
Method Article
Simultaneous Interference Reflection and Total Internal Reflection Fluorescence Microscopy for Imaging Dynamic Microtubules and Associated Proteins
In This Article
Summary
We present a protocol for implementing interference-reflection microscopy and total-internal-reflection-fluorescence microscopy for the simultaneous imaging of dynamic microtubules and fluorescently labeled microtubule-associated proteins.
Abstract
Several techniques have been employed for the direct visualization of cytoskeletal filaments and their associated proteins. Total-internal-reflection-fluorescence (TIRF) microscopy has a high signal-to-background ratio, but it suffers from photobleaching and photodamage of the fluorescent proteins. Label-free techniques such as interference reflection microscopy (IRM) and interferometric scattering microscopy (iSCAT) circumvent the problem of photobleaching but cannot readily visualize single molecules. This paper presents a protocol for combining IRM with a commercial TIRF microscope for the simultaneous imaging of microtubule-associated proteins (MAPs) and dynamic microtubules in vitro. This protocol allows for high-speed observation of MAPs interacting with dynamic microtubules. This improves on existing two-color TIRF setups by eliminating both the need for microtubule labeling and the need for several additional optical components, such as a second excitation laser. Both channels are imaged on the same camera chip to avoid image registration and frame synchronization problems. This setup is demonstrated by visualizing single kinesin molecules walking on dynamic microtubules.
Introduction
Total-internal-reflection-fluorescence (TIRF) microscopy is commonly employed for the visualization of single fluorescent molecules. Compared to epifluorescence imaging, TIRF achieves superior background suppression, allowing for high-resolution localization and tracking of single fluorophores. For this reason, TIRF is the preferred method for visualizing fluorescently labeled microtubule-associated proteins and is frequently used to image microtubules1,2.
To investigate the regulation of microtubule dynamics by MAPs, it is often necessary to image both microtubules and MAPs simultaneously. Most existing methods for this purpose are expensive or suffer from technical drawbacks. Simultaneous two-color TIRF, for example, requires two excitation lasers and two cameras. In addition to the high cost, the need for separate cameras poses frame-synchronization and image-registration problems. This need can be circumvented if a rotating filter cube is used to physically switch between excitation lasers in consecutive frames3. In such a setup, a single camera chip can be used, and the frames alternate between images of microtubules and MAPs. This technique, however, is limited by the speed of the filter change, which typically restricts the frame rate to less than 0.5 frames per second3 (fps). Such a frame rate is insufficient to resolve fast dynamic processes, such as the shrinkage of a microtubule occurring at a velocity of up to 500 nm/s, the walking of a kinesin at a velocity on the order of 800 nm/s, or the diffusion of a MAP occurring with diffusion coefficients exceeding 0.3 μm2/s4. This is particularly problematic when tracking the relative positions of two moving targets in each channel, such as the position of a MAP relative to the position of a moving microtubule tip5.
In addition to these optical constraints, two-color TIRF microscopy requires MAPs and microtubules to be labeled with different fluorophores whose emission spectra are sufficiently separated. Fluorescent labeling of tubulin can alter microtubule dynamics6, and the photobleaching of fluorophores limits the imaging speed7. Because of these issues, label-free imaging techniques have been developed to visualize microtubules. These include interferometric scattering microscopy (iSCAT)8,9, rotating-coherent-scattering microscopy (ROCS)10, spatial light interference microscopy (SLIM)11, and interference reflection microscopy (IRM)12,13. These techniques allow fast label-free imaging of microtubules without the drawbacks of fluorescence imaging, but they cannot be used to visualize single MAPs.
Of these label-free techniques, IRM stands out for its low cost and its modest demands on instrumentation. We have recently presented a protocol for combining IRM with a commercial TIRF microscope, allowing microtubules and fluorescent MAPs to be imaged in alternating frames3,13. This paper presents a protocol for modifying this setup to simultaneously capture TIRF and IRM images on a single camera chip. This involves the addition of an inexpensive beam splitter in the excitation path to simultaneously illuminate the sample with a TIRF laser and an IRM LED light source. A modified commercial image splitter is used to spectrally separate the TIRF and IRM signals and project them onto separate halves of the same camera chip. We also employ a microfluidic system that allows the rapid exchange of reagents during imaging. This protocol describes how this setup can be used to image dynamic microtubules and MAPs. The capability of the apparatus is demonstrated by presenting the first visualization of kinesin-1 proteins walking on shrinking microtubules, which is captured at a frame rate of 10 s-1.
