
Method Article
Generation of Zebrafish Larval Xenografts and Tumor Behavior Analysis
In This Article
Summary
Here, we provide a step-by-step protocol, with tips to generate xenografts and guidelines for tumor behavior analysis, whole-mount immunofluorescence, and confocal imaging quantification.
Abstract
Zebrafish larval xenografts are being widely used for cancer research to perform in vivo and real-time studies of human cancer. The possibility of rapidly visualizing the response to anti-cancer therapies (chemo, radiotherapy, and biologicals), angiogenesis and metastasis with single cell resolution, places the zebrafish xenograft model as a top choice to develop preclinical studies.
The zebrafish larval xenograft assay presents several experimental advantages compared to other models, but probably the most striking is the reduction of size scale and consequently time. This reduction of scale allows single cell imaging, the use of a relatively low number of human cells (compatible with biopsies), medium-high-throughput drug screenings, but most importantly enables a significant reduction of the time of the assay. All these advantages make the zebrafish xenograft assay extremely attractive for future personalized medicine applications.
Many zebrafish xenograft protocols have been developed with a wide diversity of human tumors; however, a general and standardized protocol to efficiently generate zebrafish larval xenografts is still lacking. Here we provide a step-by-step protocol, with tips to generate xenografts and guidelines for tumor behavior analysis, whole-mount immunofluorescence, and confocal imaging quantification.
Introduction
Zebrafish (Danio rerio) is emerging as a powerful vertebrate model organism to study development and disease. Zebrafish shares highly conserved genetic (~70% genetic homology and ~84% disease-related genes) and basic organ morphological features with humans1,2. This conservation allows the use of zebrafish to model several human diseases, including cancer3,4.
The handling and maintenance of zebrafish is much easier and more cost effective than mice due to their small size, high fecundity all year round and external fertilization3,5. Zebrafish embryos do not require live feeding during their first 5-7 days of life and have been used as an effective model for development, infection, and cancer1,4,6,7. Zebrafish embryos hatch at 48 hours post-fertilization (hpf) and are free-swimming animals with all organs formed, a beating heart and functional circulatory system, liver, brain, kidney marrow, etc.1,3. Also, at this stage of development only innate immunity is at play, adaptive immunity is still developing, allowing a general efficient engraftment of human cells with no need for use of immunocompromised mutants7,8. Nevertheless, it is important to note that not all human cells engraft equally9 and that, for instance, for leukemia cells it was shown that phagocytes (neutrophils and macrophages) need to be depleted for efficient engraftment10.
The zebrafish genetic tractability and the optical transparency of its early embryonic stages allow for single cell intravital imaging at a high resolution and thus, for the establishment of state-of-the art imaging techniques in diverse fields of biology. Furthermore, in the context of cancer, these features are useful for real-time studies of the earliest stages of host-tumor interactions, like studying angiogenic and metastatic potential, as well as interactions with the innate immune system8,9,11,12,13.
Although in the short xenograft assay there is no time for metastatic "evolution"- it is possible to analyze the metastatic capacity of the tumor cells (i.e., their efficiency to go through metastatic steps like invasion, intravasation, survival in circulation, extravasation, and colonization, and therefore study these processes in vivo and in real-time8,11,13,14).
The characteristics of its life cycle place the zebrafish as a unique model for personalized medicine in cancer. Assays can be performed in a shorter time span and results obtained in a few weeks7,8,9,11,12,15,16. The celerity and feasibility of these assays provide doctors and researchers the possibility of obtaining translational results that can be useful for cancer patients, for whom time is an essential need.
Despite the increasing attempts at generating successful zebrafish embryo xenografts, there is still a need for the standardization of the injection procedure as well as the evaluation of cell viability and tumor behavior after injection.
In this protocol we provide researchers with a clear and detailed step-by-step guide for the injection of human cancer cell lines in zebrafish embryos and subsequent fixation, immunostaining, imaging, and quantification of tumor cell behavior.
Protocol
The zebrafish (Danio rerio) model was handled and maintained according to the standard protocols of the European Animal Welfare Legislation, Directive 2010/63/EU (European Commission, 2016) and Champalimaud Fish Platform. All protocols were approved by the Champalimaud Animal Ethical Committee and Portuguese institutional organizations-ORBEA (Órgão de Bem-Estar e Ética Animal/Animal Welfare and Ethics Body) and DGAV (Direção Geral de Alimentação e Veterinária/Directorate General for Food and Veterinary).
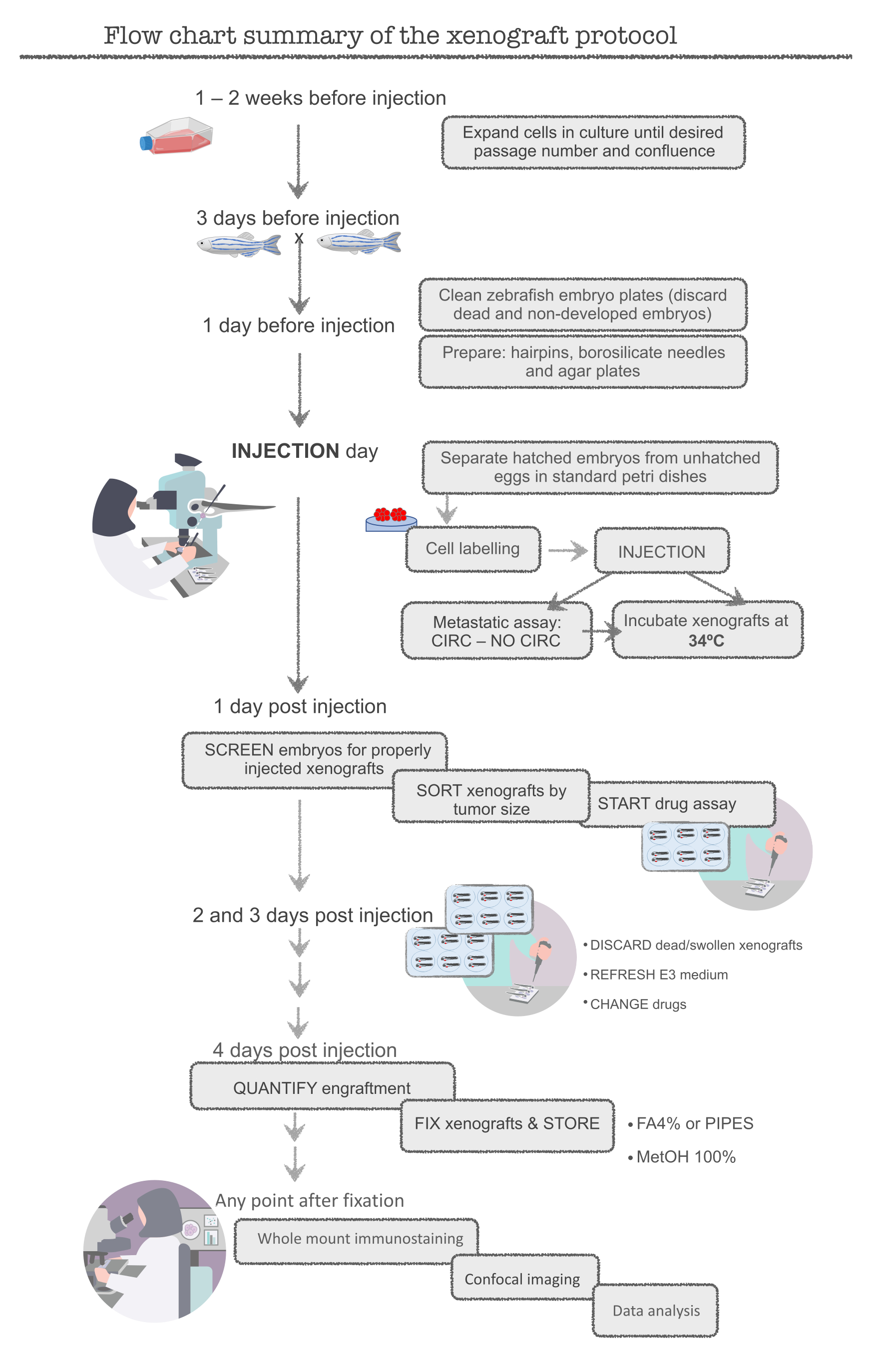
NOTE: Before starting the main experiment, practice with the human colorectal cancer (CRC) cell line HCT116. This cell line is easy to prepare (highly proliferative), easy to inject and engrafts very efficiently (around 95-100%). Start with cells in excess (~12x106 cells, T-75 flask) and excess fish (400 fish) until becoming proficient in the technique, since many cells and fish will be lost during training. Experimenters are ready once the engraftment of ~95% is achieved in HCT116 xenografts. See Figure 1 for a schematic of the complete protocol.
1. Setting up for injection
- Two weeks before injection, expand cells in culture (see Table 1 for a detailed guide of the optimal in vitro confluence for injection of several cell lines).
- Three days before the injection, cross the zebrafish of the desired background.
2. 24 h before injection
- Clean the zebrafish embryo plates (discard all dead and non-developed embryos) and refresh E3 medium.
- In the cell culture room, discard the cell culture medium from the flasks planned for injection, wash once with 1x phosphate-buffered saline (PBS) to remove the dead cells and add fresh medium.
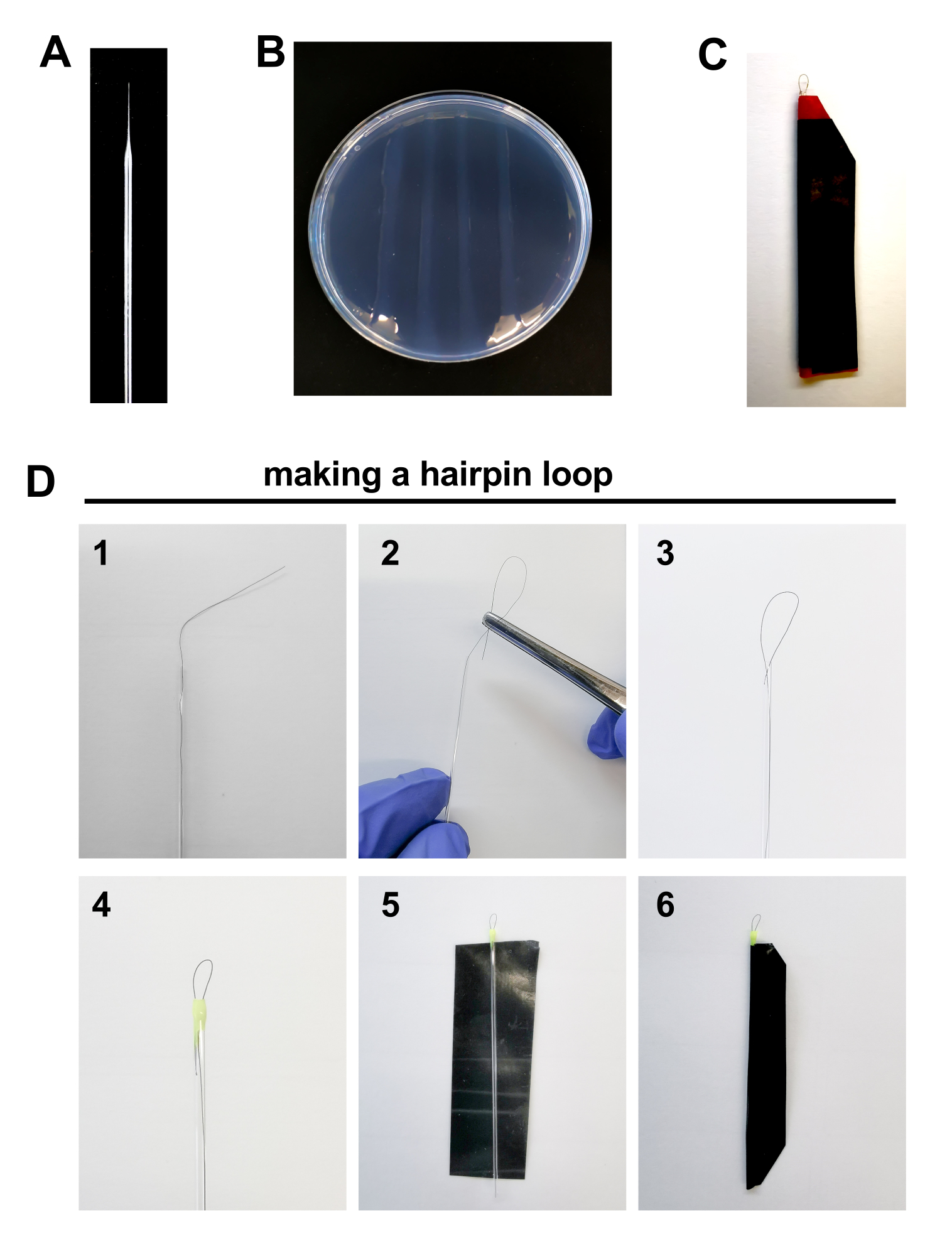
- Prepare the tools for the injection procedure, such as: microinjection needles, agarose plates and hairpins to align embryos for injection (Figure 2A-D, detailed below).
- Microinjection needles (Figure 2A)
- Use borosilicate glass capillaries (4 inches, OD 1.0 mm, No Filament in a micropipette puller (heat: ≈500; fil: 10; vel: 50; dec: 60; pull: 100).
- Plate preparation (Figure 2B)
- Prepare 2% agarose in H2O, heat it up and pour one layer of dissolved agarose in the lid of a clean Petri dish. Let it polymerize and with the help of a ruler, make three to four straight agarose lines for the alignment of the embryos.
- Hairpin assembly (Figure 2C-D)
- Place 1 hair inside a glass capillary tube leaving approximately 1 centimeter of hair outside the tube.
- Curl the outside tip of the hair with the help of forceps into the glass capillary tube forming a loop of ~0.5 mm length.
- Seal the edge of the capillary tube with a drop of nail polish. This will also help fix the loop in place. Let it dry. Repeat the procedure on the other edge of the tube.
- Cut a piece of electrical tape (more resistant, impermeable, and rigid than regular adhesive tape).
- Seal the tape around the capillary to protect it from breaking.
- Microinjection needles (Figure 2A)
3. Injection day
- Separate the hatched embryos from unhatched eggs. Adding 1x pronase (0.6 mg/mL, Table 3) to the embryo medium at this stage can boost hatching. Place the embryos into the incubator (at 28 °C) until injection.
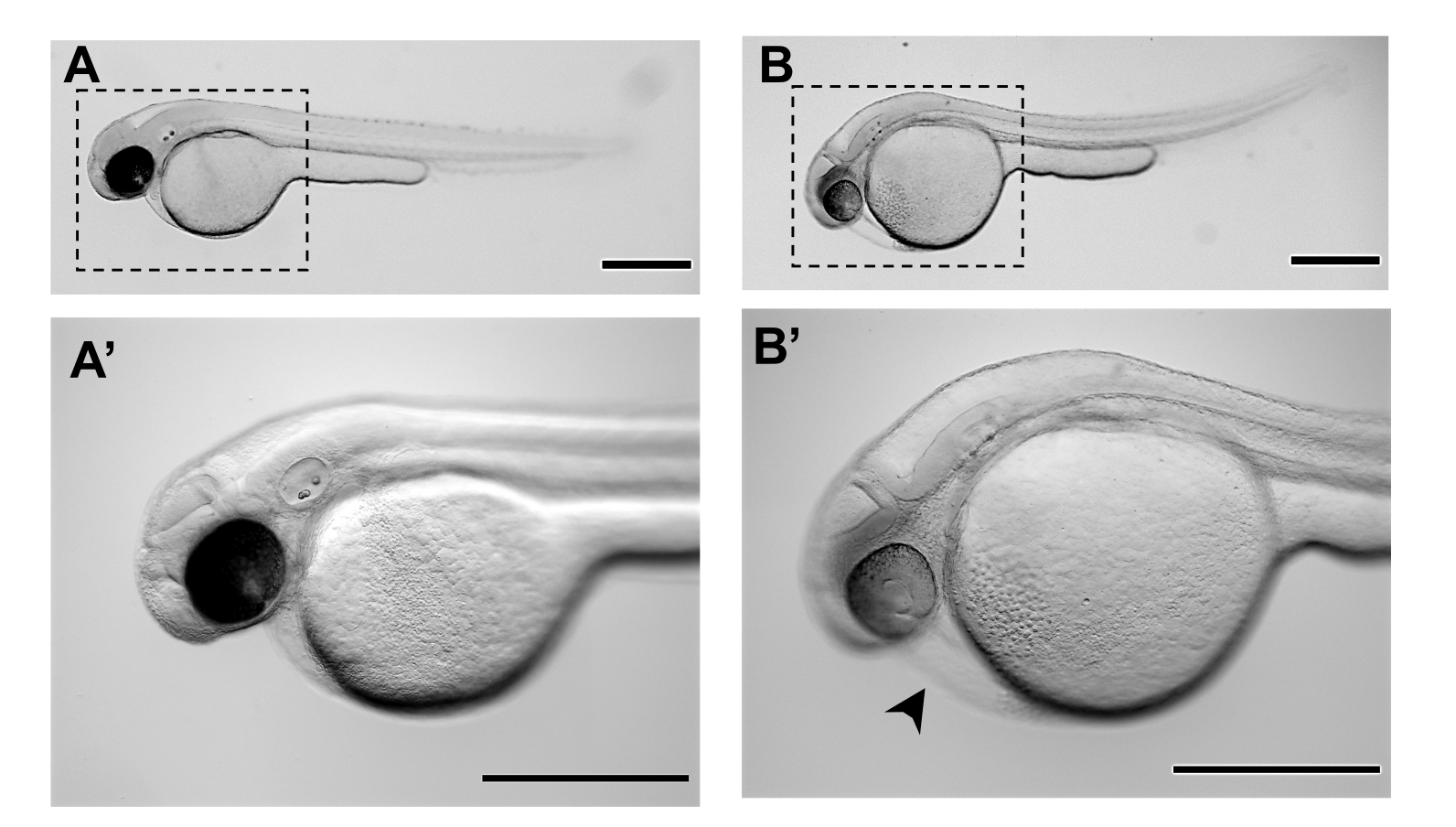
NOTE: Do not leave the embryos in the pronase solution for longer than 1 hour, since the enzyme will act on the hatched embryos increasing their risk of mortality. Ensure that the developmental stage of the embryos is the one corresponding to 48 hpf (Figure 3A, A') to avoid the risk of edema and mortality. - Prepare 1x Tricaine (from a 25x stock).
NOTE: A detailed recipe can be found in Table 3 and at the Zebrafish Information Network - ZFIN webpage17.
4. Cell labeling for injection
NOTE: Labelling of cells can be performed either directly in a flask or in a 1.5 mL microcentrifuge tube after enzymatic detachment. See Discussion for more information.
- Remove the cell culture medium and wash the flask twice (2x) with 1X PBS.
- Label the cells with a lipophilic dye of choice either directly in the flask (2 mL solution/T75 flask) or in a 1.5 mL microcentrifuge tube after enzymatic detachment. Avoid exposure to light and incubate cells at 37 °C (see Table 1 and Table 2 for conditions/solutions).
- If labelling in the flask
- Remove the dye, wash with 1x PBS and detach the cells with EDTA and a cell scraper.
- Transfer cells to 1.5 mL microcentrifuge tubes. Centrifuge for 5 minutes at 300 x g then go to step 4.5.
- If labelling in the 1.5 mL microcentrifuge tube:
- Centrifuge 5 minutes at 300 x g to remove the dye and discard the supernatant. Resuspend in 1x PBS to wash.
- Centrifuge 5 minutes at 300 x g and then go to step 4.5.
- Discard the supernatant and resuspend the pellet with cell culture medium (for a 50 µL pellet add ~ 150-200 µL of medium).
- Quantify cell viability using a Neubauer chamber with Trypan blue exclusion or other method of choice.
- Centrifuge for 4 minutes at 300 x g and discard the supernatant. Resuspend the cells in the injection medium.
NOTE: The recommended cell concentration (in general from 0.25-0.5x106 cells/µL) and medium can be found in Table 1. - From this point onwards, keep the cells on ice.
5. Injection procedure
- Anesthetize the embryos in 1x Tricaine for 5 minutes.
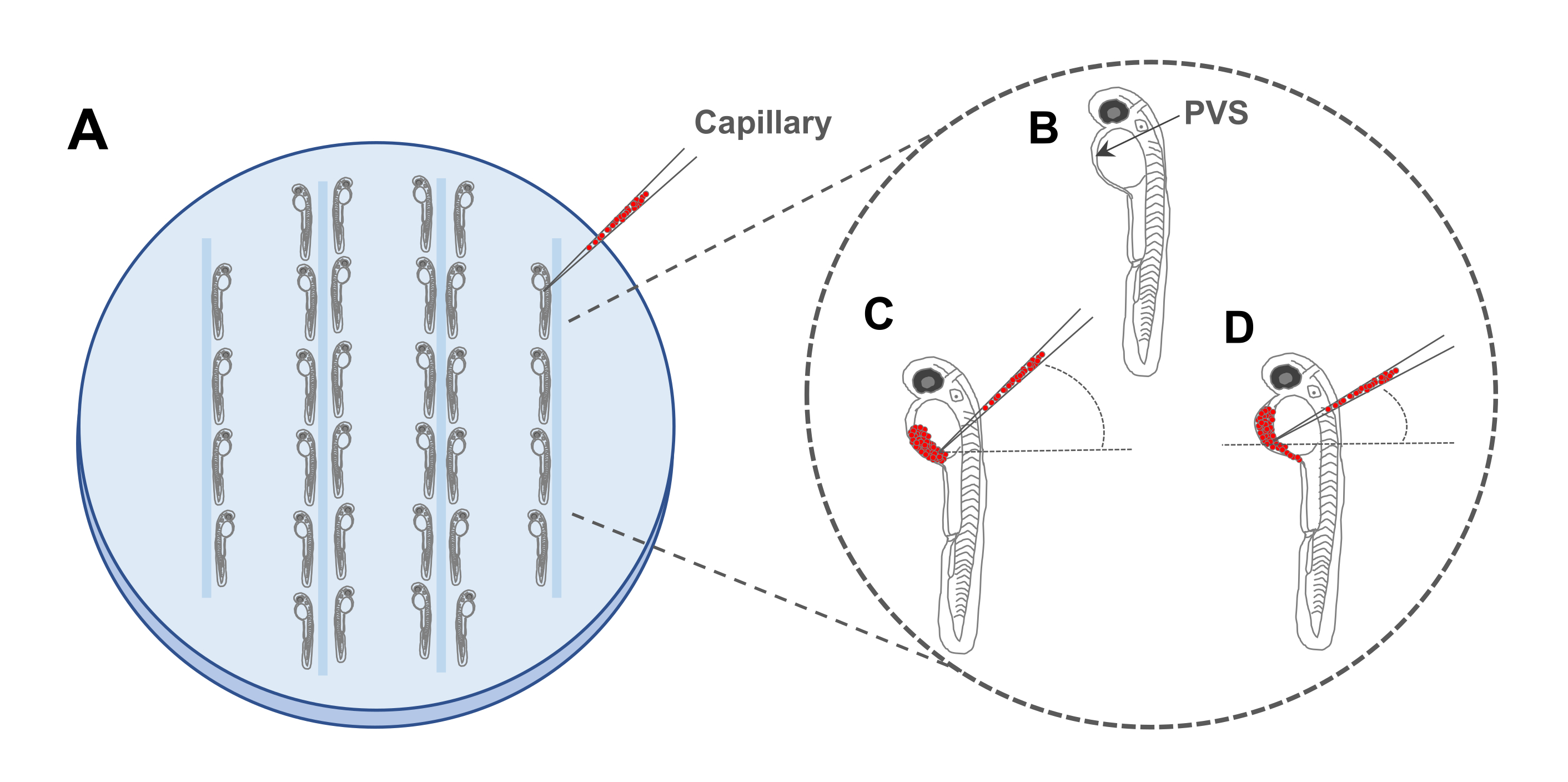
- With a plastic Pasteur pipette, transfer a small amount (~50) of anesthetized embryos to the agarose plate and carefully align them with the help of a hairpin loop. Make sure to maintain distance between the embryos, especially between the yolk of one and the head of the next one (Figure 4A).
NOTE: The number of embryos to align will vary according to the level of expertise of the researcher performing the injections. Start with a few (~10-20). For a schematic of the correct positioning of the embryos in the agar/agarose plate see Figure 4A. - Ensure the aligned embryos do not dry out in the agarose plate to prevent mortality, by carefully adding 1-3 drops of 1x Tricaine solution to the plate.
- Lightly tap the microcentrifuge tube to resuspend the cells. Backload the injection needle with the cell suspension using a microloader tip avoiding air bubbles, as they can compromise the integrity of the embryos.
- Open the air pressure valve (40 psi), set up the microinjector and carefully place the microinjection needle into the holder.
NOTE: Use the following recommended microinjector settings: Hold pressure - vent (3 psi); Eject pressure - vent; Range - 100 ms. - Cut the microinjection needle close to the tip with Dumont forceps #5 or similar under the stereomicroscope.
NOTE: The tip must be blunt and thin enough to allow the cells to pass without clogging as well as to avoid damaging the embryos and losing cells. A thick microinjection needle tip will injure the embryo and promote the formation of edema or zebrafish death. Graticule is not used for needle calibration. See Discussion for a detailed explanation. - Before injecting the embryos, test the microinjector pressure starting by the lowest eject pressure until in ~1-3 pulses a volume similar to the size of the zebrafish embryo's eye is achieved.
NOTE: The use of a fluorescence stereomicroscope whenever possible at the beginning of the training is recommended. This will allow an easier identification of fluorescently labelled cells. - Carefully pierce in the middle of the embryo's yolk, lowering the angle of the needle and cautiously push until the tip of the needle reaches the perivitelline space (PVS) (Figure 4B-D).
- Press the microinjector pedal and inject the cells into the PVS. Use the eye of the embryo as a guide. Try to inject a volume of cells similar to the size of the embryo's eye and as far as possible from the heart to prevent cardiac edema.
- Carefully remove the needle and move onto the next embryo.
- Adjust the microinjector pressure if needed.
NOTE: Cells tend to start clogging, so pressure may be increased. If needed, it is possible to cut the capillary (to increase the diameter) while reducing the pressure. - Transfer the injected embryos to a clean Petri dish (Table 4) with 1x Tricaine solution and leave them to rest for 5-10 minutes. This will give time for the wound to close.
NOTE: To transfer the embryos from the agar plate to the Petri dish add a few drops of 1x Tricaine solution on top of the embryos and carefully collect them with a plastic Pasteur pipette. Drop the collected embryos in a new Petri dish with 1x Tricaine solution. - Remove the 1x Tricaine solution and add fresh E3 medium.
- Incubate the xenografts at 34 °C (a compromised temperature between human cell lines survival and zebrafish development8).
6. Metastatic assay
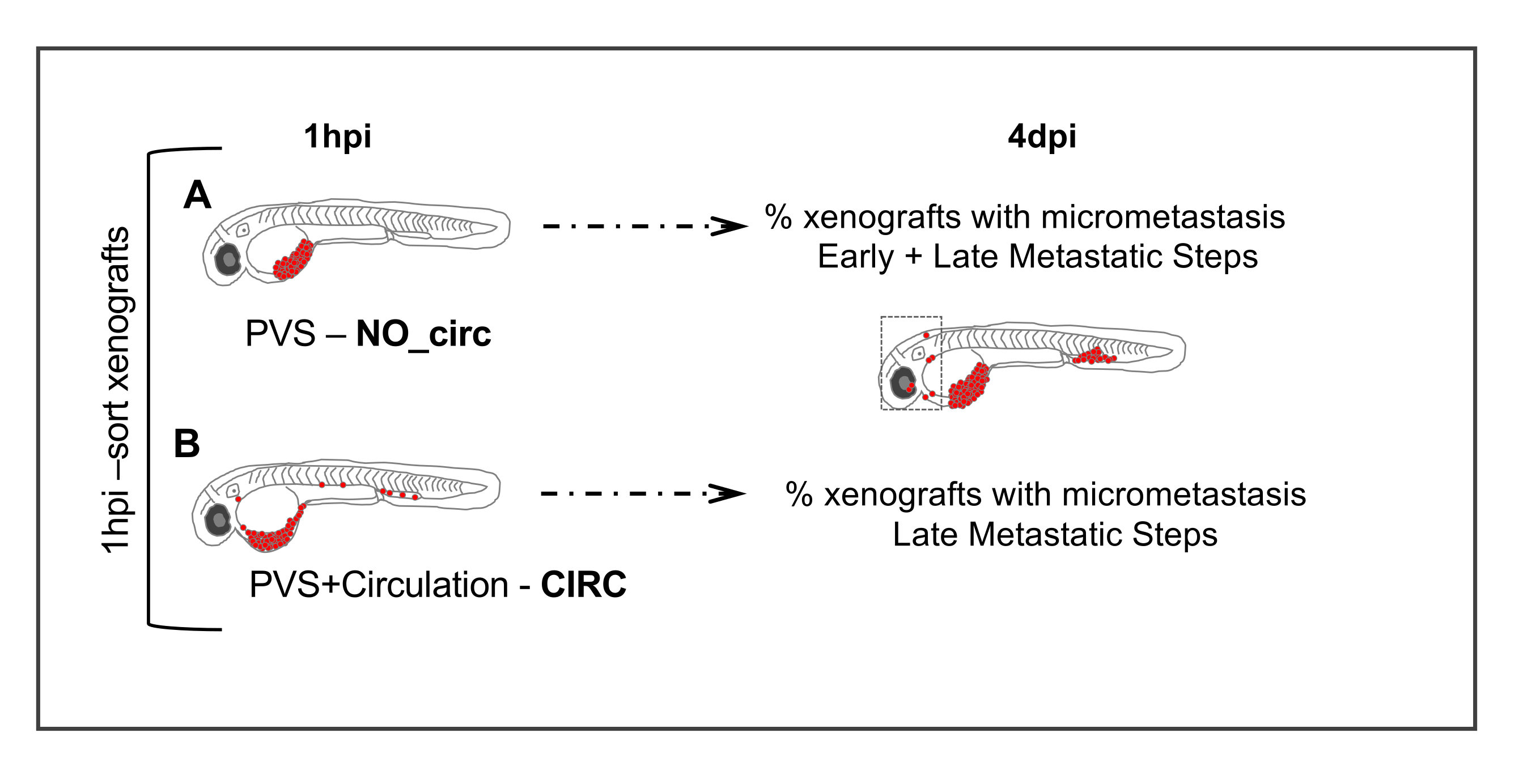
- At approximately 1-hour post-injection (hpi), screen the injected embryos on a fluorescent stereomicroscope and sort the xenografts into 2 groups, according to the absence (Figure 5A) or presence (Figure 5B) of cells in circulation.
NOTE: Even if only one cancer cell is detected in the heart or circulation, include these xenografts in the group of xenografts with cells in circulation. Alternatively, cells can be directly injected into circulation to increase the numbers of xenografts in this group.
7. 1 day post injection
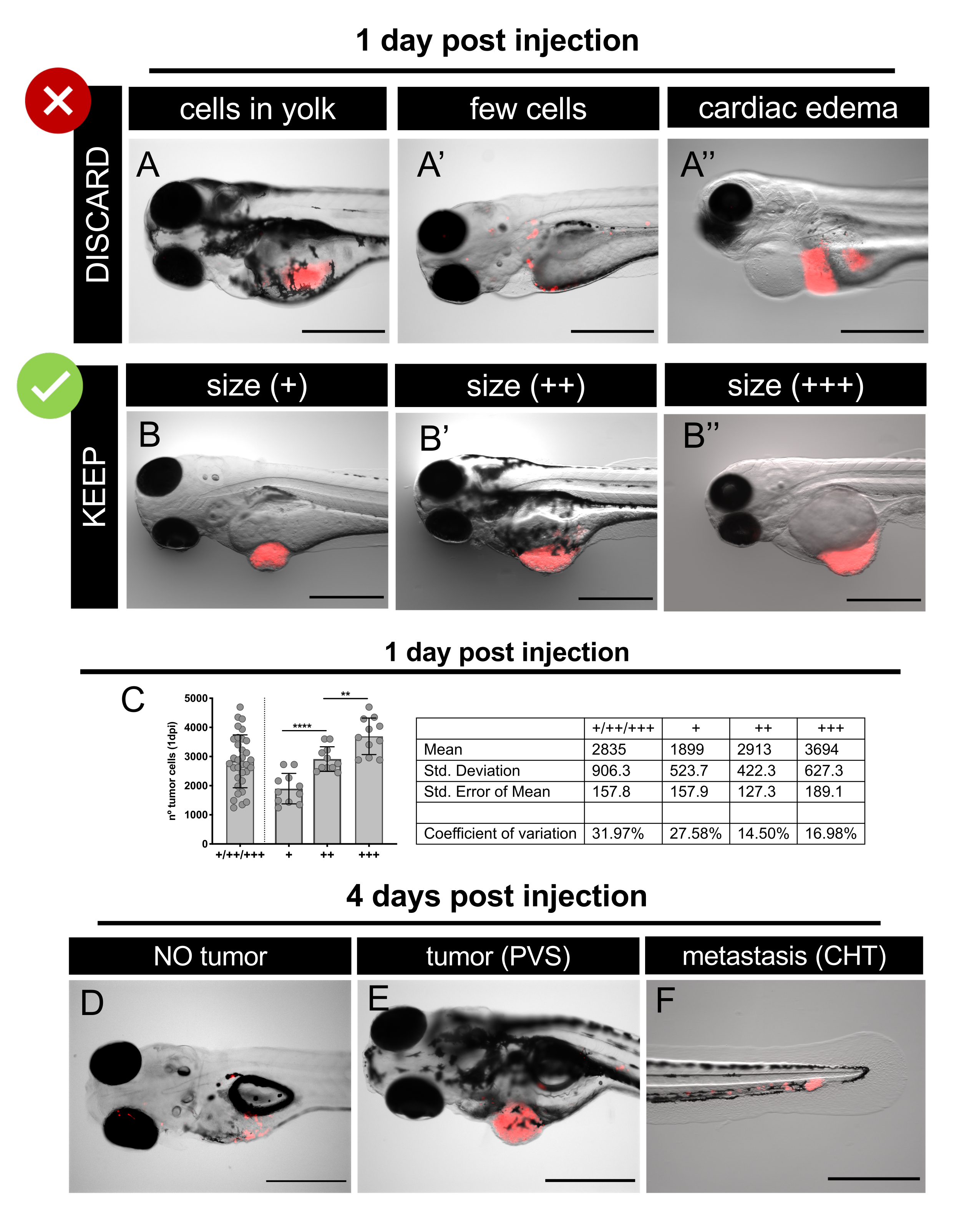
- On a fluorescent stereomicroscope carefully analyze each embryo and select those with properly injected tumors (Figure 6).
NOTE: If needed, anesthetize the injected embryos with Tricaine 1X solution before screening. - Discard the following embryos/xenografts (Figure 6A-A''):
• without tumor/abnormal morphology/dead,
• with cardiac and/or yolk edema,
• with tumor cells only in the yolk,
• with very few cancer cells. - Sort selected xenografts according to their tumor size. Use the size of the eye for comparison (Figure 6B-B'', 6C).
• Tumors smaller than the size of the eye (+),
• Tumors the same size as the eye (++),
• Tumors larger than the size of the eye (+++). - Distribute the xenografts according to the desired experimental layout and start the drug assay (control vs drug, etc.). Replace the drugs and E3 medium daily (Table 4).
- Incubate the xenografts maintaining the temperature of 34°C until the end of the assay.
8. 4 days post injection
- On the final day of the assay, anesthetize the xenografts with 1x Tricaine solution and carefully align them on the agar plate.
NOTE: Discard any dead or swollen xenografts. Drug treatments and some tumor cells can induce toxicity and eventually cause xenograft mortality. These xenografts are not considered for engraftment quantification. - To determine percentage of engraftment: on a fluorescent stereomicroscope analyze each live xenograft and assess the absence (Figure 6D) / presence (Figure 6E) of tumors in the PVS.

- To determine percentage of metastasis: on a fluorescent stereomicroscope analyze each xenograft and assess the presence/absence of micrometastasis in the caudal hematopoietic tissue (CHT) of the 2 previously defined groups (CIRC and NO CIRC, Figure 6F)

- According to the experimental set up, select the xenografts of interest and euthanize them with 25x Tricaine (Table 3).
- Fix them in 4% formaldehyde (FA) for at least 4 hours at room temperature (RT) or overnight at 4 °C.
NOTE: Use methanol-free formaldehyde (16% FA) diluted at 4% in PBS/0.1% Triton. Fill the tubes to the top with fixative. Position the tubes horizontally to ensure a homogeneous fixation of all xenografts, increasing permeability and preventing the zebrafish from aggregating at the bottom. - Alternatively fix them with PIPES (Per 1 mL: 100 µL of 1 M PIPES sodium salt (4 °C); 1 µL of 1 M MgSO4 (RT); 4 µL of 0.5 M EGTA (RT); 93.7 µL of 16% FA (RT); 801.3 µL of ddH2O).
NOTE: PIPES preserves the fluorescence of RFP and mCherry transgenic lines better than 4% FA. - If the immunostaining will be performed on a different day, replace the FA with 100% methanol (MetOH). Xenografts fixed in 100% methanol can be stored at -20 °C indefinitely.
NOTE: MetOH can impair the efficiency of some stainings (i.e., phalloidin) and quench some fluorescent labelling. Confirm the efficiency of the antibodies in MetOH fixed samples beforehand.
9. Whole mount immunostaining for confocal imaging
NOTE: The whole mount immunofluorescence technique takes 3 days divided as follows: The first day is for permeabilization of the xenografts and primary antibody incubation. The second day for washing and secondary antibody incubation and the third day for washing, fixation of xenografts and storage in the mounting media.
- Day 1
- If the xenografts were stored in 100% MetOH, rehydrate them by a series of decreasing MetOH concentrations (75%, 50%, 25% MetOH diluted in PBS/0.1% Triton). If fixed in FA, replace it by PBS/0.1% Triton.
- Wash 4x for 5 minutes in PBS/0.1% Triton.
- Wash 1x for 5 minutes in H2O.
NOTE: The tubes must be positioned horizontally always in fixation, permeabilization and washing steps unless stated otherwise. - Replace the H2O for ice cold acetone and incubate at -20 °C for 7 minutes.
NOTE: Place a 50 mL tube with acetone at -20 °C so it is ready to use. Microcentrifuge tubes must be positioned vertically on a rack so that the acetone does not leak out. - Wash 2x for 10 minutes in PBS/0.1%Triton.
- Incubate with PBDX_GS blocking solution for 1 h at RT (PBDX_GS blocking buffer: 50 mL of 1x PBS; 0.5 g of bovine serum albumin - BSA; 0.5 mL of DMSO; 250 µL of 10% Triton; 750 µL of goat serum - GS (15 µL/1 mL)).
- Remove PBDX_GS and add ~40 µL of primary antibody dilution (generally 1:100).
NOTE: The volume of the primary antibody dilution varies depending on the number of xenografts present in the microcentrifuge tube. Ensure that all the xenografts are submerged. - Incubate for 1 hour at RT and then at 4 °C overnight. Position tubes vertically.
- Day 2
- Remove primary antibody and wash 2x for 10 minutes in PBS/0.1% Triton.
- Wash 4x for 30 min in PBS/0.05% Tween.
NOTE: The following steps must be performed in the dark (use aluminum foil to protect tubes from light). - Remove PBS/0.05% Tween and add ~50-100 µL of secondary antibody dilution (generally 1:200 - 1:400) + DAPI (50 µg/mL) diluted in PBDX_GS.
- Incubate for 1 hour at RT and then at 4 °C overnight. Position tubes vertically and protect from light.
- Day 3 (use aluminum foil to protect tubes from light)
- Remove secondary antibody dilution and wash 4x for 15 minutes in PBS/0.05% Tween.
- Fix at room temperature for 20 minutes in 4% FA.
- Wash 1x for 5 minutes in PBS/0.05% Tween.
- Remove PBS/0.05% Tween and add 1 drop of aqueous mounting medium to each microcentrifuge tube. Position the tubes vertically.
- Mount or store at 4 °C protected from light until mounting. Position the tubes vertically.
10. Mounting of xenografts
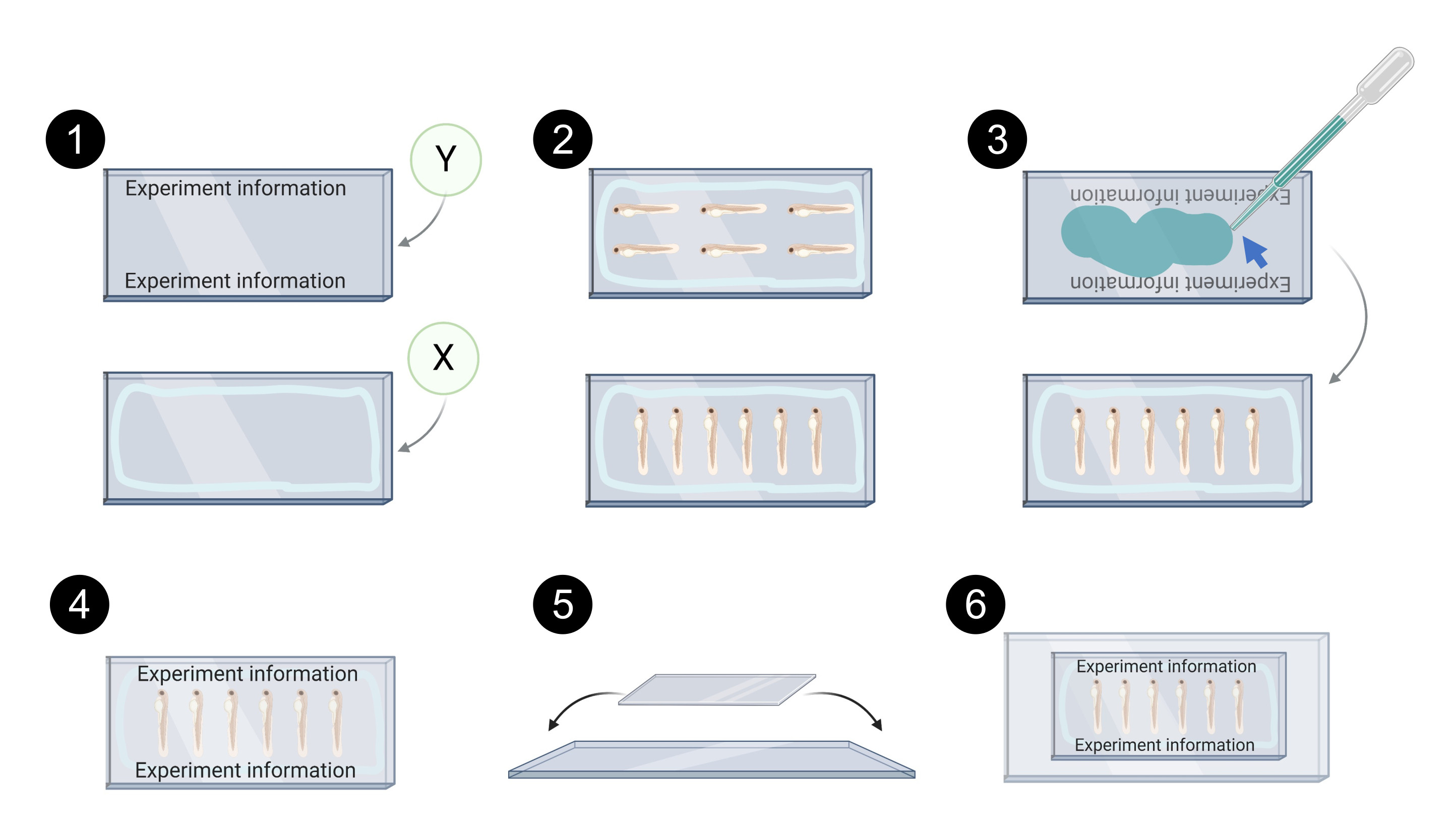
NOTE: Protect microcentrifuge tubes from the light throughout the process. Zebrafish xenografts are mounted between 2 coverslips (24 x 60 mm # 1.5). This allows to flip the mounted xenografts during confocal imaging so that both sides of the tumor (top and bottom) are accessible. Do not use plastic pipettes with mounting medium - the xenografts may get caught in the pipette. See Figure 7 for the schematic representation of the following steps.
- Label the coverslip Y and seal the edges of the coverslip X with petroleum jelly or silicone grease to avoid the leakage of the mounting media.
- Transfer the xenografts with a glass Pasteur pipette to the coverslip X.
- Carefully align them with a hairpin and remove the excess aqueous mounting media.
- Add aqueous mounting media to coverslip Y.
- Carefully place coverslip Y on top of coverslip X. Do not press the coverslips as this can potentially disrupt the xenografts.
- Place the assembled coverslips on top of a microscope slide and secure them with transparent adhesive tape. This allows an easier manipulation for confocal imaging and storage.
11. Confocal imaging
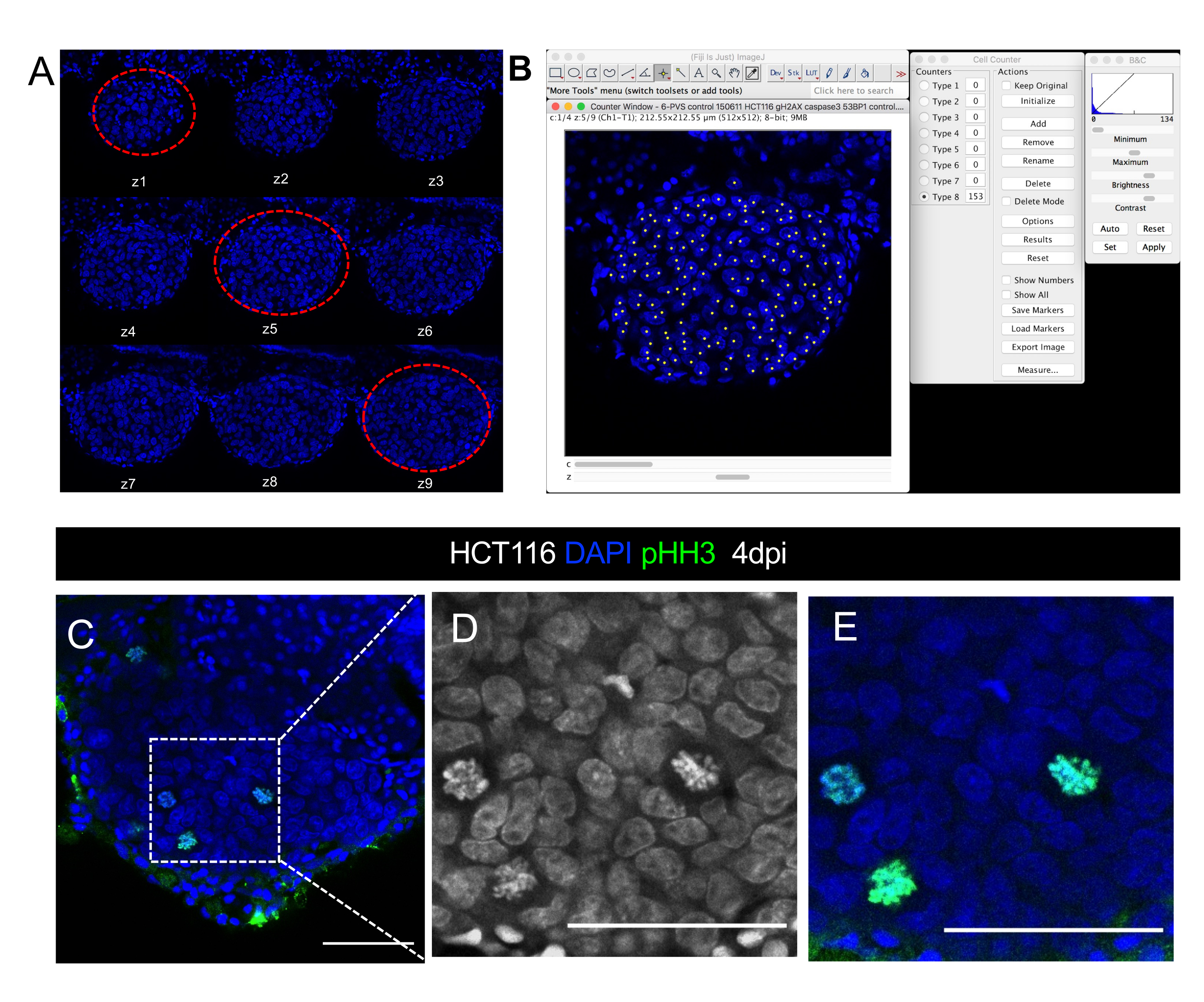
NOTE: An Apochromatic 25x immersion objective lens with water correction is optimal for imaging PVS tumors with single cell resolution (see Figure 8C-C" and Figure 9A for examples).
- Acquire samples using the z-stack function with an interval of 5µm between each slice. For images aimed at 3D reconstruction, in particularly vessels, use an interval of 1-3µm between slices (Figure 8A).
12. Analysis and quantification
- Use FIJI/ImageJ software or similar for confocal image processing and analysis.
- Open the raw data (.czi, .lsm, etc) in FIJI software.
- To select all or just a single channel in composite mode, click: Image > Colors > Channels Tool.
- To adjust brightness and contrast levels, click: Image > Adjust > Color Balance.
- To quantify tumor size
- Select three representative slices of the tumor, from the top, middle and bottom, per z-stack per xenograft (Figure 8 A).
NOTE: Confocal resolution achieves ~60-70 µm of tumor depth. If the tumor is large, it may not be possible to image its total volume. - Open a spreadsheet to annotate the data.
- Count every DAPI nuclei that corresponds to the tumor cells in the 3 selected slices (Figure 8 A, B). To do so:
- Open the cell counter plugin from FIJI/ImageJ by clicking Plugins > Analyze > Cell counter.
- In the cell counter tool, click Initialize, select a counter type, and click on the image to start the counting mode manually. For every click, the counter adds how many cells are counted (number of clicks).
- After counting one full slice, save the cell number in the corresponding excel document.
- Back to Fiji, click Reset from the Cell Counter window to delete the information if the same counter will be used. Otherwise, the information can be kept, and other counters can be used with other slices (or cells).
- Move to the second representative slice and repeat the previous steps. Gather all data.
NOTE: DAPI counterstaining is commonly used to count cell numbers due to the clear definition of individual cells, however, other cell specific staining can be used.
- To obtain the total number of cells in the tumor use the following formula:

NOTE: The 1.5 correction number was estimated for cells that have average nucleus diameter of ~10-12 µm. This correction may need adjustment if the cells are larger/smaller. See Dicussion for more details about this method.
- Select three representative slices of the tumor, from the top, middle and bottom, per z-stack per xenograft (Figure 8 A).
- To quantify other markers (immune cells, mitotic figures, PPH3, activated Caspase3, Ki67, etc.), quantify all slices using the same plugin (see Figure 8C-C'' for mitotic figures visualization). Divide the total number of counted cells by the corresponding tumor size and multiply by 100 to get the percentage.
NOTE: Beware that some cells will be located between two slices, go back and forth in the z-stack to make sure one cell is not being counted twice.
Results
Zebrafish xenograft as a tool to study anti-cancer therapies.
HCT116 colorectal cancer cell line was labelled with CM-DiI and injected in the PVS of 2 days post fertilization (dpf) embryos. After injection, xenografts were incubated at 34 °C, a temperature that allows the growth of tumor cells without compromising zebrafish development. The next day, xenografts were screened according to the presence or absence of a tumor mass in the PVS (not properly injected zebrafish were discarded and euthanized) (Figure 6A-A''). Xenografts were grouped according to their tumor size (Figure 6B-B'') and randomly distributed (in a 6-well plate: ~12 xenografts per well) in non-treated controls and FOLFIRI chemotherapy (0.18 mM Folinic acid, 0.08 mM Irinotecan and 4.2 mM Fluorouracil (5-FU)).
Control E3 and drugs were replaced daily, and dead zebrafish discarded. 4 dpi, and three days post treatment (dpt), the engraftment was quantified as in step 8.2 of the protocol. Engraftment is considered as the frequency of xenografts that present a tumor mass in the PVS at 4 dpi. For example, if at the end of the experiment there are 40 live larvae and 35 out of the 40 present a tumor in the PVS, the engraftment rate is 87.5%. Xenografts were euthanized and fixed to evaluate tumor size and apoptosis by immunofluorescence and confocal microscopy.
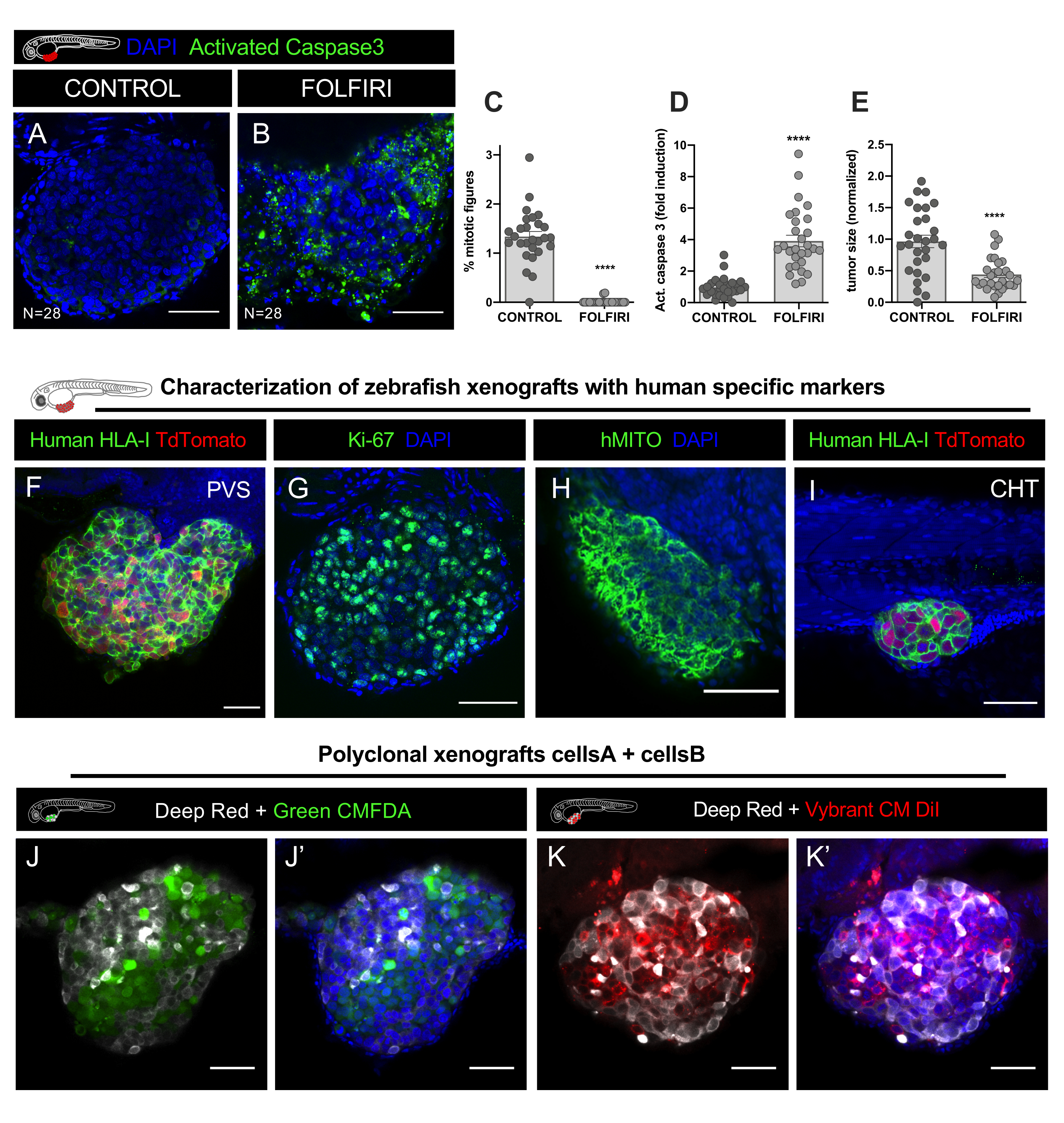
Immunofluorescence was performed to detect apoptotic cells, using anti-cleaved Caspase 3 antibody (Asp175) (rabbit, 1:100, #CST 9661) and DAPI (50 µg/mL) for nuclei counterstaining. Image stack datasets (every 5um) were acquired in a LSM710 confocal microscope and data analysis performed with FIJI/ImageJ software as explained in step 12. Quantification of the mitotic index, apoptosis (% of Activated Caspase3) and tumor size, revealed that FOLFIRI induces a significant decrease of mitosis (Mann Whitney Test, P<0.0001) and a significant induction of apoptosis (Mann Whitney test, P<0.0001), accompanied by a 54% reduction of tumor size (P<0.001) (Figure 8C-E, Figure 9A-E).
These features are useful in high throughput phenotypic drug screens as well as to test the cell intrinsic and physiological effects of several cancer treatments in a short time frame.
Characterization of human-zebrafish xenografts with human-specific antibodies
As in all xenograft models, there is a risk of misidentifying the cells. For instance, macrophages can phagocytose human cancer cells, becoming labelled with the lipophilic dye, and then travel along the zebrafish host and thus, these cells can be mistaken for tumor micrometastasis. Therefore, labelling xenografts with specific human antibodies such as human HLA, ki-67 or human mitochondria (hMITO) is crucial for initial characterization and also to become acquainted with the morphology of the tumor cells (Figure 9F-I).
Zebrafish xenograft to study cell-cell interactions.
Another great advantage of the zebrafish xenograft model is that it is possible to study the interactions of different tumor cells and analyze how each type of cell can influence the behavior of the other. Different human cancer cells (different clones from the same tumor or from different tumors) can be co-injected. In this example, two CRC cell lines derived from the same patient were labelled with different lipophilic dyes and mixed in a ratio of 1:1 for injection (Figure 9J-K').
When mixing human cell lines (to generate polyclonal tumors) on the same graft, avoid using the DiO dye as this results in non-specific double staining (Table 2). Instead use for example, CM-DiI (cell line#1) with green CMFDA (cell line#2), or CM-DiI (cell line#1) with Deep red (cell line#2) (Table 2, Figure 9J-K'), to get an easy discrimination of the populations in the end of the experiment. To quantify the frequency of each clone within the tumor, use 2 different counter types in FIJI to identify each clone and then divide by the total cell number (sum of all clones) to get the relative fraction of each one (%).

Figure 1. Flow chart summary of the zebrafish xenograft protocol Please click here to view a larger version of this figure.
{kind=link}

Figure 2. Materials for zebrafish embryo injection: A. Borosilicate needle B. 2% Agarose plate. C Hairpin loop D. Steps to make a hairpin loop: 1. Place 1 hair inside a glass capillary tube leaving approximately 1 centimeter of hair outside the tube. 2-3. Curl the outside tip of the hair with the help of forceps into the glass capillary tube forming a loop of ~0.5 mm length. 4. Seal the edge of the capillary tube with a drop of nail polish. 5-6. Seal a piece of electrical tape around the capillary to protect it from breaking. Please click here to view a larger version of this figure.
{kind=link}

Figure 3. Representative stereomicroscope images of zebrafish embryos at 48 hours post-fertilization (48 hpf): A-A'. Normal morphology of a zebrafish embryo at 48 hpf of development, ready for microinjection B-B'. Morphology of a zebrafish embryo that has not achieved the adequate developmental stage for microinjections at 48 hpf and presents already some degree of cardiac edema (black arrowhead) and distended yolk. A' and B' are magnifications of A and B respectively. Scale bars represent 500 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4. Schematic representation of zebrafish microinjection plate set up: A. Alignment of anesthetized embryos in 3% Agar/2% Agarose plate. B. Graphic representation of a 2 days post-fertilization zebrafish embryo, with a black arrow indicating the perivitelline space (PVS). C and D. Cancer cells can be injected with different angles into the PVS injected in the perivitelline space (PVS) of a 48 hours post-fertilization embryo. Please click here to view a larger version of this figure.
{kind=link}

Figure 5. Metastatic assay. 1 hour post injection (1hpi) injected embryos are sorted according to the absence (NO_circ) or presence (CIRC) of tumor cells in circulation. At 4 days post injection the number of xenografts in both groups that present micrometastasis are quantified. Cells in the (A) NO_circ group had to undergo all the metastatic steps to be able to form a micrometastasis, whereas cells in the CIRC group (B) only undergo the last steps of the metastatic cascade. Please click here to view a larger version of this figure.
{kind=link}

Figure 6. Representative fluorescent stereomicroscope images of xenografts at 1 day post-injection and 4 days post-injection. Human cancer cells expressing fluorescent protein TdTomato (red) were microinjected into 2 dpf zebrafish embryos. At 1 dpi, screen the injected embryos and discard the badly injected ones or embryos with an edema (A-A'') sort the well injected embryos according to tumor sizes (B-B''). C. Representative quantification of the total number of cells present in the different tumor size categories at 1 dpi in SW620 xenografts. Each dot represents a xenograft quantified as explained in section 12.At 4 dpi, screen the larvae and quantify the different classes: no tumor (D); with tumor in the PVS (E) and with a micrometastasis in the CHT (F). Scale bars represent 500 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 7. Schematic representation of xenograft mounting for confocal imaging. 1. Example of labelling in coverslip Y, light blue line in coverslip X represents the perimeter in which petroleum jelly/silicone grease is applied to prevent leakage of the mounting media. 2. Examples of xenograft alignment on coverslip X for confocal imaging. 3. Aqueous mounting media (blue arrow) is used to bind coverslip Y on top of coverslip X. 4. Example of properly mounted coverslips. 5. Coverslips are then placed on top of a glass slide and secured with transparent tape. 6. Mounted xenografts ready for confocal imaging. Created with BioRender.com Please click here to view a larger version of this figure.
{kind=link}

Figure 8. Visual representation of confocal microscope image quantification of tumor size. Confocal images of a HCT116 xenograft at 4 days post-injection A. Series of z-stack slices in the DAPI channel acquired with a 5 µm interval. Red dashed lines circle the three representative slices used for cell counting. B. Illustration of DAPI nuclei quantification with the Cell Counter Plugin in ImageJ/Fiji software. C-E. Example of single cell resolution within the tumor in which it is possible to visualize/quantify mitotic figures in DAPI or using a phospho-histone H3 antibody (green). D and E are magnifications of C. Scale bars represent 50 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 9. Representative results. A-E. HCT116 colorectal cancer cell line was labelled with Vybrant CM-DiI and injected in the PVS of 2 days post fertilization (dpf) embryos. After injection, xenografts were incubated at 34 °C. At 1 dpi, xenografts were screened and randomly distributed in non-treated controls and FOLFIRI, treated for 3 consecutive days and fixed at 4 dpi. A. Immunofluorescence was performed to detect apoptotic cells, using anti-cleaved Caspase 3 antibody and DAPI for nuclei counterstaining. Quantification of the mitotic index C, apoptosis (% of Activated Caspase3) D, and tumor size E, revealed that FOLFIRI induces a significant decrease of mitosis (Mann Whitney Test, P<0.0001) and a significant induction of apoptosis (Mann Whitney test, P<0.0001), accompanied by a 54% reduction of tumor size (P<0.001). The number of xenografts analyzed is indicated in the figure and each dot represents one xenograft. F-H. Representative images of colorectal cancer xenografts at 4 days post-injection labelled with human specific markers in green (human HLA, ki67 and hMITO) in the PVS and CHT I. J-J'. Confocal images of polyclonal xenografts, injected with two different human colorectal cancer cell lines labelled with lipophilic staining CellTracker Deep Red (Cy5 - white), CellTracker Green CMFDA (488 - green) and DAPI counterstaining. K-K'. Confocal images of two different human colorectal cancer cell lines labelled with lipophilic staining CellTracker Deep Red (Cy5 - white), Vybrant CM DiI (594 - red) and DAPI counterstaining. Scale bars represent 50 µm. Please click here to view a larger version of this figure.
{kind=link}
| Cell line | Tissue | Species | Morphology | Growth mode | Ideal confluence for injection | Dissociating agent for injection | Dissociation time | Protocol for labelling | Injection medium | Divide total number of cells by (to achieve ideal concentration for injection) | |||||||
| SW480 | Colorectal adenocarcinoma (primary) | Human | Epithelial | Adherent | 70% - 75 % | PBS/EDTA 2 mM | 5 minutes | Flask | PBS 1x | 0,25 | |||||||
| SW620 | Colorectal adenocarcinoma (metastasis) | Human | Epithelial | Adherent | 70% - 75 % | PBS/EDTA 2 mM | 5 minutes | Flask | PBS 1x | 0,25 | |||||||
| HCT116 | Colorectal carcinoma (primary), Kras mutant | Human | Epithelial | Adherent | 70% - 75 % | PBS/EDTA 2 mM | 5 minutes | Flask | PBS 1x | 0,25 | |||||||
| HKE3 | Colorectal carcinoma, Kras WT | Human | Epithelial | Adherent | 70% - 75 % | PBS/EDTA 2 mM | 5 minutes | Flask | PBS 1x | 0,25 | |||||||
| HT-29 | Colorectal adenocarcinoma (primary) | Human | Epithelial | Adherent | 70% - 75 % | PBS/EDTA 2 mM | 5 minutes | Flask | PBS 1x | 0,2 | |||||||
| CACO-2 | Colorectal adenocarcinoma (primary) | Human | Epithelial | Adherent | 70% - 75 % | PBS/EDTA 1 mM | 5 minutes | 1.5 mL microcentrifuge tube | Complete medium | 0,25 | |||||||
| MCF-7 | Breast adenocarcinoma (metastasis) | Human | Epithelial | Adherent | 70% - 80 % | PBS/EDTA 1 mM | 5 minutes | 1.5 mL microcentrifuge tube | 60% FBS + 40% complete medium | 0,5 | |||||||
| Hs578T | Breast carcinoma (primary) | Human | Epithelial | Adherent | 70% - 75 % | PBS/EDTA 1 mM | 2 minutes | 1.5 mL microcentrifuge tube | 60% FBS + 40% complete medium | 0,5 | |||||||
| MDA-MB-468 | Breast adenocarcinoma (metastasis) | Human | Epithelial | Adherent | 70% - 75 % | PBS/EDTA 1 mM | 8 minutes | 1.5 mL microcentrifuge tube | 60% FBS + 40% complete medium | 0,5 | |||||||
| MDA-MB-231 | Breast adenocarcinoma (metastasis) | Human | Epithelial | Adherent | 70% - 75 % | PBS/EDTA 1 mM | 5 minutes | 1.5 mL microcentrifuge tube | Complete medium | 0,5 | |||||||
| RT112 | Urinary bladder transitional cell carcinoma (primary) | Human | Epithelial | Adherent | 85% - 90% | TrypLE | 6 minutes + cell scrapper | 1.5 mL microcentrifuge tube | Complete medium | 0,5 | |||||||
| BFTC905 | Urinary bladder transitional cell carcinoma (primary) | Human | Epithelial | Adherent | 75% - 85% | TrypLE | 10 minutes | 1.5 mL microcentrifuge tube | 80% complete medium + 20% PBS/EDTA 2 mM | 0,5 | |||||||
| J82 | Urinary bladder transitional cell carcinoma (primary) | Human | Epithelial | Adherent | 85% - 90% | TrypLE | 10 minutes | 1.5 mL microcentrifuge tube | Complete medium | 0,5 | |||||||
| RT4 | Urinary bladder transitional cell carcinoma (primary) | Human | Epithelial | Adherent | 85% - 90% | TrypLE | 6 minutes | 1.5 mL microcentrifuge tube | Complete medium | 0,5 | |||||||
| MIA PaCa-2 | Pancreatic epitheliod carcinoma (primary) | Human | Epithelial | Adherent | 80% - 90% | TrypLE | 3 minutes | 1.5 mL microcentrifuge tube | 60% FBS + 40% complete medium | 0,5 | |||||||
| PANC-1 | Pancreatic epitheliod carcinoma (primary) | Human | Epithelial | Adherent | 80% | TrypLE | 5 minutes | 1.5 mL microcentrifuge tube | PBS/EDTA 2mM | 0,5 | |||||||
| VC8 | Lung fibroblasts, BRCA2 mutant, HR-deficient | Chinese hamster | Epithelial | Adherent | 70% - 80 % | PBS/EDTA 1 mM | 5 minutes | 1.5 mL microcentrifuge tube | Complete medium | 0,25 | |||||||
| VC8-B2 | Lung fibroblasts, human BRCA2, BRCA2 −/− HR-deficient | Chinese hamster | Epithelial | Adherent | 70% - 80 % | PBS/EDTA 1 mM | 5 minutes | 1.5 mL microcentrifuge tube | Complete medium | 0,25 | |||||||
Table 1: Optimal in vitro confluence for injection of several cell lines
| Cell dye | Catalog number | Fluorescence spectrum (Exc. - Em.) | Stock dilution | Working dilution to stain in a flask | Working dilution to stain in an 1.5 mL microcentrifuge | Incubation time | Observations | ||||||
| Vybrant CM-DiI | V22888 | 549 nm - 569 nm | Already diluted | 4:1000 in PBS 1x | 4:1000 in PBS 1x | 15 min @ 37 ºC + 4 min @ 4 ºC | |||||||
| Vybrant DiO | V22886 | 484 nm - 501 nm | Already diluted | 5:1000 in PBS 1x | 5:1000 in PBS 1x | 15 min @ 37 ºC + 4 min @ 4 ºC | Not good resolution in confocal imaging at 4 days post injection | ||||||
| CellTracker Deep Red | C34565 | 630 nm - 660 nm | 1 mM in DMSO | 0.5 - 2.5 µM in PBS 1x | 0.5 - 2.5 µM in PBS 1x | 15 min @ 37 ºC | |||||||
| CellTracker Green CMFDA | C7025 | 492 nm - 517 nm | 10 mM in DMSO | 0.5 - 2.5 µM in PBS 1x | 0.5 - 2.5 µM in PBS 1x | 15 min @ 37 ºC | Product leaks to the abdominal cavity 48 hours post injection, but ideal for co-injection studies | ||||||
Table 2: Dye and conditions
| Size | # of larvae | E3 1x medium Volume |
| 100 mm x 15 mm (standard) | Up to 50 | 20-25 mL |
| 60 mm x 15 mm | Up to 20 | 10 mL |
| 6-well plate | Up to 15 per well | 3-4 mL per well |
Table 3: Petri dish options
| E3 medium 50x - stock | For 10 liters of sterile water: |
| 146.9 g of NaCl | |
| 6.3 g of KCl | |
| 24.3 g of CaCl2·2H2O | |
| 40.7 g of MgSO4·7H2O | |
| E3 medium 1x – ready to use | 400 mL of E3 medium 50x |
| 60 mL of 0.01% Methylene Blue Solution | |
| Fill up to 20 liters of fish system water | |
| Tricaine 25x – stock and euthanasia | 2 g of Tricaine powder |
| 500 mL of reverse osmosis water | |
| 10 mL of 1 M Tris (pH 9) | |
| Adjust to pH 7 | |
| Tricaine 1x - Anaesthesia | 20 mL of Tricaine 25x |
| Fill up to 500 mL of system water | |
| 60 mg/mL Pronase - stock 100x | 1 g of pronase |
| 16.7 mL sterile water | |
| 0.6 mg/mL Pronase – 1x ready to use | 100 µL pronase 100x |
| 9.9 mL of E3 medium 1x |
Table 4: Solution compositions
Discussion
The increasing importance of the zebrafish as a model for cancer development and drug screening has resulted in numerous publications3,4,7,13,14,16,18,19,20,21. However, the injection of cancer cells in zebrafish embryos is a technique that requires a high level of dexterity which can be challenging for researchers. In this protocol we aim at providing practical information and some tips that can help overcome the initial challenges of setting up zebrafish embryo xenografts.
Cell handling prior injection
This optimized protocol for the generation of zebrafish xenografts with cell lines can be adapted to various types of (cancer) cells with different morphologies. We recommend that all the cell lines used for zebrafish xenografts are mycoplasma free. Unlike other bacterial contaminations, the presence of mycoplasma in cell culture does not generate changes that can be easily detected under the microscope22. Mycoplasma contamination may affect the engraftment potential of the cell lines, their sensitivity to drugs, as well as the viability of the zebrafish embryos.
Although cells may continue to proliferate for an extended period, their phenotype and genotype can be prone to changes. It is important to become familiar with the morphology and behavior of the cell lines in culture. We recommend keeping the number of cell passages after thawing between 3-12 to get reproducible results. Thus, regular mycoplasma tests should be carried out.
Cells should be at their log phase (exponential growth phase before reaching confluence ~70%) on the day of the injection. This will enable an adequate engraftment and proper development of the distinctive hallmarks of the tumor. To prevent variation in the phenotype of cells within the xenograft, it is critical to maintain the confluence for injection constant between experiments. The number of injected cells can be adapted to the characteristics of each cell line as some may require higher densities of injection in order to thrive in the zebrafish embryos.
Cell labelling considerations
To better visualize human tumor cells for injection and future analysis, tumor cells can be labelled with fluorescent dyes. Due to differences in cellular size, the total number of cells/cm2 in adherent cultures varies among cell lines. This will impact the efficacy of the staining protocol as well as the number of cells harvested for injection. Large cells that yield low numbers per flask (i.e., Hs578T) or grow in clusters (i.e., BFTC905) will require the pooling of several flasks for a single experiment. In this case, the staining of cells shouldn't be performed directly in the flask as this will cause the use of excessive amounts of dye (high cost). On the other hand, cells that are highly sensitive to excessive cycles of centrifugation as well as those that yield high numbers per flask (i.e., HCT116) can be stained directly in the flask and then detached with EDTA/cell scraper (For more information see Table 1).
Whenever possible, instead of using an enzymatic approach, use EDTA to detach the cells on the day of injection, so that cells recover their cell-cell junctions more rapidly and are subjected to less centrifugation steps. Nevertheless, if the cells are sensitive to EDTA, very adherent or grow in clusters - an enzymatic method can be applied. The optimization of the ideal concentration for injection as well as the injection medium is dependent on the characteristics of each cell line, thus it might need some adjustments (Table 1).
Microinjection calibration
Contrary to the delivery of oligonucleotides or drugs into the zebrafish embryo, a graticule is not used to calibrate the needle when working with cell lines for xenografts. After some time during injection, cells will start to clog, and it is necessary to cut the tip of the needle to increase its diameter or change the needle altogether. This procedure hinders the graticule calibration.
To address this issue, the number of cells dispensed is regulated by eject pressure and time needed to reach a size similar to the embryo eye within 1-3 pulses. Then, to further control the tumor size, at 1 dpi, xenografts are sorted according to the tumor size as shown in Figure 6 B-B". As represented in Figure 6C example, this method of sorting is efficient in reducing the variation of tumor sizes: if we pool them all together (+, ++, +++) the STEV is ~double of the ++class (~906 cells to ~422 cells) and the coefficient variation is ~31.9% compared to 14,5% in the ++class. Since the reference for injection is volume, the total number of cells varies a lot between cell types - being dependent on the size and shape of the cells. For example, big cells with a lot of cytoplasm like breast cancer Hs578T, produce much smaller tumors (~600 cells). Also, each cell line requires different number of cells. For instance, the HT29 CRC and RT112 urinary bladder cancer cell lines have shown that the higher the number of cells injected, the higher the zebrafish mortality. Therefore, a period of optimization while developing the xenografts is needed to test if the cell line has toxic effects in the embryo or requires higher/lower densities of injection.
Injection site
One of the most common discrepancies when generating zebrafish embryo xenografts is the site of injection. The yolk is usually the place of choice for injection due to its easy accessibility. However, we have observed that cells injected in the yolk have a higher tendency to die. Although technically more difficult, we recommend injecting in the PVS and as far as possible from the heart. Within the PVS, cells can aggregate, recruit vessels and immune cells, and migrate, intravasate, extravasate and form micrometastasis, if they display metastatic characteristics8,11.
Engraftment efficiency
Differences in the engraftment efficiency and tumor size among cell lines at 4 dpi are expected due to their distinct degrees of basal cell death/survival/proliferation but also due to the innate immunogenicity that each cell line may display9.
Metastasis
Metastasis comprises a multistep cascade of events that can be divided into two arbitrary stages. In the first stage, tumor cells have to detach from the primary site, migrate and invade adjacent tissues and then intravasate into the bloodstream. In the second stage, tumor cells must survive in circulation, extravasate from the blood or lymphatic vessels, and finally colonize at secondary sites23. To distinguish between these early and late events and address the potential/proficiency of the different tumor cells to perform these steps, we designed a simple assay.
In general, when injected into the PVS, tumor cells can enter directly into circulation and then get physically trapped in the caudal hematopoietic tissue (CHT) (tail region). However, according to the characteristics of each tumor cell - we have seen that some tumor cells remain at the CHT 4 dpi and are able to form micrometastasis while other tumor cells disappear (cleared after getting caught in the CHT).
Therefore, by comparing the micrometastasis efficiency (at 4 dpi) when cells were placed directly into circulation - CIRC (cells only have to go through the late steps of metastasis) vs. when not - NO CIRC (cells need to go through early and late steps to be able to form a micrometastasis) we can assess their early or late metastatic potential. We have observed tumor cells that can efficiently form micrometastasis in the CHT in both groups (CIRC and NO CIRC), suggesting that these cells have the capacity to undergo all steps of the metastatic cascade (SW480 and MDA-MB-468 for example)8,11. In contrast, other tumor cells have a very low metastatic potential in both groups, hardly ever making micrometastasis, even when injected into circulation (i.e., visible in the CHT at 24 hpi, but at 4 dpi they are no longer there, Hs578T for example)8. However, we have clearly found another group - one that is only able to form micrometastasis when injected into circulation (we can only observe micrometastasis in the CIRC group). This suggests that these cells have a low efficiency in performing the first steps of the metastatic cascade but on the other hand are able to survive in circulation, extravasate and colonize a distant site.
Immunostaining and imaging
Before fixation, this injection protocol can be used for other live imaging approaches like live differential interference contrast (DIC) microscopy, spinning disk microscopy, high-resolution live confocal imaging and light sheet microscopy, etc.
Dead cells and cellular debris appear bright when observed through the fluorescent stereomicroscope and can be mistaken for live cells, especially if the aim of the study is assessing the metastatic potential of the cell lines. We would like to stress the importance of performing confocal imaging with specific viability markers and DAPI to assess the survival state of the tumor and micrometastasis. Also, it is fundamental to use specific human antibodies to detect human cells such as anti-human mitochondria or anti-human HLA. When implementing the protocol, train experimenter eyes by comparing the staining in the stereomicroscope with the confocal images. After some time, experimenters can clearly distinguish debris from live cells in the fluorescent stereomicroscope.
Although other methods to quantify tumor burden such as whole fluorescence area are widely used, we recommend performing whole mount immunostaining and confocal imaging as a more accurate method. Not only the efficiency of lipophilic dye staining is very variable (i.e., some cells are very well stained whereas others are not -probably due to the lipid content of their membranes), but also many times lipophilic dyes form aggregates, and dead cells tend to be brighter - creating several artifacts that can be mistaken for live cells.
Cells can be transduced with fluorescent proteins to aid in their tracking and to skip cell labelling. However, make sure that the transduced and non-transduced cells produce the same results in the zebrafish xenografts.
In addition, macrophages can phagocyte these fluorescent cell debris becoming fluorescently labelled and migrate, generating false positive metastatic cells. Thus, we recommend a series of analytic tools, which of course can be extended to many other readouts for a more accurate interpretation of tumor behavior:
- Proliferation - quantification of mitotic figures with DAPI or anti-pHH3Ser10 antibody (Merck Millipore Cat. #06-570),
- Cell death by apoptosis- antibody anti-activated Caspase3Asp175 (Cell Signalling Technologies Cat. #9661) or equivalent,
- Tumor size - DAPI counting- human tumor cells show a very distinct chromatin organization, so they are easily distinguished from the zebrafish cells and is always possible to double check with the dye (when you are training your eye),
- For metastatic studies, to unambiguously detect human cells, - anti-human HLA (Abcam EP1395Y Cat. #ab52922), anti-human mitochondria (Merck Millipore Cat. #MAB1273- Clone 113-1).
For a confocal acquisition with 5 µm interval between slices in the Z-stack of the control cancer cells HCT116 (average nuclear size of 10-12 µm of diameter) with DAPI counterstaining, we have observed that ~50% of the cells are shared between two consecutive slices. Therefore, if every slice is counted, there is a high risk of counting the same cells twice. Going back and forth between slices to avoid problems in quantification results in a time consuming and error prone technique. To ease the quantification of the total number of cells and allow for more reproducibility between researchers, we created the tumor size formula previously described in this protocol8.
We included a correction number (1.5) to account for the ~50% cells shared between slices. We found that the average error of manual counting of the whole tumor between researchers was 20%. Two researchers using the formula had a 2% error. The use of this formula has a 93% accuracy rate and 98% reproducibility rate. We also tested automated methods, but they demonstrated an error higher than 50% caused by threshold settings.
Due to the characteristics of apoptotic cells, the quantification of activated Caspase 3 cells is more difficult. To reduce the number of mistakes and variation in results, we recommend that the control and experimental samples are counted by the same researcher. Additionally, when learning this technique, a new researcher must count images that were already quantified by more experienced researchers to compare results and train.
The length of the assay can be extended if required. However, it is important to consider that zebrafish larvae require live feeding starting from ~7 days post-fertilization (5 days post-injection). Additionally, the animal welfare guidelines and regulations applied to larvae older than 6 days post-fertilization may vary.
This protocol provides useful tools to enable a single researcher to inject approximately ~200-300 zebrafish larvae per hour; and results for the complete assay, including analysis and statistical interpretation, obtained in three weeks. We hope that this protocol can help researchers become experts in generating zebrafish xenografts. It is not easy; you need to practice but you will get there. Good Luck!
Disclosures
None
Acknowledgements
We thank to Champalimaud Foundation, Congento (LISBOA-01-0145-FEDER-022170, co-financed by FCT/Lisboa2020) and FCT-PTDC/MEC-ONC/31627/ 2017 for funding. FCT fellowships for VP (SFRH/BD/118252/2016), MML (PD/BD/138203/2018). All members of the Fior Lab for critical discussions; B. Costa and C. Rebelo de Almeida for sharing data; and our Lab members C. Rebelo de Almeida, M. Barroso and L. Leite for their participation in the video. We would like to thank the CF Fish Facility (C. Certal, J. Monteiro et al) and Champalimaud Communication, Events and Outreach team in particular Alexandre Azinheira for the fantastic film making and Catarina Ramos and Teresa Fernandes for their help.
Materials
| Name | Company | Catalog Number | Comments |
| Agar for bacteriology | VWR | 97064-336 | Agar plate |
| anti-Caspase3Asp175 (Rabbit monoclonal) | Cell Signalling Technologies | 9661 | Primary antibody for whole mount immuno staining (Dilution 1:100) |
| anti-human HLA (Rabbit monoclonal) | Abcam EP1395Y | ab52922 | Primary antibody for whole mount immuno staining (Dilution 1:100) |
| anti- 488 (Rabbit monoclonal) | ThermoFisher Scientific | 35552 | Secondary antibody for whole mount immuno staining (Dilution 1:200) |
| anti- 594 (Rabbit monoclonal) | ThermoFisher Scientific | 35560 | Secondary antibody for whole mount immuno staining (Dilution 1:200) |
| CellTracker Deep Red Dye | ThermoFisher Scientific | C34565 | Lipophilic dye (Dilution 1:1000) |
| CellTracker Green CMFDA Dye | ThermoFisher Scientific | C2925 | Lipophilic dye (Dilution 1:1000) |
| Conical Centrifuge tube 50mL | VWR | 525-0610 | |
| Conical Centrifuge tube 15mL | VWR | 525-0604 | |
| DAPI | Nuclear and chromosome counterstain | ||
| Laser-Based Micropipette Puller P-2000 | Sutter-Instrument | Micropipette Puller | |
| Microcentrifuge tube 1.5mL | Abdos | P10202 | |
| Microscope slides, cut edge | RS France | BPB016 | Slides for mounting |
| Mowiol | Sigma-Aldrich | 81381 | Mounting medium |
| Pneumatic Picopump | World Precision Instruments | PV820 | Microinjector |
| Rectangular cover glasses, Menzel Gläser | ThermoFisher Scientific | 631-9430 | Coverslips for mounting |
| SeaKem LE Agarose | Lonza | 50004 | Agar plate |
| Thin Wall Glass Capillaries | World Precision Instruments | TW100-4 | Borosilicate capillaries |
| TrypLE | Gibco | 12605036 | Enzymatic detachment solution |
| Vaseline | Petroleum jelly for slide sealing | ||
| Vybrant CM-DiI Dye | ThermoFisher Scientific | V22888 | Lipophilic dye (Dilution 1:1000) |
| Vybrant DiO Cell-Labeling Solution | ThermoFisher Scientific | V22886 | Lipophilic dye (Dilution 1:1000) |
| ZEISS Axio Zoom.V16 for Biology | ZEISS | Fluorescence Stereo Zoom Microscope | |
| Zeiss LSM 710 | ZEISS | Confocal microscope |
References
- Gut, P., Reischauer, S., Stainier, D. Y. R., Arnaout, R. Little Fish, Big Data: Zebrafish as a Model for Cardiovascular and Metabolic Disease. Physiological Reviews. 97, 889-938 (2017).
- Howe, K., et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 496, 498-503 (2013).
- Santoriello, C., Zon, L. I. Hooked! Modeling human disease in zebrafish. Journal of Clinical Investigation. 122, 2337-2343 (2012).
- Stoletov, K., Klemke, R. Catch of the day: zebrafish as a human cancer model. Oncogene. 27, 4509-4520 (2008).
- Weintraub, A. All eyes on zebrafish. Lab Animals. 46, 323-326 (2017).
- Novoa, B., Figueras, A., Lambris, J. D., Hajishengallis, G. Zebrafish: Model for the Study of Inflammation and the Innate Immune Response to Infectious Diseases. Current Topics in Innate Immunity II. , 253-275 (2012).
- Fazio, M., Ablain, J., Chuan, Y., Langenau, D. M., Zon, L. I. Zebrafish patient avatars in cancer biology and precision cancer therapy. Nature Reviews Cancer. 20, 263-273 (2020).
- Fior, R., et al. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proceedings of the National Academy of Sciences of the United States of America. 114, 8234-8243 (2017).
- Póvoa, V., et al. Innate immune evasion revealed in a colorectal zebrafish xenograft model. Nature Communications. 12, 1156 (2021).
- Galletti, G., et al. Targeting Macrophages Sensitizes Chronic Lymphocytic Leukemia to Apoptosis and Inhibits Disease Progression. Cell Reports. 14, 1748-1760 (2016).
- Rebelo de Almeida, C., et al. Zebrafish xenografts as a fast screening platform for bevacizumab cancer therapy. Communications Biology. 3, 1-13 (2020).
- Varanda, A. B., Martins-Logrado, A., Godinho Ferreira, M., Fior, R. Zebrafish Xenografts Unveil Sensitivity to Olaparib beyond BRCA Status. Cancers. 12, 1769 (2020).
- Osmani, N., Goetz, J. G. Multiscale Imaging of Metastasis in Zebrafish. Trends in Cancer. 5, 766-778 (2019).
- Hyenne, V., et al. Studying the Fate of Tumor Extracellular Vesicles at High Spatiotemporal Resolution Using the Zebrafish Embryo. Developmental Cell. 48, 554-572 (2019).
- Costa, B., et al. Developments in zebrafish avatars as radiotherapy sensitivity reporters - towards personalized medicine. EBioMedicine. 51, 102578 (2020).
- Costa, B., Estrada, M. F., Mendes, R. V., Fior, R. Zebrafish Avatars towards Personalized Medicine-A Comparative Review between Avatar Models. Cells. 9, 293 (2020).
- TRICAINE - Protocols. ZFIN Community Wiki Available from: https://wiki.zfin.org/display/prot/TRICAINE (2021)
- Zhao, C., et al. A Novel Xenograft Model in Zebrafish for High-Resolution Investigating Dynamics of Neovascularization in Tumors. Plos One. , (2011).
- Veinotte, C. J., Dellaire, G., Berman, J. N. Hooking the big one: the potential of zebrafish xenotransplantation to reform cancer drug screening in the genomic era. Disease Models & Mechanisms. 7, 745-754 (2014).
- Haldi, M., Ton, C., Seng, W. L., McGrath, P. Human melanoma cells transplanted into zebrafish proliferate, migrate, produce melanin, form masses and stimulate angiogenesis in zebrafish. Angiogenesis. 9, 139-151 (2006).
- Zon, L. I., Peterson, R. The New Age of Chemical Screening in Zebrafish. Zebrafish. 7, 1 (2010).
- Mycoplasma Contamination of Cell Cultures. InvivoGen Available from: https://www.invivogen.com/review-mycoplasma (2016)
- van Zijl, F., Krupitza, G., Mikulits, W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutation Research. 728, 23-34 (2011).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved