
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Isolierung von Maus-Megakaryozyten-Vorläufern
In diesem Artikel
Erratum Notice
Zusammenfassung
Diese Methode beschreibt die durch Durchflusszytometrie gereinigte Reinigung von MEP und MKp von Mäuse femuren, Tibias und Beckenknochen.
Zusammenfassung
Knochenmark-Megakaryozyten sind große polyploide Zellen, die die Produktion von Blutplättchen sicherstellen. Sie entstehen aus hämatopoetischen Stammzellen durch Megakaryopoese. Die Endstadien dieses Prozesses sind komplex und umfassen klassischerweise die bipotenten Megakaryozyten-Erythrozyten-Vorläufer (MEP) und die unipotenten Megakaryozyten-Vorläufer (MKp). Diese Populationen gehen der Bildung von echten Megakaryozyten voraus, und als solche könnte ihre Isolierung und Charakterisierung eine robuste und unvoreingenommene Analyse der Megakaryozytenbildung ermöglichen. Dieses Protokoll stellt detailliert das Verfahren zur Sammlung hämatopoetischer Zellen aus dem Knochenmark der Maus, die Anreicherung hämatopoetischer Vorläufer durch magnetische Depletion und schließlich eine Zellsortierungsstrategie vor, die hochreine MEP- und MKp-Populationen ergibt. Zuerst werden Knochenmarkzellen aus dem Femur, der Tibia und auch dem Beckenkamm, einem Knochen, der eine hohe Anzahl hämatopoetischer Vorläufer enthält, gesammelt. Die Verwendung von Beckenkammknochen erhöht drastisch die Gesamtzellzahl pro Maus und trägt somit zu einer ethischeren Verwendung von Tieren bei. Eine magnetische Linienverarmung wurde unter Verwendung von 450 nm magnetischen Kügelchen optimiert, was eine sehr effiziente Zellsortierung mittels Durchflusszytometrie ermöglicht. Schließlich präsentiert das Protokoll die Markierungs- und Gating-Strategie für die Sortierung der beiden hochreinen Megakaryozyten-Vorläuferpopulationen: MEP (Lin-Sca-1-c-Kit+CD16/32-CD150+CD9dim) und MKp (Lin- Sca-1-c-Kit+CD16/32-CD150+CD9hell ). Diese Technik ist einfach zu implementieren und liefert genügend zelluläres Material, um i) molekulare Charakterisierung für ein tieferes Wissen über ihre Identität und Biologie, ii) In-vitro-Differenzierungsassays, die ein besseres Verständnis der Mechanismen der Reifung von Megakaryozyten liefern, oder iii) In-vitro-Modelle der Interaktion mit ihrer Mikroumgebung durchzuführen.
Einleitung
Blutplättchen werden von Megakaryozyten produziert. Diese großen polyploiden Zellen befinden sich im Knochenmark und stammen wie alle Blutzellen aus hämatopoetischen Stammzellen (HSC)1. Der klassische Weg der Produktion von Megakaryozyten im Knochenmark stammt von HSC und beinhaltet die Erzeugung verschiedener Vorläufer, die ihr Differenzierungspotenzial progressiv einschränken2. Der erste Vorläufer, der das Engagement für die megakaryozytäre Linie unterzeichnet, ist der Megakaryocyte-Erythrocyte Progenitor (MEP), ein bipotenter Vorläufer, der sowohl erythroide Zellen als auch Megakaryozyten produzieren kann3,4,5. Der MEP produziert dann einen unipotenten Vorläufer / Vorläufer (MKp), der sich zu einem reifen Megakaryozyten differenziert, der in der Lage ist, Blutplättchen zu produzieren. Die Mechanismen, die an der Erzeugung dieser Vorläufer beteiligt sind, sowie ihre Differenzierung und Reifung zu Megakaryozyten sind komplex und nur teilweise verstanden. Darüber hinaus sind die Heterogenität der MEP-Population in Bezug auf das Differenzierungspotenzial und das intrinsische Engagement dieser Zellen noch unklar. Um diese Prozesse zu entschlüsseln, ist es wichtig, gereinigte Populationen von MEP und MKp für feine molekulare und Einzelzellanalysen zu erhalten (oder Zugang zu ihnen zu haben).