Protocol
1. Preparation of flow chambers
NOTE: Microfluidic flow chambers will be constructed by adhering polydimethylsiloxane (PDMS) microchannels to a cleaned and functionalized cover glass. The microchannels will be cast in a master mold.
- Preparation of PDMS channels
- Prepare cleaned and/or functionalized cover glasses with surface chemistry appropriate to the specific assay being imaged.
NOTE: For kinesin motility assays, piranha-cleaned silanized cover glasses are used. These are prepared as described in Gell et. al.1 Alternatively, a simpler procedure is to sonicate cover glasses in isopropanol followed by methanol for 3x 20 min cycles. - To prepare a master mold, cut strips of single-sided stationary tape to the desired flow channel size. Adhere the strips to the bottom of a 10 cm Petri dish, arranging them side-to-side with at least 1 cm spacing between the strips.
NOTE: If precise channel dimensions are required, the mold can be fabricated on silicon wafers using photolithography14. - To prepare the PDMS polymer, combine the curing agent and the base elastomer in a 1:10 mass ratio. Mix for 2 min.
NOTE: This mixing ratio can be varied to tune the stiffness of the polymer. A higher ratio of base to curing agent results in a softer polymer. - Degas the mixture in a vacuum chamber until all bubbles disappear.
- Pour the mixture onto the master mold in a ~0.5 cm thick layer, taking care to avoid creating bubbles.
- Bake the mixture in a preheated oven at 70 °C for 40 min.
NOTE: A longer curing time may be required if the PDMS block is thick (>1 cm). Continue heating in 5 min increments until it is fully cured. - Cut out the structured regions of the polymer. Punch holes at each end of the channel using a PDMS puncher.
NOTE: In this protocol, the hole diameter is set to 0.75 mm and can be adjusted according to the required flow rates. PDMS channels can be stored in dry environments for extended periods but should be cleaned prior to use. - Clean the structured side of the PDMS block. Use stationary tape to remove large particles. Rinse with isopropanol and then methanol. Repeat these rinses 3x, rinse with ultrapure water, and blow-dry the surface.
- Plasma clean the PDMS using oxygen or air plasma.
NOTE: In this protocol, 18 W air plasma is used. - Place the plasma cleaned PDMS on an appropriately cleaned cover glass and heat on a hot plate at 80 °C for 15 min.
NOTE: Epoxy resin can be applied to the sides of the PDMS block to better adhere it to the cover glass. - Insert appropriately sized low-density polyethylene (LDPE) tubing into the holes. Connect the outlet tubing to a 0.5 mL syringe.
NOTE: In this protocol, tubes with an inner diameter of 0.023 in are connected to the PDMS via a 0.025 in outer diameter metal adaptor. - Flow solutions into the microchannels by immersing the inlet tube in the solution and drawing the required volume with the syringe.
- Prepare cleaned and/or functionalized cover glasses with surface chemistry appropriate to the specific assay being imaged.
2. Optical setup
- Modification of the microscope (Figure 1)
- Modify a TIRF microscope to enable IRM imaging13. Replace the 50/50 (Reflectance (R)/Transmission (T)) beam splitter used in the filter wheel of the microscope with a 10/90 (R/T) beam splitter.
- Insert an image splitter between the camera and the microscope.
- In the filter cube of the image splitter, insert a 10/90 (R/T) beam splitter. Place a 600 nm long-pass filter in front of the reflected beam and an appropriate fluorescence emission filter in front of the transmitted beam.
NOTE: Beam splitters with different R/T ratios can be used to tune the fractions of the IRM and TIRF emission light that are collected. - Align the image splitter according to the manufacturer's specifications to spatially separate the TIRF and IRM signals on the camera chip.
NOTE: Spatial separation of the signals is required because the TIRF signal of typical fluorophores is much weaker than the IRM signal of a microtubule and would not be detectable if the two signals were overlayed.
3. Imaging dynamic microtubules and single Kinesin molecules
- Surface functionalization and passivation
- Flow BRB80 buffer (80 mM Na-PIPES, 1 mM EGTA, 1 mM MgCl2, titrated to pH 7.8 with NaOH) into the reaction chamber.
- Flow antibiotin solution diluted to 0.025 mg/mL in BRB80 and incubate at room temperature for 10 min.
- Wash the channel with BRB80.
- Flow in F-127 solution (1% Pluronic F-127 (w/v) dissolved in BRB80 overnight) and incubate for at least 20 min for surface passivation.
NOTE: If easy-cleaned cover glasses are used instead of silanized cover glasses, passivate using 2 mg/mL casein in BRB80 for >20 min at room temperature. - Wash the channel with BRB80.
- Preparation for imaging
- If temperature control is required, use an objective heater and set it to the correct temperature.
NOTE: In this protocol, all the experiments are performed at 28 °C. - Place the sample on the microscope stage and turn on the epiillumination light source (>600 nm) for IRM imaging.
NOTE: In this protocol, a white LED light source is used with a 600 nm long-pass filter. - Focus the microscope on the sample surface. Look for the correct focal plane near the PDMS-solution interface.
NOTE: In IRM, the aqueous interior of the channel should appear much brighter than the PDMS polymer. - Pick a field of view near the center of the channel.
- Prepare a solution of 0.1 µm fluorescent microbeads in BRB80 (density: 109 beads/mL, corresponding to a 200-fold dilution for the beads used in this protocol).
- Flow in at least one channel volume of the microbead solution.
- Monitor the reaction via IRM. Wait for the microbeads to gradually "land" onto the surface. When the desired density of microbeads is achieved, wash out the excess with BRB80.
- If temperature control is required, use an objective heater and set it to the correct temperature.
- Growing biotinylated guanylyl 5'-α,β-methylenediphosphonate (GMPCPP)-stabilized microtubule seeds
- In a 0.6 mL centrifuge tube, prepare 50 μL of a solution containing 1 mM GMPCPP, 1 mM MgCl2, and 2 μM biotinylated tubulin (5-10% labeling stoichiometry) in BRB80.
NOTE: The correct labeling stoichiometry can be achieved by combining high-density biotinylated tubulin with unlabeled bovine tubulin in the correct ratio. - Incubate the solution on ice for 5 min, then incubate at 37 °C for 12.5 min.
NOTE: The length of the seeds can be controlled by tuning the polymerization time. - Stop the polymerization by adding 100 μL of room temperature BRB80.
- Spin down the solution in an ultracentrifuge at room temperature (126,000 x g, 5 min). Discard the supernatant by using a pipette to remove unpolymerized tubulin.
NOTE: In this protocol, an air-driven ultracentrifuge is used. - Add 200 μL of room temperature BRB80 to the pellet. Resuspend the pellet by pipetting gently but thoroughly. Use a 200 μL pipette with a cut tip to reduce the shearing of the microtubules.
- In a 0.6 mL centrifuge tube, prepare 50 μL of a solution containing 1 mM GMPCPP, 1 mM MgCl2, and 2 μM biotinylated tubulin (5-10% labeling stoichiometry) in BRB80.
- Growing dynamic guanosine diphosphate (GDP)-tubulin "extensions"
NOTE: The seeds will be immobilized on the antibiotin surface of the flow channel. Dynamic GTP/GDP "extensions" will be grown from the ends of the immobilized seeds.- Dilute the seeds 20x in BRB80. Flow the diluted seeds into the reaction chamber.
- Monitor the reaction via IRM. Wait for the seeds to gradually "land" onto the surface and bind to it. When the desired density of seeds is achieved, wash out the excess with BRB80.
- Prepare a microtubule extension mixture: 12 μM unlabeled tubulin, 1 mM GTP, 5 mM dithiothreitol (DTT) in BRB80 buffer.
- Flow in at least one channel volume of the extension mixture. Ensure that the reaction temperature is 28 °C.
- Wait for the microtubule extensions to grow from the seeds over time.
NOTE: The steady-state length is usually achieved in less than 20 min. - Use the dynamic microtubules for imaging. Add and visualize fluorescent MAPs on the microtubules, as described in step 3.5 for kinesin-1.
NOTE: Because the microtubules are surface-bound, they are easily visible by TIRF.
- Kinesin motility assay
NOTE: This step describes a protocol for visualizing motile green fluorescent protein (GFP)-labeled kinesin-1 on shrinking microtubules. A truncated rat kinesin-1 construct fused to eGFP (rKin430-eGFP) was expressed and purified, as described previously15,16.- Prepare motility buffer: 1 mM ATP and 0.2 mg/mL casein in BRB80.
- Dilute kinesin-eGFP in motility buffer to 10 nM.
- Prepare a 2x oxygen scavenger solution to counteract oxidative photobleaching (80 mM glucose, 80 mg/mL glucose oxidase, 32 mg/mL catalase, 0.2 mg/mL casein, 20 mM DTT) supplemented with 2 mM ATP.
- Combine 10 parts of oxygen scavenger, 9 parts of BRB80, and 1 part of 10 nM kinesin-eGFP for a final kinesin concentration of 0.5 nM.
NOTE: The total volume should be at least 1.5-fold larger than the channel volume. - Set the imaging settings on the microscope software.
NOTE: In this protocol, videos are recorded at 10 fps with 100 ms exposure time. The laser intensity is ~0.05 kW/cm2. - Begin imaging and flow the kinesin solution into the chamber.
- After the measurement is complete, record a short (~5 s) video in which the stage is slowly translated in a circular or lateral motion. Use the median projection of this video as a background image and subtract it from the raw measurements.
4. Image processing and analysis
NOTE: Image processing was carried out using NIH ImageJ2 (imagej.nih.gov/ij/). A macro was developed to automate the splitting and alignment of the TIRF and IRM channels. This macro requires the GaussFit_OnSpot plugin to be installed (available on the ImageJ plugins repository).
- From the background recording, create a median projection in ImageJ by clicking on Image | Stacks | Z-project.
- Subtract the median background projection from the raw image data by clicking on Process | Image Calculator.
NOTE: Make sure to check the “32-bit (float) result" option. - For image registration, pick a collection of microbeads near the microtubule of interest and use them to align the TIRF and IRM images.
- For each bead in this collection, use the multipoint selection tool to mark the approximate location in the TIRF channel and then the corresponding location in the IRM channel.
NOTE: For example, if there are two beads (1 and 2), the multipoint selection would have four points in the following order: (1) bead 1 in TIRF, (2) bead 1 in IRM, (3) bead 2 in TIRF, (4) bead 2 in IRM. - Run the provided ImageJ macro (ImageSplitterRegistration.ijm).
NOTE: This automates the following steps: i) estimating the center location of each bead by fitting to a Gaussian; ii) for each bead, computing the displacement vector separating the center in the TIRF channel from the center in the IRM channel; iii) averaging this displacement vector across all beads; iv) splitting the TIRF and IRM channels into separate images; v) translating the TIRF image by the average displacement vector computed in iii; vi) overlaying the TIRF and IRM images in a multichannel .tiff file.
Results
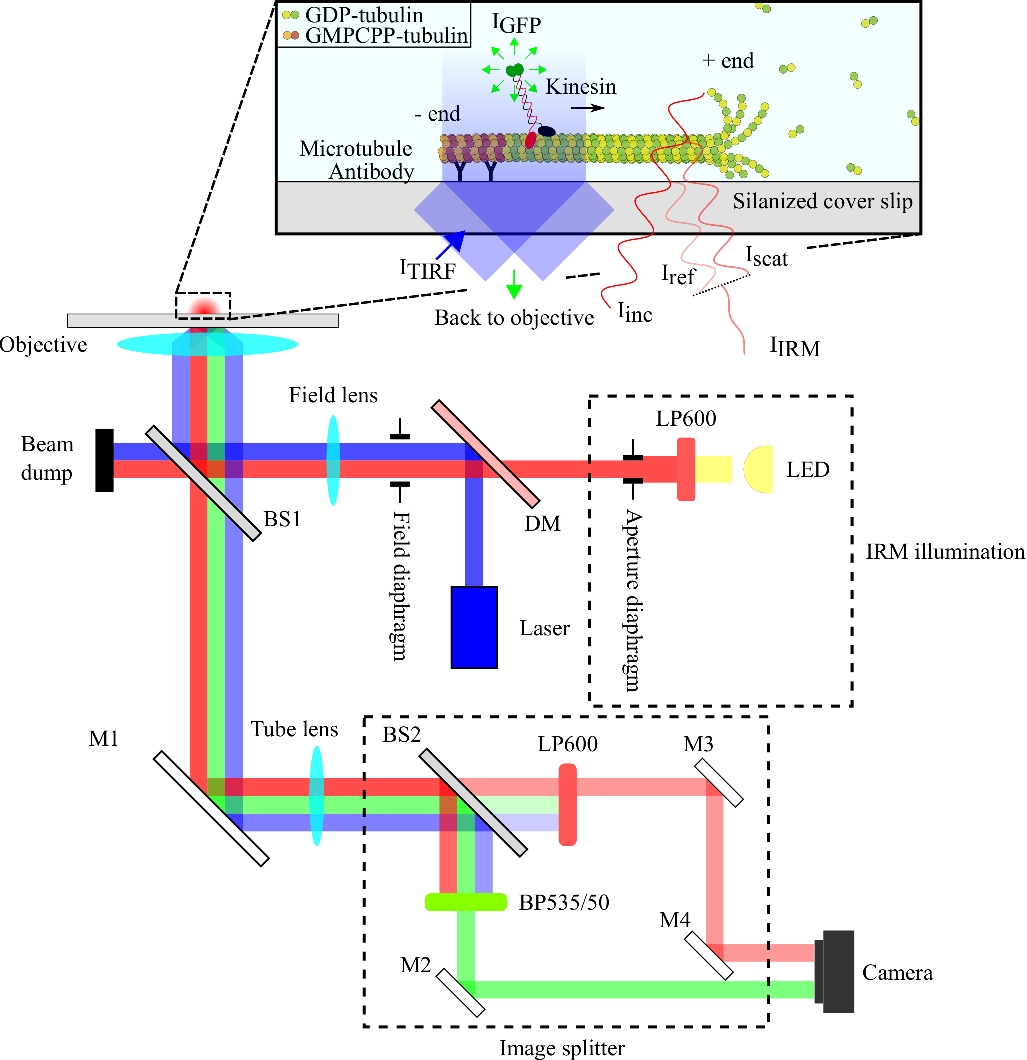
The optical setup is schematized in Figure 1. Both IRM illumination and TIRF excitation light are directed to the back aperture of the objective (100x, NA: 1.49) via a 10/90 (R/T) beam splitter (BS1). The emitted signal passes through the same beam splitter (BS1) and is reflected to the image splitter via a mirror (M1). The components of the image splitter (enclosed with dashed lines in Figure 1) separate the IRM and TIRF signals via a 90/10 (R/T) beam splitter (BS2) together with appropriate spectral filters. Finally, the split images are projected onto the camera chip for visualization. The alignment of the image splitter is such that the TIRF and IRM signals are projected on separate halves of the chip.
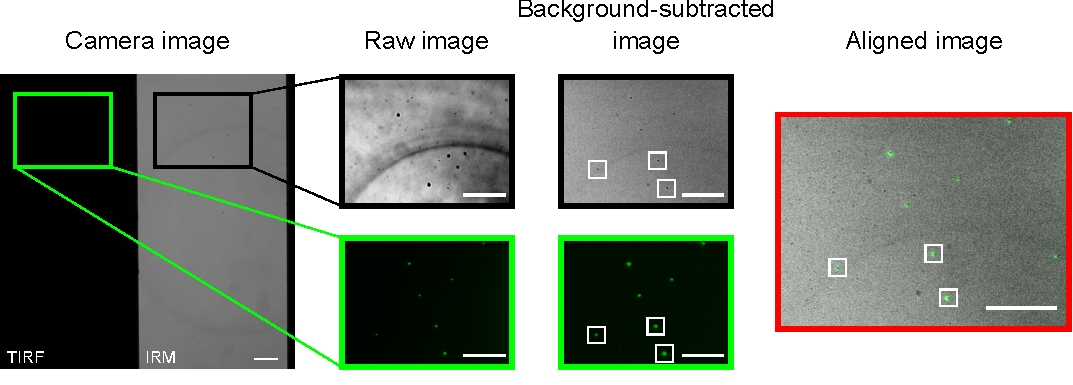
In a well-aligned microscope, the camera image should display a halfway split image, as presented in Figure 2. Surface-bound microtubules should be easily visible in the IRM channel13, and fluorescent kinesin should be visible in the TIRF channel.
The microbeads used to align and register the two channels appear as bright spots in the TIRF images and dark spots in the IRM images. Although the beads are visible in the raw data, background subtraction improves the contrast significantly (Figure 2). The background image used for the subtraction is the temporal median of a video recorded with a moving stage. As described in the protocol, image alignment was performed by selecting a collection of beads near the region of interest and executing the provided macro (imageSplitterRegistration.ijm). The macro fits the points to Gaussians and aligns the images by minimizing the average distance between the center points of the fits in each channel. This process is represented in Figure 2, which shows a good alignment of the fluorescent microbeads (green in the TIRF channel, black in the IRM channel).
Finally, the capabilities of this simultaneous imaging setup are demonstrated by observing single kinesin molecules walking towards the shrinking ends of microtubules. Figure 3 shows a kymograph of eGFP-labeled kinesin molecules (green) walking on a shrinking microtubule (gray). Also presented is a series of snapshots from the recording from which the kymograph was generated.

Figure 1: Schematic representation of the optical setup for simultaneous IRM and TIRF imaging of kinesin motility. Epiillumination from an LED light source passes through the aperture diaphragm and reaches the 10/90 (R/T) beam splitter (BS1). The beam splitter partially reflects the red IRM illumination light and the 488 nm TIRF excitation light up to the objective to illuminate the sample. The signal from the sample is collected by the same objective and directed to the image splitting assembly where IRM and TIRF images are spatially separated by the 90/10 (R/T) beam splitter (BS2). The signals are then spectrally filtered before reaching the camera chip. Abbreviations: IRM = interference reflection microscopy; TIRF = total-internal-reflection-fluorescence; LED = light-emitting diode; ITIRF = TIRF illumination; IGFP = GFP fluorescence; Iinc = IRM illumination; Iref = scattered light at the glass/water interface; Iscat = scattered light from the microtubule; IIRM = IRM signal (Interference of Iref and Iscat); R/T = reflected/transmitted; LP600: long pass filter (600 nm); DM = dichroic mirror; BS1 and BS2 = beam splitters 1 and 2; M1, M2, M3, M4 = mirrors; BP535/50 = band pass (535/50 nm); GFP = green fluorescent protein; GMPCPP = guanylyl 5'-α,β-methylenediphosphonate; GDP = guanosine diphosphate. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Background subtraction and image alignment. The TIRF (left half) and IRM (right half) images appear simultaneously on two halves of the same camera chip (camera image). Temporal median background subtraction increases the contrast of the beads (background-subtracted image), which appear dark in IRM and bright in TIRF images. IRM and TIRF images are aligned by translation (right) based on the localization of selected beads (white rectangles). Scale bars = 10 μm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Kymograph and snapshots of Kinesins' movement during microtubule shrinkage. The kymograph (left) shows eGFP-kinesin-1 (green) walking towards the plus end of the microtubule (dark grey). Snapshots from the corresponding time series are shown (right). White arrows show single kinesin-1 molecules. Please click here to view a larger version of this figure.
{kind=link}
ImageSplitterRegistration File: Please click here to download this File.
Discussion
Studying the regulation of microtubule dynamics by microtubule-associated proteins (MAPs) often requires simultaneous imaging of microtubules and MAPs. Fluorescence microscopy techniques such as TIRF are typically employed for this purpose. However, they are limited by the drawbacks of fluorescence imaging, which include photobleaching, photodamage, and the need for fluorophore labeling. Label-free methods, such as IRM, are suitable for visualizing microtubules but are not capable of imaging single fluorophores. This protocol combines label-free IRM imaging and TIRF microscopy for the simultaneous imaging of dynamic microtubules and MAPs.
The IRM setup employs an LED illumination source filtered to >600 nm, while the TIRF setup utilizes a 488 nm laser. We used an inexpensive plate beam splitter to reflect the illumination light onto the sample and transmit the collected signal to the detector (Figure 1). A beam splitter with 10% reflectance and 90% transmission was chosen to minimize the loss of the single-molecule signal. The 90% loss in the illumination light intensity is compensated for by increasing the power of the illumination laser and LED.
Spectral separation of the signals was achieved using a 90/10 (R/T) beam splitter and two spectral filters (long-pass 600 nm for IRM and band-pass 535/50 nm for TIRF). The spectrally separated IRM and TIRF signals are projected onto two halves of a single camera chip using an image splitter assembly. The use of a 90/10 beam splitter sacrifices 90% of the IRM signal, but this is compensated for by increasing the intensity of the LED illumination source. A dichroic mirror could also be used here to separate the IRM and TIRF signals more efficiently. Fluorescent microbeads included in the assays enable accurate alignment of the TIRF and IRM images and serve as a reference for focusing the objective.
The most critical optical element in this protocol is the high numerical aperture (NA) objective. This is essential not only to achieve total internal reflection but also to maximize the collection efficiency and image contrast. The quality of the obtained images also depends on the cleanliness of the glass surface and the acquisition of a clear background image to correct for nonuniform illumination and remove static features. For IRM imaging, we recommend using long-wavelength illumination (>600 nm) to minimize the photodamage of microtubules and proteins. This is particularly important if a white LED light source is used, in which case a long-pass filter should be included to remove any UV light.
This protocol allows for label-free, high-speed imaging of dynamic microtubules and simultaneous high-resolution visualization of fluorescent MAPs17. Compared to the filter cube switching technique, which alternates between capturing images of microtubules and MAPs, this setup is capable of much higher frame rates because it does not depend on the physical rotation of a filter cube. Compared to two-color TIRF imaging techniques, this technique employs a less demanding optical setup and circumvents the need for fluorophore labeling of microtubules. The primary limitations of this setup are due to the TIRF imaging of the MAPs; the frame rate is limited by the exposure time of a fluorophore, and photobleaching of fluorophores remains a possibility. Nonetheless, this protocol improves upon existing techniques because it uses TIRF only when necessary (i.e., to visualize MAPs but not microtubules) and achieves the highest speed possible within the limits of TIRF. Further improvements are possible only if both the microtubules and the MAPs are visualized via an interferometric technique, but this requires labeling MAPs with metal nanoparticles, which has its limitations and experimental challenges.
To demonstrate the capabilities of this technique, we simultaneously visualized two fast dynamical processes via IRM and TIRF: the shrinkage of a microtubule and the walking of a fluorescent kinesin molecule. This technique has been previously employed to visualize the fast diffusion of spastin on shrinking microtubules5. Beyond this application to MAPs and microtubules, this protocol can be used to visualize single fluorescent molecules simultaneously with any macromolecular structure massive enough to be visualized via IRM, such as a cell membrane or actin filament.
Disclosures
The authors have no conflicts of interest to disclose.
Acknowledgements
We thank Thierry Emonet's lab for sharing cleanroom equipments. We thank Yin-Wei Kuo for preparing the purified eGFP-kinesin used in this study. Y.T. acknowledges the support of the Alexander von Humboldt Foundation through the Feodor Lynen Research Fellowship. This work was supported by NIH Grant R01 GM139337 (to J.H.).
Materials
| Name | Company | Catalog Number | Comments |
| 10/90 (R/T) beam splitter | Thorlabs | BSN10R | Plane beam splitter used in the excitation beam path |
| 90/10 (R/T) beam splitter | Thorlabs | BSX10R | Plane beam splitter used in the image splitter |
| Anti-biotin antibody | Sigma-Aldrich | B3640 | Used to functionalize surface for bonding biotinylated microtubules |
| ATP | Sigma-Aldrich | FLAAS | Used for preparing the motility buffer |
| Band-pass filter | Newport | HPM535-50 | Hard-coated band-pas filter is used in image splitter to image GFP signal |
| Biotinylated tubulin | Cytoskeleton, Inc. | T333P-A | Used to bind microtubule seeds to the surface of the flow channel |
| Casein | Sigma-Aldrich | C8654 | Casein is used to block nonspesific interactions |
| Catalase | Sigma-Aldrich | C9322 | Used for preparing the oxygen scavenger solution |
| Desiccator chamber | Southern Labware | 55207 | Desiccator is used for degasing the resin |
| DTT | Sigma-Aldrich | D0632 | Used for preparing the oxygen scavenger solution |
| EGTA | Sigma-Aldrich | E4378 | Used for preparing the BRB80 buffer |
| Glucose | Sigma-Aldrich | G7528 | Used for preparing the oxygen scavenger solution |
| Glucose oxidase | Sigma-Aldrich | G7016 | Used for preparing the oxygen scavenger solution |
| GMPCPP nucleotides | Jena Bioscience | NU-405L | Used for the polymerization of stabilized microtubules |
| Image splitter | Teledyne-Photometrics Imaging | OptoSplit II | An image splitter is used to split the images spatially. When buying, make sure about the compatibility with the microscope |
| ImajeJ2 | NIH | ImageJ2 is used for image analysis | |
| Kinesin | prepared in house (see references in text) | ||
| LDPE tubing | Thomas Scientific | 9565S22 | Non-toxic, lower density polyethylene micro bore tubing is used for fluid transfers |
| LED light source | Lumencor | Lumencor sola light engine | Used for IRM imaging |
| Long-pass filters | Thorlabs | FELH0600 | Hard-coated long pass filters. One is used as an excitation filter, other is used in image splitter to image IRM signal |
| Magnesium chloride | Sigma-Aldrich | 63068 | Used for preparing the BRB80 buffer |
| Micoscope objective heater | okolab | H401-T-DUAL-BL | Used to keep sample temperature constant via heating the objective |
| Microscope | Nikon | Ti-Eclipse | An inverted microscope that is used in the experiments |
| Na-PIPES | Sigma-Aldrich | P2949 | Used for preparing the BRB80 buffer |
| Nikon CFI Apochromat TIRF 100XC Oil objective | Nikon | MRD01991 | The imaging objective has 1.49 numerical aperture |
| PDMS and curing agent | Electron Microscopy Sciences | Sylgard 184 (24236-10) | Used for contructing the flow channels |
| PDMS puncher | World Precision Instruments LLC | 504529 | Used to punch hole into the PDMS |
| Plasma cleaner | Harrick Plasma | DPC-32G | The air plasma is used to remove organic contamination from the PDMS surface |
| Poloxamer 407 (commercial name Pluronic F-127) | Sigma-aldrich | P2443 | Used for channel surface passivation to minimize nonspecific binding |
| Sodium hydroxide | Sigma-Aldrich | 567530 | Used for preparing the BRB80 buffer |
| Stabilized microtubules | prepared in house (see references in text) | ||
| Table-top ultracentrifuge | Beckman Coulter | 340400 | Used to spin down microtubule seeds |
| TetraSpeck beads | ThermoFisher Scientific | T7279 | Used as a reference for aligning images |
| Zyla 4.2 camera | Andor | Zyla 4.2 | Scientific CMOS camera with spesifications: 2048 x 2048 pixels (6.5 μm pixel size) with quantum efficiency of 72% and 16 bit dynamic range |
References
- Gell, C., et al. Microtubule dynamics reconstituted in vitro and imaged by single-molecule fluorescence microscopy. Methods in Cell Biology. 95, 221-245 (2010).
- Axelrod, D. Chapter 7: Total internal reflection fluorescence microscopy. Methods in Cell Biology. 89, 169-221 (2008).
- Kuo, Y. -. W., Trottier, O., Mahamdeh, M., Howard, J. Spastin is a dual-function enzyme that severs microtubules and promotes their regrowth to increase the number and mass of microtubules. Proceedings of the National Academy of Sciences of the United States of America. 116 (12), 5533-5541 (2019).
- Hinrichs, M. H., et al. Tau protein diffuses along the microtubule lattice. The Journal of Biological Chemistry. 287 (46), 38559-38568 (2012).
- Al-Hiyasat, A., Tuna, Y., Kuo, Y. -. W., Howard, J. Herding of proteins by the ends of shrinking polymers. arXiv:. , (2021).
- Guo, H., et al. Mechanism and dynamics of breakage of fluorescent microtubules. Biophysical Journal. 90 (6), 2093-2098 (2006).
- Moerner, W. E., Fromm, D. P. Methods of single-molecule fluorescence spectroscopy and microscopy. Review of Scientific Instruments. 74 (8), 3597-3619 (2003).
- Taylor, R. W., Sandoghdar, V., Astratov, V. . Interferometric scattering (iSCAT) microscopy and related techniques. in Label-free super-resolution microscopy. , 25-65 (2019).
- Young, G., Kukura, P. Interferometric scattering microscopy). Annual Review of Physical Chemistry. 70 (1), 301-322 (2019).
- Koch, M. D., Rohrbach, A. Label-free imaging and bending analysis of microtubules by ROCS microscopy and optical trapping. Biophysical Journal. 114 (1), 168-177 (2018).
- Kandel, M. E., Teng, K. W., Selvin, P. R., Popescu, G. Label-free imaging of single microtubule dynamics using spatial light interference microscopy. ACS Nano. 11 (1), 647-655 (2017).
- Mahamdeh, M., Simmert, S., Luchniak, A., Schäffer, E., Howard, J. Label-free high-speed wide-field imaging of single microtubules using interference reflection microscopy. Journal of Microscopy. 272 (1), 60-66 (2018).
- Mahamdeh, M., Howard, J. Implementation of interference reflection microscopy for label-free, high-speed imaging of microtubules. Journal of Visualized Experiments: JoVE. (150), e59520 (2019).
- Voldman, J., Gray, M. L., Schmidt, M. A. Microfabrication in biology and medicine. Annual Review of Biomedical Engineering. 1, 401-425 (1999).
- Rogers, K. R., et al. KIF1D is a fast non-processive kinesin that demonstrates novel K-loop-dependent mechanochemistry. The EMBO Journal. 20 (18), 5101-5113 (2001).
- Leduc, C., Ruhnow, F., Howard, J., Diez, S. Detection of fractional steps in cargo movement by the collective operation of kinesin-1 motors. Proceedings of the National Academy of Sciences of the United States of America. 104 (26), 10847-10852 (2007).
- Tuna, Y., Al-Hiyasat, A., Howard, J. Imaging dynamic microtubules and associated proteins by Simultaneous Interference-Reflection and Total-Internal-Reflection-Fluorescence Microscopy. arXiv. , (2022).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved