É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Predição computacional de preferências de aminoácidos de domínios de ligação peptídica potencialmente multiespecíficos envolvidos em interações proteína-proteína
Neste Artigo
Resumo
Descrevemos uma metodologia baseada na diversificação de sequências para estimar as preferências de aminoácidos de sítios de ligação multiespecíficos em interações proteína-proteína (PPIs). Nessa estratégia, milhares de potenciais ligantes peptídicos são gerados e rastreados in silico, superando assim algumas limitações dos métodos experimentais disponíveis.
Resumo
Muitas interações proteína-proteína envolvem a ligação de segmentos curtos de proteínas a domínios de ligação a peptídeos. Normalmente, tais interações requerem o reconhecimento de motivos lineares com conservação variável. A combinação de regiões altamente conservadas e mais variáveis nos mesmos ligantes geralmente contribui para a multiespecificidade da ligação, uma propriedade comum de enzimas e proteínas de sinalização celular. A caracterização das preferências de aminoácidos dos domínios de ligação a peptídeos é importante para o projeto de mediadores de interações proteína-proteína (PPIs). Os métodos computacionais são uma alternativa eficiente às técnicas experimentais muitas vezes caras e complicadas, permitindo o projeto de potenciais mediadores que podem ser posteriormente validados em experimentos a jusante. Aqui, descrevemos uma metodologia usando a aplicação Pepspec do pacote de modelagem molecular Rosetta para prever as preferências de aminoácidos dos domínios de ligação a peptídeos. Essa metodologia é útil quando a estrutura da proteína receptora e a natureza do ligante peptídico são conhecidas ou podem ser inferidas. A metodologia começa com uma âncora bem caracterizada do ligante, que é estendida pela adição aleatória de resíduos de aminoácidos. A afinidade de ligação dos peptídeos gerados dessa maneira é então avaliada por docking de peptídeos de backbone flexível para selecionar os peptídeos com as melhores pontuações de ligação previstas. Esses peptídeos são então usados para calcular as preferências de aminoácidos e, opcionalmente, calcular uma matriz posição-peso (PWM) que pode ser usada em estudos posteriores. Para ilustrar a aplicação dessa metodologia, usamos a interação entre subunidades do fator regulador de interferon humano 5 (IRF5), anteriormente conhecido por ser multiespecífico, mas globalmente guiado por um motivo conservado curto chamado pLxIS. As preferências estimadas de aminoácidos foram consistentes com o conhecimento prévio sobre a superfície de ligação do IRF5. As posições ocupadas por resíduos de serina fosforilável exibiram uma alta frequência de aspartato e glutamato, provavelmente porque suas cadeias laterais carregadas negativamente são semelhantes à fosfoserina.
Introdução
A interação entre duas proteínas geralmente envolve a ligação de segmentos curtos de aminoácidos a domínios de ligação a peptídeos, assemelhando-se a interfaces proteína-peptídeo. As proteínas receptoras envolvidas em tais interações proteína-proteína (PPI) geralmente têm a capacidade de reconhecer um certo conjunto de sequências de ligantes sobrepostas, mas divergentes, uma propriedade conhecida como multiespecificidade 1,2. O reconhecimento multiespecífico é uma característica de muitas proteínas celulares, mas é particularmente notável em enzimas e proteínas de sinalização celular3. As proteínas que interagem com sítios de ligação multiespecíficos geralmente têm uma combinação de regiões mais e menos conservadas em sua sequência 4,5,6. Nesse cenário, os motivos de sequência mais conservados estão envolvidos em interações moleculares rigorosas. Por outro lado, as sequências mais variáveis interagem com superfícies de alguma forma permissivas no local de ligação ao receptor. Normalmente, esses segmentos menos conservados, mas ainda funcionalmente relevantes, são loops sem padrões de estrutura secundária definidos ou têm conformações ainda mais dinâmicas, como as típicas de proteínas intrinsecamente desordenadas7.
A identificação de potenciais ligantes peptídicos de sítios de ligação é geralmente o primeiro passo no projeto de mediadores capazes de interferir nos PPIs correspondentes8. No entanto, muitas vezes é improvável encontrar um único resíduo de aminoácido mais frequente na maioria das posições de sequência em ligantes de sítios de ligação multiespecíficos. Em vez disso, esses locais podem ter preferências particulares por uma classe específica de aminoácidos de acordo com suas propriedades químicas, por exemplo, aminoácidos ácidos e carregados negativamente, como aspartato ou glutamato, aminoácidos aromáticos volumosos, como fenilalanina, ou resíduos mais hidrofóbicos, como aminoácidos alifáticos alanina, valina, leucina ou isoleucina3. Vários métodos experimentais podem fornecer informações sobre as preferências de aminoácidos dos locais de ligação às proteínas, incluindo evolução dirigida9, mutagênese de varredura de múltiplos códons10 e varredura mutacional profunda11. Todos esses métodos seguem a abordagem de diversificação de sequência, que se baseia na introdução de mutações nos ligantes originais e na análise de seu efeito na função da proteína receptora (ver Bratulic e Badran12 para uma revisão abrangente). No entanto, esses métodos geralmente exigem o levantamento de grandes bibliotecas de sequências, o que os torna mais complicados, caros e demorados.
Métodos computacionais para inferir as preferências de aminoácidos de sítios de ligação multiespecíficos têm o potencial de contornar as limitações dos métodos de laboratório úmido. Dentre estes, a abordagem de diversificação da sequência in silico avalia o impacto energético de uma ampla gama de substituições de aminoácidos na sequência do ligante como forma de caracterizar a plasticidade estrutural do PPI13. Este método começa com a estrutura ou modelo do ligante peptídico ligado ao local de ligação do receptor e, posteriormente, introduz mutações na sequência do ligante. As funções estatísticas e de pontuação de energia são então usadas para avaliar o impacto dessas mutações na estabilidade e na afinidade de ligação. O conjunto de sequências de ligantes de melhor pontuação resultantes da fase de avaliação pode então ser usado para calcular as preferências de aminoácidos. Essa estratégia tem o potencial de processar um número muito alto de sequências de ligantes de maneira eficiente. Portanto, ele pode fornecer uma inferência mais completa e consistente das preferências de aminoácidos em comparação com aquelas calculadas a partir do número mais limitado de sequências que geralmente podem ser processadas em abordagens de laboratório úmido.
A aplicação Pepspec do conjunto de modelagem molecular Rosetta14 é uma ferramenta que realiza a diversificação de sequências como uma etapa fundamental de seu modo de design de peptídeos. Esta aplicação requer uma estrutura ou modelo da proteína receptora com um peptídeo ligado a um único resíduo de aminoácido de comprimento, que é usado como âncora para as próximas etapas. A sequência do peptídeo ligado é então estendida (se necessário) e diversificada para gerar um grande número de ligantes peptídicos putativos. A afinidade de ligação desses peptídeos é então avaliada por acoplamento de peptídeos de backbone flexível para selecionar aqueles com as melhores pontuações de ligação previstas. Embora a principal saída desta aplicação sejam os melhores candidatos a peptídeos selecionados no final da fase de projeto, o conjunto muito maior de peptídeos aceitos durante esta fase também pode ser usado para calcular as preferências de aminoácidos do local de ligação alvo. As preferências de aminoácidos são calculadas como a frequência de cada resíduo de aminoácido por posição da sequência de ligantes representada como uma matriz de peso de posição (PWM) ou como um logotipo de sequência mais visual.
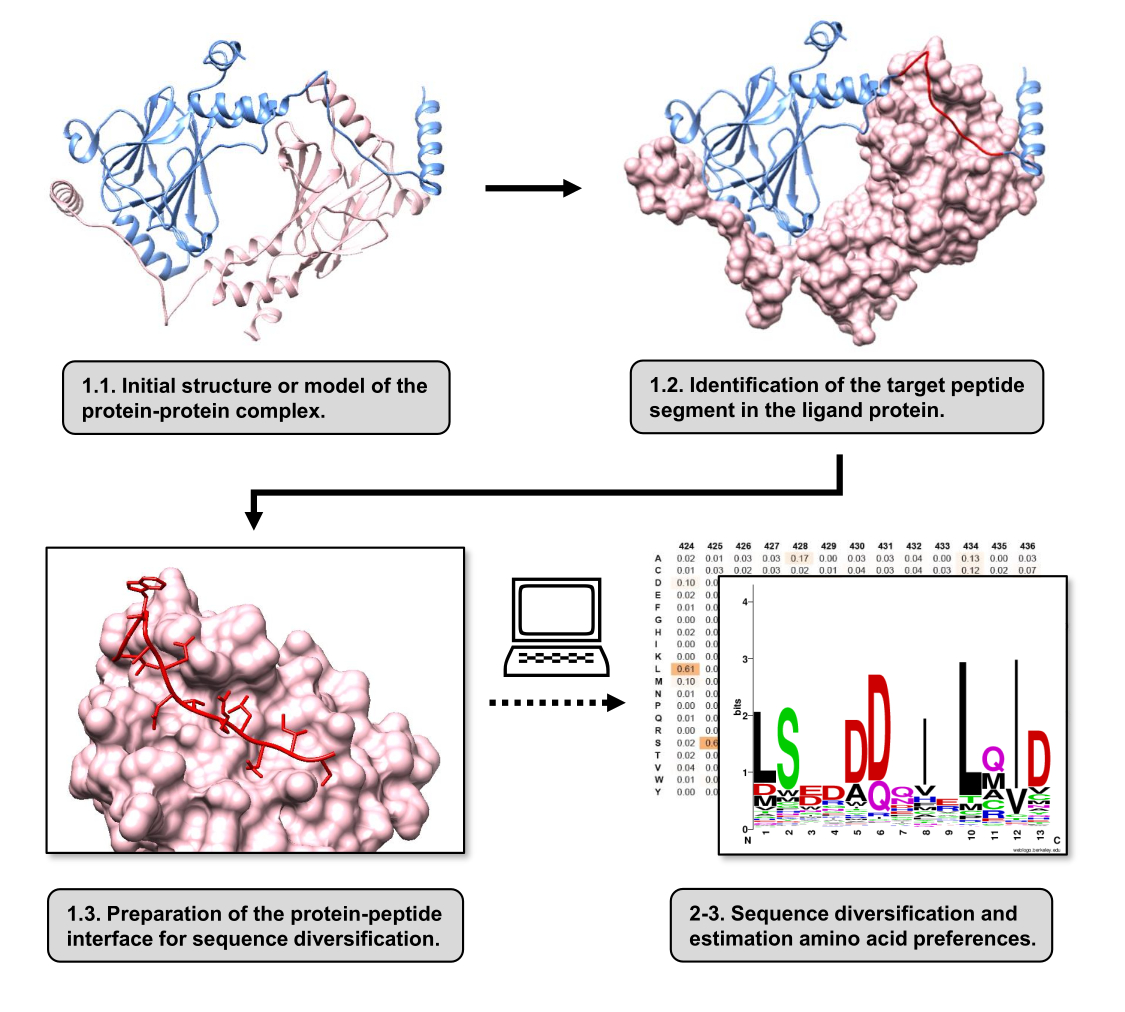
Neste artigo, descrevemos um protocolo para estimar as preferências de aminoácidos da superfície de ligação de uma proteína receptora envolvida em um PPI. O protocolo é focado em PPIs nos quais um segmento linear do ligante de proteína é conhecido por se ligar à proteína receptora, de modo que o cenário pode ser modelado como uma interface proteína-peptídeo. Nesse cenário, os motivos conservados do ligante normalmente interagem com bolsas definidas no local de ligação do receptor, embora todo o segmento do ligante envolvido no PPI possa conter regiões menos conservadas. Um fluxograma resumindo as principais etapas do protocolo é mostrado na Figura 1. O protocolo começa com a estrutura 3D do complexo proteína-proteína e reduz ainda mais a proteína ligante para o potencial segmento de melhor interação, deixando a proteína receptora intacta. O segmento de melhor interação é inferido usando o servidor BUDE Alanine Scan15, que conduz mutagênese computacional de varredura de alanina para identificar resíduos de pontos quentes entre as duas proteínas que interagem. Nesta abordagem, os resíduos do ligante são substituídos individualmente por alanina, e a mudança estimada na energia livre ou estabilidade do complexo (ΔΔG) é então usada para inferir a relevância do resíduo correspondente para o PPI alvo. Uma vez inferido o segmento de melhor interação, seu complexo com a proteína receptora é usado como a estrutura de base submetida ao Pepspec para realizar a diversificação de sequências.

Figura 1: Visão geral das principais etapas do protocolo proposto neste trabalho. Os números correspondem aos números das etapas na seção de protocolo. As figuras foram feitas com o complexo proteína-proteína usado como exemplo descrito no texto. Neste complexo, a cadeia de proteínas considerada como o receptor é mostrada em rosa, enquanto a cadeia considerada como o ligante é mostrada em azul claro com seu segmento de melhor interação previsto destacado em vermelho. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Uma das limitações do protocolo sugerido é a necessidade de uma estrutura resolvida da interface proteína-peptídeo. O protocolo pode, alternativamente, começar com um modelo da interface proteína-peptídeo alvo, embora as etapas específicas de modelagem não sejam descritas aqui. Além disso, embora o protocolo possa ser conduzido em um computador pessoal executando qualquer sistema operacional, um ambiente Linux é necessário para as etapas que envolvem os aplicativos Rosetta. Um cluster de computadores também é altamente recomendado para a etapa de diversificação de sequências devido ao grande número de iterações normalmente executadas pelo Pepspec.
A aplicação do protocolo sugerido é ilustrada com a estimativa das preferências de aminoácidos da superfície de ligação do IRF5, um membro da família do fator regulador do interferon humano (IRF). Escolhemos essa proteína como exemplo porque, durante sua ativação, duas subunidades se ligam para formar um dímero cuja estrutura é bem caracterizada16. Nos dímeros IRF, a ligação pode ser modelada como uma interface proteína-peptídeo na qual uma subunidade fornece a superfície de ligação e a outra interage através de uma região contendo um motivo conservado curto chamado pLxIS17,18. Além disso, a ligação às subunidades IRF é multiespecífica; portanto, eles podem formar homodímeros, heterodímeros e complexos com outras proteínas celulares conhecidas como coativadores18.
Protocolo
1. Preparação inicial da interface proteína-peptídeo
- Baixando a estrutura do complexo proteína-proteína
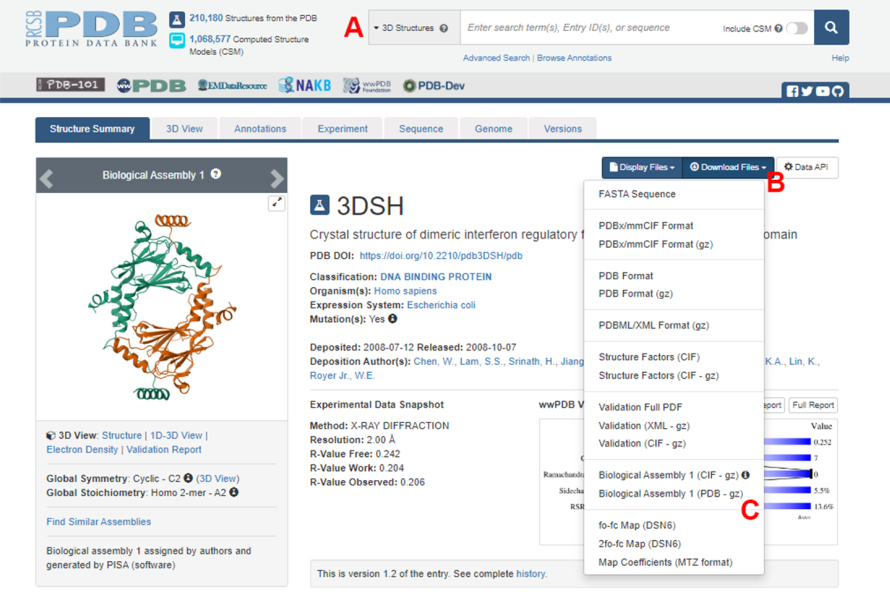
- Navegue até a página inicial do Protein Data Bank (PDB) (https://www.rcsb.org/) e digite o ID do PDB para a estrutura do complexo proteína-proteína na caixa de pesquisa principal (Figura 2A). O ID PDB para a estrutura do dímero IRF5, usado como exemplo neste trabalho, é 3DSH19.
- Na página principal da estrutura desejada, clique em Download Files (Figura 2B) e depois em Biological Assembly 1 (PDB - gz) (Figura 2C).
NOTA: No banco de dados PDB, estruturas de muitos complexos proteicos formados por monômeros idênticos são representadas como conjuntos biológicos, nos quais apenas a estrutura de um monômero (unidade assimétrica) é armazenada no arquivo PDB. A estrutura do multímero, neste caso, o dímero IRF5, deve ser baixada como o conjunto biológico contendo duas instâncias da unidade assimétrica. Para facilitar as próximas etapas deste protocolo, os dois monômeros são primeiro separados e diferentes IDs de cadeia são atribuídos a eles. - Abra a estrutura baixada no UCSF Chimera20 e clique em Ferramentas > Edição de Estrutura > IDs da Cadeia de Alterações. Neste exemplo, ambas as cadeias na montagem biológica são nomeadas A. Renomeie a segunda cadeia (rotulada como #0.2) para B e clique em OK.
- Clique em Favoritos > Painel Modelo e selecione o modelo que contém as duas cadeias. Clique no botão Agrupar/Desagrupar para separar cada cadeia em um modelo diferente. Em seguida, selecione os dois modelos e clique no botão Copiar/Combinar. Insira um novo nome para o modelo combinado, marque Fechar modelos de origem e clique em OK.
- Clique em Selecionar > Cadeia e confirme que cada corrente no dímero agora é identificada por uma letra diferente, ou seja, A e B.
- Use File > Save PDB para salvar a estrutura editada em um arquivo PDB diferente, que será usado nas próximas etapas do protocolo (aqui, o nome IRF5_dimer.pdb foi usado).

Figura 2: A página do Banco de Dados de Proteínas (PDB) para a estrutura usada como exemplo representativo neste trabalho. (A) Caixa de pesquisa para introduzir o código de acesso PDB da estrutura alvo. (B) Menu para baixar a estrutura em vários formatos. (C) Opções para baixar montagens biológicas quando a estrutura tiver sido salva como uma unidade assimétrica (consulte a etapa 1.1.2 para obter mais detalhes). Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Identificando o segmento alvo na proteína ligante
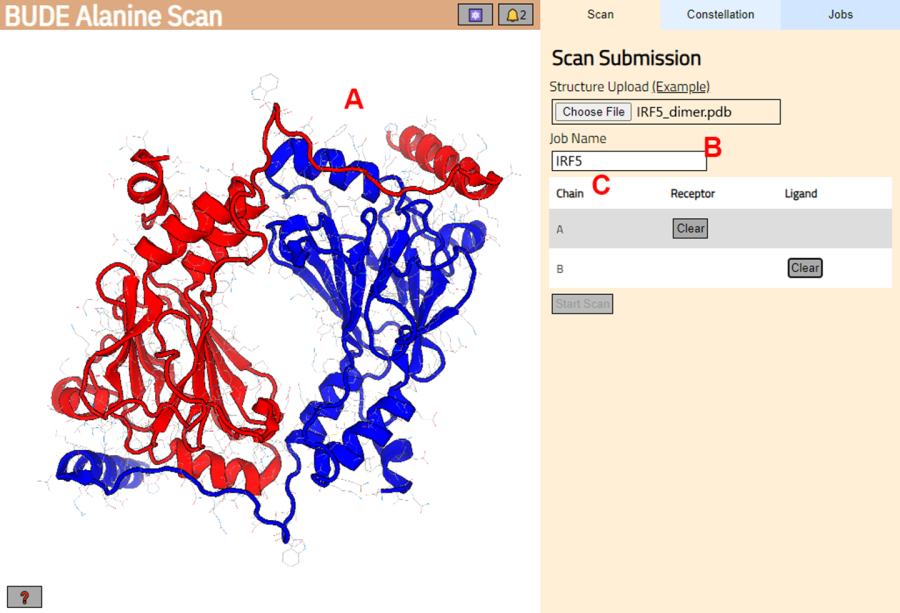
- Navegue até o servidor de varredura de alanina BUDE (https://pragmaticproteindesign.bio.ed.ac.uk/balas/). Clique no botão Escolher arquivo em Upload de estrutura e selecione o arquivo PDB salvo na etapa 1.1.6.
- Na próxima página, verifique se a estrutura foi carregada corretamente (Figura 3A) e insira um nome para o trabalho no servidor (Figura 3B).
- Defina as cadeias do PDB que serão tratadas como receptor (A) e ligante (B) (Figura 3C). Em seguida, clique no botão Iniciar digitalização para enviar o trabalho.
- Quando o trabalho estiver concluído, clique em Mostrar resultados para abrir a página de resultados (Figura 4).
NOTA: Na página de resultados, os resíduos da estrutura do ligante são coloridos de acordo com sua mudança estimada na energia livre (ΔΔG), e aqueles com valores mais altos são coloridos em vermelho. - Na lista de resíduos, selecione o trecho de resíduos previsto para interagir melhor com a superfície de ligação alvo. Certifique-se de que esses resíduos agrupem os valores mais altos para a diferença de energia livre (ΔΔG). Neste exemplo, o segmento entre os resíduos Leu424 e Ser436 foi selecionado (destacado com uma caixa vermelha no painel direito da Figura 4).
- Preparando a interface proteína-peptídeo para diversificação de sequências
- Abra o arquivo PDB salvo na etapa 1.1.6 no Chimera e verifique se não há átomos ou ligações ausentes na estrutura das subunidades alvo.
- Exclua todas as pequenas moléculas, íons e solventes que foram co-cristalizados com a estrutura original. Para fazer isso, clique em Selecionar resíduos > e, em seguida, selecione todas as moléculas, exceto os aminoácidos padrão. Em seguida, clique em Ações > Átomos/Ligações e Excluir.
- Corte a cadeia de ligantes para o segmento de melhor interação escolhido na etapa 1.2.5. Para fazer isso, clique em Favoritos e Sequência e, em seguida, clique na cadeia considerada como o ligante (B). No painel Sequência , arraste o mouse para selecionar todos os resíduos, exceto aqueles entre as posições 424 e 436. Para excluir esses resíduos, clique em Ações > Átomos/Ligações e Excluir.
- Use File > Save PDB para salvar a estrutura editada em um arquivo PDB diferente, que é usado nas próximas etapas do protocolo (aqui, o nome IRF5_interface.pdb foi usado).

Figura 3: Seleção do receptor e do ligante no servidor BUDE Alanine Scan. (A) Representação gráfica do complexo proteína-proteína. (B) Caixa de texto para inserir o nome do trabalho no servidor. (C) Painel para selecionar interativamente as cadeias que serão consideradas como receptor e ligante (consulte a etapa 1.2 para obter mais detalhes). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Página de resultados do servidor BUDE Alanine Scan. O segmento potencial de melhor interação na sequência de ligantes é indicado com uma caixa vermelha. No painel esquerdo, o resíduo com a maior contribuição de energia prevista (Leu433) é destacado em verde. Clique aqui para ver uma versão maior desta figura.
{kind=link}
2. Diversificação de sequências
NOTA: Nas etapas a seguir, rosetta_main refere-se ao diretório principal de instalação do Rosetta, que normalmente está localizado em /opt/rosetta_src__bundle/main/, onde indica a versão instalada do Rosetta. Além disso, presume-se que os aplicativos Rosetta sejam acessíveis em todo o sistema; Se esse não for o caso, o caminho completo para os executáveis deve ser fornecido. Quando compilados a partir da origem, esses executáveis estão localizados no diretório /rosetta_main/source/bin/ .
- Otimização inicial das cadeias laterais de aminoácidos
- Copie a estrutura editada salva na etapa 1.3.4 para um local Linux acessível pelos aplicativos Rosetta.
- Use o aplicativo FixBB da Rosetta para realizar um reempacotamento de todas as cadeias laterais de aminoácidos da estrutura de base antes da diversificação da sequência. Nesta operação, a orientação de todas as cadeias laterais de aminoácidos é otimizada para minimizar a energia e melhorar a estabilidade do complexo. Para fazer isso, execute o seguinte comando:

NOTA: Este comando gera um arquivo PDB com o nome da estrutura original com um sufixo numérico adicional (IRF5_interface_0001.pdb neste exemplo). - Para facilitar a próxima etapa do protocolo, renomeie o arquivo PDB reempacotado com o sufixo _repack usando o seguinte comando:
MV IRF5_interface_0001.PDB IRF5_repack.PDB
- Diversificação de sequências
- Execute Pepspec no modo de design para executar a etapa de diversificação de sequência real usando o seguinte comando:

A seguir estão as opções gerais:- -s indica o arquivo de entrada (o arquivo PDB reempacotado gerado na etapa 2.1.3).
- -o indica o prefixo para nomear os arquivos de saída.
- - database indica o caminho para o banco de dados principal do Rosetta 3.
- -ex1, -ex2 e extrachi_cutoff são opções de biblioteca de rotâmeros (consulte a documentação do Pepspec para obter mais detalhes).
- -overwrite informa ao aplicativo para substituir possíveis saídas pré-existentes geradas por iterações anteriores.
- -pepspec:pep_chain indica as cadeias PDB consideradas como ligante ('b' neste exemplo).
- -pepspec:native_pep_anchor indica o resíduo de aminoácido usado como âncora (neste exemplo, o resíduo de Leu na posição 10 do peptídeo ligante).
- -pepspec:n_peptides indica o número de estruturas peptídicas a serem emitidas.
- -pepspec:no_prepack_prot informa ao aplicativo para ignorar o reempacotamento na estrutura base de entrada (já que isso foi executado anteriormente na etapa 2.1).
NOTA: A saída principal do Pepspec é um diretório que contém os arquivos PDB para peptídeos resultantes da fase de design, nomeado usando o prefixo de saída com o sufixo .pdbs (IRF5.pdbs no exemplo). Além disso, o Pepspec gera todas as sequências de peptídeos aceitas testadas como parte da etapa de diversificação de sequências e suas pontuações de energia Rosetta correspondentes em um arquivo de texto delimitado por tabulação com o nome do prefixo de saída, com o . spec (IRF5.spec no exemplo). Como o protocolo descrito neste trabalho visa estimar as preferências de aminoácidos em vez do design real do peptídeo, os próximos passos usam IRF5.spec em vez das estruturas PDB no diretório .pdbs .
- Execute Pepspec no modo de design para executar a etapa de diversificação de sequência real usando o seguinte comando:
3. Estimativa das preferências de aminoácidos
- Calculando um PWM
- Para gerar um PWM, use o script gen_pepspec_pwm.py incluído no pacote Rosetta. Para executar esse script, use o seguinte comando:

onde:- IRF5.spec é o arquivo de saída Pepspec gerado na etapa 2.2.
- -1 indica que não há resíduos N-terminais adicionais na sequência e, portanto, as posições no PWM são baseadas em 1.
- 0,2 diz ao script para considerar apenas os 20% dos peptídeos com melhor pontuação da saída Pepspec (o valor padrão é 0,1, correspondente aos 10%)
- interface_score diz ao script para classificar os peptídeos com base na pontuação da interface, que é uma das várias pontuações da Rosetta incluídas no arquivo de saída do Pepspec.
NOTA: Este script gera dois arquivos de saída, um para o PWM calculado (com o sufixo .pwm ) e outro para as sequências do subconjunto de peptídeos usados para calcular o PWM (com o sufixo .seq ). Os nomes desses arquivos também incluem a pontuação e a fração de peptídeos usados para classificação. Neste exemplo, esses arquivos são nomeados respectivamente IRF5_interface_score_0.2.pwm e IRF5_interface_score_0.2.seq.
- Para gerar um PWM, use o script gen_pepspec_pwm.py incluído no pacote Rosetta. Para executar esse script, use o seguinte comando:
- Gerando um logotipo de sequência
- Navegue até o servidor WebLogo (https://weblogo.berkeley.edu/logo.cgi)21 e clique no botão Escolher arquivo ao lado de Carregar dados de sequência. Carregue o arquivo com sequências de peptídeos geradas na etapa 3.1.1 (IRF5_interface_score_0.2.seq neste exemplo).
- Escolha o formato e o tamanho desejados do logotipo de acordo com o comprimento de entrada. O exemplo usa o formato PDF e um tamanho de 15 cm x 12 cm. Clique em Criar logotipo.
Resultados
Neste artigo, descrevemos um protocolo para prever as preferências de aminoácidos da superfície de ligação do IRF5, um membro de uma família de fatores de transcrição conhecidos como fatores reguladores do interferon humano. Essas proteínas são reguladoras das respostas imunes inatas e adaptativas e participam da diferenciação e ativação de várias células imunes. As subunidades IRF possuem superfícies de ligação altamente plásticas e multiespecíficas, sendo capazes d...
Discussão
O presente artigo descreve um protocolo para estimar as preferências de aminoácidos de sítios de ligação potencialmente multiespecíficos com base na diversificação de sequências in silico. Poucas ferramentas computacionais foram desenvolvidas para estimar as preferências de aminoácidos das interfaces proteína-peptídeo 14,25,26. Essas ferramentas têm natureza preditiva, mas diferem ...
Divulgações
Os autores não têm nada a divulgar.
Agradecimentos
O apoio financeiro do Sistema Nacional de Investigación (SNI) (processos SNI-043-2023 e SNI-170-2021), Secretaría Nacional de Ciencia, Tecnología e Innovación (SENACYT) do Panamá e Instituto para la Formación y Aprovechamiento de Recursos Humanos (IFARHU) são agradecidos. Os autores gostariam de agradecer ao Dr. Miguel Rodríguez pela revisão cuidadosa do manuscrito.
Materiais
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
Referências
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627 (2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267 (2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33 (2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -. M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999 (2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451 (2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614 (2017).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados