes son positivas (rojas) para BrdU y anti-pKi67 (rojas) en la columna izquierda y las células no proliferantes, de hecho, las células senescentes en la Figura 2 se muestran en la columna de la derecha. Las señales verdes son telómeros individuales revelados con un kit PNA FISH/FITC de telómeros. La combinación de inmunofluorescencia con halos de ADN permite el análisis durante diferentes estados celulares, como se muestra en la Figura 2 cuando se investigan células proliferantes, quiescentes y senescentes. Dependiendo del anticuerpo elegido, se pueden examinar otras condiciones, como la diferenciación, el daño del ADN por irradiación, etc.
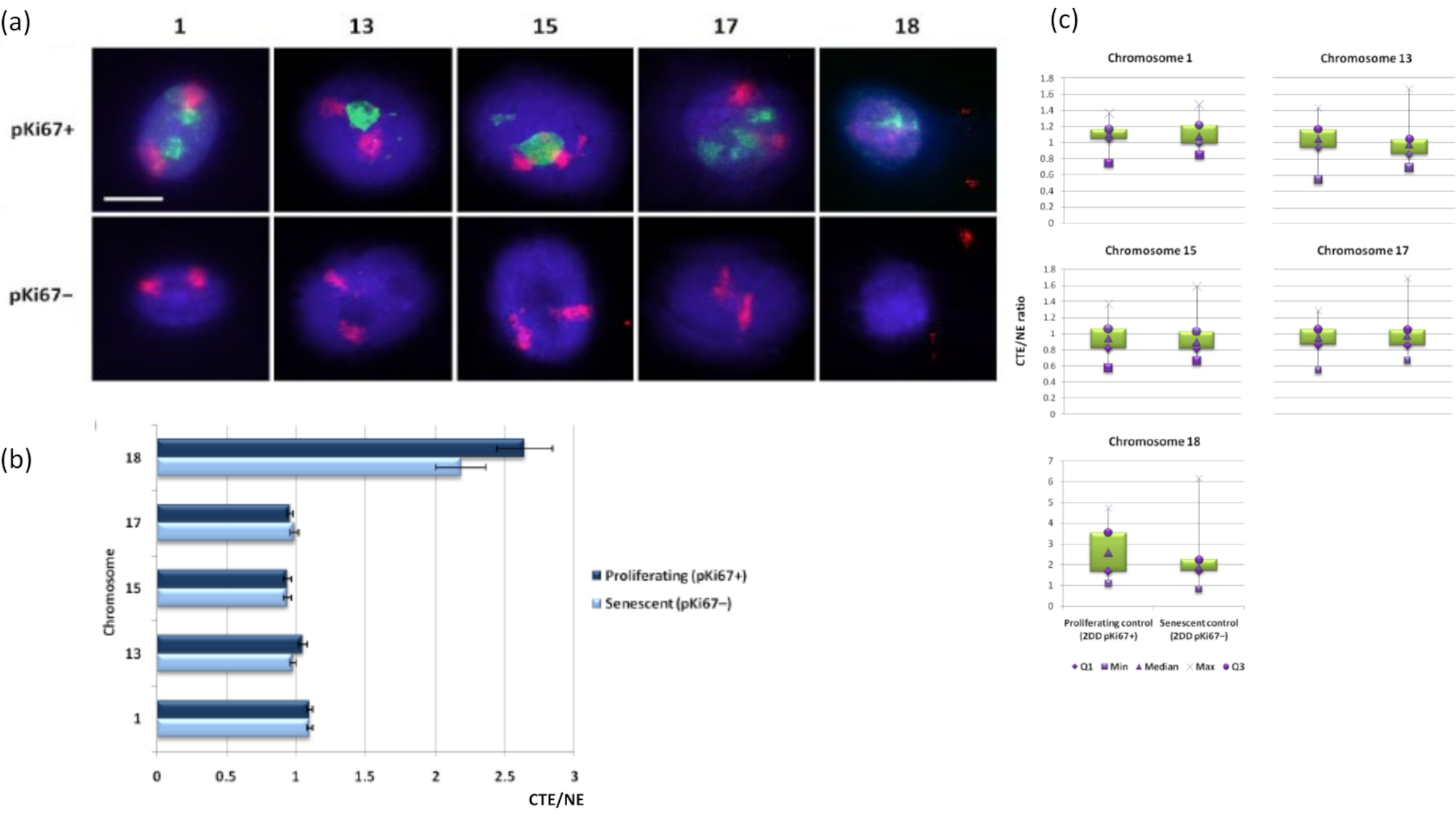
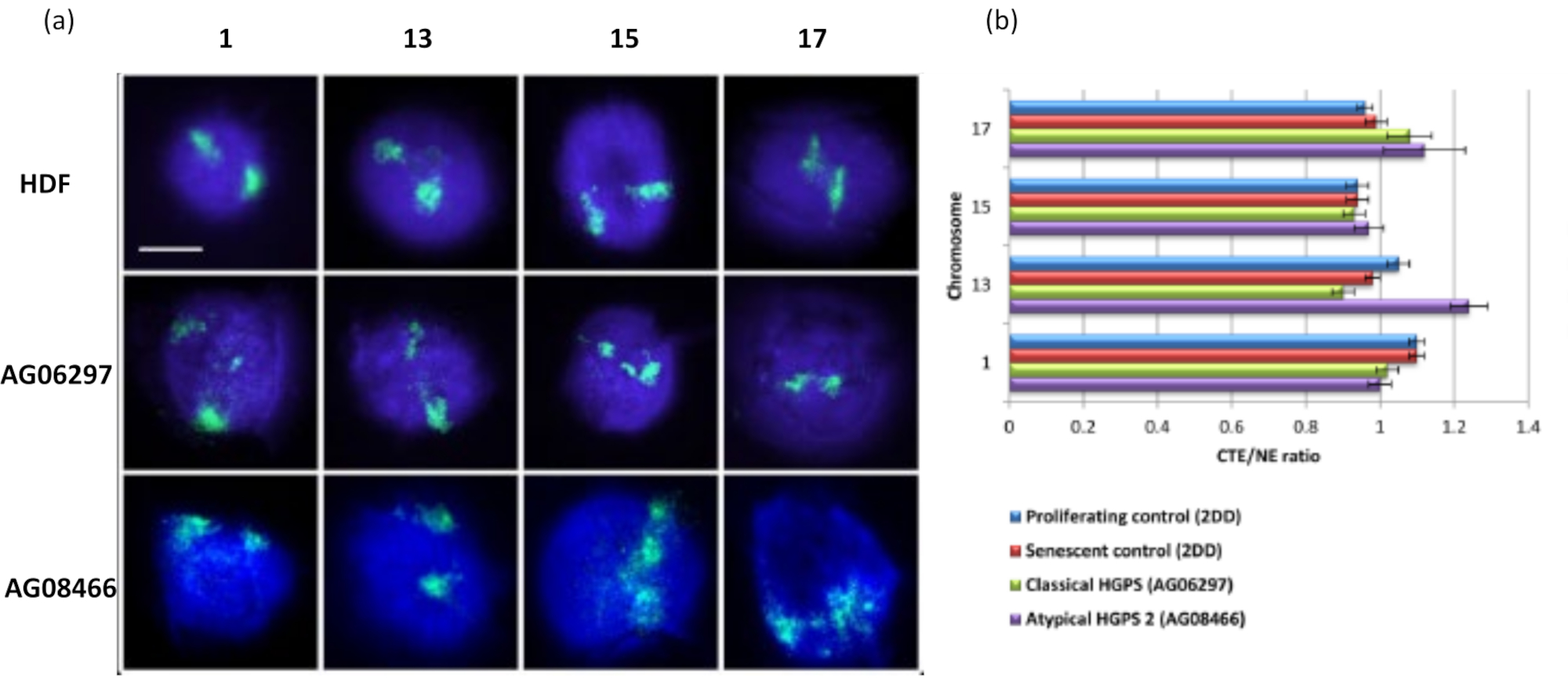
Los territorios cromosómicos también se pueden visualizar dentro de los halos de ADN utilizando FISH. Debido a que la preparación permite la bobina de ADN fuera de los núcleos, la forma del territorio cromosómico puede ser alterada, con cantidades más pequeñas o mayores del cromosoma encontradas en el halo de ADN, dependiendo del anclaje del genoma dentro del núcleo residual y sus estructuras. La Figura 3 revela un panel de halos de ADN mediante los cuales se han revelado cromosomas individuales con sondas específicas de pintura cromosómica de brazo entero (rojo) para los cromosomas 1, 13, 17 y 18. Anti-pKi67 (verde) se ha utilizado para marcar células en proliferación y su ausencia dentro del mismo cultivo, en el mismo portaobjetos, denotando células senescentes. Es muy obvio a partir de las imágenes y los datos presentados como CTE / NE que el pequeño cromosoma 18 pobre en genes es un cromosoma que tiene pocas uniones y se enrolla más lejos en el halo de ADN lejos de los núcleos residuales y está significativamente más lejos del centro de los núcleos residuales que los otros cromosomas. Sin embargo, esto también es cierto para el cromosoma 1. Utilizando el marcador proliferativo anti-pKi67 también ha sido posible comparar la proliferación con células senescentes, dentro del mismo cultivo, y en el mismo portaobjetos, y este análisis ha revelado que los cromosomas dentro de estos dos estados celulares muy diferentes no son significativamente diferentes entre sí, con respecto a la unión con las estructuras nucleares residuales.
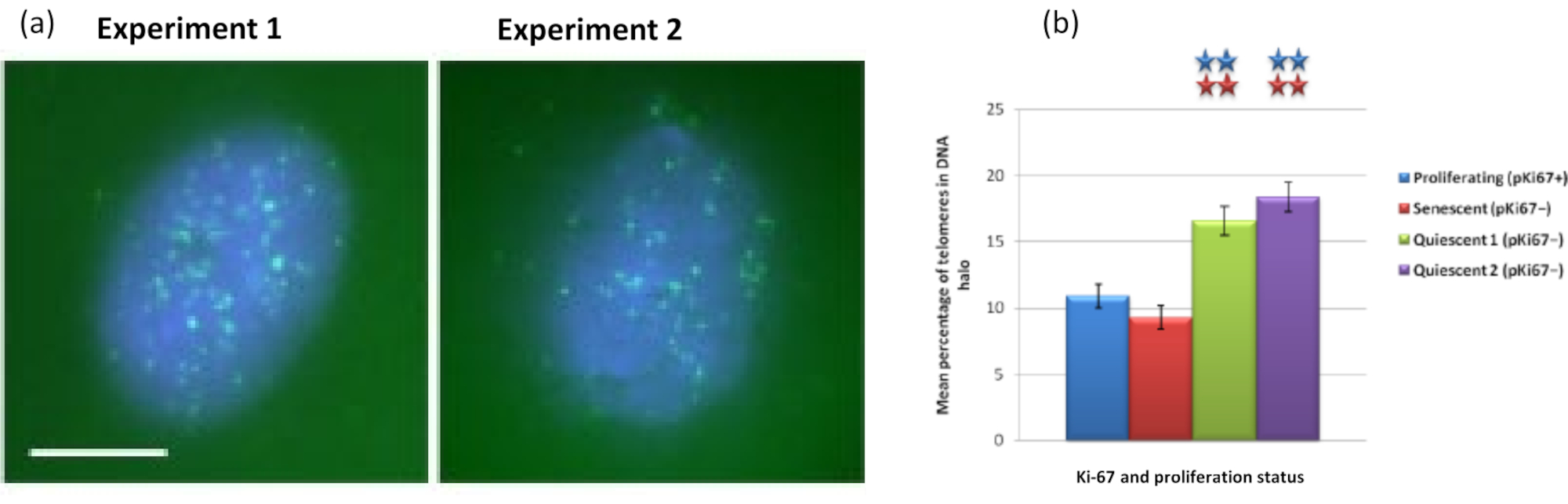
Curiosamente, los genes también muestran diferencias estadísticamente significativas entre las células proliferantes y senescentes con respecto a permanecer dentro de un núcleo residual o estar ubicadas en el halo de ADN. La Figura 4 demuestra esto con loci de genes delineados por sondas BAC marcadas en rojo y anti-Ki67 en verde. No hay diferencias significativas entre las ubicaciones de los genes en las células proliferantes frente a las senescentes, después de una preparación de Halo de ADN. Sin embargo, hay significativamente más loci de catenina alfa 1 CTNNA1 dentro del halo de ADN que los loci de ciclina D1 CNDD1 , donde hay muy pocos. La Figura 5 muestra preparaciones de halo de ADN con telómeros en verde. El fondo se deja deliberadamente alto para permitir que las señales de los telómeros se visualicen dentro del halo de ADN. En este conjunto de células quiescentes de datos, es decir, se han incluido células que han estado hambrientas de suero durante 7 días y, curiosamente, hay significativamente más telómeros no unidos y ubicados dentro de los halos de ADN en las células quiescentes que para las células proliferantes y senescentes. En la Figura 5a se puede observar la proporción de telómeros en el halo de ADN, particularmente para la imagen 'Experimento 2'. Esto se corresponde con la Figura 5b, donde el porcentaje medio de telómeros en el halo de ADN es de aproximadamente el 17% en las células quiescentes. Existe cierta evidencia de que no todos los telómeros en las células senescentes pueden verse como algunos de ellos tal vez muy cortos.
Este método de halo de ADN ha sido exitoso para nosotros para investigar alteraciones de la interacción del genoma dentro de los núcleos en células enfermas15. La Figura 6 muestra diferencias en la unión cromosómica en fibroblastos de control primario y en células enfermas con síndrome de progeria de Hutchinson-Gilford típico (mutación lamina A) y atípico, expresando una isoforma SUN1 diferente y sin mutación en laminaA 19. Los cromosomas 1 y 13 muestran diferencias estadísticamente significativas en su unión dentro de los núcleos residuales en comparación con los halos de ADN de control. La figura 6b correlaciona la posición de todo el territorio cromosómico con el núcleo residual y el halo de ADN. Los valores de 1 o menos indican que el cromosoma se encuentra dentro del núcleo residual y los valores superiores a 1 muestran cromosomas o porciones de cromosomas dentro del halo de ADN.
En general, esto resalta la utilidad de HALO-FISH en la investigación de las interacciones genómicas de cromosomas completos, genes específicos y telómeros bajo una variedad de condiciones que afectan el ciclo celular (proliferación, quiescencia y senescencia) o dentro de las células de la enfermedad, por ejemplo, progeria y líneas celulares cancerosas. De hecho, las diferencias en las interacciones entre estos estados implican que el nucleoesqueleto tiene un papel importante en la regulación de procesos clave dentro del núcleo.

Figura 1: Núcleo extraído de HDF que muestra el núcleo residual y el halo de ADN y una descripción general del método de análisis. (a) Un núcleo HDF preparado mediante ensayo de halo de ADN y teñido con DAPI. El núcleo residual brillantemente teñido muestra ADN anclado al nucleoesqueleto y este está rodeado por el ADN no unido que forma un halo de ADN. Ampliación = x 100; barra de escala 10 μm. (b) El canal azul captura el núcleo teñido de DAPI y el ADN circundante. El núcleo residual se selecciona y se elimina mediante ImageJ. La flecha representa la distancia desde el centro nuclear hasta el borde nuclear residual (NE). (c) El canal rojo muestra la señal de la sonda. d) La imagen denominada «Resultado» es el resultado de la superposición del canal rojo sobre la imagen del canal azul; esto permite la distancia desde el centro nuclear hasta el borde del territorio cromosómico más lejano (CTE). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Preparación del halo de ADN con telómeros PNA FISH en HDFs proliferantes y senescentes. Telómero PNA FISH en HDFs sometidos a ensayo de halo de ADN. Las señales de los telómeros se visualizan en verde (FITC), el ADN residual y de halo se teñó con DAPI (azul) y los núcleos proliferantes se detectaron utilizando anticuerpos anti-BrdU o anti-pKi67 a través de inmunofluorescencia indirecta en rojo (TRITC). Ampliación = x 100; Barra de escala 10 μm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Interacciones nucleoesqueleto-cromosoma y análisis mediante ensayo de halo de ADN. (a) Se realizó 2D-FISH con sondas específicas para los cromosomas 1, 13, 15, 17 y 18 en HDF sometidos a preparación de halo de ADN. Se pintaron cromosomas enteros en rojo (Cy3) y los núcleos se sondearon con pKi67 para determinar si estaban proliferando o eran senescentes. Las células en proliferación (pKi67+) se delinearon en verde (FITC), mientras que las células senescentes permanecieron sin teñir (pKi67-), es decir, no se detectó ninguna señal verde. Ampliación = x 100; barra de escala 10 μm. (b) Anclaje cromosómico por el nucleoesqueleto en HDF proliferantes y senescentes que habían sido sometidos a HALO-FISH. Las mediciones muestran la relación entre el borde del territorio cromosómico más lejano (CTE) y el borde nuclear respectivo (NE) para los cromosomas 1, 13, 15, 17 y 18 en células en proliferación (pKi67+) y senescentes (pKi67-). Las barras de error representan ± SEM. (c) Representación modificada del diagrama de caja del borde del territorio cromosómico (CTE) al borde nuclear respectivo (NE) de cromosomas específicos en núcleos pKi67+ y pKi67-. Q1 = cuartil inferior; Min = valor más bajo registrado; Med = mediana; Max = valor máximo registrado; Q3 = cuartil superior. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Interacciones específicas de genes en HDFs usando HALO-FISH. (a) Los núcleos extraídos del halo de ADN se sondearon con sondas específicas de genes (CCND1 y CTNNA1) para investigar su anclaje al NM en células proliferantes y senescentes. Las señales genéticas se muestran en rojo (Cy3) y anti-pKi67 representa células en proliferación y la señal se visualiza en verde (FITC). Para la imagen CCND1 en proliferación, el núcleo residual está encerrado dentro del círculo blanco, y el espacio entre el círculo blanco y verde representa el halo de ADN. Ampliación = x 100; barra de escala 10 μm. (b) Las señales específicas de genes para CCND1 y CTNNA1 se comparan entre el núcleo residual y el halo de ADN, y también, entre las células en proliferación y senescentes. Las barras de error representan ± SEM. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Ensayo de halo de ADN en HDF quiescentes sondeados con telómeros PNA-FISH. (a) La quiescencia de las HDF fue inducida por cultivo en medio sérico bajo durante 7 días. Se realizó el ensayo de halo de ADN, y PNA-FISH permitió la visualización de los telómeros por señal FITC (verde) y el núcleo residual y el halo de ADN circundante se contrastaron con DAPI (azul). Las células también se tiñeron con anticuerpos anti-pKi67 para garantizar que los núcleos no proliferaran. Esto se repitió en dos ocasiones distintas. Ampliación = x 100; barra de escala 10 μm. (b) Comparación del porcentaje medio de telómeros localizados dentro del halo de ADN en células HDF proliferantes, senescentes y quiescentes. Las barras de error representan ± SEM. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Examen del anclaje cromosómico completo al nucleoesqueleto en células HGPS utilizando HALO-FISH26. (a) Los núcleos de control HDF (2DD), HGPS clásico (AG06297) y HGPS tipo 2 atípico (AG08466) se sometieron a la preparación del halo de ADN y luego a 2D-FISH utilizando pinturas cromosómicas completas para los cromosomas 1, 13, 15 y 17. Los cromosomas enteros se representan en verde (FITC) y el ADN se teñó con DAPI (azul). Ampliación = x 100; barra de escala 10 μm. (b) El posicionamiento de los cromosomas dentro de los núcleos extraídos se determinó midiendo la relación entre el borde del territorio cromosómico medio (CTE) y el borde nuclear (NE). Una relación por encima de 1 demuestra que el CTE más lejano se encuentra fuera del NE correspondiente dentro del halo de ADN, mientras que una relación por debajo de 1 significa que el CTE más lejano se encuentra dentro del NE dentro del núcleo residual. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Componentes | Volumen (μL) |
| 5XDOP-PCRbuffer | 10 |
| dNTPmix(sinTTP)(2mM) | 5 |
| dTTP (2mM) | 2 |
| Biotina-16-dUTPorDigoxigenina-11-dUTP | 10 |
| DOPprimer (20μM) | 5 |
| TaqDNAPolimerasa (1U/μL) | 1 |
| PCRgradoagua | 12 |
| Plantilla | 5 |
Tabla 1: Tabla que muestra los componentes y volúmenes de DOP-PCR para una reacción 1x
| Paso | Ciclos | Temp (grado centígrado) | Hora |
| Desnaturalización inicial | 1 | 95 | 3 minutos |
| Desnaturalización | 34 | 98 | 20 s |
| Recocido de imprimación | 62 | 1 minuto | |
| Extensión | 72 | 30 s | |
| Extensión final | 1 | 72 | 5 minutos |
| Enfriamiento | 4 | Sostener |
Tabla 2: Tabla que muestra el ciclo DOP-PCR, la temperatura y el perfil de tiempo.
| Constituyente | Volumen (μL) |
| Tampón NT 10x (0.5M Tris-HCl pH 8,50 mM MgCl2, 0.5 mg/ml BSA) | 5 |
| 0,1 M de beta-mercaptoetanol | 5 |
| Stock de nucleótidos 10X (0,5 mM dATP, 0,5 mM dCTP, 0,5 mM dGTP, 0,5 mM dTTP, 0,5 mg/ml biotina-16-dUTP) | 5 |
| Dnase I (1 ng/ml) | 2 |
| DNApolimerasa I | 5U por μg de ADN |
| DNAtemplate (1 μg) | 1 |
| Agua tratada con DEPC | Hasta 50 μL |
Tabla 3: Tabla que muestra los componentes y volúmenes de traducción de nick para una sonda.
Discusión
El método del halo de ADN es un excelente método de elección cuando se analizan las interacciones entre el nucleoesqueleto y el genoma, sin embargo, hay algunos pasos críticos que también deben cumplirse. Uno de los parámetros más importantes es la optimización de la densidad de siembra celular. Si las células se vuelven demasiado confluentes, entonces los halos de ADN se superpondrán con las células vecinas, lo que hará imposible realizar el análisis. El CSK y los tampones de extracción deben hacerse siempre frescos el día de su uso, añadiéndose espermina, espermidina y digitonina al tampón de extracción al final del proceso de preparación para mantener su actividad biológica. Si se realiza Halo-FISH, es extremadamente importante utilizar la temperatura de desnaturalización correcta de los halos de ADN para permitir que la sonda o la pintura se hibriden posteriormente.
La microscopía electrónica se ha utilizado para visualizar la matriz nuclear, identificándose estructuras filamentosas20. Sin embargo, la microscopía electrónica es limitada ya que las asociaciones matriciales con la cromatina no se pueden deducir fácilmente. De hecho, el método DNA Halo es más versátil en comparación con la microscopía electrónica, ya que se pueden examinar genes específicos, cromosomas y estados celulares. Además, se está estudiando el análisis proteómico de proteínas de la matriz nuclear21,22. Este método es bueno para comparar componentes de la matriz nuclear, particularmente cuando se comparan células enfermas, sin embargo, no proporciona la distribución espacial y las uniones resaltadas por la técnica estándar de ADN Halo.
Los ensayos de ADN Halo tienen limitaciones. En primer lugar, a medida que se extrae la matriz, esto solo se puede realizar en células fijas, por lo que no es posible obtener imágenes en vivo. Aunque el método DNA Halo es relativamente rápido y fácil de realizar, el proceso general puede llevar mucho tiempo cuando se tiene en cuenta el cultivo celular, la generación de sondas, Halo-FISH y el análisis.
La captura de imágenes de halos de ADN y HALO-FISH utilizando microscopía de súper resolución mejoraría en gran medida la resolución de sondas y anticuerpos específicos de ADN. Además, como los fluorocromos se pueden resolver espectralmente más fácilmente, puede ser posible utilizar varias sondas de ADN en un solo experimento, proporcionando aún más información. Las mejoras en las técnicas de biología molecular, como la captura de conformación cromosómica (3C), se han utilizado para determinar las interacciones de los loci de los genes y analizar la organización espacial de la cromatina en la célula. Los ensayos DNA Halo y 3C se pueden combinar, un término conocido como M3C23, demostrando nuevamente la adaptabilidad de la técnica DNA Halo.
Los datos originales presentados aquí son para demostrar las posibilidades de interrogación del comportamiento del genoma y cómo presentar esos datos. Con estos datos hemos demostrado que es posible determinar diferencias significativas en la unión del genoma utilizando (1) sondas de pintura cromosómica, revelando en este estudio que el cromosoma 18 es el cromosoma menos unido de los analizados (Figura 3); (2) Loci de genes con diferencias significativas entre dos loci de genes y (Figura 4) (3) Telómeros, que están menos fuertemente unidos en las células quiescentes en comparación con las células proliferantes y senescentes (Figura 5). Somos capaces de diferenciar entre células proliferantes y no proliferantes a través de la presencia del marcador de proliferación del antígeno Ki67, que es una proteína insoluble, por lo que permanece con los núcleos residuales o utilizando la incorporación de nucleótidos para resaltar las células que han pasado por la fase S dentro de un período de tiempo específico (Figura 2). Esta técnica también nos ha permitido analizar el comportamiento del genoma en células que están comprometidas en sus nucleoesqueletos, es decir, células laminopatías, y aquí y en Bikkul et al., 2018 revelamos que el genoma puede estar menos unido en comparación con las células de control y puede restaurarse cuando se trata con medicamentos específicos que mejoran el efecto de la mutación lamin A en células HGPS clásicas15. Sin embargo, mostramos nuevos datos aquí para las células HGPS AGO8466 atípicas, que carecen de una mutación de lamina A pero que contienen una forma inusual de la proteína del complejo LINC SUN1 19 en la que el cromosoma13 está menos unido (Figura 6).
HALO-FISH es un método único que permite el estudio de las interacciones genómicas con el nucleoesqueleto en combinación con inmunofluorescencia indirecta para resolver proteínas no eliminadas del procedimiento de extracción. Se ha demostrado que el nucleoesqueleto está modificado en diversas enfermedades como ciertos tipos de cáncer19 y la importancia de algunas proteínas asociadas al nucleoesqueleto como biomarcadores diagnósticos24,25. Así, esta técnica tiene un papel importante en el examen del efecto del nucleoesqueleto sobre la organización/desorganización de la cromatina en la enfermedad 15,24,25,27 y no está restringida a células humanas, con sondas de pintura cromosómica de otros animales, el mismo protocolo ADN-halo podría ser empleado 28.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Nos gustaría agradecer al profesor Michael Bittner por el amable regalo de las sondas de pintura de brazos cromosómicos. LG recibió el apoyo del proyecto financiado por la UE EURO-Laminopathies y el Brunel Progeria Research Fund.
Materiales
| Name | Company | Catalog Number | Comments |
| 10X PBS | Thermo Fisher Scientific | 10388739 | Used to create DNA halos |
| 5-bromo-2′-deoxy-uridine | Sigma-Aldrich | B5002-100MG | Labelled nucleotide |
| 5-Fluoro-2′-deoxyuridine | Sigma-Aldrich | F0503-100MG | Labelled nucleotide |
| Agar Technical | Thermo Fisher Scientific | 15562141 | DNA isolation of BAC clones |
| Agarose | Sigma-Aldrich | A939-50G | Check product size of DOP-PCR and nick translation |
| Atypical type 2 HGPS fibroblasts (AG08466) | Coriell Institute | AG08466 | Cell line |
| Bacto tryptone | Thermo Fisher Scientific | 16269751 | DNA isolation of BAC clones |
| Biotin-16-dUTP | Roche Diagnostics | 11093711103 | Labelled nucleotides |
| Chloramphenicol | Sigma-Aldrich | C0378-25G | DNA isolation of BAC clones |
| Classical Hutchinson-Gilford progeria syndrome (HGPS) fibroblasts (AG06297) | Coriell Institute | AG0297 | Cell line |
| Coplin jar | Thermo Fisher Scientific | 12608596 | Holds 5 slides or 8 slides back to back |
| Cot-1 DNA | Thermo Fisher Scientific | 15279011 | Block nonspecific hybridization in HALO FISH |
| DEPC-treated water | Sigma-Aldrich | 693520-1L | DNA isolation of BAC clones |
| Dextran sulphate | Sigma-Aldrich | S4030 | Hybridisation mixture |
| Digitonin | Sigma-Aldrich | D141 | Component of extraction buffer |
| Digoxigenin-11-dUTP | Sigma-Aldrich | 11093088910 | Labelled nucleotides |
| Donkey anti-mouse Cy3 | Jackson Laboratory | 715-165-150 | Secondary antibody |
| EDTA | Sigma-Aldrich | E6758 | Component of extraction buffer |
| Ethanol | Component of extraction buffer | ||
| Ethanol | Sigma-Aldrich | 443611 | Probe precipitation and HALO FISH |
| Fetal bovine system | Thermo Fisher Scientific | 26140079 | Cell culture serum |
| Formamide | Thermo Fisher Scientific | 10523525 | 2D FISH of DNA halos |
| Glass wool | Sigma-Aldrich | 18421 | Spin column |
| Herring sperm | Sigma-Aldrich | D7290 | Probe precipitation |
| HXP™ Lamp (metal halide microscope lamp) | OSRAM | HXP-R120W45C VIS | Image capture of DNA halos |
| Hydrochloric acid | Thermo Fisher Scientific | 10313680 | Cleaning microscope slides |
| Isopropanol | Sigma-Aldrich | I9516-25ML | DNA isolation of BAC clones |
| KAPA HiFi PCR Kit | KAPA Biosystems | KK2103 | PCR Kit |
| Leica DM4000 fluorescent microscope with DFC365 FX camera and LAS AF (Version: 4.5.0) image acquisition software. | Leica Microsystems | Image capture of DNA halos | |
| Luria-Bertani agar | Thermo Fisher Scientific | 13274843 | DNA isolation of BAC clones |
| Magnesium chloride | Sigma-Aldrich | M8266 | Component of CSK buffer |
| Methanol | Thermo Fisher Scientific | 10284580 | Cleaning and sterilizing microscope slides |
| Mouse anti-BrdU antibody | BD Pharmingen | B2531-100UL | BrdU visualisation |
| Newborn calf serum | Thermo Fisher Scientific | 16010159 | Cell culture serum and blocking reagent |
| Nick translation kit | Invitrogen | ||
| PCR grade water | Sigma-Aldrich | 693520-1L | PCR and DNA isolation of BAC clones |
| PCR Primers | Sigma-Aldrich | ||
| PIPES | Sigma-Aldrich | P1851 | Component of CSK and extraction buffers |
| Potassium acetate | Sigma-Aldrich | P1190-100G | DNA isolation of BAC clones |
| QuadriPERM® 4 X 12 | SARSTEDT | 94.6077.307 | Square cell culture dish, polysterene with four compartments. This has hydrophobic surface, is sterile, non-pyrogenic/endotoxin-fee and non-cytotoxic. |
| Rabbit Anti-Ki67 antibody | Sigma-Aldrich | ZRB1007-25UL | Proliferation marker |
| Rnase A | Sigma-Aldrich | R6513 | DNA isolation of BAC clones |
| Rubber cement | Halford's | 101836 | 2D FISH of DNA halos |
| Sephadex G-50 | Sigma-Aldrich | S6022-25G | Spin column |
| Sodium acetate | Sigma-Aldrich | S2889 | Probe precipitation |
| Sodium chloride | Sigma-Aldrich | S5886 | Component of CSK, extraction and SSC buffers |
| Sodium citrate | Sigma-Aldrich | C8532 | Component of SSC buffer |
| Sodium dodecyl sulphate | L3771-100G | DNA isolation of BAC clones | |
| Sodium hydroxide | Sigma-Aldrich | S8045-500G | DNA isolation of BAC clones |
| Spermidine | Sigma-Aldrich | S2626 | Component of extraction buffer |
| Spermine | Sigma-Aldrich | S4264 | Component of extraction buffer |
| Streptavidin-Cy3 | Amersham Life Sciences Ltd, Scientific Laboratory Supplies | pa43001 | Probe antibody |
| Sucrose | Sigma-Aldrich | S0389 | Component of CSK buffer |
| Sucrose | Sigma-Aldrich | S0389 | CSK buffer+A66:D68 |
| SuperFrost™ microscope slides | Thermo Fisher Scientific | 12372098 | Microscope slides: 1 mm thickness, 76 mm length, 26 mm width. Uncoated. |
| Swine anti-rabbit TRITC | Dako | ||
| TELO-PNA FISH KIT | Agilent Dako | K532511-8 | Delineation of telomeres |
| Tris-HCl | Sigma-Aldrich | T3253-100G | Column buffer |
| Triton™ X-100 | Sigma-Aldrich | T9284 | Component of CSK buffer |
| Tryptone | Thermo Fisher Scientific | 10158962 | DNA isolation of BAC clones |
| Tween-20 | Sigma-Aldrich | P9416- 100ML | Detergent |
| Vectashield mountant containing DAPI | Vector Laboratories | H-1200 | 2D FISH of DNA halos |
| Whole human chromosome probes | Calbiochem | 2D FISH of DNA halos | |
| Yeast extract | Thermo Fisher Scientific | 10108202 | DNA isolation of BAC clones |
Referencias
- Berezney, R., Coffey, D. S. Identification of a nuclear protein matrix. Biochemical Biophysical Research Communications. 60 (4), 1410-1417 (1974).
- Haaf, T., Ward, D. C. High resolution ordering of YAC contigs using extended chromatin and chromosomes. Human Molecular Genetics. 3 (4), 629-633 (1994).
- Parra, I., Windle, B. High resolution visual mapping of stretched DNA by fluorescent hybridization. Nature Genetics. 5 (1), 17-21 (1993).
- Senger, G., et al. Released chromatin: linearized DNA for high resolution fluorescence in situ hybridization. Human Molecular Genetics. 3 (8), 1275-1280 (1994).
- Florijn, R. J., et al. High-resolution DNA Fiber-FISH for genomic DNA mapping and colour bar-coding of large genes. Human Molecular Genetics. 4 (5), 831-836 (1995).
- Elcock, L. S., Bridger, J. M. Fluorescence in situ hybridization on DNA halo preparations and extended chromatin fibres. Methods Molecular Biology. 659, 21-31 (2010).
- Heiskanen, M., et al. Visual mapping by fiber-FISH. Genomics. 30 (1), 31-36 (1995).
- Bensimon, A., et al. Alignment and sensitive detection of DNA by a moving interface. Science. 265 (5181), 2096-2098 (1994).
- Michalet, X., et al. Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science. 277 (5331), 1518-1523 (1997).
- Wilson, R. H., Coverley, D. Relationship between DNA replication and the nuclear matrix. Genes Cells. 18 (1), 17-31 (2013).
- Wilson, R. H. C., Coverley, D. Transformation-induced changes in the DNA-nuclear matrix interface, revealed by high-throughput analysis of DNA halos. Science Reports. 7 (1), 6475 (2017).
- Iarovaia, O. V., Akopov, S. B., Nikolaev, L. G., Sverdlov, E. D., Razin, S. V. Induction of transcription within chromosomal DNA loops flanked by MAR elements causes an association of loop DNA with the nuclear matrix. Nucleic Acids Research. 33 (13), 4157-4163 (2005).
- Tandara, M., et al. Sperm DNA integrity testing: big halo is a good predictor of embryo quality and pregnancy after conventional IVF. Andrology. 2 (5), 678-686 (2014).
- Repping, S., et al. The use of spermHALO-FISH to determine DAZ gene copy number. Mol Human Reproduction. 9 (4), 183-188 (2003).
- Bikkul, M. U., et al. Farnesyltransferase inhibitor and rapamycin correct aberrant genome organisation and decrease DNA damage respectively, in Hutchinson-Gilford progeria syndrome fibroblasts. Biogerontology. 19 (6), 579-602 (2018).
- Telenius, H., et al. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 13 (3), 718-725 (1992).
- Bridger, J. M., et al. Association of pKi-67 with satellite DNA of the human genome in early G1 cells. Chromosome Research. 6, 13-24 (1998).
- Sales Gil, R., Vagnarelli, P. Ki-67: More Hidden behind a 'Classic Proliferation Marker'. Trends in Biochemical Sciences. 43 (10), 747-748 (2018).
- Bikkul, M. U., et al. Telomere elongation through hTERT immortalization leads to chromosome repositioning in control cells and genomic instability in Hutchinson-Gilford progeria syndrome fibroblasts, expressing a novel SUN1 isoform. Genes Chromosomes Cancer. 58 (6), 341-356 (2019).
- Jackson, D. A., Cook, P. R. Visualization of a filamentous nucleoskeleton with a 23 nm axial repeat. EMBO Journal. 7, 3667-3677 (1988).
- Albrethsen, J., et al. Unravelling the nuclear matrix proteome. Journal of Proteomics. 72, 71-81 (2009).
- Mika, S., Rost, B. NMPdb: Database of nuclear matrix proteins. Nucleic Acids Research. 33, 160-163 (2005).
- Gavrilov, A. A., et al. of the nuclear matrix-bound chromatin hubs by a new M3C experimental procedure. Nucleic Acids Research. 38, 8051-8060 (2010).
- Sjakste, N., et al. Role of the nuclear matrix proteins in malignant transformation and cancer diagnosis. Experimental Oncology. 26 (3), 170-178 (2004).
- Leman, E. S., Getzenberg, R. H. Nuclear structure as a source of cancer specific biomarkers. Journal of Cellular Biochemistry. 104 (6), 1988-1993 (2008).
- Volpi, E. V., Bridger, J. M. FISH glossary: an overview of the fluorescence in situ hybridization technique. Biotechniques. 45 (4), 385-386 (2008).
- Bridger, J. M., Foster, H. A. Senescence and the Genome. Human Interphase Chromosomes. , (2021).
- Foster, H. A., Griffin, D. K., Bridger, J. M. Interphase chromosome positioning in in vitro porcine cells and ex vivo porcine tissues. BMC Cell Biology. 13 (1), 30 (2012).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados