
Method Article
Isolation of Adeno-Associated Viral Vectors Through a Single-Step and Semi-Automated Heparin Affinity Chromatography Protocol
* These authors contributed equally
In This Article
Summary
This manuscript describes a detailed protocol for the generation and purification of adeno-associated viral vectors using an optimized heparin-based affinity chromatography method. It presents a simple, scalable, and cost-effective approach, eliminating the need for ultracentrifugation. The resulting vectors exhibit high purity and biological activity, proving their value in preclinical studies.
Abstract
Adeno-associated virus (AAV) has become an increasingly valuable vector for in vivo gene delivery and is currently undergoing human clinical trials. However, the commonly used methods to purify AAVs make use of cesium chloride or iodixanol density gradient ultracentrifugation. Despite their advantages, these methods are time-consuming, have limited scalability, and often result in vectors with low purity. To overcome these constraints, researchers are turning their attention to chromatography techniques. Here, we present an optimized heparin-based affinity chromatography protocol that serves as a universal capture step for the purification of AAVs.
This method relies on the intrinsic affinity of AAV serotype 2 (AAV2) for heparan sulfate proteoglycans. Specifically, the protocol entails the co-transfection of plasmids encoding the desired AAV capsid proteins with those of AAV2, yielding mosaic AAV vectors that combine the properties of both parental serotypes. Briefly, after the lysis of producer cells, a mixture containing AAV particles is directly purified following an optimized single-step heparin affinity chromatography protocol using a standard fast protein liquid chromatography (FPLC) system. Purified AAV particles are subsequently concentrated and subjected to comprehensive characterization in terms of purity and biological activity. This protocol offers a simplified and scalable approach that can be performed without the need for ultracentrifugation and gradients, yielding clean and high viral titers.
Introduction
Adeno-associated virus (AAV) vector is conquering its way as one of the most promising delivery systems in current gene therapy studies. Initially identified in 19651, AAV is a small, non-enveloped virus, with an icosahedral protein capsid of about 25 nm in diameter, harboring a single-stranded DNA genome. AAVs belong to the Parvoviridae family and to the Dependoparvovirus genus owing to their unique dependence on co-infection with a helper virus, such as herpes simplex virus or, more frequently, adenovirus, to complete their lytic cycle2,3.
The 4.7 kilobase genome of AAVs consists of two open reading frames (ORFs) flanked by two inverted terminal repeats (ITRs) that form characteristic T-shaped hairpin ends4. ITRs are the only cis-acting elements critical for AAV packaging, replication, and integration, therefore being the only AAV sequences retained in recombinant AAV (rAAV) vectors. In this system, the genes necessary for vector production are supplied separately, in trans, allowing the gene of interest to be packaged inside the viral capsid5,6.
Each viral gene codifies different proteins through alternative splicing and start codons. Within the Rep ORF, four non-structural proteins (Rep40, Rep52, Rep68, and Rep78) are encoded, playing crucial roles in replication, site-specific integration, and encapsidation of viral DNA7,8. The Cap ORF serves as a template for the expression of three structural proteins differing from each other at their N-terminus, (VP1, VP2, and VP3), that assemble to form a 60-mer viral capsid at a ratio of 1:1:104,9. Additionally, an alternative ORF nested in the Cap gene with a nonconventional CUG start codon encodes an assembly-activating protein (AAP). This nuclear protein has been shown to interact with the newly synthesized capsid proteins VP1-3 and promote capsid assembly10,11.
Differences in the amino acid sequence of the capsid account for the 11 naturally occurring AAV serotypes and over 100 variants isolated from humans and non-human primate tissues7,12,13. Variations in the conformation of structurally variable regions govern the distinct antigenic properties and receptor-binding specificities of capsids from different strains. This results in distinct tissue tropisms and transduction efficiencies across different mammalian organs14.
Early production methods of rAAVs relied on adenovirus infection for helper purposes15,16,17,18,19. Despite being efficient and usually easy to produce on a large scale, several problems arise from this infection. Even after the purification and a heat-denaturing step for inactivation, adenoviral particles may still be present in AAV preparations, constituting an unwanted safety issue20. Moreover, the presence of denatured adenoviral proteins is unacceptable for clinical use. Other production strategies take advantage of recombinant herpes simplex virus strains engineered to bring the Rep/Cap and transgene into the target cells21 or of the baculovirus-insect cell system22. Although these systems offer advantages in terms of scalability and GMP compatibility, they still face similar problems.
The triple transfection method for rAAV production has been commonly adopted to easily overcome these issues. Briefly, the rAAV assembly relies on the transient transfection of cells with three plasmids encoding for: 1) the transgene expression cassette packed between the ITRs from the wild-type AAV2 genome (pITR); 2) the Rep/Cap sequences necessary for replication and virion assembly (pAAV-RC); and 3) the minimal adenoviral proteins (E1A, E1B, E4, and E2A) along with the adenovirus virus-associated RNAs required for the helper effect (pHelper)6,20,23. While plasmid transfection methods provide simplicity and flexibility for rAAV production in preclinical studies, these procedures have limitations in terms of scalability and reproducibility when applied to large-scale production. As an alternative approach, rAAV production can be achieved through the use of AAV producer cell lines (of both adherent and suspension growth), stably expressing either AAV Rep/Cap genes or Rep/Cap in combination with vector constructs. In these systems, the adenoviral helper genes are introduced through plasmid transfection. Even though this strategy improves the scalability of the cell culture process, it is technically complex and time-consuming21,24,25.
In either case, the producer cells are then lysed and subjected to one or multiple purification steps. Currently, the principal methods for purifying rAAVs include the use of cesium chloride (CsCl) or iodixanol ultra-high speed density gradient centrifugation followed, or not, by chromatography techniques26. The original purification scheme for viral precipitation used ammonium sulfate, followed by two or three rounds of ultracentrifugation through a CsCl gradient. The main advantages of this process include the possibility to purify all serotypes and the ability to physically separate full particles from empty capsids based on their different densities. This method, however, is elaborate, time-consuming, and has limited scalability, often resulting in poor yield and low sample quality27,28,29,30. Moreover, dialysis against a physiological buffer is often necessary before in vivo studies due to the toxic effects that CsCl can exert on mammals.
Iodixanol has also been used as an alternative iso-osmotic gradient medium to purify rAAV vectors, with advantages over CsCl from both safety and vector potency points of view. However, like CsCl, the iodixanol method presents some drawbacks related to the loading capacity of cell culture lysate (and thus the scalability of rAAV purification) and it remains a time-consuming and expensive method30,31.
To overcome these constraints, researchers turned their attention to chromatography techniques. In this regard, several purification approaches were developed that either incorporate affinity, hydrophobic, or ion-exchange chromatography methods. These methods rely on the biochemical properties of a particular serotype, including their natural receptors, or the charge characteristics of the viral particle32. For instance, the discovery that AAV2, AAV3, AAV6, and AAV13 preferably bind to heparan sulfate proteoglycans (HSPG), opened the possibility of using the closely related heparin in affinity chromatography purification. However, the binding sites to HSPG can differ among serotypes, mediating AAV attachment and infection of target cells in different ways2,33,34,35,36. On the other hand, AAV1, AAV5, and AAV6 bind to N-linked sialic acid (SA), while AAV4 uses O-linked SA2,14,34. Following the same rationale, a single-step affinity chromatography protocol for the purification of rAAV5 has also been developed based on the use of mucin, a mammalian protein highly enriched in SA37. Like heparin-based techniques, this purification is also dependent on the specific serotype being generated. Apart from heparin and mucin, other ligands have been explored for affinity chromatography, such as the A20 monoclonal antibody and camelid single-domain antibodies (AVB Sepharose and POROS CaptureSelect)22,23,38,39,40,41. Other innovative strategies to improve the previously existing purification methods involve the introduction of small modifications in the rAAV capsid to present specific binding epitopes. For instance, hexa-histidine-tagged or biotinylated rAAVs can be purified using ligands that target those epitopes (nickel nitrilotriacetic acid and avidin resins, respectively)42,43,44.
In an effort to broaden the desired characteristics of rAAVs, investigators have cross-dressed the virions by mixing their capsids. This is accomplished by supplying the capsid gene from two distinct AAV serotypes in equimolar or different ratios during production, giving rise to a capsid structure composed of a mixture of capsid subunits from different serotypes34,45,46,47,48,49,50. Previous studies provide physical evidence that co-expressing capsid proteins from AAV2 with AAV1 (1:1 ratio) and AAV2 with AAV9 (1:1 ratio) results in the generation of mosaic rAAV1/2 and rAAV2/9 vectors, respectively45,46,48. A major benefit of the generation of mosaic rAAVs is the capacity to integrate advantageous traits from different AAV serotypes, resulting in synergistic improvements in transgene expression and tropism, while maintaining other properties useful during rAAV production. Interestingly, certain mosaic variants even exhibit novel properties different from either parental virus46,47,49. By taking advantage of the heparin-binding ability of AAV2, mosaic rAAV vectors could potentially be generated and purified by mixing AAV2 with other natural or new AAV capsids generated by directed evolution and/or rational design. Nonetheless, previous studies have highlighted the importance of capsid subunit compatibility when attempting to assemble mosaic vectors. For instance, Rabinowitz and colleagues demonstrated that although the transcapsidation of AAV1, AAV2, and AAV3 led to an efficient co-assembly of mosaic capsids, the cross-dressing of these serotypes with AAV4 hindered the generation of stable virions34,45,47. Additionally, AAV1, AAV2, and AAV3 showed low compatibility with AAV5, given the reduced viral titers obtained when mixing these capsids at different ratios. Interestingly, mosaic rAAV2/5 showed decreased heparin-binding properties, while maintaining mucin-binding ability like parental AAV5. However, rAAV3/5 at a 3:1 ratio preserved the dual binding to heparin and mucin. Overall, the generation of new mosaic rAAVs with enhanced transduction, specific tropism, or low immunogenicity could greatly benefit from our understanding of capsid assembly and receptor interactions, with specific combinations still requiring thorough investigation and optimization.
In the present work, we describe a step-by-step protocol for the production and purification of rAAVs using an optimized heparin affinity chromatography method. rAAVs are produced by transient transfection and are purified using a fast protein liquid chromatography (FPLC) system. After the concentration of selected purified fractions, the resulting viral stocks are characterized in terms of titer, purity, intrinsic physical properties, and biological activity in vitro and in vivo. As a proof of concept, we demonstrate the improvements and applicability of this protocol for the generation of mosaic rAAV1/2 and rAAV2/9 vectors. The choice of each serotype was based on their strikingly different tropisms, potentially conferring their unique characteristics to the mosaic versions as well. AAV serotype 1, with an overall moderate tropism for the central nervous system (CNS), has the ability to transduce neurons and glia (to a lesser extent) and undergoes axonal transport in the anterograde and retrograde directions in vivo2,7,8. Additionally, AAV serotype 9 was chosen for its natural ability to cross the blood-brain barrier and target the CNS in both neonatal and adult mice51,52. Finally, AAV serotype 2 was selected given its ability to bind to heparin, allowing affinity chromatography33. The purified rAAV1/2 and rAAV2/9 particles combine the properties of both parental AAV serotypes and, therefore, constitute suitable vectors for the transduction of the CNS45,46,48,49.
Protocol
NOTE: See Figure 1 for an illustration summarizing the protocol. See the Table of Materials for details about all materials, instruments, and reagents used in this protocol. All work involving cells and viruses should be performed in dedicated biosafety cabinets and incubators, separated from those regularly used for the maintenance of cell lines. The equipment and reagents coming in contact with cultured cells and viruses should be sterile. It is essential that the disposal of hazardous reagents and materials contaminated with viruses is performed in accordance with the material safety data sheets and in compliance with the national laws and guidelines provided by each institution's environmental health and safety office. As of April 2019, the NIH guidelines for research involving recombinant or synthetic nucleic acid molecules categorize as Risk Group 1 agents (not associated with disease in healthy adult humans) all AAV serotypes, as well as recombinant or synthetic AAV constructs. This classification applies when the transgene does not encode a potentially tumorigenic gene product or a toxin, and the constructs are produced without an helper virus.
All experiments involving animals were carried out in compliance with the European Union Community directive (2010/63/EU) for the care and use of laboratory animals, transposed into the Portuguese law in 2013 (Decree Law 113/2013). Additionally, animal procedures were approved by the Responsible Organization for the Animal Welfare of the Faculty of Medicine and Center for Neuroscience and Cell Biology of the University of Coimbra licensed animal facility. The researchers received adequate training (FELASA-certified course) and certification from the Portuguese authorities (Direcção Geral de Alimentação e Veterinária, Lisbon, Portugal) to perform the experiments.
1. Plasmid constructs
- Follow the manufacturer's instructions of a maxiprep endotoxin-free kit to isolate and purify substantial DNA quantities of the following plasmids: i) pITR: the transfer vector of interest; ii) pAAV-RC plasmid: pRV1, containing the AAV2 Rep and Cap sequences36; iii) pAAV-RC plasmid: pH21, containing the AAV1 Rep and Cap sequences36; iv) pAAV-RC plasmid: pAAV2/9n, containing the AAV2 Rep and AAV9 Cap sequences; v) pHelper: pFdelta6, Adenovirus-helper plasmid36.
- Screen the integrity of the generated plasmids by performing the recommended enzymatic restrictions36. Monitor the integrity of pITR plasmids by digestion with SmaI, a restriction endonuclease, which cuts twice within an unstable portion of the ITRs53,54.
NOTE: Since the ITRs are highly unstable and susceptible to deletions, the use of SURE 2 supercompetent cells is recommended to minimize recombination in these sites.
2. Cell culture
- Culture human embryonic kidney 293, stably expressing the SV40 large T antigen (HEK293T) cell line in Dulbecco's Modified Eagle Medium (DMEM) high glucose, supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin, at 37 °C, under a humidified atmosphere containing 5% CO2, as a starting point for subsequent steps.
- Subculture the cells using sterile 1x Phosphate Buffered Saline (PBS), pH 7.4, to wash the cells before adding 0.05% trypsin-ethylenediaminetetraacetic acid (EDTA).
NOTE: Avoid utilizing cells that have undergone an excessive number of passages (maximum of 20). Regularly test cell cultures for mycoplasma contamination.
3. rAAV production by transient transfection
- Day 1: Cell plating
- For each viral production, plate HEK293T cells into 10 treated culture dishes (15 cm diameter) the day before transfection, at a density of 10.5 × 106 cells per dish in 22 mL of supplemented culture medium and incubate for 24 h until the cells are 70%-80% confluent and ready for transfection.
- Day 2: Cell transfection using polyethylenimine (PEI)
- For each viral production (equivalent to 10.5 dishes), set up the following transfection mixture in a microcentrifuge tube: 54.6 µg of pITR; 45.675 µg of pRV1; 45.675 µg of pH21 or pAAV2/9n; 109.2 µg of pFdelta6. Mix by tapping.
- Add the mixture to 4.557 mL of non-supplemented DMEM in a 50 mL centrifuge tube. Mix by tapping.
- Add 1.365 mL of sterile PEI solution at 1 mg/mL (pH 7.4), drop by drop. Mix by tapping. Incubate for 10 min at room temperature to allow the formation of DNA-PEI complexes.
- Add this mixture to 231 mL of prewarmed supplemented DMEM. Replace the entire culture medium of each dish with 22 mL of this transfection mixture. Incubate the cells for 48 h.
NOTE: This step must be performed with care to avoid cell detachment.
- Day 4: Cell harvest
- When the pITR encodes a fluorescent reporter, visualize the transfected cells under a fluorescence microscope.
- Collect the medium from each dish into 50 mL centrifuge tubes and centrifuge them at 800 × g for 10 min. Discard the supernatant.
NOTE: This step is optional and aims to recover transfected cells that may have detached due to a very high confluency. - Add 10 mL of prewarmed PBS to each plate. Gently remove the cells with a cell scraper and collect the suspension into the 50 mL centrifuge tubes from step 3.3.2.
- Wash 5 dishes at a time with an extra 10 mL of PBS and transfer the suspension into the 50 mL centrifuge tubes from step 3.3.3. Pellet the cells at 800 × g for 10 min and discard the supernatant.
- Freeze the cell pellets at -80 °C.
NOTE: Cell pellets may be stored for several months (pause point).
4. rAAV extraction and FPLC purification
- Day 5: Cell lysate preparation
- Thaw the cell pellets at room temperature. Resuspend the cells collected from the 10 dishes in 100 mL of a sterile buffer containing 150 mM sodium chloride (NaCl) and 20 mM Tris, pH 8.0, in ultrapure (type I) water. Mix the suspension by pipetting up and down, to ensure a homogeneous suspension.
- Add 12.5 mL of a freshly prepared sterile solution of 10% sodium deoxycholate in ultrapure water to induce cell lysis. Mix by pipetting up and down.
NOTE: Dispose of sodium deoxycholate, as well as the materials in contact with it, in accordance with the material safety data sheet and the guidelines provided by the institution's environmental health and safety office. It is also recommended to wear a face mask while handling this powder. After mixing, the solution becomes highly viscous. - Add 27 µL of benzonase nuclease to the previous mixture. Mix thoroughly by pipetting up and down until the sample is no longer viscous. Incubate at 37 °C for 1 h, performing a vortex every 10 min.
NOTE: This endonuclease is capable of efficiently degrading all forms of DNA and RNA without exhibiting any proteolytic activity. - Remove cellular debris by centrifuging the mixture at 3,000 × g for 60 min at 25 °C. Filter the supernatant with a 0.45 µm sterile polyvinylidene difluoride (PVDF) syringe filter and transfer it into a new sterile container.
NOTE: This important step ensures that most of the cellular debris is removed, therefore preventing the clogging of chromatography columns. Save a small aliquot of this mixture for analysis (optional step).
- Day 5 (continued): Heparin column purification of rAAVs
NOTE: Sample application can be performed using a sample pump or a 50 mL or 150 mL superloop as part of the system. Since more air is dissolved at lower temperatures, it is important to allow sufficient time for the buffers and solutions (usually stored at 4 °C) to acclimate to room temperature before their use in the FPLC system.- Optional: If the system has been stored for long periods, fill the system and all inlets with freshly prepared storage solution (20% ethanol) using manual instructions or a predefined system cleaning in place (system CIP) method.
- Completely wash the liquid flow path with sterile ultrapure water using manual instructions or a predefined system CIP method.
- Connect a 1 mL prepacked heparin column, set the pressure alarm, and wash with five column volumes (CVs) of ultrapure water at a flow rate of 1 mL/min.
- Switch the solutions in the buffer tray from ultrapure water to buffer A (sterile solution of 100 mM NaCl and 20 mM Tris, pH 8, in ultrapure water) for inlet A (system pump A) and to buffer B (sterile solution of 500 mM NaCl and 20 mM Tris, pH 8, in ultrapure water) for inlet B (system pump B). If the system has a sample pump, place the buffer inlet of the sample inlet valve in buffer A.
- Wash the system pump B with buffer B and fill the rest of the liquid flow path with buffer A.
NOTE: If necessary, disconnect the column from the flow path and reconnect it afterward. - Insert a sample inlet tubing from the sample inlet valve, for example, S1, in the container with the viral preparation obtained in step 4.1.4. (from cell lysate preparation). Prime the flow path from sample inlet S1 to the injection valve with the sample solution. Alternatively, fill a 50 mL or 150 mL superloop with the sample containing rAAVs using a 50 mL syringe.
- Equilibrate the column with a total volume of five CVs using 12.5% of buffer B at a rate of 1 mL/min.
- Apply the total volume of the sample into the column using the sample pump (select inject all sample using air sensor) or the superloop at 0.5 mL/min and collect the flowthrough using the outlet port in a new sterile container.
NOTE: When the flow control to prevent overpressure feature is enabled, the flow will automatically decrease in case of column clogging. If the flow rate drops significantly below 0.5 mL/min, halt the sample application, perform a wash with 2-5 CVs of buffer A, and then resume the sample application. - Wash the column at 1 mL/min with 20 CVs of buffer A, collecting the flowthrough using the outlet port.
- Elute the sample at 1 mL/min with the following scheme: i) linear gradient with a target of 50% of buffer B for five CVs; ii) step with a target of 90% of buffer B during five CVs; iii) step with a target of 100% of buffer B during five CVs.
- Collect the eluted sample in 1 mL fractions using a fraction collector and low-retention microcentrifuge tubes (2 mL) and store them at -20 °C.
NOTE: The rAAV fractions may be stored for several weeks (pause point). - Re-equilibrate the column at 1 mL/min with 12.5% of buffer B for five CVs.
- Switch the inlets from the buffer solutions to ultrapure water and wash the column at 1 mL/min for five CVs.
- Switch the inlets from ultrapure water to 20% ethanol and wash the column at 1 mL/min for five CVs. Disconnect the column and store it at 4 °C.
NOTE: Columns can be reused several times without any other major cleaning and sanitization procedures if the same rAAV serotype and transgene are used. - Completely wash the liquid flow path with 20% ethanol using manual instructions or a predefined system CIP method.
5. Concentration of purified rAAVs
- Day 6: Concentration step 1
- Concentrate rAAVs using a 15 mL centrifugal filter unit with a 100 kDa molecular weight cutoff. Load the desired fractions containing rAAVs (FPLC fractions 7 to 16) into the 15 mL centrifugal filter unit and centrifuge it at 2,000 × g for 2 min at room temperature. Make sure that the concentrated volume in the filter unit is approximately 500 µL. If the concentrated volume largely exceeds 500 µL, repeat the centrifugation steps in 1 min intervals until it reaches the desired volume.
- Day 6 (continued): Buffer exchange
- Add 1 mL of sterile PBS to the centrifugal filter unit containing the rAAVs. Carefully pipette up and down to wash the filter. Centrifuge at 2,000 × g in 1 min intervals until the final volume of 500 µL is reached.
- Day 6 (continued): Concentration step 2
- Transfer the 500 µL of concentrated rAAVs obtained in the previous step to a 0.5 mL centrifugal filter unit with a 100 kDa molecular weight cutoff and centrifuge at 6,000 × g for 1 min. If necessary, repeat the centrifugation step until a final volume of less than 100 µL is reached.
- Day 6 (continued): Recovery
- To recover the concentrated rAAV, place the filter device upside down in a new microcentrifuge collection tube. Place the tube in a microcentrifuge with the cap towards the center and perform a prolonged spin inside the flow chamber to transfer concentrated rAAVs from the device to the microcentrifuge tube. Alternatively, centrifuge at 1,000 × g for 2 min.
- Supplement with sterile Pluronic F-68 0.001% (optional).
NOTE: Pluronic F-68 is a non-ionic surfactant approved for human use by the Food and Drug Administration that is able to mitigate losses of rAAVs by preventing their interactions with the surfaces of materials (plastics) used during dilution preparation, syringe loading, and delivery equipment55,56. - Aliquot rAAVs into low-retention microcentrifuge tubes and store at -80 °C (pause point).
6. Quantification of purified rAAVs
- Day 6 (continued): Determine the titer of rAAV preparations, expressed in viral genomes/µL (vg/µL), by real-time quantitative polymerase chain reaction (RT-qPCR) using a commercial kit and following the manufacturer's instructions.
- Incubate the rAAV particle solution with DNase I at 37 °C for 20 min.
NOTE: This procedure promotes the digestion of free genomic DNA and plasmid DNA derived from host cells, thus ensuring that only the nucleic acid sequence inside intact rAAV particles is preserved. - Heat-inactivate DNase I at 95 °C for 10 min.
- Add Lysis Buffer and incubate for 10 min at 70 °C to promote heat-denaturation of the proteins of rAAV particles.
- Dilute the obtained rAAV genome solution in dilution buffer before proceeding to the RT-qPCR. Prepare a set of serially diluted standards of the positive control (from 2 × 107 vg/µL to 2 × 102 vg/µL) provided with the kit.
- Perform a reaction mixture containing 12.5 µL of Taq II mix, 0.5 µL of diluted primer mix, 7 µL of water, and 5 µL of the diluted rAAV DNA (unknown AAV sample and standards from step 6.1.4.).
- Perform the RT-qPCR in a real-time PCR detection system, using the following protocol: 1 cycle at 95 °C for 2 min (initial denaturation) and 40 cycles at 95 °C for 5 s (denaturation) and 60 °C for 30 s (annealing, extension, and plate reading), followed by a melt curve analysis.
- Calculate the absolute sample concentration from the standard curve (linear regression line), considering the dilution factor that results from the rAAV sample preparation.
NOTE: The quantification of the number of viral genomes is achieved through the amplification of the ITR sequence of AAV2 (target sequence of the primers provided by the kit).
- Incubate the rAAV particle solution with DNase I at 37 °C for 20 min.
7. SDS-PAGE, Coomassie blue staining, and western blot
- Denature 40 µL of each sample (each FPLC fraction; the flowthrough; the precolumn samples, and a total of 2.3 × 1010 vg of the final concentrated product) by adding 6x sample buffer (0.5 M Tris-HCl/0.4% sodium dodecyl sulfate (SDS) pH 6.8, 30% glycerol, 10% SDS, 0.6 M dithiothreitol (DTT), 0.012% bromophenol blue) and incubating the samples for 5 min at 95 °C.
- Load the denatured samples (48 µL) into an SDS-polyacrylamide gel (4% stacking and 10% resolving gel) and perform the electrophoretic separation at 100 V for 70 min, adjacent to a protein ladder.
- Protein analysis
NOTE: Either Coomassie blue staining or western blotting can be performed.- Coomassie blue staining
- To visualize protein bands, stain the gel for 10 min with a 0.25% Coomassie blue G250 solution dissolved in 50% methanol and 10% acetic acid glacial.
- Perform gel destaining by washing it several times with a solution containing 25% methanol and 5% acetic acid glacial until clear bands with low background become visible.
- Capture images using an appropriate imaging system.
- Western blot
- Transfer the proteins onto a PVDF membrane according to standard protocols.
- Block the membrane by incubation in 5% non-fat milk diluted in TBS-T (0.1% Tween 20 in Tris-buffered saline) for 1 h at room temperature.
- Use the following primary antibodies (diluted in blocking solution) for the overnight incubation at 4 °C: mouse monoclonal anti-AAV, VP1, VP2, VP3 antibody (B1, 1:1,000), or mouse monoclonal anti-AAV, VP1, VP2 antibody (A69, 1:1,000).
- Wash the membranes for 3 x 15 min in TBS-T and incubate with an alkaline phosphatase-linked goat anti-mouse secondary antibody (1:10,000), for 2 h at room temperature.
- Wash the membranes for 3 x 15 min in TBS-T. Add enhanced chemifluorescent substrate (ECF) and visualize protein bands by chemifluorescence imaging.
- Coomassie blue staining
8. Transmission electron microscopy (TEM)
- Place a Formvar-carbon-coated 200 mesh grid upside down on top of a drop of an rAAV sample and allow it to settle for 1 min.
- Wash the grids in a drop of water and dry the liquid excess with filter paper.
- Negatively stain the grids with 1% uranyl acetate solution (pH 7) for 1 min to fix and contrast the viral particles.
- Wash the grids in a drop of water and dry the liquid excess with filter paper.
- Examine the samples in a transmission electron microscope.
NOTE: High-salt concentrations may directly impact the binding of rAAVs to the grid and lead to the visualization of crystal-like structures.
9. Consecutive ultraviolet-visible light absorption, static light scattering, and dynamic light scattering analysis
- On a 96-well quantification plate, load 2 µL of an rAAV sample and 2 µL of PBS to be used as buffer blank (perform this in duplicates).
- Use the AAV Quant application in the Client analysis software, place the names of the samples in the correct location of the plate, select the AAV serotype, and click Next.
- Load the 96-well quantification plate into the dedicated equipment and proceed with plate reading for data acquisition.
10. In vitro transduction assays
- Different cell lines can be used to quickly analyze the transduction efficiency of rAAVs.
- Evenly seed HEK293T cells in 24-well plates (at a density of 137,500 cells/well) and a mouse neuroblastoma-2A (Neuro2a) cell line in either 24-well plates (50,000 cells/well) or in an 8-well chamber slide (27,000 cells/well), using DMEM high glucose, supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin, as described above. Allow the cells to adhere overnight at 37 °C in a humidified atmosphere containing 5% CO2.
- Collect conditioned medium from each well (250 µL from 24-well plates and 50 µL from the 8-well chamber slide) and store it at 4 °C for later use.
- Add the following rAAV preparations to each well and incubate the cells for 24 h at 37 °C in a 5% CO2 atmosphere.
- Add 50 µL of the FPLC-collected fractions F2-F16 and flowthrough to HEK293T cells plated in 24-well plates.
- Add a total of 5.5 × 109 vg of the concentrated rAAVs diluted in 50 µL of PBS to Neuro2a cells plated in 24-well plates (include a negative control well, by adding 50 µL of PBS to the cells).
- Add a total of 2.75 × 109 vg of the concentrated rAAVs diluted in 25 µL of PBS to Neuro2a cells plated in an 8-well chamber slide (include the negative control condition, by adding 50 µL of PBS to the cells).
- Add the previously stored conditioned medium (step 10.1.2) to each well and incubate for 24 h.
- Discard the medium and wash the cells 2x with PBS.
- Add a 4% paraformaldehyde (PFA) solution, supplemented with 4% sucrose in PBS, prewarmed to 37 °C, to each well and incubate at room temperature for 20 min.
- Wash 2x with PBS and store at 4 °C until imaging is performed (pause point).
- Acquire images on an inverted fluorescence microscope equipped with a 10x/0.30 objective, or an inverted confocal microscope equipped with a 40x/1.4 Oil DIC objective.
- To have a more relevant and reflective model of the in vivo environment, use primary neuronal cultures as follows:
- Prepare primary cultures of cortical neurons as previously described by Santos et al.57. Briefly, seed 200,000 cells/mL in 12-well plates and maintain them in culture until day in vitro 16.
- Collect conditioned medium from each well (100 µL) and store it at 4 °C for later use.
- Add the rAAVs to be tested to each well: a total of 2.75 × 109 vg of the concentrated rAAVs diluted in 25 µL of PBS (include the negative control: 25 µL of PBS). Incubate for 24 h at 37 °C in a 5% CO2 atmosphere.
- Add the conditioned medium previously stored and incubate for 24 h.
- Discard the medium in each well and wash 2x with PBS.
- Fix the cells with 4% PFA/4% sucrose in PBS, as described in step 10.1.6. Wash 2x with PBS.
- Incubate each well with 5 µg/mL of wheat germ agglutinin (WGA) conjugated with Alexa Fluor 633 for 10 min at room temperature (optional step: perform immunocytochemistry instead). Wash 2x with PBS.
- Incubate in 0.25% Triton X-100 in PBS for 5 min at room temperature. Wash with PBS.
- Incubate with 4',6-diamidino-2-phenylindole (DAPI) for 5 min at room temperature. Wash 2x with PBS.
- Acquire images on an inverted fluorescence microscope equipped with a 40x/0.95 objective, or an inverted confocal microscope equipped with a 40x/1.4 Oil DIC objective.
11. In vivo experiments
NOTE: The animals were housed in a temperature-controlled room, maintained on a 12 h light/dark cycle. Food and water were provided ad libitum. All efforts were made to minimize animal suffering.
- Stereotaxic injection in the cerebellum
- Anesthetize 9-week-old C57BL/6 animals by inhalation of 2% isoflurane in the presence of oxygen (0.8 L/min) in a chamber connected to a vaporizer.
- Place the anesthetized animal in the stereotaxic apparatus (on a 35 °C warmed pad) and place the isoflurane mask into the animal's nose. Turn the isoflurane level down to 1.3-1.7%.
NOTE: Make sure that the animal is correctly anesthetized before proceeding (loss of the reflex for flexion in both hind limbs). - Apply lubricant eye ointment to avoid drying of the corneas and inject the animal with an approved analgesic.
NOTE: All subsequent steps must be performed in sterile conditions. - After shaving the fur of the animal's head and disinfecting the surgical area, expose the skull and place the tip of a 30 G blunt-tip injection needle, connected to a 10 µL Hamilton syringe, directly over bregma (use bregma as zero for the calculation of stereotaxic coordinates).
- Move the needle to the intended coordinate and drill a hole through the skull where the needle will enter.
NOTE: In the framework of this study, a single injection was performed centrally in the cerebellum. - Inject 4 µL of a solution of rAAVs containing a total of 8 × 109 vg, diluted in PBS, at an infusion rate of 0.5 µL/min using an automatic injector. Use the following coordinates, calculated from bregma, to perform a single injection centrally in the cerebellum of an adult C57BL/6 mouse: antero-posterior: -6.5 mm; lateral: 0 mm; ventral: -2.9 mm.
NOTE: These coordinates may vary according to the mouse strain, sex, and age of the animals in use. - To minimize backflow and allow the viral vector diffusion, once the infusion is complete, leave the syringe needle at these coordinates for 3 min, then slowly retract it by 0.3 mm, and allow it to remain in place for 2 additional min prior to its complete removal from the mouse brain.
- Close the incision and clean it with a disinfectant agent (e.g., 10% povidone-iodine).
- Allow the animals to recover from anesthesia before returning them to their home cages.
- Tissue collection and preparation
NOTE: In this experiment, the transduction levels were observed 12 weeks post injection, but the same procedure could be evaluated as soon as 4 weeks post injection.- Terminally anesthetize the animals by intraperitoneal administration of an overdose of xylazine/ketamine (8/160 mg/kg body weight).
- Transcardially perfuse the animals with ice-cold PBS for 6 min at a rate of 2.5 mL/min, followed by the perfusion with a freshly prepared ice-cold 4% PFA solution for 10 min at the same rate.
- Postfix the excised brains in 4% PFA overnight at room temperature and then transfer them to a 25% sucrose/PBS solution for cryoprotection. Once the brains sink (approximately 48 h later), store them at -80 °C.
- Cut serial sagittal sections with a thickness of 30 µm using a cryostat at -21 °C. For each animal, collect 96 sagittal sections of a brain hemisphere in anatomical series as free-floating sections in PBS supplemented with 0.05% sodium azide. Store at 4 °C until further processing.
- Standard fluorescence immunohistochemistry
- Select eight sagittal sections per animal, at a distance of 240 µm from each other.
- Incubate the free-floating sections in blocking/permeabilization solution (0.1% Triton X-100 containing 10% normal goat serum (NGS) in PBS) for 1 h at room temperature.
- Incubate the sections overnight at 4 °C with chicken polyclonal anti-GFP primary antibody (1:1,000).
- Wash for 3 x 15 min in PBS and incubate the sections for 2 h at room temperature with the secondary antibody goat polyclonal anti-chicken antibody conjugated to Alexa Fluor 488 fluorophore (1:200).
- Wash for 3 x 15 min in PBS. Incubate with DAPI for 5 min at room temperature.
- Wash for 3 x 15 min in PBS. Place the sections in gelatin-coated slides and coverslip with fluorescence mounting medium.
- Acquire images on a slide scanner fluorescence microscope equipped with a 20x/0.8 objective.
Results
In this work, we present a detailed protocol for the production, purification, and characterization of mosaic rAAVs (summarized in Figure 1), which have the potential to target and transduce the CNS (e.g. AAV1 and AAV9), being simultaneously suitable for heparin affinity chromatography purification (AAV2). To achieve that, capsids from natural AAV serotypes 1, 2, and 9 were used to develop mosaic rAAV1/2 and rAAV2/9 vectors.
Before starting, plasmid preparations were screened for structural integrity. In addition to the digestions necessary to validate the correct insertion of cloning fragments, it is essential to consistently screen pITR plasmids to detect potential ITR deletions/insertions. As an example, the integrity of ITRs in different clones of a pITR plasmid was monitored after the plasmid digestion with the restriction enzyme SmaI (Supplementary Figure S1).
Both types of mosaic vectors were generated by the co-transfection of the respective AAV capsid plasmids in a 1:1 ratio, according to standard transfection methods6. Briefly, HEK293T cells were transfected with i) a plasmid containing the transgene of interest packed between the ITR (pITR) sequences, ii) a plasmid containing the wild-type AAV genome Rep and Cap ORFs of AAV2 and AAV1 or AAV9 (pAAV-RC plasmids) and iii) a plasmid codifying the adenoviral proteins (E1A, E1B, E4, and E2A) as well as the adenovirus virus-associated RNAs essential for helper functions (pHelper). Forty-eight hours later, the cells were harvested6,36, and rAAVs were purified from the cell homogenate by affinity chromatography using an FPLC system. As depicted in Figure 2A, after column equilibration (equilibration step), the cell lysate containing the rAAVs was applied to the column (sample loading). Due to the natural affinity of rAAV2 for heparin33, rAAVs bound to the column's resin, while other components were carried out in the running buffer and detected by the UV monitor (flowthrough), resulting in an increase in the absorbance. The column was subsequently washed (washing step) and rAAVs were finally eluted by an increase of NaCl concentration (elution step). The eluted viruses were detected by the UV monitor and collected in 1 mL fractions.
A representative elution peak profile of rAAV1/2 and rAAV2/9 is shown in Figure 2B and Supplementary Figure S2A, respectively, with different viral batches consistently presenting a single peak starting at fraction F7 up to F16. Peak height is variable among rAAV productions, with higher peaks usually leading to higher rAAV yields. Each fraction of the produced rAAV1/2 and rAAV2/9 was subsequently characterized by RT-qPCR to assess viral titers (Figure 2C and Supplementary Figure S2B).
To characterize the purity of the eluted material, 40 µL of each fraction and of the respective flowthrough were examined by 10% SDS-polyacrylamide gel electrophoresis (Figure 2D for rAAV1/2 and Supplementary Figure S2C for rAAV2/9). Coomassie blue staining revealed three major bands in fractions F7-F16, with molecular weights corresponding to the VP1 (87 kDa), VP2 (72 kDa), and VP3 (62 kDa) capsid proteins of AAVs at the appropriate ratios 1:1:10, as previously described by Van Vliet and colleagues14. In both cases, and based on UV absorbance, RT-qPCR, and gel band intensity, it is clear that the majority of mosaic rAAVs is present in fractions F7 and F8 and starts to gradually decrease in fractions F9-F16. In addition to the three viral capsid proteins, another protein (or proteins) of approximately 17 kDa in size was/were detected in fractions F8-F16.
To eliminate this co-purifying protein(s), fractions F7-16 were subsequently filtered and concentrated using 100 KDa centrifugal filter units and the final rAAV titer was determined by RT-qPCR (as shown in Figure 3A,B for rAAV1/2). The final yield of an rAAV production is dependent on the length and complexity of the pITR, the integrity of ITR sequences, cell culture conditions (e.g., number of cell passages), and transfection efficiency24,58,59,60,61. Nonetheless, the final titer can be adjusted by performing multiple centrifugations of the rAAV preparation using 0.5 mL centrifugal filter units (concentration step 2). Following this protocol, for a final volume in the range of 50 to 100 µL, concentrations are usually comprised between 2 × 109 and 5 × 1010 vg/µL (quantification performed using the referenced titration kit).
The purity of the final rAAV preps was then evaluated on a 10% SDS-polyacrylamide gel. As depicted in Figure 3C, only three bands representing the rAAVs capsid proteins were observed for the rAAV1/2 preparation and no detectable co-purifying proteins were identified. These results were consistent with those obtained for rAAV2/9 (Supplementary Figure S2C). To confirm the identity and further characterize the purity of rAAV1/2 and rAAV2/9 vectors, viral fractions and concentrated stocks were analyzed by western blot, with the specific antibodies B1 (Supplementary Figure S3A and Supplementary Figure S4A) and A69 (Supplementary Figure S3B and Supplementary Figure S4B). While the antibody B1 recognizes a C-terminal epitope common to all VP proteins of most AAV serotypes62, the clone A69 only recognizes epitopes of VP1 and VP263. Nonetheless, some faint bands with molecular weights lower than VP3 (<62 kDa) can also be detected upon B1 and A69 labeling.
To characterize the structural morphology and further evaluate the purity of rAAVs, the viral particles were directly visualized by TEM. This technique has been the standard procedure to assess sample integrity and purity in viral samples, as it allows the quantification of empty and full rAAV particles, as well as the assessment of contamination in a sample29,64,65,66,67. As shown in Figure 3D, large amounts of rAAV particles, ~25 nm in diameter, could be observed on a clean background. Empty particles (black arrow) with an electron-dense center, as well as full vectors (white arrow) could also be observed throughout the sample field.
We also performed quality control of the purified rAAVs using Stunner, a platform that combines ultraviolet-visible (UV-Vis) spectroscopy, static light scattering (SLS), and dynamic light scattering (DLS)68. For each sample, the total amount of protein, ssDNA, as well as absorbing impurities and background turbidity, were measured by UV-Vis spectroscopy (Figure 3E and Supplementary Figure S5A). SLS and DLS were then applied to assess the light-scattering behavior of rAAV capsids. Given that AAVs have a mean diameter of 25 nm, particles within a 15-45 nm diameter range are considered intact. Larger particles typically represent viral aggregates, and everything smaller comprises most likely small particles, including unassembled capsid proteins68. For rAAV1/2, a single peak corresponding to intact capsid particles was observed at 30 nm (Figure 3F), with 0% of aggregate intensity and 0% of small particle intensity. For the rAAV2/9 preparation, a peak at 30 nm was also detected representing a 78% capsid intensity (Supplementary Figure S5B). Even though the small particle intensity was 0%, for this sample, an aggregate intensity of 22% was measured (depicted in grey), with the major contribution (19.9%) from large aggregates with a mean diameter of 620 nm (Supplementary Figure S5B). Through the combination of UV-Vis spectroscopy with SLS and DLS information, Stunner revealed the overall total capsid titer, full capsid titer, free and aggregated protein, as well as free and aggregated ssDNA for the two viral preparations, shown in Figure 3G and Supplementary Figure S5C (specific values indicated in each figure legend).
In parallel, to evaluate the biological activity of the developed mosaic AAV vectors, HEK293T cells were infected with 50 µL of each FPLC-obtained fraction (F2-F16) of either the rAAV1/2 or rAAV2/9 preparation. Since the rAAV1/2 vector encodes a single-strand green fluorescent protein (GFP), under the control of a CMV promoter (pAAV-CMV-ssGFP), and the rAAV2/9 vector encodes a self-complementary GFP, under the control of CMV promoter (pAAV-CMV-scGFP53), direct GFP fluorescence was examined in these cells 48 hours post-infection (Supplementary Figure S6 and Supplementary Figure S7). Consistently with the previous observations for RT-qPCR, Coomassie blue, and western blot, the highest infectivity level was achieved for viral fractions F7 and F8, gradually decreasing in fractions F9 to F16.
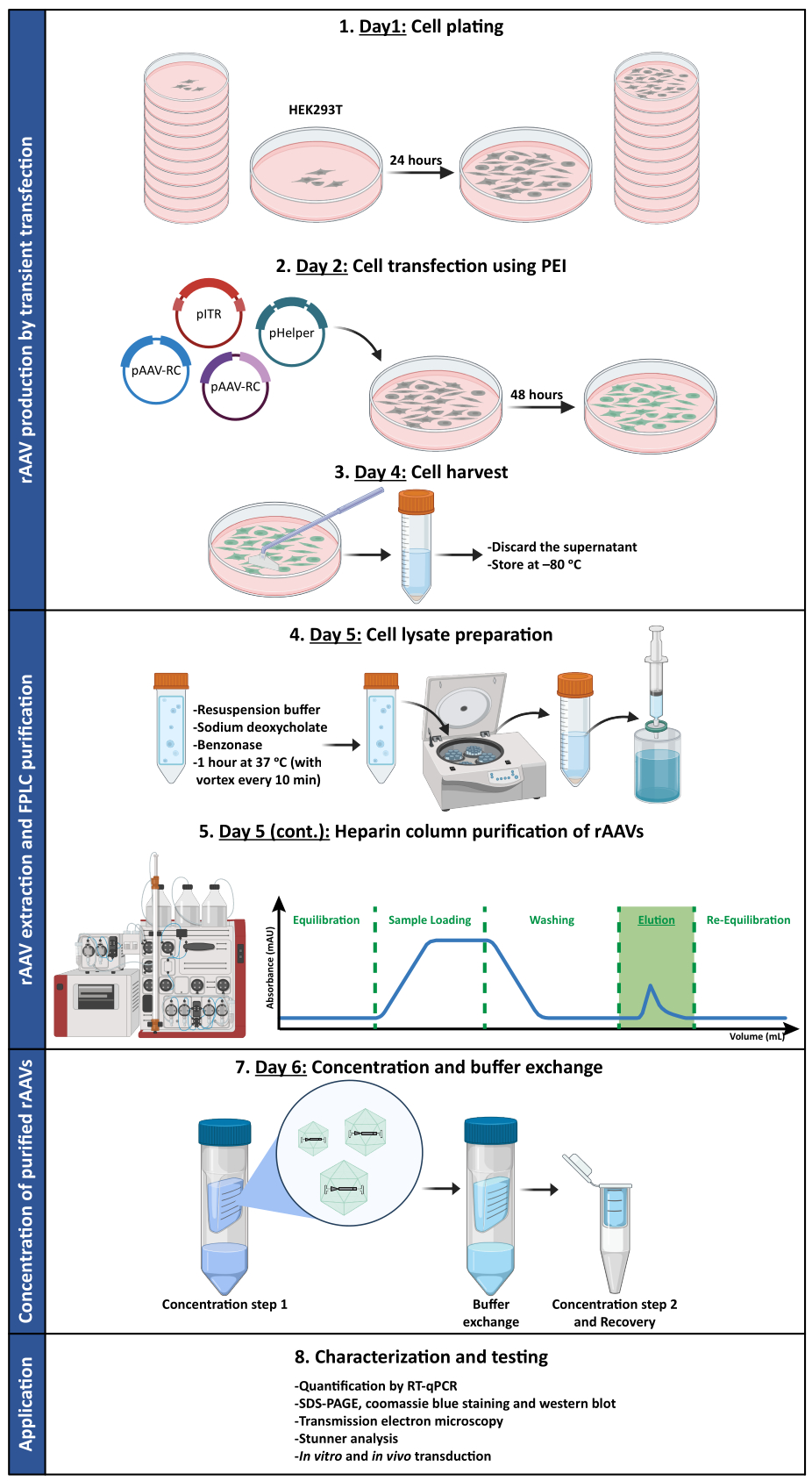
To confirm whether the biological activity of rAAVs was maintained after ultrafiltration and concentration steps, Neuro2A cells, plated in both 24-well plates and an 8-well chamber slide, were infected with the concentrated rAAV1/2 vector, encoding scGFP under the control of CMV promoter (pAAV-CMV-scGFP53). Brightfield and fluorescence images were acquired 48 h post-infection (Figure 4A,B for higher resolution images).
Aiming to explore the infectious capacity of the produced rAAVs in a more relevant and reflective cell model, semi-dense primary neuronal cultures from the cortex were seeded on a 12-well plate and infected with the previously used rAAV1/2 - CMV-scGFP. Forty-eight hours after infection, cells were fixed and labeled with DAPI and WGA conjugated with Alexa Fluor 633, a widely used lectin to label fixed cells. The images shown in Figure 4C,D were acquired with a Zeiss Axio Observer Z1 and on a Zeiss confocal LSM 710. As depicted in these figures by direct GFP fluorescence, concentrated mosaic viruses preserve their gene transfer properties for neuronal cells.
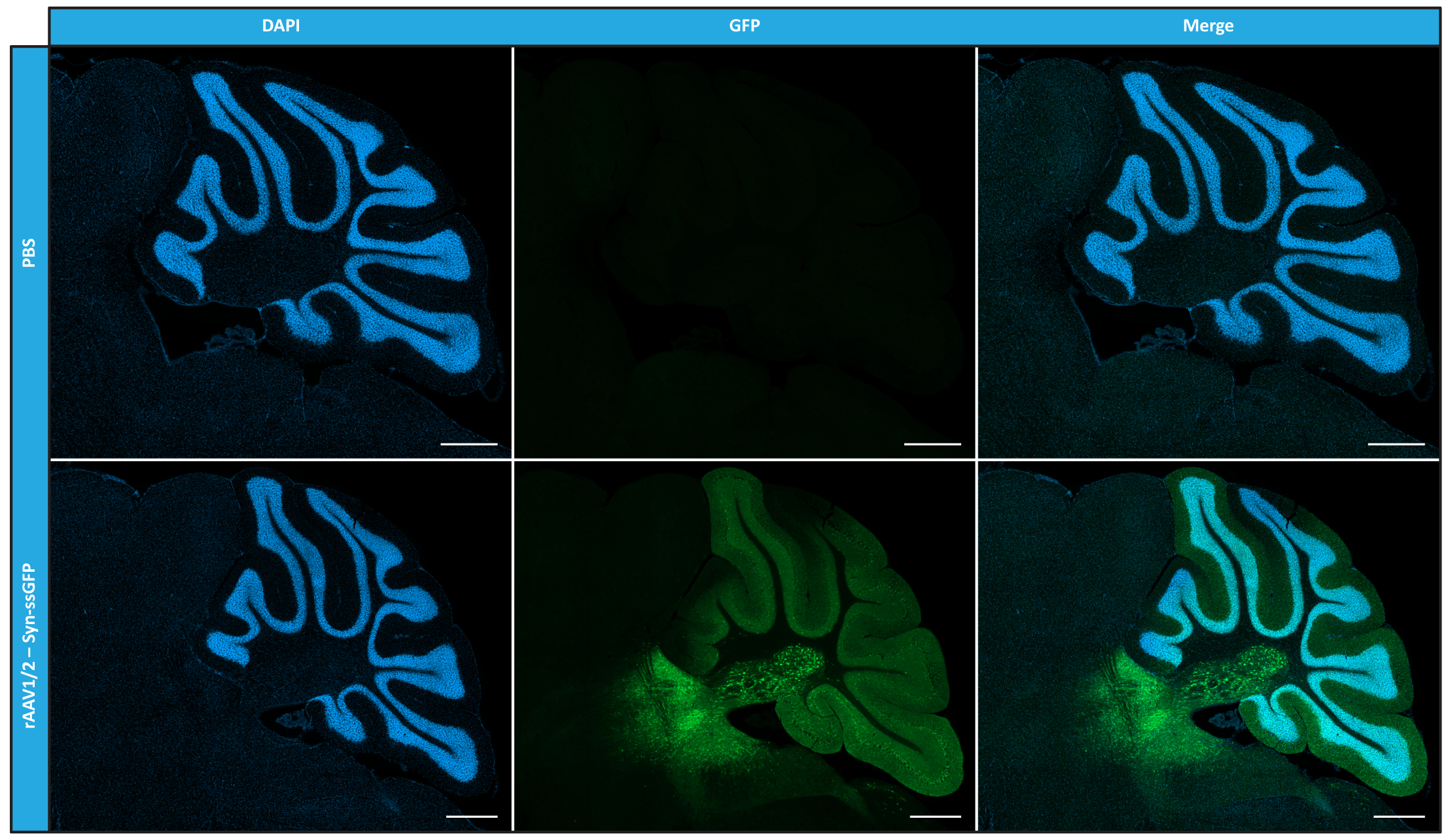
Having characterized mosaic rAAVs in terms of purity, physical properties, and functionality in vitro, we next evaluated the possibility of using the purified rAAV1/2 mosaic vectors to transduce the cerebellum of C57BL/6 mice. For that, a stereotaxic injection was performed in 9-week-old mice and the GFP expression was evaluated 12 weeks later. As anticipated, animals injected with PBS exhibited no fluorescence upon GFP immunolabeling. Epifluorescence images from mice injected with rAAV1/2 vectors encoding GFP under the control of synapsin 1 promoter (rAAV1/2 - Syn-ssGFP) revealed that rAAV1/2 vectors successfully transduced several regions of the cerebellum, namely the deep cerebellar nuclei (DCN) region, as well as the different lobules of the cerebellum (Figure 5). These results demonstrate the prolonged expression of the transgene in the mammalian brain (12 weeks).

Figure 1: Schematic representation of the rAAV production and purification protocol. rAAVs are produced by transient transfection of HEK293T cells using polyethylenimine (PEI). Subsequently, cells are harvested and lysed, and rAAVs are purified from the cell homogenate via affinity chromatography. The collected fractions containing rAAVs are then concentrated, and the final viral stocks are characterized in terms of titer, purity, morphological features, and biological activity. Abbreviations: rAAV = recombinant adeno-associated virus; PEI = polyethylenimine; RT-qPCR = real-time quantitative polymerase chain reaction; SDS-PAGE = sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: FPLC purification protocol and representative elution profile of rAAV1/2. (A) Schematic representation of a complete chromatogram profile, showing the different stages of the rAAV purification process. After a column equilibration step, the sample is applied. The column is then washed, and the elution is performed with increasing concentrations of NaCl. The unbound material (flowthrough) and 1 mL fractions of the eluted viruses are collected for analysis. The absorbance at 280 nm is expressed in mAU and the x-axis indicates the volume in mL. (B) Enlarged partial chromatogram showing an rAAV1/2 elution peak (in black), with the corresponding fraction numbers (F2-F16) and waste (indicated in red). The issued concentration of buffer B and conductivity (expressed in mS/cm) are also shown in green and purple, respectively. (C) RT-qPCR of each fraction collected during affinity purification (F2-F16) and flowthrough. The titer in vg/µL is represented on a logarithmic scale. (D) SDS-PAGE analysis of the collected viral fractions. Equal volumes (40 µL) of each fraction from the elution step (F2-F16), and respective flowthrough were loaded and resolved on a 10% SDS-polyacrylamide gel. Protein bands were visualized by Coomassie blue staining. Bands corresponding to AAV capsid proteins VP1, VP2, and VP3 are indicated. Standard protein size ladder is designated as (L) and the corresponding molecular weights are also indicated. Abbreviations: rAAV = recombinant adeno-associated virus; RT-qPCR = real-time quantitative polymerase chain reaction; SDS-PAGE = sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Characterization of the concentrated rAAV1/2 vectors. (A) Amplification curves of a concentrated rAAV1/2 sample (in blue), serially diluted standards from 2 × 107 vg/µL to 2 × 102 vg/µL (in black) and a no-template control (in green), obtained during RT-qPCR. (B) Standard curve (linear regression) for the determination of the titer of an rAAV sample in vg/µL. (C) SDS-PAGE analysis of the concentrated viral particles. A total of 2.3 × 1010 vg of the concentrated stock were pooled on the gel. (D) Transmission electron microscopy image of rAAV1/2 particles with ~25-30 nm in diameter. Empty particles with an electron-dense center (evidenced by black arrows) can be distinguished from full capsids (evidenced by white arrows). Scale bar = 100 nm. (E) Absorbance spectrum of an rAAV1/2 preparation measured by Stunner (in black). The contribution of proteins (in blue), ssDNA (in green), other UV-absorbing compounds or impurities (in purple), and background turbidity (in grey) are also shown. (F) DLS intensity distribution of rAAV1/2 with a single peak at 30 nm, measured by Stunner. A capsid scattering intensity of 100% was determined by measuring the area under the curve from 15 to 45 nm (shaded green). (G) Stunner analysis of an rAAV1/2 vector preparation exhibiting a total capsid titer of 1.19 × 1014 cp/mL (dark blue) and a full capsid titer of 1.73 × 1013 vg/mL (dark green). A free and aggregated protein of 7.16 × 1012 cp/mL equivalents (light blue), as well as a free and aggregated ssDNA of 1.04 × 1012 vg/mL equivalents (light green), were also measured. Abbreviations: rAAV = recombinant adeno-associated virus; RT-qPCR = real-time quantitative polymerase chain reaction; SDS-PAGE = sodium dodecyl sulfate-polyacrylamide gel electrophoresis; ssDNA = single-stranded DNA; DLS = dynamic light scattering. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: In vitro infectivity assessment of a concentrated rAAV1/2 sample. (A) Neuro2A cells were infected with rAAV1/2 - CMV-scGFP or incubated with an equivalent volume of PBS, as a negative control. Brightfield and fluorescence images of cells imaged 48 h post-infection. Images were acquired in a Zeiss Axio Observer Z1 (10x objective). Scale bars = 100 µm. (B) Detailed images of Neuro2A cells 48 h post-infection with rAAV1/2 - CMV-scGFP. Images were acquired in a Zeiss LSM 710 (40x objective). Scale bars = 20 µm. (C) Semi-dense primary neuronal cultures infected with rAAV1/2 - CMV-scGFP or incubated with an equivalent volume of PBS, serving as a negative control. Cells were labeled with a nuclear stain (DAPI in blue) and a membrane stain (WGA in white). Images were acquired in a Zeiss Axio Observer Z1 (40x objective). Scale bars = 20 µm. (D) Detailed images of semi-dense primary neuronal cultures 48 h post-infection with rAAV1/2 - CMV-scGFP. Images were acquired in a Zeiss LSM 710 (40x objective). Scale bars = 20 µm. Abbreviations: rAAV = recombinant adeno-associated virus; CMV = cytomegalovirus; scGFP = self-complementary green fluorescent protein; PBS = phosphate-buffered saline; DAPI = 4',6-diamidino-2-phenylindole; WGA = wheat germ agglutinin. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: In vivo transduction efficiency of rAAV1/2 following an intraparenchymal injection. Representative immunofluorescence images showing the widespread GFP expression (in green) throughout the cerebellum upon a central injection of rAAV1/2 - Syn-ssGFP in the cerebellum. Nuclei were stained with DAPI (in blue). Scale bars = 500 µm. Abbreviations: rAAV = recombinant adeno-associated virus; Syn = Synapsin 1; ssGFP = single-strand green fluorescent protein; DAPI = 4',6-diamidino-2-phenylindole; PBS = phosphate buffered saline. Please click here to view a larger version of this figure.
{kind=link}
Supplementary Figure S1: Agarose gel analysis of an rAAV vector plasmid digested with SmaI. Six clones (C1-C6) of a pITR were digested with SmaI restriction enzyme (lanes 2, 4, 6, 8, 10, and 12), which cuts twice within each inverted terminal repeat. In this case, a complete digestion of this pITR would be expected to generate two bands (3,796 bp and 3,013 bp). In successful preparations (C1, C3, C4, and C5) a band of 6809 bp, resulting from partial digestion is still visible (~5% of the total). In preparations with ITR recombination, the proportions are reversed (C2), or the digestion did not occur (C6). The respective non-digested clones are also presented (lanes 3, 5, 7, 9, 11, 13). Abbreviations: rAAV = recombinant adeno-associated virus; ITR = inverted terminal repeat. Please click here to download this File.
Supplementary Figure S2: rAAV2/9 purification by heparin-based affinity chromatography. (A) Elution profile of rAAV2/9 exhibiting a single peak (in black), following an increase in the concentration of NaCl. The collected fractions are indicated by numbers (2-16) in red at the bottom of the graph, the absorbance at 280 nm is expressed in mAU, conductivity is expressed in mS/cm, and the x-axis indicates the volume in mL. (B) rAAV titers quantified by RT-qPCR for each pooled fraction (F2-F16) and flowthrough. Values are represented on a logarithmic scale. (C) Purity assay by SDS-PAGE and Coomassie blue staining. Equal volumes (40 µL) of each fraction (F2-F16) and the respective flowthrough were loaded and resolved on a 10% SDS-PAGE. Concentrated stock was quantified by RT-qPCR and 2.3 × 1010 vg were diluted in 40 µL of PBS and pooled on the gel. Protein bands were visualized by Coomassie blue staining. The AAV capsid proteins (VP1, VP2, and VP3) are indicated. Standard protein size ladder is designated with (L) and the corresponding molecular weights are also indicated. Abbreviations: rAAV = recombinant adeno-associated virus; RT-qPCR = real-time quantitative polymerase chain reaction; SDS-PAGE = sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Please click here to download this File.
Supplementary Figure S3: Western blot analysis of rAAV1/2 vectors purified by FPLC. (A) The collected fractions and concentrated rAAV1/2 vectors were resolved on an SDS-PAGE gel and probed with mouse monoclonal anti-AAV antibody (B1) that recognizes VP1, VP2, and VP3 capsid proteins. (B) The collected fractions and concentrated rAAV1/2 vectors were resolved on an SDS-PAGE gel and probed with mouse monoclonal anti-AAV antibody (A69) that recognizes VP1 and VP2 capsid proteins. Abbreviations: rAAV = recombinant adeno-associated virus; FPLC = fast protein liquid chromatography; SDS-PAGE = sodium dodecyl sulfate-polyacrylamide gel electrophoresis; L = standard protein size ladder. Please click here to download this File.
Supplementary Figure S4: Western blot analysis of rAAV2/9 vectors purified by FPLC. (A) The collected fractions and concentrated rAAV2/9 vectors were resolved on an SDS-PAGE gel and probed with mouse monoclonal anti-AAV antibody (B1) that recognizes VP1, VP2, and VP3 capsid proteins. (B) The collected fractions and concentrated rAAV2/9 vectors were resolved on an SDS-PAGE gel and probed with mouse monoclonal anti-AAV antibody (A69) that recognizes VP1 and VP2 capsid proteins. Abbreviations: rAAV = recombinant adeno-associated virus; FPLC = fast protein liquid chromatography; SDS-PAGE = sodium dodecyl sulfate-polyacrylamide gel electrophoresis; L = standard protein size ladder. Please click here to download this File.
Supplementary Figure S5: rAAV2/9 vector quantification and characterization via Stunner. (A) Absorbance spectrum (black) of an rAAV2/9 vector measured by Stunner. The contribution of proteins (blue), ssDNA (green), other UV-absorbing compounds or impurities (purple), and background turbidity (grey) are also depicted. (B) DLS intensity distribution of rAAV2/9 with a major peak at 30 nm corresponding to a capsid scattering intensity of 78%, as determined by measuring the area under the curve from 15 to 45 nm (shaded green). A total aggregate intensity of 22% (shaded in grey) was also measured with a main contribution from large aggregates (19.9%) with a mean diameter of 620 nm. (C) Stunner analysis of an rAAV2/9 vector preparation exhibiting a total capsid titer of 2.18 × 1014 cp/mL (dark blue) and a full capsid titer of 2.35 × 1013 vg/mL (dark green). A free and aggregated protein of 2.92 × 1013 cp/mL equivalents (light blue), as well as a free and aggregated ssDNA of 3.14 × 1012 vg/mL equivalents (light green), were also measured in this preparation. Abbreviations: rAAV = recombinant adeno-associated virus; ssDNA = single-stranded DNA; DLS = dynamic light scattering. Please click here to download this File.
Supplementary Figure S6: In vitro transduction efficiency and viability of the purified fractions of rAAV1/2. HEK293T cells expressing GFP (direct fluorescence) 48 h after the transduction with 50 µL of FPLC fractions of an rAAV1/2 vector encoding ssGFP (rAAV1/2 - CMV-ssGFP). Scale bars = 100 µm. Abbreviations: rAAV = recombinant adeno-associated virus; FPLC = fast protein liquid chromatography; ssGFP = single-strand green fluorescent protein. Please click here to download this File.
Supplementary Figure S7: In vitro transduction efficiency and viability of the purified fractions of rAAV2/9. HEK293T cells were infected with 50 µL of each FPLC fraction (F2-F16) or flowthrough of an rAAV2/9 vector encoding scGFP under the control of the CMV promoter. The GFP-expressing cells were visualized 48 h post-infection. Scale bars = 100 µm. Abbreviations: rAAV = recombinant adeno-associated virus; FPLC = fast protein liquid chromatography; scGFP = self-complementary green fluorescent protein; CMV = cytomegalovirus. Please click here to download this File.
Discussion
The rapidly expanding AAV vector toolkit has become one of the most promising gene delivery systems for a wide range of cell types through different routes of administration. In this work, we aimed to develop an improved protocol for the production, purification, and characterization of mosaic rAAV vectors that could prove their worth in preclinical studies. For that purpose, the generation of rAAV1/2 and rAAV2/9 mosaic vectors is described here, but the procedure can also be applied to purify standard rAAV2 vectors (data not shown).
Mosaic rAAVs were produced following an optimized transfection method using PEI as a transfection reagent. A transient transfection method was selected due to its greater flexibility and speed, considerable advantages in early-stage preclinical studies. Once a particular transgene and serotype have been validated, the production system can be fine-tuned to achieve better scalability and cost-effectiveness by establishing a stable transfected cell line that expresses a subset of the specific Rep/Cap genes, with additional genes provided by an infection process24. Compared to calcium-phosphate transfection, PEI presents several advantages. It is a stable and cost-effective transfection reagent that operates effectively within a broader pH range. Additionally, it eliminates the requirement to change the cell medium after transfection, resulting in a significant reduction in both cost and workload69.
In an attempt to circumvent some of the limitations imposed by CsCl or iodixanol gradients, the produced rAAVs were harvested and purified by affinity chromatography. This strategy offers a simplified and scalable approach that can be performed without the need for ultracentrifugation and gradients, yielding clean and high viral titers. Indeed, chromatography techniques using an FPLC system can be automated and scaled up by packing more resin volume in a column with a higher bed height. The protocol described herein can be easily adapted to incorporate 5 mL HiTrap Heparin HP columns (data not shown). Additionally, heparin columns may be reused several times, thus contributing to the cost-effectiveness of this method.
The purified rAAVs were then characterized in terms of titer, purity, morphological features, and biological activity. Interestingly, in Coomassie blue staining, a band with approximately 17 kDa was detected in fractions F8-F16 in addition to the three typical viral capsid proteins. However, this band is no longer present after the concentration step of rAAVs. Moreover, some faint bands with molecular weights lower than VP3 (<62 kDa) can also be detected upon B1 and A69 labeling, suggesting that these could be fragments of the VP1, VP2, and VP3 capsid proteins70. Another possibility is that these are in fact other co-purifying proteins such as ferritin or other cellular proteins with polypeptides that share similar protein fingerprints with the AAV capsid proteins and could be involved in the AAV biology, as previously suggested26,71,72.
TEM and stunner analysis also revealed the presence of empty particles at variable levels across different productions. Similarly, other studies previously reported the generation of variable and high levels (>65%) of empty capsids for rAAVs prepared by transfection or infection methods24,73. The mechanism behind rAAV generation starts with the rapid formation of empty capsids from newly synthesized VP proteins, followed by a slow rate-limiting step of genome packaging into the preformed capsids mediated by Rep proteins74,75. Therefore, empty capsids are generated in rAAV productions, although the proportion of empty and full capsids can vary depending on the size and sequence of the transgene of interest and cell culture conditions58,73. Empty capsids raise some concerns since, by lacking the genome of interest, they are unable to provide a therapeutic effect and may also potentially increase an innate or adaptive immune response. However, some reports have also shown that, by adjusting their ratio, empty AAV capsids may serve as highly effective decoys for AAV-specific neutralizing antibodies and therefore, increase transduction efficiencies60,76,77. If the presence of empty capsids is critically detrimental and given the slightly less anionic character of empty particles compared to full vectors, a potential solution could involve conducting a second polishing purification step using anion exchange chromatography techniques64.
This study also provides compelling evidence that the generated mosaic rAAVs are able to efficiently transduce not only in vitro neuronal cultures but also the CNS upon intracranial injection of rAAV1/2. Overall, these results suggest that the described production and purification protocol renders highly pure and biologically active rAAVs ready to use in 6 days, presenting itself as a versatile and cost-effective method for the generation of rAAVs in preclinical studies.
Disclosures
The authors declare no conflicts of interest.
Acknowledgements
We are grateful for the collaboration, insights, and technical assistance provided by Dr. Mónica Zuzarte, at the Coimbra Institute for Clinical and Biomedical Research (iCBR) and Center for Innovative Biomedicine and Biotechnology (CIBB), regarding the TEM analysis of rAAVs. We extend our appreciation to Dr. Dominique Fernandes, at the Center for Neuroscience and Cell Biology of the University of Coimbra (CNC-UC) and the Institute for Interdisciplinary Research of the University of Coimbra (IIIUC), for her invaluable technical assistance and insights regarding the primary neuronal culture experiments. The pRV1, pH21, and pFdelta6 plasmids, essential for this study, were generously provided by Dr. Christina McClure, at the School of Medical Sciences, College of Life Sciences and Medicine, University of Aberdeen, for which we are grateful. This work was funded by the European Regional Development Fund (ERDF), through the Centro 2020 Regional Operational Program; through the COMPETE 2020 - Operational Programme for Competitiveness and Internationalisation, and Portuguese national funds via FCT - Fundação para a Ciência e a Tecnologia, under the projects: UIDB/04539/2020, UIDP/04539/2020, LA/P/0058/2020, ViraVector (CENTRO-01-0145-FEDER-022095), Imagene (PTDC/BBB-NAN/0932/2014 | POCI-01-0145-FEDER-016807), ReSet - IDT-COP (CENTRO-01-0247-FEDER-070162), Fighting Sars-CoV-2 (CENTRO-01-01D2-FEDER-000002), BDforMJD (CENTRO-01-0145-FEDER-181240), ModelPolyQ2.0 (CENTRO-01-0145-FEDER-181258), MJDEDIT (CENTRO-01-0145-FEDER-181266); by the American Portuguese Biomedical Research Fund (APBRF) and the Richard Chin and Lily Lock Machado-Joseph Disease Research Fund, ARDAT under the IMI2 JU Grant agreement No 945473 supported by EU and EFPIA; GeneT- Teaming Project 101059981 supported by the European Union's Horizon Europe program. M.M.L. was supported by 2021.05776.BD; C.H. was supported by 2021.06939.BD; A.C.S. was supported by 2020.07721.BD; and D.D.L. was supported by 2020.09668.BD. Figure 1 was created using BioRender.com.
Materials
| Name | Company | Catalog Number | Comments |
| 10% povidone-iodine | Medline | MDS093943 | |
| 12-well plates | Thermo Scientific | 11889684 | |
| 24-well plates | VWR | 734-2325 | |
| 4’,6-diamidino-2-phenylindole (DAPI) | Invitrogen | D1306 | |
| 96-well Stunner plate | Unchained Labs | 701-2025 | 96-well quantification plate for the consecutive ultraviolet-visible light absorption, static light scattering, and dynamic light scattering analysis of rAAV samples |
| AAVpro Titration Kit (for Real-Time PCR) Ver.2 | Takara | 6233 | For determining the titer of AAV using RT-qPCR. This kit contains DNase I, Lysis Buffer, Dilution Buffer, positive control, Taq II mix, primer forward, primer reverse, water |
| Acetic acid glacial | Fisher Chemical | A/0360/PB17 | |
| ÄKTA pure 25 | Cytiva | 29018224 | FPLC system controlled by UNICORN software, version 6.3 |
| Alkaline phosphatase-linked goat anti-mouse | Invitrogen | 31328 | |
| Amicon ultra-0.5 centrifugal filter unit | Merck Millipore | UFC5100 | |
| Amicon ultra-15 centrifugal filter unit | Merck Millipore | UFC9100 | |
| Benzonase Nuclease | Merck Millipore | E1014 | |
| Bromophenol blue | Sigma-Aldrich | B0126 | |
| CFX96 Real-Time PCR detection system | Biorad | 184-5096 | |
| ChemiDoc Touch Imaging System | Bio-Rad Laboratories | 1708370 | |
| Chicken polyclonal anti-GFP primary antibody | Abcam | ab13970 | |
| Coomassie Blue G250 | Fisher Chemical | C/P541/46 | |
| Dithiothreitol (DTT) | Fisher Bioreagents | BP17225 | |
| DMEM | Sigma-Aldrich | D5796 | |
| ECF Substrate for Western Blotting | Cytiva | RPN5785 | |
| FastDigest SmaI | Thermo Scientific | FD0663 | |
| FEI-Tecnai G2 Spirit Biotwin | FEI | Biotwin | Transmission electron microscope |
| Fetal bovine serum | Biowest | S1810 | |
| Fluorescence mounting medium | Dako | S3023 | |
| Formvar-carbon coated 200 mesh grid | TAAB Laboratories Equipment | F077/025 | |
| Gas evacuation apparatus | RWD | R546W | |
| Glycerol | Fisher BioReagents | 10021083 | |
| Goat polyclonal anti-chicken antibody, Alexa Fluor 488 | Invitrogen | A-11039 | |
| Hamilton needle 30G, Small Hub RN Needle, 25 mm, PST3 | Hamilton | 7803-07 | |
| Hamilton syringe (10 µL) | Hamilton | 7653-01 | |
| HEK293T | American Type Culture Collection | CRL-11268 | |
| HiTrap Heparin HP 1 x 5 mL | Cytiva | 17040701 | Pre-packed heparin column |
| HiTrap Heparin HP 5 x 1 mL | Cytiva | 17040601 | Pre-packed heparin column |
| Immobilon-P PVDF Membrane | Merck Millipore | IPVH00010 | |
| Isoflurane | Braun | 469860 | |
| Ketamine | Dechra Pharmaceuticals | N/A | Nimatek |
| Low-retention microcentrifuge tubes (2 mL) | Fisher Scientific | 11906965 | |
| Lunatic & Stunner Client software | Unchained Labs | N/A | Client analysis software version 8.0.1.235. Software for the consecutive ultraviolet-visible light absorption, static light scattering, and dynamic light scattering analysis of rAAV samples |
| Methanol | Fisher Chemical | M/4000/FP21 | |
| Mouse monoclonal anti-AAV, VP1, VP2 antibody (A69) | American Research Products | 03-61057 | |
| Mouse monoclonal anti-AAV, VP1, VP2, VP3 antibody (B1) | American Research Products | 03-61058 | |
| Neuro2a | American Type Culture Collection | CCL-131 | |
| Normal goat serum | Gibco | 16210064 | |
| NucleoBond Xtra Maxi EF | Macherey-Nagel | 12738422 | |
| NZYColour Protein Marker II | NZYtech | MB09002 | |
| pAAV-CMV-scGFP | Addgene | 32396 | Addgene plasmid # 32396; http://n2t.net/addgene:32396; RRID:Addgene_32396 |
| pAAV-CMV-ssGFP | Addgene | 105530 | Addgene plasmid # 105530; http://n2t.net/addgene:105530; RRID:Addgene_105530 |
| pAAV2/9n | Addgene | 112865 | Addgene plasmid # 112865; http://n2t.net/addgene:112865; RRID:Addgene_112865 |
| Paraformaldehyde | Acros Organics | 10342243 | |
| PBS | Fisher BioReagents | BP2438 | |
| Penicillin-streptomycin | Gibco | 15140-122 | |
| Pluronic F-68 Non-ionic Surfactant (100x) | Gibco | 24040032 | |
| Polyethylenimine MAX, MW 40,000 | Polysciences Europe | 24765 | |
| R500 Series Compact Small Animal Anesthesia Machine - Isoflurane | RWD | N/A | |
| Sample Inlet Valve V9-IS | Cytiva | 29027746 | |
| Sample pump P9-S | Cytiva | 29027745 | |
| Sodium azide | Sigma-Aldrich | S2002 | |
| Sodium chloride | Fisher Scientific | 10428420 | |
| Sodium deoxycholate | Sigma-Aldrich | D6750 | |
| Sodium dodecyl sulfate (SDS) | Fisher Bioreagents | BP166 | |
| Sterile PVDF syringe filter | Fisher Scientific | 15191499 | |
| Stunner Platform | Unchained Labs | 700-2002 | Equipment for the consecutive ultraviolet-visible light absorption, static light scattering, and dynamic light scattering analysis of rAAV samples |
| Superloop 150 mL | Cytiva | 18-1023-85 | |
| Superloop 50 mL | Cytiva | 18-1113-82 | |
| SURE 2 supercompetent cells | Stratagene, Agilent Technologies | HPA200152 | |
| Treated culture dishes | Corning | 734-1711 | |
| Tris base | Fisher BioReagents | BP152 | |
| Tris hydrochloride | Fisher BioReagents | BP153 | |
| Triton X-100 | Sigma-Aldrich | T8787 | |
| Trypsin-EDTA | Gibco | 25200-072 | |
| Wheat Germ Agglutinin (WGA) conjugated with Alexa Fluor 633 | Invitrogen | W21404 | |
| Xylazine | Dechra Pharmaceuticals | N/A | Sedaxylan |
| Zeiss Axio Observer Z1 | Carl Zeiss Microscopy GmbH | N/A | Inverted fluorescence microscope equiped with an EC Plan-Neofluar 10x/0.30 objective and a Plan-Apochromat 40x/0.95 objective |
| Zeiss Axio Scan.Z1 | Carl Zeiss Microscopy GmbH | N/A | Slide scanner fluorescence microscope equipped with a Plan-Apochromat 20x/0.8 objective |
| Zeiss LSM 710 | Carl Zeiss Microscopy GmbH | N/A | Inverted confocal microscope equipped with a Plan-Apochromat 40x/1.4 Oil DIC objective |
| µ-Slide 8 well Ibidi | Ibidi | 80826 | 8-well chamber slide |
References
- Atchison, R. W., Casto, B. C., Hammon, W. M. Adenovirus-associated defective virus particles. Science. 149 (3685), 754-756 (1965).
- Murlidharan, G., Samulski, R. J., Asokan, A. Biology of adeno-associated viral vectors in the central nervous system. Front Mol Neurosci. 7, 1-9 (2014).
- Muzyczka, N. Use of Adeno-Associated Virus as a General Transduction Vector for Mammalian Cells. Viral Expression Vectors. 158, 97-129 (1992).
- Goncalves, M. A. F. V Adeno-associated virus: from defective virus to effective vector. Virol J. 2, 43 (2005).
- Flotte, T. R. Gene therapy progress and prospects: recombinant adeno-associated virus (rAAV) vectors. Gene Ther. 11 (10), 805-810 (2004).
- Grieger, J. C., Choi, V. W., Samulski, R. J. Production and characterization of adeno-associated viral vectors. Nat Protoc. 1 (3), 1412-1428 (2006).
- Ojala, D. S., Amara, D. P., Schaffer, D. V Adeno-Associated Virus Vectors and Neurological Gene Therapy. Neuroscientist. 21 (1), 84-98 (2014).
- Saraiva, J., Nobre, R. J., Pereira de Almeida, L. Gene therapy for the CNS using AAVs: The impact of systemic delivery by AAV9. Journal of Controlled Release. 241, 94-109 (2016).
- Agbandje-McKenna, M., Kleinschmidt, J. AAV capsid structure and cell interactions. Methods Mol Biol. 807, 47-92 (2011).
- Sonntag, F., Schmidt, K., Kleinschmidt, J. A. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc Natl Acad Sci U S A. 107 (22), 10220-10225 (2010).
- Sonntag, F., et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J Virol. 85 (23), 12686-12697 (2011).
- Gao, G., et al. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 78 (12), 6381-6388 (2004).
- Gao, G., Vandenberghe, L. H., Wilson, J. M. New recombinant serotypes of AAV vectors. Curr Gene Ther. 5 (3), 285-297 (2005).
- Van Vliet, K. M., Blouin, V., Brument, N., Agbandje-McKenna, M., Snyder, R. O. The role of the adeno-associated virus capsid in gene transfer. Methods Mol Biol. 437, 51-91 (2008).
- Clark, K. R., Voulgaropoulou, F., Fraley, D. M., Johnson, P. R. Cell lines for the production of recombinant adeno-associated virus. Hum Gene Ther. 6 (10), 1329-1341 (1995).
- Inoue, N., Russell, D. W. Packaging cells based on inducible gene amplification for the production of adeno-associated virus vectors. J Virol. 72 (9), 7024-7031 (1998).
- Liu, X., Voulgaropoulou, F., Chen, R., Johnson, P. R., Clark, K. R. Selective Rep-Cap gene amplification as a mechanism for high-titer recombinant AAV production from stable cell lines. Mol Ther. 2 (4), 394-403 (2000).
- Mathews, L. C., Gray, J. T., Gallagher, M. R., Snyder, R. O. [23] Recombinant adeno-associated viral vector production using stable packaging and producer cell lines. Methods Enzymol. 346, 393-413 (2002).
- Gao, G., et al. Purification of recombinant adeno-associated virus vectors by column chromatography and its performance in vivo. Hum Gene Ther. 11 (15), 2079-2091 (2000).
- Xiao, X., Li, J., Samulski, R. J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 72 (3), 2224-2232 (1998).
- Aponte-Ubillus, J. J., et al. Molecular design for recombinant adeno-associated virus (rAAV) vector production. Appl Microbiol Biotechnol. 102 (3), 1045-1054 (2018).
- Smith, R. H., Levy, J. R., Kotin, R. M. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol Ther. 17 (11), 1888-1896 (2009).
- Grimm, D., Kern, A., Rittner, K., Kleinschmidt, J. A. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum Gene Ther. 9 (18), 2745-2760 (1998).
- Wright, J. F. Transient transfection methods for clinical adeno-associated viral vector production. Hum Gene Ther. 20 (7), 698-706 (2009).
- Yuan, Z., Qiao, C., Hu, P., Li, J., Xiao, X. A versatile adeno-associated virus vector producer cell line method for scalable vector production of different serotypes. Hum Gene Ther. 22 (5), 613-624 (2011).
- Strobel, B., Miller, F. D., Rist, W., Lamla, T. Comparative analysis of cesium chloride- and iodixanol-based purification of recombinant adeno-associated viral vectors for preclinical applications. Hum Gene Ther Methods. 26 (4), 147-157 (2015).
- Zolotukhin, S., et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 6 (6), 973-985 (1999).
- Anderson, R., Macdonald, I., Corbett, T., Whiteway, A., Prentice, H. G. A method for the preparation of highly purified adeno-associated virus using affinity column chromatography, protease digestion and solvent extraction. J Virol Methods. 85 (1-2), 23-34 (2000).
- Okada, T., et al. Scalable purification of adeno-associated virus serotype 1 (AAV1) and AAV8 vectors, using dual ion-exchange adsorptive membranes. Hum Gene Ther. 20 (9), 1013-1021 (2009).
- Guo, P., et al. Rapid and simplified purification of recombinant adeno-associated virus. J Virol Methods. 183 (2), 139-146 (2012).
- Lock, M., et al. Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum Gene Ther. 21 (10), 1259-1271 (2010).
- Wang, L., Blouin, V., Brument, N., Bello-Roufai, M., Francois, A. Production and Purification of Recombinant Adeno-Associated Vectors. Adeno-Associated Virus: Methods and Protocols. 807, 361-404 (2011).
- Summerford, C., Samulski, R. J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 72 (2), 1438-1445 (1998).
- Wu, Z., Asokan, A., Samulski, R. J. Adeno-associated Virus Serotypes: Vector Toolkit for Human Gene Therapy. Molecular Therapy. 14 (3), 316-327 (2006).
- Mietzsch, M., Broecker, F., Reinhardt, A., Seeberger, P. H., Heilbronn, R. Differential adeno-associated virus serotype-specific interaction patterns with synthetic heparins and other glycans. J Virol. 88 (5), 2991-3003 (2014).
- McClure, C., Cole, K. L. H., Wulff, P., Klugmann, M., Murray, A. J. Production and titering of recombinant adeno-associated viral vectors. JoVE. (57), e3348 (2011).
- Auricchio, A., O’Connor, E., Hildinger, M., Wilson, J. M. A single-step affinity column for purification of serotype-5 based adeno-associated viral vectors. Mol Ther. 4 (4), 372-374 (2001).
- Wang, Q., et al. Identification of an adeno-associated virus binding epitope for AVB sepharose affinity resin. Mol Ther Methods Clin Dev. 2, 15040 (2015).
- Mietzsch, M., et al. OneBac: platform for scalable and high-titer production of adeno-associated virus serotype 1–12 vectors for gene therapy. Hum Gene Ther. 25 (3), 212-222 (2014).
- Mietzsch, M., et al. Characterization of AAV-specific affinity ligands: consequences for vector purification and development strategies. Mol Ther Methods Clin Dev. 19, 362-373 (2020).
- Florea, M., et al. High-efficiency purification of divergent AAV serotypes using AAVX affinity chromatography. Mol Ther Methods Clin Dev. 28, 146-159 (2023).
- Koerber, J. T., Jang, J. -. H., Yu, J. H., Kane, R. S., Schaffer, D. V. Engineering adeno-associated virus for one-Step purification via immobilized metal affinity chromatography. Hum Gene Ther. 18 (4), 367-378 (2007).
- Zhang, H. -. G., et al. Addition of six-His-tagged peptide to the C terminus of adeno-associated virus VP3 does not affect viral tropism or production. J Virol. 76 (23), 12023-12031 (2002).
- Arnold, G. S., Sasser, A. K., Stachler, M. D., Bartlett, J. S. Metabolic biotinylation provides a unique platform for the purification and targeting of multiple AAV vector serotypes. Mol Ther. 14 (1), 97-106 (2006).
- Rabinowitz, J. E., et al. Cross-dressing the virion: the transcapsidation of adeno-associated virus serotypes functionally defines subgroups. J Virol. 78 (9), 4421-4432 (2004).
- Hauck, B., Chen, L., Xiao, W. Generation and characterization of chimeric recombinant AAV vectors. Mol Ther. 7 (3), 419-425 (2003).
- Choi, V., McCarty, D., Samulski, R. AAV hybrid serotypes: improved vectors for gene delivery. Curr Gene Ther. 5 (3), 299-310 (2005).
- Nonnenmacher, M., van Bakel, H., Hajjar, R. J., Weber, T. High capsid–genome correlation facilitates creation of AAV libraries for directed evolution. Mol Ther. 23 (4), 675-682 (2015).
- Kimura, K., et al. A mosaic adeno-associated virus vector as a versatile tool that exhibits high levels of transgene expression and neuron specificity in primate brain. Nat Commun. 14 (1), 4762 (2023).
- Issa, S. S., Shaimardanova, A. A., Solovyeva, V. V., Rizvanov, A. A. Various AAV serotypes and their applications in gene therapy: an overview. Cells. 12 (5), 785 (2023).
- Foust, K. D., et al. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 27 (1), 59-65 (2009).
- Lopes, M. M., et al. A new protocol for whole-brain biodistribution analysis of AAVs by tissue clearing, light-sheet microscopy and semi-automated spatial quantification. Gene Ther. 29 (12), 665-679 (2022).
- Gray, J. T., Zolotukhin, S. Design and construction of functional AAV vectors. Methods Mol Biol. 807, 25-46 (2011).
- Choi, V. W., Asokan, A., Haberman, R. A., Samulski, R. J. Production of recombinant adeno-associated viral vectors. Curr Protoc Hum Genet. , (2007).
- Bennicelli, J., et al. Reversal of blindness in animal models of leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol Ther. 16 (3), 458-465 (2008).
- Fischer, M. D., Hickey, D. G., Singh, M. S., MacLaren, R. E. Evaluation of an optimized injection system for retinal gene therapy in human patients. Hum Gene Ther Methods. 27 (4), 150-158 (2016).
- Santos, S. D., et al. Contactin-associated protein 1 (Caspr1) regulates the traffic and synaptic content of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors. J Biol Chem. 287 (9), 6868-6877 (2012).
- Sommer, J. M., et al. Quantification of adeno-associated virus particles and empty capsids by optical density measurement. Mol Ther. 7 (1), 122-128 (2003).
- Dong, B., Nakai, H., Xiao, W. Characterization of genome integrity for oversized recombinant AAV vector. Mol Ther. 18 (1), 87-92 (2010).
- Wright, J. F. AAV empty capsids: For better or for worse. Mol Ther. 22 (1), 1-2 (2014).
- Asaad, W., et al. AAV genome modification for efficient AAV production. Heliyon. 9 (4), e15071 (2023).
- Wobus, C. E., et al. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J Virol. 74 (19), 9281-9293 (2000).
- Wistuba, A., Kern, A., Weger, S., Grimm, D., Kleinschmidt, J. A. Subcellular compartmentalization of adeno-associated virus type 2 assembly. J Virol. 71 (2), 1341-1352 (1997).
- Qu, G., et al. Separation of adeno-associated virus type 2 empty particles from genome containing vectors by anion-exchange column chromatography. J Virol Methods. 140 (1-2), 183-192 (2007).
- Brument, N., et al. A versatile and scalable two-step ion-exchange chromatography process for the purification of recombinant adeno-associated virus serotypes-2 and -5. Mol Ther. 6 (5), 678-686 (2002).
- Potter, M., et al. A simplified purification protocol for recombinant adeno-associated virus vectors. Mol Ther Methods Clin Dev. 1, 14034 (2014).
- Dobnik, D., et al. Accurate quantification and characterization of adeno-associated viral vectors. Front Microbiol. 10, 1570 (2019).
- Yang, Q. -. E., et al. Rapid quality control assessment of adeno-associated virus vectors via Stunner. GEN Biotechnology. 1 (3), 300-310 (2022).
- Huang, X., et al. AAV2 production with optimized N/P ratio and PEI-mediated transfection results in low toxicity and high titer for in vitro and in vivo applications. J Virol Methods. 193 (2), 270-277 (2013).
- Salganik, M., et al. Evidence for pH-dependent protease activity in the adeno-associated virus capsid. J Virol. 86 (21), 11877-11885 (2012).
- Dong, B., et al. Proteomics analysis of co-purifying cellular proteins associated with rAAV vectors. PLoS One. 9 (2), e86453 (2014).
- Grieger, J. C., Soltys, S. M., Samulski, R. J. Production of recombinant adeno-associated virus vectors using suspension HEK293 cells and continuous harvest of vector from the culture media for GMP FIX and FLT1 clinical vector. Mol Ther. 24 (2), 287-297 (2016).
- Wright, J. Product-related impurities in clinical-grade recombinant AAV vectors: characterization and risk assessment. Biomedicines. 2 (1), 80-97 (2014).
- Bennett, A., Mietzsch, M., Agbandje-McKenna, M. Understanding capsid assembly and genome packaging for adeno-associated viruses. Future Virol. 12 (6), 283-297 (2017).
- Myers, M. W., Carter, B. J. Assembly of adeno-associated virus. Virology. 102 (1), 71-82 (1980).
- Mingozzi, F., et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci Transl Med. 5 (194), (2013).
- Hoffman, B. E., Herzog, R. W. Covert warfare against the immune system: decoy capsids, stealth genomes, and suppressors. Mol Ther. 21 (9), 1648-1650 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved