Method Article
Hybridation in situ par fluorescence sur des préparations de halo d’ADN pour révéler des chromosomes entiers, des télomères et des loci de gènes
Dans cet article
Résumé
La combinaison de préparations de halo d’ADN avec l’hybridation in situ par fluorescence permet une analyse à haute résolution des interactions génomiques avec le nucléosquelette. Le génome attaché conduit à des signaux fluorescents hybridés situés dans les noyaux extraits résiduels, tandis que le génome non attaché est dans le halo d’ADN entourant les noyaux résiduels.
Résumé
Le génome est associé à plusieurs structures à l’intérieur des noyaux cellulaires, afin de réguler son activité et de l’ancrer à des endroits spécifiques. Ces structures sont collectivement connues sous le nom de nucléosquelette et comprennent la lame nucléaire, les nucléoles et les corps nucléaires. Bien qu’il existe de nombreuses variantes de l’hybridation in situ en fluorescence (FISH) pour étudier le génome et son organisation, elles sont souvent limitées par la résolution et fournissent des informations insuffisantes sur l’association du génome avec les structures nucléaires. La méthode du halo d’ADN utilise des concentrations élevées de sel et des détergents non ioniques pour générer des boucles d’ADN qui restent ancrées aux structures des noyaux par le biais de régions d’attachement dans le génome. Ici, les protéines nucléaires solubles, telles que les histones, les lipides et l’ADN qui ne sont pas étroitement liés à la matrice nucléaire, sont extraites. Cela conduit à la formation d’un halo d’ADN non attaché entourant un noyau résiduel qui contient lui-même de l’ADN étroitement associé à des structures nucléaires internes et à des protéines résistantes à l’extraction. Ces brins d’ADN étendus permettent une résolution accrue et peuvent faciliter la cartographie physique. En combinaison avec FISH, cette méthode présente l’avantage supplémentaire d’étudier les interactions génomiques avec toutes les structures par lesquelles le génome est ancré. Cette technique, appelée HALO-FISH, est très polyvalente où les halos d’ADN peuvent être couplés à des sondes d’acide nucléique pour révéler les loci de gènes, les chromosomes entiers, les satellites alpha, les télomères et même l’ARN. Cette technique donne un aperçu de l’organisation et de la fonction nucléaires dans les cellules normales et de la progression de la maladie comme avec le cancer.
Introduction
La « matrice nucléaire » a été décrite pour la première fois par Berezney et Coffey en 19741. Après avoir effectué des extractions avec des molarités salines élevées et un traitement par nucléase sur des noyaux de foie de rat, ils ont identifié un cadre structurel protéique. La procédure du halo d’ADN a ensuite été adaptée à partir de cette méthode et implique l’élimination des protéines solubles de sorte que seules la matrice nucléaire (NM) et les protéines et chromosomes associés au NM persistent. Les régions d’attachement de l’ADN sont situées à la base des boucles d’ADN et sont appelées régions attachées à la matrice (MAR) ou régions d’attache d’échafaudage (SAR), qui sont résistantes à l’extraction avec des concentrations élevées de sel et de détergent ionique lithium-3,5-diiodosalicylate (LIS) respectivement. Dans les halos d’ADN, l’ADN associé aux MAR/SAR est lié dans le noyau résiduel tandis que les boucles d’ADN s’étendent loin de ces sites et forment le halo d’ADN. Nous savons maintenant que le génome est ancré via des domaines associés à la lame nucléaire (LAD) à la lame nucléaire et à travers des régions associées nucléolaires (NAD) et éventuellement à travers d’autres structures nucléaires telles que des corps nucléaires spécifiques.
La méthode du halo d’ADN peut être utilisée pour la cartographie physique de l’ADN, des gènes et des régions chromosomiques car l’ADN étendu et la chromatine offrent une plus grande résolution car la chromatine est dépouillée des histones et l’ADN est étiré 2,3,4,5,6. Cependant, il existe certaines limitations lors de l’utilisation de halos d’ADN pour cette application. Par exemple, l’ADN étroitement associé aux noyaux résiduels des halos d’ADN peut être inaccessible aux sondes, ce qui l’empêche de procéder à l’analyse et à la cartographie physique6. D’autres techniques telles que la fibre-FISH 2,4,5,7 et le peignage moléculaire 8 permettent également une cartographie physique et ont l’avantage d’être relativement rapides et faciles à réaliser. Les deux sont utilisés de préférence pour la cartographie de l’ADN des gènes sur les halos d’ADN. Ces méthodes extraient les fibres de chromatine via l’utilisation d’extractions par solvant ou sel du noyau, cependant, le peignage moléculaire tend à avoir une meilleure reproductibilité 8,9.
Il existe de plus en plus de preuves que le nucléosquelette joue un rôle dans le soutien de processus nucléaires clés, tels que les sites de fixation de l’ADN, le remodelage de la chromatine, la transcription de l’ADN, la réparation de l’ADN et la réplication de l’ADN11,12. En tant que telle, la technique du halo d’ADN a été développée pour étudier les interactions entre le nucléosquelette et le génome au cours de ces activités cellulaires et a été couramment utilisée et rapportée dans la recherche. Cette technique a également été utilisée pour étudier les interactions entre le génome et le nucléosquelette en relation avec la progression de la maladie avec des changements associés à la malignité dans la structure nucléaire identifiés11.
La technique du halo d’ADN a également été utilisée pour étudier la relation entre le génome et le nucléosquelette au cours du développement et de la différenciation12. Un certain nombre d’études ont utilisé une variante de la technique du halo d’ADN connue sous le nom d’halosperme13 ou SpermHalo-FISH si elle est couplée à FISH14. La chromatine des spermatozoïdes est étroitement liée aux protéines connues sous le nom de protamines et cette technique a été développée pour améliorer l’accès à l’ADN des spermatozoïdes. L’halosperme a été utilisé pour étudier l’intégrité de l’ADN des spermatozoïdes et déterminer si des dommages à l’ADN sont présents. Les spermatozoïdes avec moins de dommages à l’ADN sont corrélés à une plus grande taille de halo d’ADN, tandis que les spermatozoïdes avec des niveaux accrus d’ADN fragmenté et endommagé avaient de petits halos ou pas du tout. Ainsi, l’halosperme peut être utilisée comme marqueur pronostique potentiel de la qualité de l’embryon et de la réussite de la grossesse avec la FIV13. Cet exemple met l’accent sur les applications cliniques potentielles de cette technique. Dans notre travail, nous avons utilisé HALO-FISH pour évaluer les changements dans le comportement du génome et l’effet de traitements médicamenteux spécifiques dans la maladie du vieillissement prématuré Syndrome de Hutchinson-Gilford Progeria (HGPS)15.
Ensemble, ces études, et d’autres, mettent en évidence l’étendue des processus / applications que la technique du halo d’ADN peut être utilisée pour étudier et l’utilité de la technique.
Protocole
1. Préparation des lames, stérilisation et culture cellulaire
- Préparer 500 mL de HCl à 10 % (v/v) et verser dans un grand bécher.
- Déposer les lames de microscope individuellement dans l’acide et incuber pendant 1 h à température ambiante sur un agitateur réglé à 2 x g.
ATTENTION : Le HCl est corrosif et irritant. Il peut causer de graves brûlures cutanées et des lésions oculaires et une irritation de la peau, des yeux et du système respiratoire. Assurez-vous que la protection individuelle appropriée est portée, y compris des gants en nitrile, une protection oculaire et une blouse de laboratoire. - L’acide décanté du bécher et des lames de lavage dix fois dans l’eau du robinet, puis dix fois de plus dans l’eau désionisée.
- Rincer deux fois les lames de méthanol et les conserver jusqu’à stérilisation par flambage.
MISE EN GARDE : Le méthanol est un liquide hautement inflammable et toxique en cas d’ingestion, de contact avec la peau ou d’inhalation. En outre, le méthanol peut causer des dommages aux organes, est corrosif et irritant. Respectez les limites d’exposition en milieu de travail et assurez-vous que la protection individuelle appropriée est portée, y compris des gants en caoutchouc butyle, une protection oculaire et une blouse de laboratoire. Dans la mesure du possible, manipuler dans une hotte de ventilation par aspiration locale (LEV). - À l’aide de pinces métalliques ou de longues pinces, retirez une lame de microscope du bécher contenant du méthanol. Flamme au-dessus d’un brûleur Bunsen pour stériliser et transférer dans un récipient de culture cellulaire rectangulaire contenant quatre compartiments pour lames, situé près du brûleur Bunsen.
ATTENTION : Le flambage permet la stérilisation immédiate des lames de microscope avant utilisation; Cependant, cette méthode comporte des dangers. Comme le méthanol est hautement inflammable, il est important que le bécher contenant les lames soit placé loin du brûleur Bunsen. Il faut utiliser de longues pinces ou des pinces qui serrent fermement les glissières. Le niveau de méthanol dans le bécher doit simplement couvrir les lames, à la fois pour minimiser la quantité de méthanol utilisée et pour que seules les extrémités des pinces / pinces soient en contact avec le méthanol. Assurez-vous toujours que le méthanol s’est évaporé des pinces ou des pinces après utilisation et que celles-ci ont refroidi avant de le remettre dans le bécher contenant les lames et le méthanol. Le bécher doit être recouvert d’un morceau de papier d’aluminium pour affamer l’oxygène si le méthanol prend feu. Ne jamais glisser de flamme à l’intérieur d’une hotte à flux laminaire de classe II où l’air circule. - Vous pouvez également effectuer les étapes 1.1 à 1.4, mais plutôt que de brûler les lames après l’incubation avec du méthanol, placez les lames sur un tissu non pelucheux pour les sécher à l’air. Une fois sec, enveloppez-le dans du papier d’aluminium et placez-le dans un four stérilisateur ou un autoclave.
- Cultiver des cellules dans le milieu approprié avec du sérum pendant au moins 48 h à 37 °C dans 5 % de CO2 jusqu’à ce que la confluence soit atteinte à 60-70 %. Ce protocole a été réalisé sur un passage précoce de fibroblastes dermiques humains (HDF) et sur des fibroblastes classiques du syndrome de Hutchinson-Gilford progeria (HGPS) (AG06297) et des fibroblastes HGPS atypiques de type 2 (AG08466). Récoltez chaque type de cellule et comptez à l’aide d’un hémocytomètre pour déterminer la densité cellulaire. Ensemencer 1 x 105 cellules dans un milieu de 10 ml par lame.
REMARQUE: La densité cellulaire est importante car les boucles d’ADN de différents noyaux peuvent converger si les cellules deviennent trop confluentes. Les densités d’ensemencement peuvent devoir être optimisées en fonction du type de cellule utilisé, car les cellules transformées peuvent proliférer plus rapidement, tandis que les cultures cellulaires de passage ultérieur peuvent prendre plus de temps pour atteindre la confluence souhaitée. - Si les cellules doivent être arrêtées dans G0 pour devenir quiescentes, alors ensemencer 1 x 105 cellules (dans un milieu de 10 ml) par lame et laisser pousser pendant 24 h. Laver les cellules deux fois avec un milieu sans sérum et incuber dans un milieu standard contenant une concentration plus faible de sérum à 0,5% (sérum de veau nouveau-né, NCS; ou sérum bovin fœtal, FBS) pendant 7 jours.
- Si l’état prolifératif des cellules est requis pour le test du halo d’ADN, déterminez les cellules qui sont passées par la phase S par incorporation de 5-bromo-2'-désoxy-uridine (BrdU) dans l’ADN pendant la réplication.
- Cellules de graines comme d’habitude et se développent pendant 24 h. Retirer le milieu de culture et le remplacer par un milieu contenant du BrdU et de la 5-fluoro-2'-désoxyuridine (3 μg/μL). Après 24 heures supplémentaires, retirez le milieu, lavez les cellules une fois avec du milieu (10% NCS), puis réalimentez-les avec du milieu frais (10% NCS). Incuber pendant 24 heures supplémentaires, puis préparer les lames pour le test du halo d’ADN.
2. Préparation de la sonde
- Sondes de peinture chromosomique entière et bras
- Fabriquer des sondes chromosomiques à partir de l’amplification de chromosomes triés ou microdisséqués par réaction en chaîne par polymérase amorcée par oligonucléotide dégénéré (DOP-PCR) en utilisant la méthode de Telenius et al.16. Utilisez la DOP-PCR pour marquer les sondes chromosomiques avec de la biotine-16-dUTP ou de la digoxigénine-11-dUTP, comme indiqué dans le tableau 1. Veuillez vérifier les instructions du fabricant pour le profil d’amplification, cependant, les conditions utilisées pour cette expérience sont indiquées dans le tableau 2.
- Préparer le bras ou la sonde chromosomique entière en additionnant 8 μL de produit PCR marqué, 7 μL d’ADN Cot-1, 3 μL de sperme de hareng, 1/20evolume d’acétate de sodium 3 M (pH 5,4) et 2 volumes d’éthanol à 100 %. Incuber la solution de sonde pendant au moins 30 min à -80 °C.
REMARQUE: Cette méthode peut être utilisée pour créer des sondes chromosomiques uniques ou plusieurs sondes chromosomiques si différentes étiquettes (c.-à-d. Biotine-16-UTP et Digoxigénine-11-dUTP) sont utilisées pour chaque chromosome d’intérêt.
ATTENTION : L’éthanol est un liquide et une vapeur hautement inflammables et peut causer une irritation oculaire grave. Tenir à l’écart de la chaleur, des surfaces chaudes et des sources d’inflammation. Respectez les limites d’exposition en milieu de travail et assurez-vous que la protection individuelle appropriée est portée, y compris des gants en caoutchouc butyle, une protection oculaire et une blouse de laboratoire. Dans la mesure du possible, manipuler dans une hotte LEV. - Solution de sonde centrifugeuse à 13 700 x g pendant 15 min à 4 °C, puis laver à l’éthanol à 70%. Répétez la procédure de centrifugation et jetez le surnageant, en prenant soin de ne pas perturber ou perdre la pastille d’ADN. Laissez sécher la pastille d’ADN.
- Ajouter 12 μL de tampon d’hybridation (50 % de formamide, 10 % de sulfate de dextrane, 10 % de citrate de sodium salin 20x (SSC; NaCl 3 M, citrate trisodique 0,3 M; pH 7,0), 1 % (v/v) de monolaurate de polyoxyéthylène sorbinal (Tween-20)) à la pastille d’ADN. Laisser à 37 °C pendant au moins 2 h pour que la pastille d’ADN se dissolve dans le tampon d’hybridation.
MISE EN GARDE : Le formamide est cancérogène et tératogène et peut donc causer de graves dommages à l’enfant à naître. Si une femme est enceinte ou soupçonne qu’elle est enceinte, elle devrait éviter de travailler avec du formamide. Le formamide doit être utilisé dans une hotte LEV. Respectez les limites d’exposition en milieu de travail et assurez-vous que la protection individuelle appropriée est portée, y compris des gants en caoutchouc butyle, une protection oculaire et une blouse de laboratoire.
- Isolement de l’ADN à partir de chromosomes artificiels bactériens (BAC)
- Étaler une petite partie du stock de glycérol du clone BAC sur une plaque de gélose Luria-Bertani (LB) (1% (P / V) NaCl; 1% (p / v) tryptone, 0,5% (p / v) extrait de levure, 1,5% (p / v) Agar Technical, 12,5 μg / mL (p / v) chloramphénicol). Incuber pendant une nuit à 37 °C.
- Choisir une seule colonie dans la plaque et inoculer 10 mL de bouillon LB (NaCl à 1 % (p/v), 1 % (p/v) bactotryptone, extrait de levure à 0,5 % (p/v), 12,5 μg/mL (p/v) de chloramphénicol). Laisser la solution dans un incubateur à agitation pendant une nuit à 37 °C.
ATTENTION : Le chloramphénicol est soupçonné de causer le cancer. Manipuler avec précaution et réduire l’exposition. - Culture par centrifugation à 1 700 x g pendant 10 min à température ambiante.
- Jeter le surnageant et ajouter 300 μL de solution P1 (15 mM Tris (pH 8), 10 mM EDTA, 100 μg/mL RNase A) à la pastille. Vortex vigoureusement et transférer les cellules dans un tube microcentrifuge de 2 mL.
- Ajouter 300 μL de solution de P2 (0,2 M NaOH, 1 % (p/v) de dodécylsulfate de sodium (SDS) goutte à goutte aux cellules. Retourner le tube de microcentrifugation fermé 5 fois et laisser à température ambiante pendant 5 min maximum.
ATTENTION : L’hydroxyde de sodium est corrosif et peut causer de graves brûlures cutanées et des lésions oculaires. Il peut être corrosif pour les métaux. Manipuler avec précaution et réduire l’exposition. Respectez les limites d’exposition en milieu de travail et assurez-vous que la protection individuelle appropriée est portée, y compris des gants en caoutchouc nitrile, une protection oculaire et une blouse de laboratoire. Dans la mesure du possible, manipuler dans une hotte LEV.
ATTENTION : Le dodécylsulfate de sodium est un solide inflammable, nocif en cas d’ingestion et pouvant causer une irritation cutanée et respiratoire. Il peut également causer de graves lésions oculaires. Assurez-vous que la protection individuelle appropriée est portée, y compris des gants en caoutchouc nitrile, une protection oculaire et une blouse de laboratoire. Dans la mesure du possible, manipuler dans une hotte LEV. - Ajouter 300 μL de P3 (acétate de potassium 3 M) lentement aux cellules et mélanger doucement. Placer le tube de microcentrifugeuse sur de la glace pendant 10 min.
- Centrifuger à 8 100 x g pendant 10 min à 4 °C et transférer le surnageant dans un tube contenant 800 μL d’isopropanol glacé. Retourner le tube plusieurs fois et incuber à -20 °C pendant une nuit.
ATTENTION: L’isopropanol est un liquide et une vapeur hautement inflammables et peut causer une irritation oculaire grave, de la somnolence ou des étourdissements. Tenir à l’écart de la chaleur, des surfaces chaudes et des sources d’inflammation. Respectez les limites d’exposition en milieu de travail et assurez-vous que la protection individuelle appropriée est portée, y compris des gants en caoutchouc nitrile, une protection oculaire et une blouse de laboratoire. Dans la mesure du possible, manipuler dans une armoire à fumée LEV. - Centrifuger à 8 100 x g pendant 15 min à 4 °C. Retirer le surnageant et transférer dans un autre tube. Ajouter 500 μL d’éthanol glacé à 70 %. Inverser le tube plusieurs fois et centrifuger à 8 100 x g pendant 5 min à 4 °C.
- Retirer le surnageant et sécher à l’air libre le granulé à température ambiante. Une fois la pastille sèche, remettez en suspension dans 40 μL d’eau traitée au pyrocarbonate de diéthyle (traitée DEPC) et laissez reposer à 4 °C pendant la nuit. Une fois complètement remis en suspension, prélever 5 μL de solution et charger sur un gel d’agarose à 1% pour vérifier la présence d’ADN.
- Préparation par sonde monogénique des BAC par traduction de nick
- Utilisez des kits d’étiquetage de traduction de pseudo disponibles dans le commerce. Vous pouvez également utiliser le protocole suivant. Voir le tableau 3 pour les constituants et les volumes.
- Ajouter les constituants du tableau 3 ensemble dans un tube microcentrifuge en ajoutant l’ADN polymérase I en dernier, mélanger doucement et centrifuger brièvement pendant quelques secondes. Incuber la solution à 15 °C pendant 2 h.
- Pour vérifier la taille des fragments, chargez 5 μL de la solution sur un gel d’agarose à 2%. La plage de taille des fragments d’ADN doit être comprise entre 200 et 600 pb. Si la taille des fragments d’ADN est plus grande, continuez à incuber la solution pendant 15 minutes supplémentaires à 15 °C et faites couler les produits sur du gel d’agarose à 2%.
- Arrêtez la réaction de traduction en ajoutant 10 mM d’EDTA, 0,1% de SDS (2,5 μL de 0,5 M EDTA, pH 8,0 dans 100 μL et 1 μL de 10% SDS dans 100 μL). Chauffer la solution à 65 °C pendant 5 min.
- Pour éliminer les nucléotides non incorporés, appliquez une sonde BAC sur une colonne de spin. Les colonnes de spin commerciales peuvent être achetées, ou elles peuvent être créées à l’aide d’une seringue comme suit:
- Ajouter 30 g de Sephadex G-50 à 500 mL de tampon colonne (10 mM Tris-HCl (pH8), 1 mM EDTA, 0,1% SDS). Autoclaver le mélange. Préparez également 500 ml de tampon colonne (sans Sephadex G-50) et d’autoclave.
- Faites tourner les colonnes en ajoutant de la laine de verre autoclavée au fond d’une seringue de 1 mL. Remplissez la seringue de 1 ml avec Sephadex G-50 dans un tampon colonne. Placer une seringue de 1 mL dans un tube à centrifuger de 15 mL muni d’un tube microcentrifuge sans couvercle au fond. Centrifuger à 1 600 x g pendant 5 min.
- Retirer la seringue et jeter le tube de microcentrifugation au fond. Ajoutez un nouveau tube de microcentrifugation dans le tube à centrifuger de 15 mL. Ajouter le tampon colonne (sans Sephadex G-50) à la seringue de 1 mL et réinsérer dans le tube à centrifuger de 15 mL. Centrifuger à 1 600 x g pendant 5 min. Répétez cette étape deux fois.
- Retirer la seringue et l’insérer dans un tube à centrifuger de 15 mL contenant un nouveau tube microcentrifuge propre. Appliquer la sonde sur la seringue et recueillir la sonde dans le tube de microcentrifugeuse.
- Pour précipiter la sonde à ADN, ajoutez 5 μL d’ADN de spermatozoïdes de hareng (10 mg/mL), 10 μL d’acétate de sodium et 2,25 volumes d’éthanol à 100 % à la solution d’ADN. Mélanger délicatement la solution et incuber à -80 °C pendant 1 h minimum. Centrifuger à 13 700 x g pendant 15 min à 4 °C.
- Jeter le surnageant et laver la pastille avec 200 μL d’éthanol à 70 % glacé pendant 15 minutes à 4 °C. Retirer le surnageant et sécher à l’air. Une fois sec, remettre en suspension les granulés dans 20 μL d’eau traitée DEPC à température ambiante pendant plusieurs heures ou toute la nuit à 4 °C. La sonde est maintenant prête à être utilisée ou peut être stockée à -20 °C.
- Pour chaque lame, mélanger 5 μL d’ADN de sonde avec 5 μL d’ADN Cot-1 et sécher à l’aide d’un concentrateur sous vide Speed Vac. Une fois la pastille séchée, re-suspendre dans 12 μL de mélange d’hybridation.
3. Préparation du halo d’ADN
- Retirez la boîte de culture carrée contenant les lames et les cellules attachées de l’incubateur. Jeter le milieu, étiqueter les lames à l’aide d’un crayon et les placer dans un pot Coplin contenant 50 mL de tampon de cytosquelette glacé (CSK) : 100 mM de NaCl, 3 mM de MgCl2, 0,3 M de saccharose, 10 mM d’acide 1 ,4-pipérazine dipéthanesulfonique (PIPES; pH 7,8), 0,5 % (v/v) de Triton X-100 composé dans de l’eau désionisée. Incuber pendant 15 min sur glace ou à 4 °C.
ATTENTION : Le Triton X-100 peut provoquer une irritation de la peau et de graves lésions oculaires. Manipuler à l’aide d’un équipement de protection individuelle approprié, y compris des gants en nitrile, des lunettes de protection et une blouse de laboratoire. - Jeter le tampon CSK et rincer rapidement les lames dans 50 mL de 1x tampon de halo d’ADN (DHB; 140 mM NaCl, 27 mM KCl, 110 mM NaHPO 4, 15 mM KH2PO 4; pH7.4) trois fois, c’est-à-dire tremper la glissière dans un pot Coplin contenant DHB et retirer.
- Transvaser les lames dans un pot Coplin contenant 50 mL de tampon d’extraction : 2 M de NaCl, 10 mM de TUYAUX (pH 6,8), 10 mM d’acide éthylènediaminetétraacétique (EDTA), 0,1 % (p/v) de digitonine, 0,05 mM (v/v) de spermine, 0,125 mM (v/v) de spermidine. Incuber pendant 4 min à température ambiante.
ATTENTION : La digitonine est toxique en cas d’ingestion ou de contact cutané et mortelle en cas d’inhalation. Assurez-vous que la digitonine est manipulée dans une armoire à fumée LEV et portez une blouse de laboratoire, des gants en nitrile (double gant), des lunettes de sécurité et un masque. La spermine et la spermidine peuvent causer de graves brûlures cutanées et des lésions oculaires, tandis que l’EDTA provoque une irritation oculaire grave, alors manipulez chaque produit chimique avec soin.
NOTE: Préparer la digitonine séparément en dissolvant la poudre dans de l’eau à une température de 60-70 °C. Ajouter la digitonine dissoute au tampon d’extraction une fois refroidi. Ajouter la spermine, la spermidine et la digitonine en dernier au tampon d’extraction pour préserver l’activité biologique. - Incuber des lames consécutivement dans 50 mL de 10x DHB (1,4 M NaCl, 270 mM KCl, 1,1 M NaHPO 4, 150 mM KH2PO4; PH7.4), 5x, 2x et 1x DHB pendant 1 min chacun.
- Trempez les glissements (droit et droit) à travers une série séquentielle d’éthanol de 50 ml d’éthanol à 10 %, 30 %, 70 % et 95 % (v/v).
- Sécher à l’air libre les lames et stocker à -80 °C jusqu’à ce qu’une hybridation in situ par fluorescence bidimensionnelle (2D FISH) soit effectuée.
4. Hybridation in situ par fluorescence bidimensionnelle
- Faire 20x SSC: 3 M NaCl, 0,3 M citrate trisodique, pH 7,0. Ce tampon peut être autoclavé, stocké à température ambiante et dilué au besoin.
- Faire 70 % (v/v) de formamide, 2x SSC pH 7,0 et chauffer à 70 °C dans un bain-marie.
- Incuber les lames, pendant 5 minutes chacune, à travers une série séquentielle d’éthanol de 50 ml d’éthanol à 70, 90 et 100 %.
- Sécher à l’air libre sur une plaque chauffante et cuire au four à 70 °C pendant 5 min.
- Dénaturer les lames en plaçant dans la solution de formamide à 70 % de 2x SSC pendant 2 min à 70 °C.
REMARQUE: La température et le moment sont critiques pour l’étape 4.5. Si la température est l’ADN ne se dénaturera pas et les sondes ne s’hybrideront pas, et aucun signal ne sera obtenu à partir du halo d’ADN FISH. - Placer la lame dénaturée dans 50 mL d’éthanol glacé à 70 % pendant 5 minutes et prendre à travers une série d’éthanol de 90 %, 95 % et 100 % à température ambiante pendant 5 minutes chacune.
- Sécher à l’air libre sur une plaque chauffante
- Manipulez directement les sondes chromosomiques humaines totales étiquetées conformément aux instructions du fabricant. Pour ces expériences, nous avons utilisé les peintures chromosomiques entières humaines 1, 13, 15, 17 et 18. De plus, dans cette expérience, des sondes de gènes CCND1 et CTNNA1 ont été utilisées.
REMARQUE: Les sondes chromosomiques entières et les sondes spécifiques au gène BAC ont été marquées avec de la biotine-11-dUTP et détectées par la streptavidine conjuguée à la cyanine 3 (Cy3). Pour les sondes de peinture chromosomique fabriquées par (DOP-PCR) et l’ADN BAC marqués par traduction de nick, ceux-ci seront appelés sondes ADN à partir de ce moment dans le protocole et traités comme suit. - Sonde ADN dénaturée (peinture chromosomique entière ou sonde spécifique au gène) à 75 °C pendant 10 min dans un bloc chaud ou un bain-marie.
- Sondes d’ADN chaudes à 37 °C pendant 30 minutes dans un bloc chaud ou un bain-marie avant de pipeter 10 μL sur la lame appropriée.
REMARQUE: Cette étape est importante pour bloquer les séquences chromosomiques répétitives. S’ils ne sont pas effectués, des signaux non spécifiques peuvent être produits dans le DNA Halo FISH. - Sonde de recouvrement avec glissement de couverture de 21 mm x 21 mm et joint à l’aide de ciment en caoutchouc.
- Incuber les lames pendant au moins 18 h à 37 °C dans une chambre d’hybridation humidifiée.
REMARQUE : Les chambres d’hybridation humidifiées peuvent être fabriquées à partir de boîtes sandwich contenant plusieurs couches de tissu humidifié et d’une plate-forme surélevée construite à partir de pipettes en plastique coupées de 10 ml sur lesquelles reposer les lames. Ceci est recouvert de papier d’aluminium pour minimiser l’exposition à la lumière. - Retirez soigneusement le ciment en caoutchouc à l’aide d’une pince.
- Incuber les lames dans 50 ml de formamide à 50 % (v/v), 2x SSC, solution de pH 7,0 qui a été préchauffée à 45 °C pendant trois incubations de 5 minutes.
REMARQUE: Laisser tomber la lamelle de couverture de la lame lors de la première incubation dans 50% (v/v) formamide, 2x SSC, solution de pH 7,0. Cela évite d’endommager la préparation DNA Halo qui pourrait être causé par le fait de « glisser » la lamelle de couverture. Les glissières peuvent être agitées dans le tampon via une pince pour aider à détacher le couvercle. - Ensuite, placez les lames dans 50 ml de solution 0,1x SSC, pH 7,0 qui a été préchauffée à 60 °C mais placée dans un bain-marie à 45 °C. Incuber pendant 5 min et remplacer le tampon deux fois de plus par 5 min d’incubation.
- Placer les lames dans un pot Coplin contenant 50 ml de 4x SSC, solution de pH 7,0 à température ambiante et incuber pendant 15 minutes avec trois changements de tampon.
- Appliquer 100 μL de BSA à 4 %, 4x solution SSC sur chaque lame et superposer avec un morceau de film de paraffine. Incuber à température ambiante pendant 10 min. Cela empêche la liaison non spécifique de l’anticorps.
- Pour détecter la sonde marquée (biotine-16-dUTP), incuber avec 100 μL d’un streptavidine-Cy3 1:200 (fabriqué dans 1% BSA, 4x SSC) pendant 1 h à température ambiante.
REMARQUE : Suivre les instructions du fabricant avec les dilutions d’anticorps et la dilution d’essai avant l’expérience pour s’assurer qu’un bon signal est produit. - Placer les lames dans un pot Coplin contenant 50 ml de 4x SSC (0,5% Tween-20) solution pH 7,0 à température ambiante et incuber pendant 15 minutes avec trois changements de tampon. Les lames peuvent être montées à ce stade, comme indiqué à l’étape 4.21, si l’immunofluorescence n’est pas nécessaire.
- Si l’état prolifératif des cellules transformées en halos d’ADN est requis, colorer avec des anticorps anti-pKi67 après les étapes FISH, avant le montage ou la coloration pour BrdU incorporé.
- Laver les lames 3 fois pendant 5 minutes chacune dans 50 mL de 1x solution saline tamponnée au phosphate (PBS), puis bloquer avec 4% de NCS dans PBS pendant 1 h à température ambiante.
- Appliquer 200 μL de l’anticorps primaire nécessaire (lapin anti-humain pKi67; souris anti-BrdU) sur la lame, recouvrir d’une bande de film de paraffine et incuber à température ambiante pendant 1 h.
- Laver les lames 3 fois pendant 5 min dans 1x PBS et incuber à température ambiante pendant 1 h dans 200 μL d’anticorps secondaire conjugué au fluorochrome (pKi67 : TRITC anti-lapin porcin ; BrdU : âne anti-souris Cy3). Effectuez 3 lavages supplémentaires de 5 minutes avec PBS. Toutes les dilutions doivent être effectuées en utilisant 1 % (v/v) de NCS dans du PBS dans la gamme de dilutions suggérées par le fabricant.
- Montez des glissières dans 20 μL de support contenant DAPI et une superposition avec une glissière de couverture de 22 mm x 50 mm.
5. Télomère PNA POISSON
- Pour détecter les télomères, utilisez le kit télomère PNA FISH - FITC; Effectuez la procédure avec les instructions du fabricant. La procédure doit être exécutée à température ambiante, sauf indication contraire.
- Immerger les lames dans une solution saline tamponnée au tris (TBS, pH 7,5) pendant 2 min, puis placer dans du formaldéhyde à 3,7% (dans TBS; v / v) pendant exactement 2 min.
ATTENTION : La solution TBS contient 10-30% de trométamol et 10-30% de chlorhydrate de 2-amino-2-(hydroxyméthyl)propane-1,3-diol. Cela peut causer une grave irritation des yeux et de la peau, alors portez des gants de protection et des lunettes de protection / protection du visage. Utiliser dans un endroit bien ventilé. - Lavez les lames dans un pot Coplin deux fois avec TBS pendant 5 minutes chacune.
- Immerger les lames dans la solution de prétraitement pendant 10 minutes, puis laver deux fois avec TBS pendant 5 minutes par lavage.
- Ensuite, prenez les lames à travers une série d’éthanol glacé comprenant 50 ml d’éthanol à 70%, 85% et 95% (v/v) pendant 2 minutes par concentration. Ensuite, laissez les toboggans sécher à l’air.
- Appliquer 10 μL de Telomere PNA Probe/FITC (ou Cy3) selon le choix de la coloration du marqueur fluorescent, sur chaque lame et couvrir la couche avec une lame. Incuber dans un four préchauffé réglé à 80 °C pendant 5 min, puis placer à l’obscurité pendant environ 1 h.
ATTENTION : Telomere PNA Probe/FITC contient 6-100% de formamide, qui provoque une irritation oculaire grave et est tératogène et peut donc causer de graves dommages à un enfant à naître. Si une femme est enceinte ou soupçonne qu’elle est enceinte, elle devrait éviter de travailler avec du formamide. Le formamide doit être utilisé dans une hotte anti-fumée LEV et une protection appropriée des yeux ou du visage doit être portée. - Pour retirer les lames de couverture, immerger les lames dans la « Solution de rinçage » pendant 1 min, puis placer dans la « Solution de lavage » pendant 5 min à 65 °C.
ATTENTION : La solution de lavage contient 1 à 5 % de polyoxyéthylène octyl phényléther et 1 à 5 % de chlorure de sodium. Ceci est corrosif et peut causer de graves lésions oculaires. Assurez-vous que des lunettes de protection ou un protecteur facial sont portés lors de la manipulation de la solution de lavage. - Incuber des lames à travers une série de 50 ml d’éthanol glacé (70 %, 85 % et 95 % (v/v)) pendant 2 minutes par concentration, puis sécher à l’air. Une fois sec, glissière de montage avec support contenant DAPI et superposition avec un couvercle.
6. Capture et analyse d’images
- Pour visualiser les halos d’ADN et les territoires chromosomiques, utilisez un microscope à épifluorescence (par exemple, le microscope Leica DM4000) pour capturer des images à l’aide d’un objectif d’huile HC PL FLUOTAR 100X / 1.30 et d’une caméra DFC365FX.
- Capturez des images en niveaux de gris et définissez la couleur de chaque couche capturée pour activer la pseudocoloration des images. Un logiciel commercial a été utilisé dans cette expérience (p. ex., le logiciel LAS AF version 4.5.0). Les canaux de couleur individuels ont été exportés au format TIFF.
- Analysez des images à l’aide du programme de traitement d’images Java Fiji ImageJ. Téléchargez l’image en appuyant sur Fichier et ouvrir.
- Chargez des couches d’image distinctes ou divisez une image composite en couches de niveaux de gris distinctes en cliquant sur Image | Couleur | Fractionner les canaux. Sélectionnez un canal d’image et cliquez sur Image | Ajustez , puis sélectionnez Luminosité et contraste. Modifiez en conséquence et répétez avec d’autres canaux.
- Créez un masque du noyau résiduel en sélectionnant le canal coloré DAPI représentant le noyau. Cliquez sur Image | Ajustez , puis sélectionnez Seuil. Une boîte de dialogue apparaîtra où le seuil peut être modifié, cochez la case Arrière-plan sombre . Modifiez jusqu’à ce que le noyau résiduel soit clair et appuyez sur Appliquer et fermez la boîte de dialogue.
REMARQUE: Cela crée un masque binaire basé sur l’intensité des pixels, avec des pixels blancs montrant les régions d’intérêt et des pixels noirs montrant l’arrière-plan. Répétez la même procédure sur le canal de sonde. - Utilisez la sélection à main levée pour délimiter la périphérie du noyau résiduel, puis cliquez sur Modifier et effacer l’extérieur. Superposez le canal de la sonde sur le noyau résiduel. Cela peut être fait en appuyant sur Image| Couleur | Fusionner les canaux.
- Pour définir l’échelle de mesure dans ImageJ, tracez une ligne sur la barre d’échelle ou entre les points de deux distances connues. Accédez à Analyser et appuyez sur Définir l’échelle. Dans la boîte de dialogue, ajoutez la longueur de distance et cliquez sur OK. Pour mesurer les distances, tracez une ligne entre les points mesurés et cliquez sur Analyser | Mesurer. Cela transférera les valeurs de distance vers une fenêtre de données.
- Mesurez l’intensité DAPI la plus brillante car elle coïncide avec le centre du noyau. À partir de là, mesurez la distance entre le centre nucléaire et le bord du territoire chromosomique (CTE) le plus éloigné. Mesurer la distance entre le centre nucléaire et le bord nucléaire (NE).
- S’assurer que les résultats sont présentés sous la forme d’un rapport CTE/EN. Ici, la distance entre le centre nucléaire et chaque bord du territoire chromosomique le plus éloigné (CTE) est divisée par la distance entre le centre nucléaire et chaque bord nucléaire respectif (NE). Cela devrait être effectué sur un minimum de 50 noyaux. Cela peut être représenté sous la forme d’un graphique à barres ou en boîte.
- Pour l’analyse des télomères, analysez un minimum de 30 noyaux par ensemble de données. Les images peuvent être analysées à l’aide de Fiji ImageJ ou manuellement pour compter le nombre de télomères dans le noyau résiduel et dans le halo d’ADN. BrdU ou pKi67 a permis de différencier les noyaux proliférants (BrdU/piK67+) et sénescents/quiescents (BrdU/pKi67-). Les données peuvent être représentées dans des diagrammes à barres avec des barres d’erreur correspondant à l’erreur type de la moyenne (MEB).
- Utilisez le test t de Student (non apparié) pour comparer statistiquement les résultats avec p> 0,05 considéré comme significatif.
Résultats
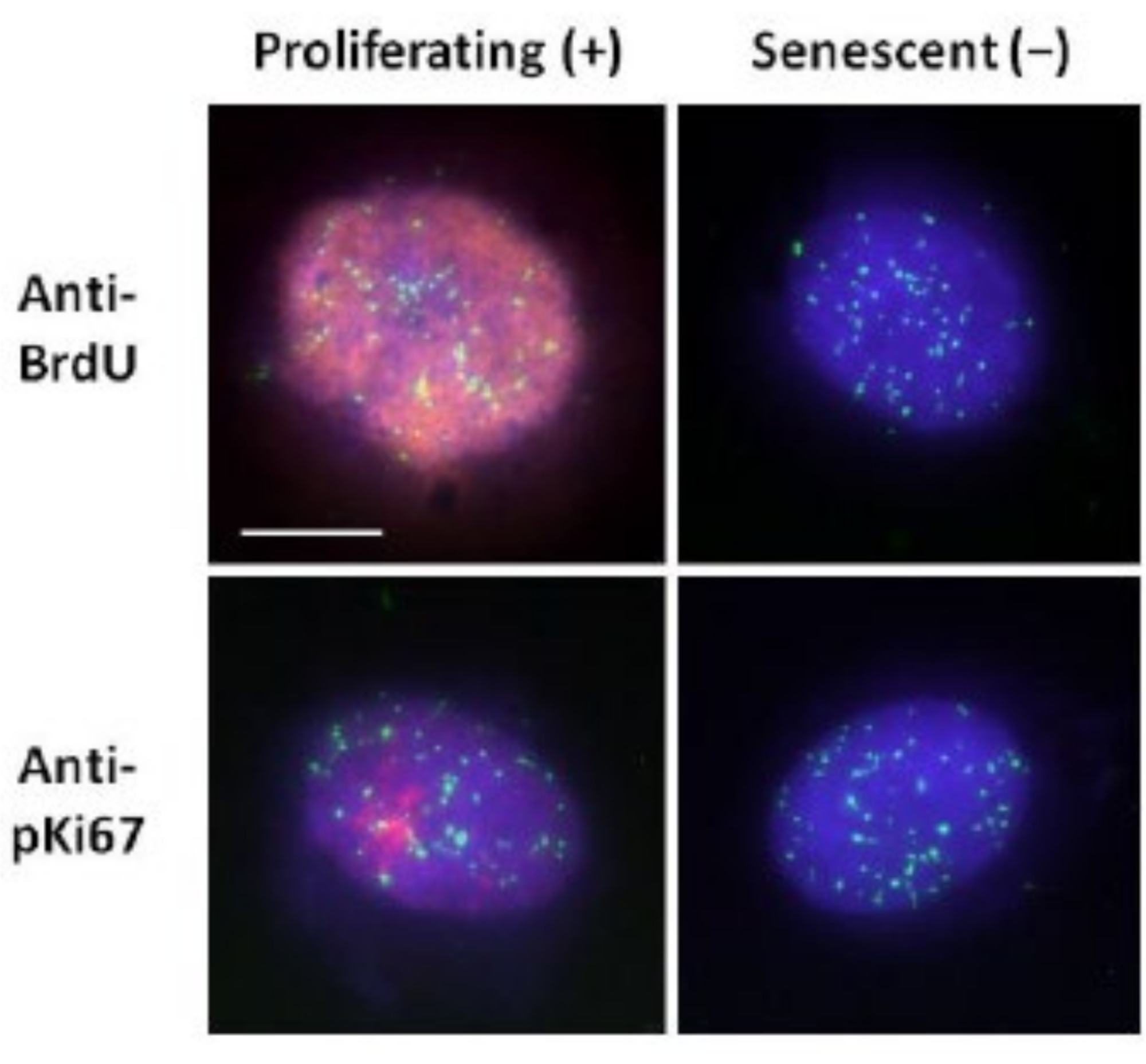
Cette méthode de préparation du halo d’ADN nous a aidés dans nos efforts pour déterminer les différences de comportement du génome au sein des cellules jeunes et âgées, mais aussi dans les cellules dérivées de maladies de vieillissement prématuré avec des protéines nucléosquelettiques aberrantes15. La figure 1 montre des exemples de halos d’ADN où il est possible de voir le bord d’un noyau résiduel, l’ADN restant dans le noyau résiduel et l’ADN non attaché qui s’est enroulé dans la zone environnante créant un halo d’ADN. Il décrit également l’analyse montrant comment le noyau résiduel est obtenu et les mesures NE et CTE. Il est possible de différencier les cellules proliférantes des cellules non proliférantes en incorporant un nucléotide marqué tel que BrdU lorsque les cellules sont en phase S ou en utilisant le marqueur de prolifération diagnostique anti-pKi67, qui révèle les nucléoles et les régions d’hétérochromatine dans les cellules G117,18. Les cellules primaires cultivées dans le sérum élevé sans atteindre la confluence, qui sont négatives pour les marqueurs de prolifération, sont supposées être sénescentes. Les cellules primaires cultivées dans le sérum bas ou devenues confluentes, c’est-à-dire inhibées par le contact qui sont négatives pour les marqueurs de prolifération, sont considérées comme quiescentes et seraient capables de réintégrer le cycle cellulaire prolifératif si les nutriments et la situation sont corrects. La capacité de différencier les cellules Ki67 positives et négatives nous a permis de déterminer les différences entre les fibroblastes dermiques humains proliférants, quiescents et sénescents. La figure 2 montre les halos d’ADN de fibroblastes dermiques humains en prolifération créés à partir de cellules où le BrdU y a été incorporé lors de la réplication de l’ADN, un mécanisme qui ne se produit pas dans les cellules non proliférantes, et ensuite coloré avec des anticorps anti-BrdU. La coloration avec le marqueur prolifératif anti-anticorps pKi67 est également visible sur la figure 2. Il s’agit d’un antigène robuste qui survit au protocole FISH et peut donc être coloré pour le post-FISH et le prémontage. Ainsi, les cellules proliférantes sont positives (rouge) pour BrdU et anti-pKi67 (rouge) dans la colonne de gauche et les cellules non proliférantes, en effet les cellules sénescentes de la figure 2 sont affichées dans la colonne de droite. Les signaux verts sont des télomères individuels révélés avec un kit PNA FISH/FITC de télomères. La combinaison de l’immunofluorescence avec des halos d’ADN permet une analyse au cours de différents états cellulaires, comme le montre la figure 2 lors de l’étude de cellules proliférantes, quiescentes et sénescentes. Selon l’anticorps choisi, d’autres conditions peuvent être examinées, telles que la différenciation, les dommages à l’ADN par irradiation, etc.
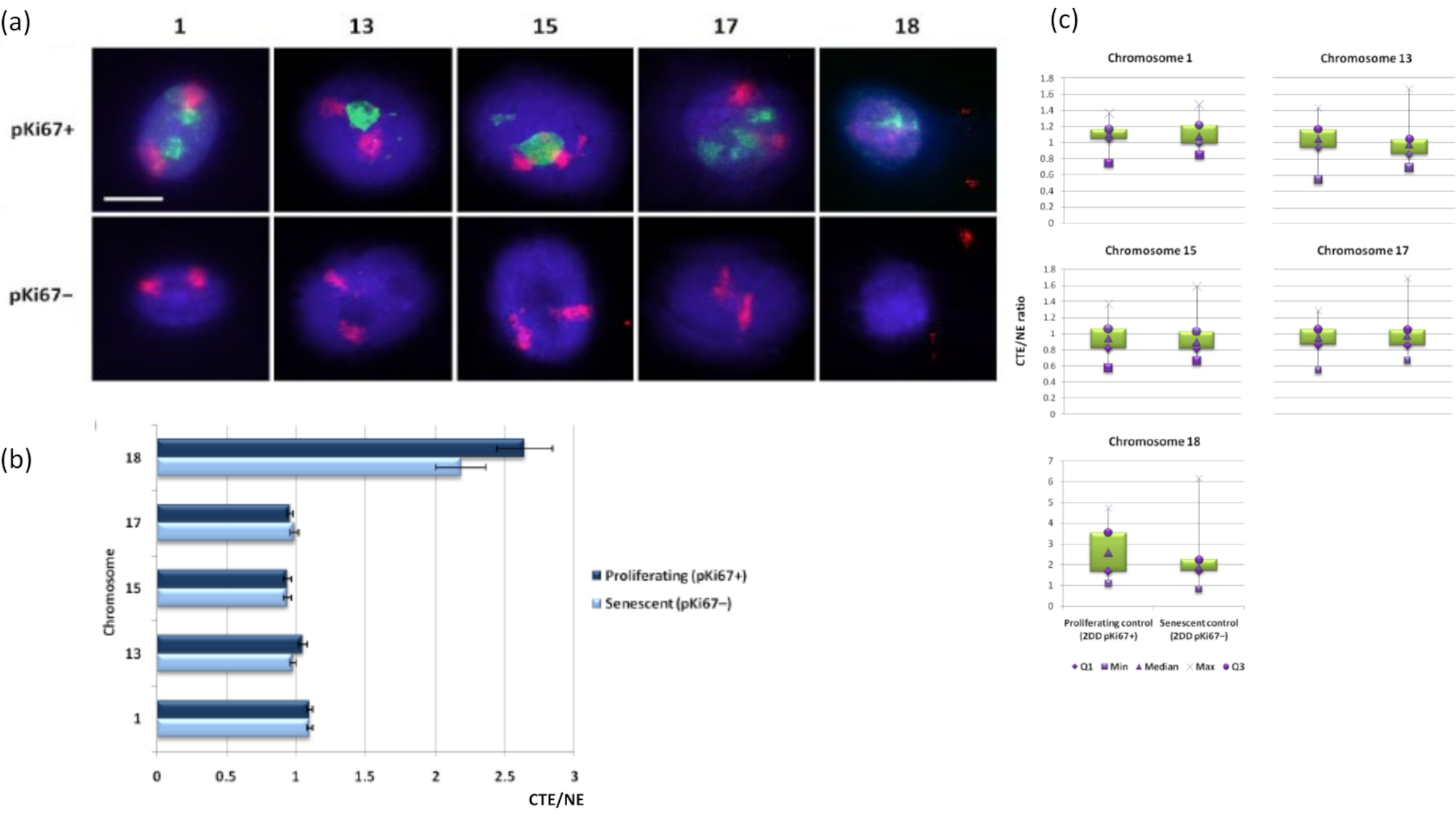
Les territoires chromosomiques peuvent également être visualisés dans des halos d’ADN à l’aide de FISH. En raison de la préparation permettant l’enroulement de l’ADN hors des noyaux, la forme du territoire chromosomique peut être perturbée, avec des quantités plus ou moins grandes du chromosome trouvées dans le halo d’ADN, en fonction de l’ancrage du génome à l’intérieur du noyau résiduel et de ses structures. La figure 3 révèle un panel de halos d’ADN dans lesquels des chromosomes individuels ont été révélés avec des sondes de peinture chromosomiques spécifiques du bras entier (rouge) pour les chromosomes 1, 13, 17 et 18. Anti-pKi67 (vert) a été utilisé pour marquer les cellules proliférantes et son absence dans la même culture, sur la même lame, désignant les cellules sénescentes. Il est très évident d’après les images et les données présentées comme CTE / NE que le petit chromosome 18 pauvre en gènes est un chromosome qui a peu d’attaches et qui s’enroule plus loin dans le halo d’ADN loin des noyaux résiduels et est significativement plus éloigné du centre des noyaux résiduels que les autres chromosomes. Cependant, cela est également vrai pour le chromosome 1. En utilisant le marqueur prolifératif anti-pKi67, il a également été possible de comparer la prolifération avec des cellules sénescentes, au sein de la même culture et sur la même lame, et cette analyse a révélé que les chromosomes au sein de ces deux statuts cellulaires très différents ne sont pas significativement différents l’un de l’autre, en ce qui concerne la fixation avec les structures nucléaires résiduelles.
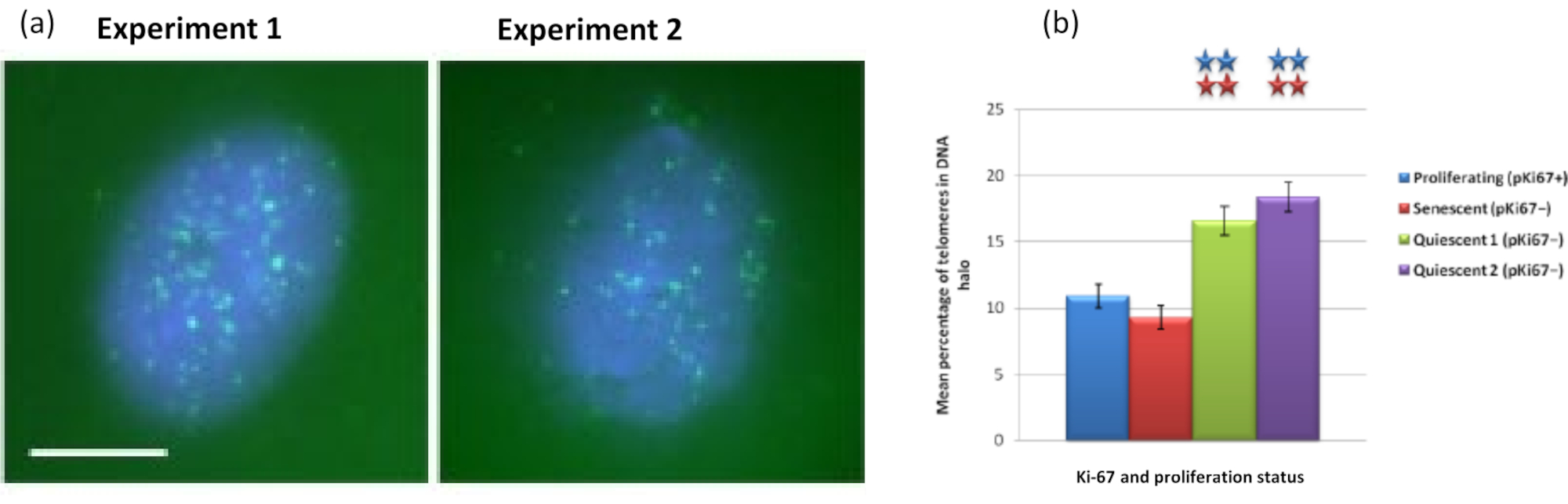
Fait intéressant, les gènes montrent également des différences statistiquement significatives entre les cellules proliférantes et sénescentes en ce qui concerne le fait de rester dans un noyau résiduel ou d’être situé dans le halo d’ADN. La figure 4 le démontre avec des loci génétiques délimités par des sondes BAC marquées en rouge et anti-Ki67 en vert. Il n’y a pas de différences significatives entre les emplacements des gènes dans les cellules proliférantes par rapport aux cellules sénescentes, après une préparation de halo d’ADN. Cependant, il y a significativement plus de loci de caténine alpha 1 CTNNA1 dans le halo d’ADN que de loci de cycline D1 CNDD1 , où il y en a très peu. La figure 5 montre les préparations de halo d’ADN avec des télomères en vert. L’arrière-plan est laissé délibérément haut pour permettre aux signaux des télomères d’être visualisés dans le halo d’ADN. Dans cet ensemble de données sur les cellules quiescentes, c’est-à-dire que les cellules qui ont été affamées de sérum pendant 7 jours ont été incluses et, il est intéressant de noter qu’il y a beaucoup plus de télomères non attachés et situés dans les halos d’ADN dans les cellules quiescentes que pour les cellules proliférantes et sénescentes. Sur la figure 5a , la proportion de télomères dans le halo d’ADN peut être observée, en particulier pour l’image « Expérience 2 ». Cela correspond à la figure 5b où le pourcentage moyen de télomères dans le halo d’ADN est d’environ 17% dans les cellules quiescentes. Il existe des preuves que tous les télomères dans les cellules sénescentes ne peuvent pas être considérés comme certains d’entre eux peut-être très courts.
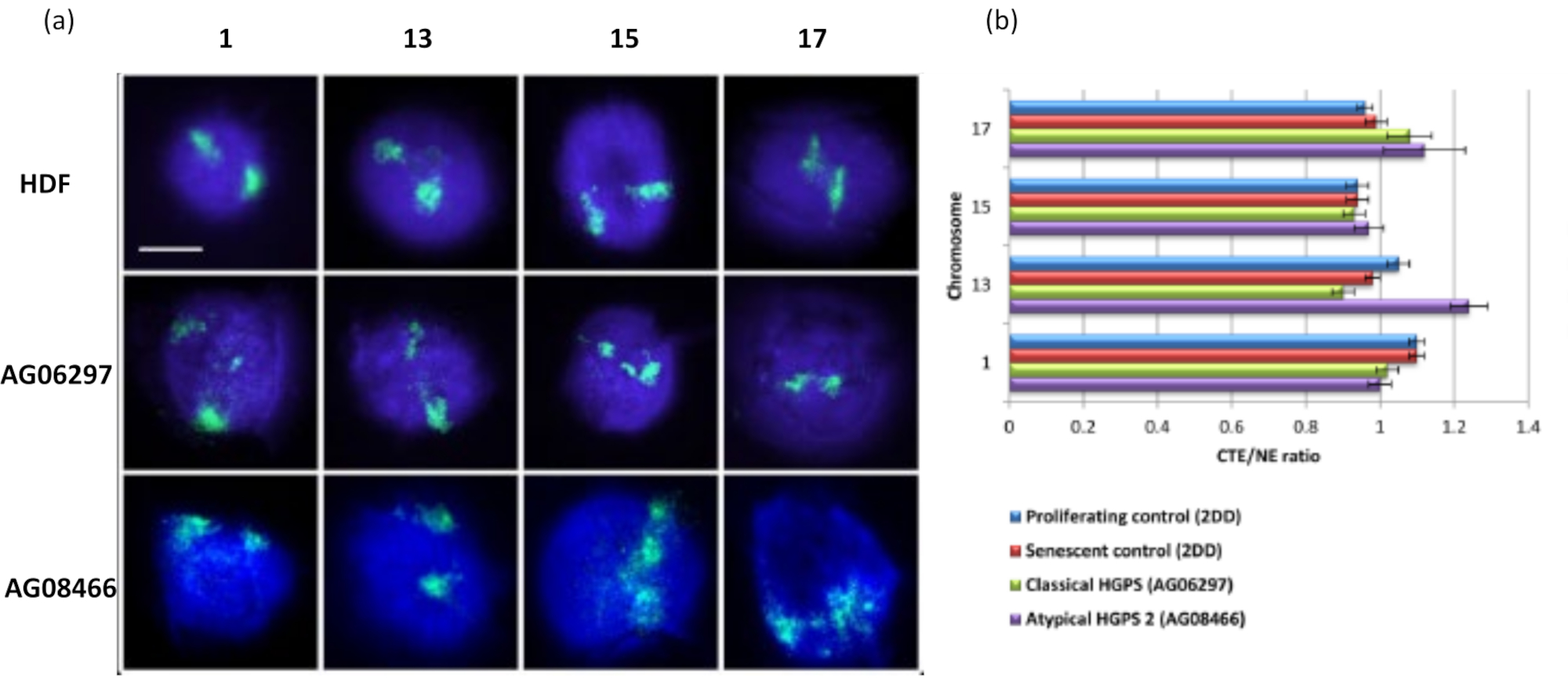
Cette méthode de halo d’ADN nous a permis d’étudier les altérations de l’interaction du génome dans les noyaux des cellules malades15. La figure 6 montre des différences dans l’attachement chromosomique dans les fibroblastes de contrôle primaire et dans les cellules malades présentant un syndrome typique (mutation de la lamine A) et atypique de Hutchinson-Gilford Progeria, exprimant une isoforme SUN1 différente et aucune mutation de la lamine A19. Les chromosomes 1 et 13 montrent des différences statistiquement significatives dans leur attachement dans les noyaux résiduels par rapport aux halos d’ADN témoins. La figure 6b met en corrélation la position de l’ensemble du territoire chromosomique au noyau résiduel et au halo d’ADN. Les valeurs de 1 ou moins indiquent que le chromosome est situé dans le noyau résiduel et les valeurs supérieures à 1 indiquent les chromosomes ou les parties de chromosomes dans le halo d’ADN.
Dans l’ensemble, cela met en évidence l’utilité de HALO-FISH dans l’étude des interactions génomiques de chromosomes entiers, de gènes spécifiques et de télomères dans diverses conditions qui affectent le cycle cellulaire (prolifération, quiescence et sénescence) ou au sein des cellules pathogènes, par exemple la progéria et les lignées cellulaires cancéreuses. En effet, les différences d’interactions entre ces états impliquent que le nucléosquelette joue un rôle important dans la régulation des processus clés au sein du noyau.

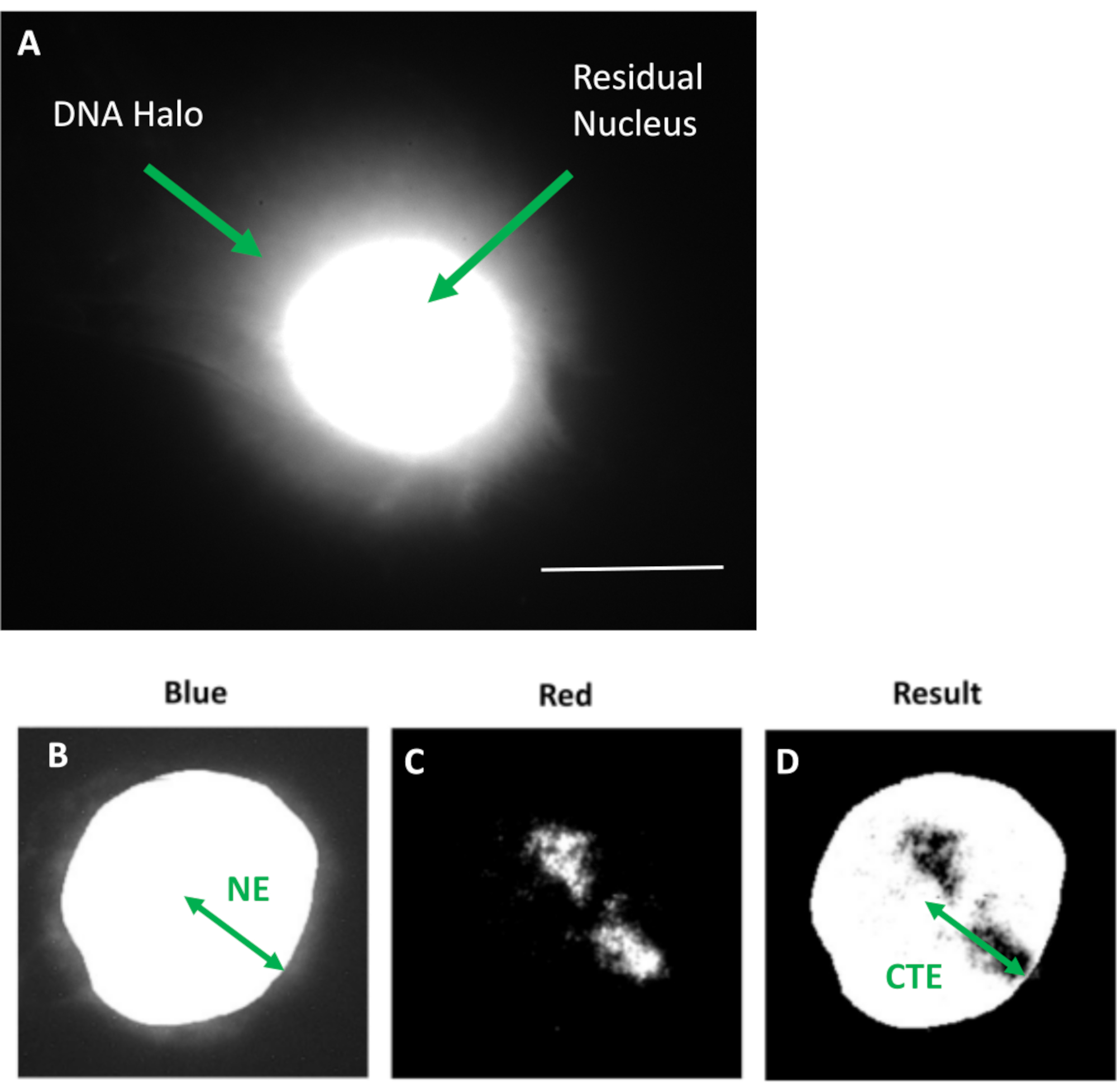
Figure 1 : Noyau extrait par HDF affichant le noyau résiduel et le halo d’ADN et aperçu de la méthode d’analyse. a) Un noyau HDF préparé par dosage du halo ADN et contre-coloré avec du DAPI. Le noyau résiduel coloré de couleurs vives montre de l’ADN ancré au nucléosquelette et celui-ci est entouré par l’ADN non attaché qui forme un halo d’ADN. Grossissement = x 100; barre d’échelle 10 μm. (b) Le canal bleu capture le noyau coloré par DAPI et l’ADN environnant. Le noyau résiduel est sélectionné et éliminé à l’aide d’ImageJ. La flèche représente la distance entre le centre nucléaire et le bord nucléaire résiduel (NE). c) Le canal rouge indique le signal de la sonde. d) L’image portant la mention « Résultat » est le résultat de la superposition du canal rouge sur l’image du canal bleu; cela permet la distance entre le centre nucléaire et le bord du territoire chromosomique (CTE) le plus éloigné. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Préparation du halo d’ADN avec le télomère PNA FISH sur les HDF proliférants et sénescents. Télomère PNA FISH sur HDF soumis à un test de halo d’ADN. Les signaux des télomères sont visualisés en vert (FITC), l’ADN résiduel et halo a été contre-coloré à l’aide de DAPI (bleu) et les noyaux proliférants ont été détectés à l’aide d’anticorps anti-BrdU ou anti-pKi67 via une immunofluorescence indirecte en rouge (TRITC). Grossissement = x 100; Barre d’échelle 10 μm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Interactions nucléosquelettique-chromosome et analyse à l’aide du test du halo de l’ADN. a) Le 2D-FISH avec des sondes spécifiques des chromosomes 1, 13, 15, 17 et 18 a été réalisé sur des HDF soumis à une préparation de halo d’ADN. Les chromosomes entiers ont été peints en rouge (Cy3) et les noyaux ont été sondés avec pKi67 pour déterminer s’ils proliféraient ou sénescents. Les cellules proliférantes (pKi67+) ont été délimitées en vert (FITC), tandis que les cellules sénescentes sont restées non colorées (pKi67-), c’est-à-dire qu’aucun signal vert n’a été détecté. Grossissement = x 100; barre d’échelle 10 μm. b) Ancrage chromosomique par le nucléosquelette dans les HDF proliférants et sénescents ayant subi HALO-FISH. Les mesures montrent le rapport entre le bord du territoire chromosomique (CTE) le plus éloigné et le bord nucléaire (NE) respectif pour les chromosomes 1, 13, 15, 17 et 18 dans les cellules proliférantes (pKi67+) et sénescentes (pKi67-). Les barres d’erreur représentent ± SEM. (c) Représentation modifiée du bord du territoire chromosomique (CTE) au bord nucléaire respectif (NE) de chromosomes spécifiques dans les noyaux pKi67+ et pKi67-. Q1 = quartile inférieur; Min = valeur la plus basse enregistrée; Med = médiane; Max = valeur maximale enregistrée; Q3 = quartile supérieur. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Interactions spécifiques aux gènes dans les HDF à l’aide de HALO-FISH. a) Les noyaux extraits du halo d’ADN ont été sondés à l’aide de sondes génétiques spécifiques (CCND1 et CTNNA1) afin d’étudier leur ancrage au NM sur des cellules proliférantes et sénescentes. Les signaux génétiques sont affichés en rouge (Cy3) et l’anti-pKi67 représente les cellules proliférantes et le signal est visualisé en vert (FITC). Pour l’image CCND1 proliférante, le noyau résiduel est enfermé dans le cercle blanc, et l’espace entre le cercle blanc et le cercle vert représente le halo d’ADN. Grossissement = x 100; barre d’échelle 10 μm. (b) Les signaux spécifiques aux gènes pour CCND1 et CTNNA1 sont comparés entre le noyau résiduel et le halo d’ADN, ainsi qu’entre les cellules proliférantes et sénescentes. Les barres d’erreur représentent ± SEM. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 5 : Test de halo d’ADN sur des HDF quiescents sondés avec le télomère PNA-FISH. a) La quiescence des HDF a été induite par culture dans un milieu sérique bas pendant 7 jours. Le test de halo d’ADN a été effectué et PNA-FISH a permis la visualisation des télomères par signal FITC (vert) et le noyau résiduel et le halo d’ADN environnant ont été contre-colorés avec DAPI (bleu). Les cellules ont également été colorées avec des anticorps anti-pKi67 pour s’assurer que les noyaux ne proliféraient pas. Cela s’est répété à deux reprises. Grossissement = x 100; barre d’échelle 10 μm. (b) Comparaison du pourcentage moyen de télomères localisés dans le halo d’ADN dans les cellules HDF proliférantes, sénescentes et quiescentes. Les barres d’erreur représentent ± SEM. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 6: Examen de l’ancrage chromosomique entier au nucléosquelette dans les cellules HGPS à l’aide de HALO-FISH26. a) Les noyaux de contrôle HDF (2DD), HGPS classique (AG06297) et HGPS atypique de type 2 (AG08466) ont subi une préparation au halo d’ADN, puis 2D-FISH en utilisant des peintures chromosomiques entières pour les chromosomes 1, 13, 15 et 17. Les chromosomes entiers sont représentés en vert (FITC) et l’ADN a été contre-coloré avec DAPI (bleu). Grossissement = x 100; barre d’échelle 10 μm. (b) Le positionnement des chromosomes dans les noyaux extraits a été déterminé en mesurant le rapport entre le bord moyen du territoire chromosomique (CTE) et le bord nucléaire (NE). Un rapport supérieur à 1 démontre que le CTE le plus éloigné se trouve à l’extérieur du NE correspondant dans le halo d’ADN, tandis qu’un rapport inférieur à 1 signifie que le CTE le plus éloigné se trouve dans le NE dans le noyau résiduel. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
| Constituants | Volume (μL) |
| 5XDOP-PCRbuffer | 10 |
| dNTPmix(sansTTP)(2mM) | 5 |
| dTTP (2mM) | 2 |
| Biotine-16-dUTPoudigoxigénine-11-dUTP | 10 |
| DOPprimer(20μM) | 5 |
| TaqDNAPolymérase (1U/μL) | 1 |
| PCRgradeeau | 12 |
| Modèle | 5 |
Tableau 1 : Tableau montrant les composants et les volumes de la DP-PCR pour une réaction 1x
| Pas | Cycles | Temp (degré centigrade) | Heure |
| Dénaturation initiale | 1 | 95 | 3 min |
| Dénaturation | 34 | 98 | 20 s |
| Recuit d’apprêt | 62 | 1 min | |
| Extension | 72 | 30 s | |
| Prolongation finale | 1 | 72 | 5 min |
| Refroidissement | 4 | Tenir |
Tableau 2 : Tableau montrant le cycle DOP-PCR, la température et le profil temporel.
| Constituant | Volume (μL) |
| 10x tampon NT (0,5M Tris-HCl pH 8,50 mM MgCl2, 0,5 mg/ml BSA) | 5 |
| 0,1 M bêta-mercaptoéthanol | 5 |
| 10X Stock de nucléotides (0,5 mM dATP, 0,5 mM dCTP, 0,5 mM dGTP, 0,5 mM dTTP, 0,5 mg/ml de biotine-16-dUTP) | 5 |
| Dnase I (1 ng/ml) | 2 |
| ADN polymérase I | 5U par μg d’ADN |
| Modèle d’ADN (1 μg) | 1 |
| Eau traitée DEPC | À 50 μL |
Tableau 3 : Tableau montrant les composants et les volumes de translation des entailles pour une sonde.
Discussion
La méthode du halo d’ADN est une excellente méthode de choix pour analyser les interactions entre le nucléosquelette et le génome, mais certaines étapes critiques doivent également être respectées. L’un des paramètres les plus importants est l’optimisation de la densité d’ensemencement cellulaire. Si les cellules deviennent confluentes, les halos d’ADN se chevaucheront avec les cellules voisines, ce qui rendra impossible l’analyse. Le CSK et les tampons d’extraction doivent toujours être renouvelés le jour de l’utilisation avec de la spermine, de la spermidine et de la digitonine ajoutées au tampon d’extraction à la fin du processus de préparation pour maintenir leur activité biologique. Si vous effectuez Halo-FISH, il est extrêmement important d’utiliser la température de dénaturation correcte des halos d’ADN pour permettre à la sonde ou à la peinture de s’hybrider par la suite.
La microscopie électronique a été utilisée pour visualiser la matrice nucléaire, les structures filamenteuses étant identifiées20. Cependant, la microscopie électronique est limitée car les associations matricielles avec la chromatine ne peuvent pas être facilement déduites. En effet, la méthode DNA Halo est plus polyvalente que la microscopie électronique car des gènes, des chromosomes et des états cellulaires spécifiques peuvent tous être examinés. En outre, l’analyse protéomique des protéines de la matrice nucléaire est à l’étude21,22. Cette méthode est bonne pour comparer les composants de la matrice nucléaire, en particulier lors de la comparaison de cellules malades, mais elle ne fournit pas la distribution spatiale et les attachements mis en évidence par la technique standard DNA Halo.
Les tests ADN Halo ont des limites. Tout d’abord, lorsque la matrice est extraite, cela ne peut être effectué que sur des cellules fixes, de sorte que l’imagerie en direct n’est pas possible. Bien que la méthode DNA Halo soit relativement rapide et facile à réaliser, le processus global peut prendre beaucoup de temps lorsque la culture cellulaire, la génération de sonde, Halo-FISH et l’analyse sont toutes prises en compte.
La capture d’images de DNA Halos et de HALO-FISH à l’aide de la microscopie à super-résolution améliorerait considérablement la résolution des sondes et des anticorps spécifiques à l’ADN. En outre, comme les fluorochromes peuvent être résolus plus facilement spectralement, il peut être possible d’utiliser un certain nombre de sondes ADN dans une seule expérience, fournissant encore plus d’informations. Des améliorations dans les techniques de biologie moléculaire telles que la capture de conformation chromosomique (3C) ont été utilisées pour déterminer les interactions des loci de gènes et analyser l’organisation spatiale de la chromatine dans la cellule. Les tests DNA Halo et 3C peuvent être combinés, un terme connu sous le nom de M3C23, démontrant à nouveau l’adaptabilité de la technique DNA Halo.
Les données originales présentées ici visent à démontrer les possibilités d’interrogation du comportement du génome et comment présenter ces données. Avec ces données, nous avons démontré qu’il est possible de déterminer des différences significatives dans l’attachement du génome en utilisant (1) des sondes de peinture chromosomique, dans cette étude révélant que le chromosome 18 est le chromosome le moins attaché parmi ceux analysés (Figure 3); (2) Loci géniques présentant des différences significatives entre deux loci géniques et (Figure 4) (3) Télomères, qui sont moins fortement attachés dans les cellules quiescentes que les cellules proliférantes et sénescentes (Figure 5). Nous sommes en mesure de différencier les cellules proliférantes et non proliférantes grâce à la présence du marqueur de prolifération antigène Ki67 qui est une protéine insoluble qui reste donc avec les noyaux résiduels ou en utilisant l’incorporation de nucléotides pour mettre en évidence les cellules qui ont traversé la phase S dans un laps de temps spécifique (Figure 2). Cette technique nous a également permis d’analyser le comportement du génome dans les cellules qui sont compromises dans leurs nucléo-squelettes, c’est-à-dire les cellules de laminopathie, et ici et dans Bikkul et al., 2018, nous révélons que le génome peut être moins étroitement attaché par rapport aux cellules témoins et peut être restauré lors du traitement avec des médicaments spécifiques qui améliorent l’effet de la mutation lamin A dans les cellules HGPS classiques15. Cependant, nous montrons ici de nouvelles données pour les cellules HGPS AGO8466 atypiques, dépourvues d’une mutation lamin A mais contenant une forme inhabituelle de la protéine complexe LINC SUN1 19 dans laquelle le chromosome13 est moins étroitement attaché (Figure 6).
HALO-FISH est une méthode unique qui permet l’étude des interactions génomiques avec le nucléosquelette en combinaison avec l’immunofluorescence indirecte pour résoudre les protéines non retirées de la procédure d’extraction. Il a été démontré que le nucléosquelette est modifié dans diverses maladies telles que certains types de cancer19 et l’importance de certaines protéines associées au nucléosquelette comme biomarqueurs diagnostiques24,25. Ainsi, cette technique a un rôle important dans l’examen de l’effet du nucléosquelette sur l’organisation/désorganisation de la chromatine dans la maladie 15,24,25,27 et n’est pas limitée aux cellules humaines, avec des sondes de peinture chromosomique d’autres animaux, le même protocole ADN-halo pourrait être utilisé 28.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous tenons à remercier le professeur Michael Bittner pour l’aimable cadeau des sondes de peinture de bras chromosomiques. LG a été soutenu par le projet EURO-Laminopathies financé par l’UE et le Fonds de recherche Brunel Progeria.
matériels
| Name | Company | Catalog Number | Comments |
| 10X PBS | Thermo Fisher Scientific | 10388739 | Used to create DNA halos |
| 5-bromo-2′-deoxy-uridine | Sigma-Aldrich | B5002-100MG | Labelled nucleotide |
| 5-Fluoro-2′-deoxyuridine | Sigma-Aldrich | F0503-100MG | Labelled nucleotide |
| Agar Technical | Thermo Fisher Scientific | 15562141 | DNA isolation of BAC clones |
| Agarose | Sigma-Aldrich | A939-50G | Check product size of DOP-PCR and nick translation |
| Atypical type 2 HGPS fibroblasts (AG08466) | Coriell Institute | AG08466 | Cell line |
| Bacto tryptone | Thermo Fisher Scientific | 16269751 | DNA isolation of BAC clones |
| Biotin-16-dUTP | Roche Diagnostics | 11093711103 | Labelled nucleotides |
| Chloramphenicol | Sigma-Aldrich | C0378-25G | DNA isolation of BAC clones |
| Classical Hutchinson-Gilford progeria syndrome (HGPS) fibroblasts (AG06297) | Coriell Institute | AG0297 | Cell line |
| Coplin jar | Thermo Fisher Scientific | 12608596 | Holds 5 slides or 8 slides back to back |
| Cot-1 DNA | Thermo Fisher Scientific | 15279011 | Block nonspecific hybridization in HALO FISH |
| DEPC-treated water | Sigma-Aldrich | 693520-1L | DNA isolation of BAC clones |
| Dextran sulphate | Sigma-Aldrich | S4030 | Hybridisation mixture |
| Digitonin | Sigma-Aldrich | D141 | Component of extraction buffer |
| Digoxigenin-11-dUTP | Sigma-Aldrich | 11093088910 | Labelled nucleotides |
| Donkey anti-mouse Cy3 | Jackson Laboratory | 715-165-150 | Secondary antibody |
| EDTA | Sigma-Aldrich | E6758 | Component of extraction buffer |
| Ethanol | Component of extraction buffer | ||
| Ethanol | Sigma-Aldrich | 443611 | Probe precipitation and HALO FISH |
| Fetal bovine system | Thermo Fisher Scientific | 26140079 | Cell culture serum |
| Formamide | Thermo Fisher Scientific | 10523525 | 2D FISH of DNA halos |
| Glass wool | Sigma-Aldrich | 18421 | Spin column |
| Herring sperm | Sigma-Aldrich | D7290 | Probe precipitation |
| HXP™ Lamp (metal halide microscope lamp) | OSRAM | HXP-R120W45C VIS | Image capture of DNA halos |
| Hydrochloric acid | Thermo Fisher Scientific | 10313680 | Cleaning microscope slides |
| Isopropanol | Sigma-Aldrich | I9516-25ML | DNA isolation of BAC clones |
| KAPA HiFi PCR Kit | KAPA Biosystems | KK2103 | PCR Kit |
| Leica DM4000 fluorescent microscope with DFC365 FX camera and LAS AF (Version: 4.5.0) image acquisition software. | Leica Microsystems | Image capture of DNA halos | |
| Luria-Bertani agar | Thermo Fisher Scientific | 13274843 | DNA isolation of BAC clones |
| Magnesium chloride | Sigma-Aldrich | M8266 | Component of CSK buffer |
| Methanol | Thermo Fisher Scientific | 10284580 | Cleaning and sterilizing microscope slides |
| Mouse anti-BrdU antibody | BD Pharmingen | B2531-100UL | BrdU visualisation |
| Newborn calf serum | Thermo Fisher Scientific | 16010159 | Cell culture serum and blocking reagent |
| Nick translation kit | Invitrogen | ||
| PCR grade water | Sigma-Aldrich | 693520-1L | PCR and DNA isolation of BAC clones |
| PCR Primers | Sigma-Aldrich | ||
| PIPES | Sigma-Aldrich | P1851 | Component of CSK and extraction buffers |
| Potassium acetate | Sigma-Aldrich | P1190-100G | DNA isolation of BAC clones |
| QuadriPERM® 4 X 12 | SARSTEDT | 94.6077.307 | Square cell culture dish, polysterene with four compartments. This has hydrophobic surface, is sterile, non-pyrogenic/endotoxin-fee and non-cytotoxic. |
| Rabbit Anti-Ki67 antibody | Sigma-Aldrich | ZRB1007-25UL | Proliferation marker |
| Rnase A | Sigma-Aldrich | R6513 | DNA isolation of BAC clones |
| Rubber cement | Halford's | 101836 | 2D FISH of DNA halos |
| Sephadex G-50 | Sigma-Aldrich | S6022-25G | Spin column |
| Sodium acetate | Sigma-Aldrich | S2889 | Probe precipitation |
| Sodium chloride | Sigma-Aldrich | S5886 | Component of CSK, extraction and SSC buffers |
| Sodium citrate | Sigma-Aldrich | C8532 | Component of SSC buffer |
| Sodium dodecyl sulphate | L3771-100G | DNA isolation of BAC clones | |
| Sodium hydroxide | Sigma-Aldrich | S8045-500G | DNA isolation of BAC clones |
| Spermidine | Sigma-Aldrich | S2626 | Component of extraction buffer |
| Spermine | Sigma-Aldrich | S4264 | Component of extraction buffer |
| Streptavidin-Cy3 | Amersham Life Sciences Ltd, Scientific Laboratory Supplies | pa43001 | Probe antibody |
| Sucrose | Sigma-Aldrich | S0389 | Component of CSK buffer |
| Sucrose | Sigma-Aldrich | S0389 | CSK buffer+A66:D68 |
| SuperFrost™ microscope slides | Thermo Fisher Scientific | 12372098 | Microscope slides: 1 mm thickness, 76 mm length, 26 mm width. Uncoated. |
| Swine anti-rabbit TRITC | Dako | ||
| TELO-PNA FISH KIT | Agilent Dako | K532511-8 | Delineation of telomeres |
| Tris-HCl | Sigma-Aldrich | T3253-100G | Column buffer |
| Triton™ X-100 | Sigma-Aldrich | T9284 | Component of CSK buffer |
| Tryptone | Thermo Fisher Scientific | 10158962 | DNA isolation of BAC clones |
| Tween-20 | Sigma-Aldrich | P9416- 100ML | Detergent |
| Vectashield mountant containing DAPI | Vector Laboratories | H-1200 | 2D FISH of DNA halos |
| Whole human chromosome probes | Calbiochem | 2D FISH of DNA halos | |
| Yeast extract | Thermo Fisher Scientific | 10108202 | DNA isolation of BAC clones |
Références
- Berezney, R., Coffey, D. S. Identification of a nuclear protein matrix. Biochemical Biophysical Research Communications. 60 (4), 1410-1417 (1974).
- Haaf, T., Ward, D. C. High resolution ordering of YAC contigs using extended chromatin and chromosomes. Human Molecular Genetics. 3 (4), 629-633 (1994).
- Parra, I., Windle, B. High resolution visual mapping of stretched DNA by fluorescent hybridization. Nature Genetics. 5 (1), 17-21 (1993).
- Senger, G., et al. Released chromatin: linearized DNA for high resolution fluorescence in situ hybridization. Human Molecular Genetics. 3 (8), 1275-1280 (1994).
- Florijn, R. J., et al. High-resolution DNA Fiber-FISH for genomic DNA mapping and colour bar-coding of large genes. Human Molecular Genetics. 4 (5), 831-836 (1995).
- Elcock, L. S., Bridger, J. M. Fluorescence in situ hybridization on DNA halo preparations and extended chromatin fibres. Methods Molecular Biology. 659, 21-31 (2010).
- Heiskanen, M., et al. Visual mapping by fiber-FISH. Genomics. 30 (1), 31-36 (1995).
- Bensimon, A., et al. Alignment and sensitive detection of DNA by a moving interface. Science. 265 (5181), 2096-2098 (1994).
- Michalet, X., et al. Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science. 277 (5331), 1518-1523 (1997).
- Wilson, R. H., Coverley, D. Relationship between DNA replication and the nuclear matrix. Genes Cells. 18 (1), 17-31 (2013).
- Wilson, R. H. C., Coverley, D. Transformation-induced changes in the DNA-nuclear matrix interface, revealed by high-throughput analysis of DNA halos. Science Reports. 7 (1), 6475 (2017).
- Iarovaia, O. V., Akopov, S. B., Nikolaev, L. G., Sverdlov, E. D., Razin, S. V. Induction of transcription within chromosomal DNA loops flanked by MAR elements causes an association of loop DNA with the nuclear matrix. Nucleic Acids Research. 33 (13), 4157-4163 (2005).
- Tandara, M., et al. Sperm DNA integrity testing: big halo is a good predictor of embryo quality and pregnancy after conventional IVF. Andrology. 2 (5), 678-686 (2014).
- Repping, S., et al. The use of spermHALO-FISH to determine DAZ gene copy number. Mol Human Reproduction. 9 (4), 183-188 (2003).
- Bikkul, M. U., et al. Farnesyltransferase inhibitor and rapamycin correct aberrant genome organisation and decrease DNA damage respectively, in Hutchinson-Gilford progeria syndrome fibroblasts. Biogerontology. 19 (6), 579-602 (2018).
- Telenius, H., et al. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 13 (3), 718-725 (1992).
- Bridger, J. M., et al. Association of pKi-67 with satellite DNA of the human genome in early G1 cells. Chromosome Research. 6, 13-24 (1998).
- Sales Gil, R., Vagnarelli, P. Ki-67: More Hidden behind a 'Classic Proliferation Marker'. Trends in Biochemical Sciences. 43 (10), 747-748 (2018).
- Bikkul, M. U., et al. Telomere elongation through hTERT immortalization leads to chromosome repositioning in control cells and genomic instability in Hutchinson-Gilford progeria syndrome fibroblasts, expressing a novel SUN1 isoform. Genes Chromosomes Cancer. 58 (6), 341-356 (2019).
- Jackson, D. A., Cook, P. R. Visualization of a filamentous nucleoskeleton with a 23 nm axial repeat. EMBO Journal. 7, 3667-3677 (1988).
- Albrethsen, J., et al. Unravelling the nuclear matrix proteome. Journal of Proteomics. 72, 71-81 (2009).
- Mika, S., Rost, B. NMPdb: Database of nuclear matrix proteins. Nucleic Acids Research. 33, 160-163 (2005).
- Gavrilov, A. A., et al. of the nuclear matrix-bound chromatin hubs by a new M3C experimental procedure. Nucleic Acids Research. 38, 8051-8060 (2010).
- Sjakste, N., et al. Role of the nuclear matrix proteins in malignant transformation and cancer diagnosis. Experimental Oncology. 26 (3), 170-178 (2004).
- Leman, E. S., Getzenberg, R. H. Nuclear structure as a source of cancer specific biomarkers. Journal of Cellular Biochemistry. 104 (6), 1988-1993 (2008).
- Volpi, E. V., Bridger, J. M. FISH glossary: an overview of the fluorescence in situ hybridization technique. Biotechniques. 45 (4), 385-386 (2008).
- Bridger, J. M., Foster, H. A. Senescence and the Genome. Human Interphase Chromosomes. , (2021).
- Foster, H. A., Griffin, D. K., Bridger, J. M. Interphase chromosome positioning in in vitro porcine cells and ex vivo porcine tissues. BMC Cell Biology. 13 (1), 30 (2012).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.