Mehrere Studien haben bestimmte Kombinationen von Zelloberflächenmarkern für die Identifizierung von Vorläufern gezeigt, die der megakaryozytären Linie in der Maus zugesetzt sind6,7,8. Aus diesen wurde eine Methode entwickelt, die die Reinigung von MEP und MKp von Mäusen ermöglicht. Diese Methode wurde optimiert, um Zellen in ausreichender Anzahl und Qualität für eine große Anzahl von Assays zu erhalten. Aus ethischen Gründen und um die Anzahl der an den Experimenten beteiligten Tiere zu minimieren, haben wir die Entnahme des Knochenmarks aus dem Femur und der Tibia sowie aus dem Beckenkamm veranlasst. Dieser Knochen enthält eine hohe Häufigkeit und Anzahl von hämatopoetischen Vorläufern und wird die meiste Zeit während der langen Knochenentnahme beschädigt. Hier wird eine detaillierte Methode zur zuverlässigen Sammlung dieses Knochens vorgestellt.
Das zweite Optimierungskriterium ist die Herstellung hochreiner Zellpopulationen. Fluorescent Activated Cell Sorting (FACS) ist eine Methode der Wahl, um gereinigte Populationen von Zellen von Interesse zu erhalten. Niedrige Erträge werden jedoch erreicht, wenn die Zellpopulation von Interesse sehr selten ist. Anreicherungsverfahren sind daher notwendig. In diesem Protokoll wurde ein negatives Selektionsverfahren unter Verwendung von Magnetperlen gewählt.
Protokoll
Die Protokolle mit Tieren wurden in Übereinstimmung mit dem CREMEAS-Ausschuss für die Ethik von Tierversuchen der Universität Straßburg (Comité Régional d'Ethique en Matière d'Expérimentation Animale Strasbourg) durchgeführt. Genehmigungsnummer: E67-482-10).
1. Sammlung von Mausknochen
- Opfern Sie das Tier in Übereinstimmung mit den institutionellen Richtlinien.
HINWEIS: Die in diesem Manuskript präsentierten Daten stammen von C57Bl/6-Mäusen im Alter von 8 bis 12 Wochen. Die Anzahl der erhaltenen Zellen und die Häufigkeit der zitierten Populationen können je nach Alter und Mausstamm variieren. - Besprühen Sie den Körper mit 70% Ethanol.
- Machen Sie mit einer Schere einen 0,5-1 cm langen Schnitt der Haut senkrecht zur Wirbelsäule und reißen Sie die Haut um den ganzen Körper herum. Ziehen Sie die Haut vom Unterkörper herunter und entfernen Sie die Haut.
- Legen Sie das Tier mit dem Gesicht nach unten auf das Sezierpad. Lokalisieren Sie die Beckenknochen, indem Sie Ihre Finger entlang der freiliegenden Wirbelsäule von oben nach unten gleiten lassen. Um den Beckenkamm zu lokalisieren, identifizieren Sie die kleine Beule in der Lendengegend in der Nähe der Hinterbeine (die anterosuperiorische Region des Beckenknochens). Abbildung 1A,B zeigt eine schematische Darstellung der Anatomie der Maus.

Abbildung 1: Anatomie der Maus. (A) Röntgenaufnahme der Maus, die die Hinterbeinknochen zeigt. Beachten Sie den Raum zwischen dem Beckenknochen und der Wirbelsäule (gelber Pfeil), wo die Schere eingeführt werden muss, um die Hinterbeine richtig vom Körper der Maus zu trennen (gelbe gepunktete Linie). (B) Schematische Darstellung der knochenmarkreichen Knochen von Interesse. Die Beckenknochen sind rot, die Oberschenkelknochen lila und die Tibias grün dargestellt. (C) Schematische Darstellung des Beckenknochens der Maus. Das Darmbein entspricht dem markreichen Teil des Beckenknochens und ist rot hervorgehoben. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Legen Sie die Schere parallel zur Wirbelsäule gegen die Wirbel und in die Nähe der Beckenkammbeule. Fahren Sie fort, die Muskeln entlang der Seite der Wirbelsäule über dem Beckenknochen zu schneiden, indem Sie die Schere entlang der Wirbel bis zum Schwanz gleiten lassen.
HINWEIS: Dieser erste Muskelabschnitt kann auch mit einer Skalpellklinge durchgeführt werden. - Legen Sie die Schere parallel zur Wirbelsäule und schneiden Sie zwischen den Wirbeln und dem Beckenkamm, wie durch die gelb gepunktete Linie in Abbildung 1Aangezeigt . Achten Sie darauf, so nah wie möglich an den Wirbeln zu bleiben. Schneiden Sie die verbleibenden Muskeln ab, um die Extremität vom Körper zu lösen.
HINWEIS: Es sollte wenig bis gar keinen Widerstand geben. - Wiederholen Sie dies auf der anderen Seite, um das zweite Glied zu lösen.
- Übertragen Sie die Gliedmaßen auf eine saubere Oberfläche und entsorgen Sie den Rest des Körpers in Übereinstimmung mit den institutionellen Richtlinien.
- Legen Sie die Becken-, Oberschenkel- und Tibiaknochen frei, indem Sie so viel umgebendes Gewebe wie möglich mit der Pinzette und den Skalpellen entfernen.
- Fahren Sie fort, den Hüftkopf vorsichtig vom Beckenknochen zu entfernen, indem Sie das distale Ende des Femurs mit der Pinzette halten, während Sie die Muskeln um die Artikulation mit den Skalpellen sanft schneiden. Wackeln Sie die Knochen, um die Luxation zu erleichtern.
- Kratzen Sie den verbleibenden Muskel vom Beckenknochen ab und schneiden Sie mit einem Skalpell in der Mitte der Höhle, die den Femurkopf hielt. Das Darmbein wird gehalten, da es reich an hämatopoetischen Vorläufern ist, während die dreieckige, sehr dünne Seite des Knochens verworfen wird, wie in Abbildung 1Cgezeigt.
- Entfernen Sie die Restgewebe um das Darmbein mit dem Skalpell und legen Sie den gereinigten Knochen in steriles PBS, ergänzt mit 2% Newborn Calf Serum (PBS-2% NBCS).
- Schneiden Sie mit einer Schere den Fuß vom Bein am Knöchel ab.
- Halten Sie den unteren Teil der Tibia mit der Pinzette und kratzen Sie den Muskel in Richtung Knie. Entsorgen Sie die Fibeln und schneiden Sie mit dem Skalpell über das Tibiaplateau. Legen Sie die Tibia in steriles PBS-2% NBCS.
- Entfernen Sie die Restgewebe um den Femur mit Skalpellen.
- Halten Sie die Oberseite des Femurs mit einer Pinzette; Legen Sie die Skalpellklinge an die Basis der Kniescheibe. Wenden Sie eine Kraft in Richtung der Kniescheibe parallel zum Femur bis zur Ablösung der Kniescheibe an. Legen Sie den Femur in steriles PBS-2% NBCS. Das Entfernen der Kniescheibe bietet einen sauberen Zugang zum Einführen der Nadel für die Markspülung.
2. Magnetische Erschöpfung von linienpositiven Zellen
- Übertragen Sie die Knochen in einem Laminar-Flow-Schrank in eine sterile Petrischale, die mit sterilem PBS-2% NBCS gefüllt ist.
- Mit einem Skalpell den Kopf der Oberschenkelknochen abschneiden.
- Füllen Sie eine 1 ml Spritze mit sterilem PBS-2% NBCS und befestigen Sie eine 21 G Nadel am Auslass.
- Füllen Sie ein 5-ml-Polypropylenröhrchen mit 2 ml sterilem PBS-2% NBCS.
- Halten Sie den Femur mit der Pinzette; Führen Sie die Nadel vorsichtig in die nach der Entfernung der Kniescheibe linke Rille ein. Wenden Sie während des Einführens eine Rotation auf die Nadel an, um ein Verstopfen der Nadel zu vermeiden. Stellen Sie sicher, dass die Nadel bis zur Fase vollständig in den Knochen eingeführt ist.
- Übertragen Sie den Knochen mit der Nadel in das Röhrchen, das 2 ml PBS-2% NBCS enthält. Geben Sie das PBS-2% NBCS aus der Spritze ab und aspirieren Sie es, bis der Knochen klar ist.
- Entfernen Sie die Nadel vom Femur und führen Sie sie in das Loch auf der gegenüberliegenden Seite ein, wo sich der Femurkopf befand. Den Puffer wieder abgeben und absaugen und den Knochen entsorgen.
- Für den Beckenkamm und die Tibia halten Sie den Knochen mit der Pinzette; Führen Sie die Nadel vorsichtig in die offene Seite ein. Wenden Sie während des Einführens eine Rotation auf die Nadel an, um ein Verstopfen der Nadel zu vermeiden. Stellen Sie sicher, dass die Nadel bis zur Fase vollständig in den Knochen eingeführt ist. Übertragen Sie den Knochen mit der Nadel in das Röhrchen, das 2 ml PBS-2% NBCS enthält. Geben Sie das PBS-2% NBCS aus der Spritze ab und aspirieren Sie es, bis der Knochen klar ist. Entsorgen Sie die Knochen.
HINWEIS: Knochen von bis zu drei Mäusen können in dieselbe Röhre gespült werden. Poolen Sie die Zellsuspensionen. - Führen Sie die gepoolte Zellsuspension durch eine 40 μm Zellsiebkappe, die auf ein steriles 5 ml Polystyrolröhrchen aufgetragen wird.
- Fahren Sie mit dem Zählen der Zellen fort.
HINWEIS: Die Zellzahl kann mit jedem Hämozytometer, mit Trypan Blue zur Beurteilung der Lebensfähigkeit oder mit jedem automatisierten Zellzähler durchgeführt werden. Eine Maus liefert typischerweise 105 ± 7 x 106 Zellen. - Nehmen Sie 100 μL der Zellsuspension als Gesamtknochenmark beiseite, fügen Sie 500 μL PBS-2% NBCS hinzu und bewahren Sie es für das Färbeverfahren auf Eis auf.
- Die filtrierte Suspension wird durch Zentrifugieren bei 400 x g für 5 min bei 4 °C pelletiert und der Überstand entsorgt.
HINWEIS: Rote Blutkörperchen können lysiert werden, indem das Pellet in frisch zubereiteter Lyselösung (1/10in dH2O) wiederverwendet wird. 5 min inkubieren, bis die Suspension klar und leuchtend rot wird, und 10 Volumen steriles PBS hinzufügen. Fahren Sie fort, die Zellen in PBS-2% NBCS durch Zentrifugation bei 400 x g für 5 Minuten bei 4 °C zu waschen. Seien Sie vorsichtig, wenn Sie den Überstand entfernen, da das Zellpellet sehr locker ist. Führen Sie eine zweite Wäsche mit PBS-2% NBCS durch Zentrifugation bei 400 x g für 5 min bei 4 °C durch und fahren Sie mit Schritt 2.13 fort. - Resuspendieren Sie das Zellpellet in frisch zubereiteten primären Antikörpercocktail mit einem Verhältnis von 100 μL pro 1 x 107 Zellen. 30-45 Min. auf Eis inkubieren.
| Antikörper | Verdünnung |
| Gr-1-Biotin | 1:500 |
| B220-Biotin | 1:500 |
| Mac-1-Biotin | 1:500 |
| CD3-Biotin | 1:500 |
| CD4-Biotin | 1:500 |
| CD5-Biotin | 1:500 |
| CD8-Biotin | 1:500 |
| TER119-Biotin | 1:1000 |
| CD127-Biotin | 1:500 |
Tabelle 1.
- 10 μL der Zellsuspension in ein steriles 5 mL Polystyrolröhrchen mit der Bezeichnung Lin-Pos Fraction geben. Fügen Sie 90 μL PBS-2% NBCS hinzu und bewahren Sie es für das Färbeverfahren auf Eis auf.
- Fahren Sie fort, die Zellen zweimal mit sterilem PBS-2% NBCS durch Zentrifugation bei 400 x g für 5 min bei 4 °C zu waschen. Stellen Sie sicher, dass Sie die letzte Wäsche in einem sterilen 5-ml-Polypropylenröhrchen durchführen.
- Bereiten Sie während der Waschschritte die Perlen auf die magnetische Erschöpfung vor.
- Resuspendieren Sie die Perlen in der Durchstechflasche, indem Sie 30 s lang gründlich wirbeln.
- Übertragen Sie ein Volumen von Perlen, das zwei Perlen pro Zielzelle entspricht, in ein 5 ml Polypropylenröhrchen.
- Waschen Sie die Perlen zweimal mit PBS-2% NBCS, indem Sie das Röhrchen auf den Magneten legen und den Waschpuffer mit einer sterilen Pasteurpipette aus Glas entfernen.
- Resuspendieren Sie die Kügelchen in 500 μL sterilem PBS-2NBCS%.
- Resuspendieren Sie das Pellet der markierten Zellen in 250 μL Perlen und mischen Sie vorsichtig für 5 min auf Eis. Fügen Sie 2 ml steriles PBS-2% NBCS hinzu und mischen Sie es vorsichtig. Schütteln Sie das Röhrchen nicht.
- Legen Sie das Rohr für 2 min auf den Magneten.
- Sammeln Sie die nichtmagnetische Fraktion mit einer sterilen Pasteurpipette aus Glas und geben Sie sie auf die verbleibenden 250 μL Magnetperlen. Verschließen Sie das Röhrchen mit Parafilm.
- Legen Sie das Rohr für 20 min bei 4 °C auf eine Rohrwalze.
- Fügen Sie 2 ml steriles PBS-2% NBCS hinzu und mischen Sie es vorsichtig. Schütteln Sie das Röhrchen nicht.
- Legen Sie das Rohr für 2 min in den Magneten.
- Sammeln Sie die nichtmagnetische Fraktion in einem sterilen 5-ml-Polypropylenröhrchen mit der Bezeichnung Lin-Neg-Fraktion mit einer sterilen Pasteurpipette aus Glas.
- Die Zellen durch Zentrifugation bei 400 x g für 5 min bei 4 °C pelletieren und den Überstand entfernen.
- Resuspendieren Sie die nichtmagnetischen Zellen in 500 μL sterilem PBS-2% NBCS.
- Fahren Sie mit dem Zählen der Zellen fort.
HINWEIS: Eine Maus liefert typischerweise 3,9 ± 1,1 x 106 Zellen. Typische Abstammungsfärbungen vor und nach der Depletion sind in Abbildung 2B dargestellt.
3. Zellsortierung von Megakaryozyten-Vorläufern mittels Durchflusszytometrie
- Nehmen Sie die Röhrchen mit der Bezeichnung Total Bone Marrow, Lin-Pos Fraction und Lin-Neg Fraction.
- Fahren Sie fort, den Inhalt des Röhrchens Total Bone Marrow gleichmäßig in sechs sterile 5 ml Polystyrolröhrchen aufzuteilen. Beschriften Sie die Tuben mit den Zahlen 1-6.
- Fahren Sie fort, die Tube Lin-Pos Fraction mit der Zahl 7 zu beschriften.
- Fahren Sie fort, den Inhalt der Tube Lin-Neg Fraction wie folgt aufzuteilen.
- 50 μL in ein steriles 5 mL Polystyrolröhrchen mit 250 μL sterilem PBS-2%NBCS überführen. Dann teilen Sie seinen Inhalt gleichmäßig in 3 sterile 5 ml Polystyrolröhrchen auf. Beschriften Sie diese Tuben mit den Nummern 8-10.
- Die restlichen 450 μL der Lin-Neg Fraction Zellsuspension entsprechen dem Röhrchen mit der Nummer 11.
- Die Antikörper werden wie in Tabelle 2beschrieben in die Röhrchen gegeben.
| Rohr | Etikett | Antikörper-Cocktail |
| Knochenmark gesamt | ||
| 1 | Unbefleckte Kontrolle | |
| 2 | Einzelne gefärbte Steuerung | CD45-FITC (1/200) |
| 3 | Einzelne gefärbte Steuerung | CD45-PE (1/200) |
| 4 | Einzelne gefärbte Steuerung | TER119-APC (1/200) |
| 5 | Einzelne gefärbte Steuerung | CD45-PECy7 (1/200) |
| 6 | Einzelne gefärbte Steuerung | CD45-APC-Cy7 Biotin (1/200) |
| Lin-Pos Fraktion | ||
| 7 | Einzelne gefärbte Steuerung | Einzelne gefärbte Kontrolle. Streptavidin-APC-Cy7 (1:500) |
| Lin-Neg-Fraktion | ||
| 8 | FMO FITC Steuerung | c-kit-APC (1/200) + Sca-1-PE (1/200) + CD16/32-PE (1/200) + CD150-PECy7 (1/200) + Streptavidin-APC-Cy7 (1/500) |
| 9 | FMO PE-Steuerung | CD9-FITC (1/200) + c-kit-APC (1/200) + CD150-PECy7 (1/200) + Streptavidin-APC-Cy7 (1/500) |
| 10 | FMO PECy7 Steuerung | CD9-FITC (1/200) + C-Kit-APC (1/200) + Sca-1-PE (1/200) + CD16/32-PE (1/200) + Streptavidin-APC-Cy7 (1/500) |
| 11 | Positives Röhrchen zum Sortieren | CD9-FITC (1/200) + C-Kit-APC (1/200) + Sca-1-PE (1/200) + CD16/32-PE (1/200) + CD150-PECy7 (1/200) + Streptavidin-APC-Cy7 (1/500) |
Tabelle 2.
- Inkubieren Sie auf Eis für 30-45 min im Dunkeln.
- Waschen Sie die Zellen mit sterilem PBS-2% NBCS durch Zentrifugation bei 400 x g für 5 min bei 4 °C.
- Resuspendieren Sie die Zellpellets wie folgt.
- Für die Röhrchen 1 bis 10 wird das Pellet in 300 μL sterilem PBS-2%NBCS, ergänzt mit 7AAD (2,5 μg/ml endg) (PBS-7AAD), resuspendiert.
ACHTUNG: 7AAD ist ein DNA-Interkalans und muss daher mit entsprechender PSA (Handschuhe) gehandhabt werden. - Für Röhrchen 11 wird das Pellet in sterilem PBS-7AAD in einer maximalen Konzentration von 5 x 10 6 Zellen pro ml undeinem Mindestvolumen von 1 ml resuspendiert.
- Für die Röhrchen 1 bis 10 wird das Pellet in 300 μL sterilem PBS-2%NBCS, ergänzt mit 7AAD (2,5 μg/ml endg) (PBS-7AAD), resuspendiert.
- Bereiten Sie zwei Polypropylen-Sammelröhrchen mit der Bezeichnung MEP und MKp vor, die 2 ml PBS-2% NBCS enthalten.
HINWEIS: Alternativ können Zellen in Abhängigkeit von der anschließenden Anwendung für die sortierten Zellen in Kulturmedium oder Zelllysepuffer gesammelt werden. Die Verwendung von Polystyrolröhrchen wird wegen möglicher Interferenzen mit den geladenen Tröpfchen, die die interessierenden Zellen enthalten, nicht empfohlen. - Halten Sie alle Röhren im Dunkeln auf Eis.
- Fahren Sie mit der Einrichtung des Zellensortierers fort.
- Verwenden Sie die Röhren 1-7, um Spannung und Kompensation einzurichten, die Röhren 7-10, um die Sortiergatter für die Zellpopulationen von Interesse zu bestimmen, und Röhre 11 für die Zellsortierung.
- Die ersten Schritte der Gating-Strategie zielen darauf ab, Dubletten und tote Zellen von der Analyse auszuschließen, wie in Abbildung 3beschrieben. Identifizieren Sie einzelne lebensfähige Zellen und zeigen Sie das SSC-vs Lin-APC-Cy7-Punktdiagramm an, um die Effizienz der Abstammungsverarmung zu bestätigen. Aus denLin-Zellen wird ein Gate gesetzt, um Zellen auszuwählen, die positiv für C-Kit und negativ oder dim für Sca-1 und CD16/32 sind. Ein CD9 vs CD150 Expression Dot Plot für die ausgewählten Zellen ermöglicht die Identifizierung von vier Populationen.
HINWEIS: MEP- und MKp-Zellen sind beide positiv für CD150. Für CD9 können drei Ausdrucksebenen definiert werden (neg, dim und high). MKp exprimiert einen hohen CD9-Gehalt und MEP-Express-CD9 bei einer mittleren Fluoreszenzintensität. MEP Population entspricht Lin- c-Kit+ Sca-1-CD16/32-/dim CD150+ CD9dim und MKp Population entspricht Lin- c-Kit+ Sca-1-CD16/32-/dim CD150+ CD9hell. Die Unterscheidung zwischen CD9 high und CD9 dim Populationen für die CD150 positiven Zellen wird basierend auf dem maximalen Niveau der CD9-Expression in der CD150 negativen Population festgelegt. Eine Maus liefert typischerweise 5,3 ± 0,6 x 103 MKp und 27,2 ± 2,4 x 103 MEP.
Ergebnisse
Die phänotypische Analyse der als MEP und MKp identifizierten Zellen wurde mittels Durchflusszytometrie durchgeführt. Die Zellen wurden mit fluoreszenzkonjugierten Antikörpern gegen CD41a und CD42c, klassische Marker der megakaryozytären und Thrombozytenlinien, markiert. Beide Marker wurden von den Zellen der MKp-Population exprimiert, während diese Marker an der Oberfläche der Zellen der MEP-Population noch nicht nachgewiesen sind (Abbildung 4Ai,4Aii). Polyploidie ist...
Diskussion
Die in diesem Artikel beschriebene Methode ermöglicht die Extraktion und Reinigung von Maus-MEP und MKp. Ein wichtiger Parameter bei der Optimierung des Protokolls war es, eine ausreichende Anzahl von Zellen zu erhalten, die mit den meisten molekularen und zellulären Assays kompatibel sind. Die allgemeine Praxis der Mausknochenentnahme für die hämatopoetische Zellextraktion besteht normalerweise darin, sowohl die Femuren als auch die Tibien jeder Maus zu ernten. Der Beckenknochen, eine weitere Quelle für hämatopoet...
Offenlegungen
Die Autoren erklären keine konkurrierenden finanziellen Interessen.
Danksagungen
Die Autoren danken Monique Freund, Catherine Ziessel und Ketty für die technische Unterstützung. Diese Arbeit wurde von ARMESA (Association de Recherche et Développement en Médecine et Santé Publique) und von Grant ANR-17-CE14-0001-01 an Henri.de la unterstützt. Salle.
Materialien
| Name | Company | Catalog Number | Comments |
| 21-gauge needles | BD Microlance | 301155 | |
| 7AAD | Sigma-Aldrich | A9400 | |
| Antibody Gr-1-biotin | eBioscience | 13-5931-85 | Magnetic depletion |
| Antibody B220-biotin | eBioscience | 13-0452-85 | Magnetic depletion |
| Antibody Mac-1-biotin | eBioscience | 13-0112-85 | Magnetic depletion |
| Antibody CD3e-biotin | eBioscience | 13-0031-85 | Magnetic depletion |
| Antibody CD4-biotin | eBioscience | 13-9766-82 | Magnetic depletion |
| Antibody CD5-biotin | eBioscience | 13-0051-85 | Magnetic depletion |
| Antibody CD8a-biotin | eBioscience | 13-0081-85 | Magnetic depletion |
| Antibody TER119-biotin | eBioscience | 13-5921-85 | Magnetic depletion |
| Antibody CD127-biotin | eBioscience | 13-1271-85 | Magnetic depletion |
| Antibody CD45-FITC | eBioscience | 11-0451-85 | Cell sorting |
| Antibody CD45-PE | eBioscience | 12-0451-83 | Cell sorting |
| Antibody TER119-APC | eBioscience | 17-5921-83 | Cell sorting |
| Antibody CD45-PECy7 | eBioscience | 25-0451-82 | Cell sorting |
| Antibody CD45-biotin | eBioscience | 13-0451-85 | Cell sorting |
| Antibody CD9-FITC | eBioscience | 11-0091-82 | Cell sorting |
| Antibody c-kit-APC | eBioscience | 17-1171-83 | Cell sorting |
| Antibody Sca-1-PE | eBioscience | 12-5981-83 | Cell sorting |
| Antibody CD16/32-PE | eBioscience | 12-0161-83 | Cell sorting |
| Antibody CD150-PECy7 | eBioscience | 25-1502-82 | Cell sorting |
| Culture medium StemSpan-SFEM | Stemcell technologies | #09650 | |
| Dissection pad | Fisher Scientific | 10452395 | |
| DPBS | Life Technologies | 14190-094 | |
| Ethanol | vWR Chemicals | 83813.360 | |
| Forceps | Euronexia | P-120-AS | |
| Glass pasteur pipette | Dutscher | 42011 | |
| Magnet : DynaMag-5 | Thermo Fisher Scientific | 12303D | |
| Magnetic beads: Dynabeads Sheep Anti-Rat IgG | Thermo Fisher Scientific | 11035 | |
| Megacult | Stemcell technologies | #04970 | |
| MethoCult SF M3436 | Stemcell technologies | #03436 | |
| Newborn Calf Serum | Dutscher | 50750-500 | |
| Red Cell Lysis solution | BD Bioscience | 555899 | |
| Scalpels | Fisher Scientific | 12308009 | |
| Scissors | Euronexia | C-165-ASB | |
| Sterile 1 mL syringes | BD Bioscience | 303172 | |
| Sterile 15mL tubes | Sarstedt | 62.554.502 | |
| Sterile 5mL polypropylene tubes | Falcon | 352063 | |
| Sterile 5mL polystyrene tubes | Falcon | 352054 | |
| Sterile tubes with 70µm cell strainer cap | Falcon | 352235 | |
| Sterile petri dish | Falcon | 353003 | |
| Streptavidin-APC-Cy7 | BD Biosciences | 554063 | Cell sorting |
| Tube roller | Benchmark Scientific | R3005 |
Referenzen
- Kaushansky, K. Historical review: megakaryopoiesis and thrombopoiesis. Blood. 111 (3), 981-986 (2008).
- Akashi, K., Traver, D., Miyamoto, T., Weissman, I. L. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 404 (6774), 193-197 (2000).
- Debili, N., et al. Characterization of a bipotent erythro-megakaryocytic progenitor in human bone marrow. Blood. 88 (4), 1284-1296 (1996).
- Forsberg, E. C., Serwold, T., Kogan, S., Weissman, I. L., Passegué, E. New evidence supporting megakaryocyte-erythrocyte potential of flk2/flt3+ multipotent hematopoietic progenitors. Cell. 126 (2), 415-426 (2006).
- Vannucchi, A. M., et al. Identification and characterization of a bipotent (erythroid and megakaryocytic) cell precursor from the spleen of phenylhydrazine-treated mice. Blood. 95 (8), 2559-2568 (2000).
- Pronk, C. J., et al. Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell. 1 (4), 428-442 (2007).
- Nakorn, T. N., Miyamoto, T., Weissman, I. L. Characterization of mouse clonogenic megakaryocyte progenitors. Proceedings of the National Academy of Sciences of the United States of America. 100 (1), 205-210 (2003).
- Ng, A. P., et al. Characterization of thrombopoietin (TPO)-responsive progenitor cells in adult mouse bone marrow with in vivo megakaryocyte and erythroid potential. Proceedings of the National Academy of Sciences of the United States of America. 109 (7), 2364-2369 (2012).
- Strassel, C., et al. Hirudin and heparin enable efficient megakaryocyte differentiation of mouse bone marrow progenitors. Experimental Cell Research. 318 (1), 25-32 (2012).
- Brouard, N., et al. A unique microenvironment in the developing liver supports the expansion of megakaryocyte progenitors. Blood Advances. 1 (21), 1854-1866 (2017).
- Boscher, J., Gachet, C., Lanza, F., Léon, C. Megakaryocyte culture in 3D methylcellulose-based hydrogel to improve cell maturation and study the impact of stiffness and confinement. Journal of Visualized Experiments:JOVE. , (2021).
- Sanjuan-Pla, A., et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. 502 (7470), 232-236 (2013).
- Haas, S., et al. Inflammation-driven fast-track differentiation of HSCs into the megakaryocytic lineage. Experimental Hematology. 42 (8), 14 (2014).
- Shin, J. Y., Hu, W., Naramura, M., Park, C. Y. High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. The Journal of Experimental Medicine. 211 (2), 217-231 (2014).
Erratum
Formal Correction: Erratum: Isolation of Mouse Megakaryocyte Progenitors
Posted by JoVE Editors on 7/28/2021. Citeable Link.
An erratum was issued for: Isolation of Mouse Megakaryocyte Progenitors. A figure was updated.
Figure 2 was updated from:
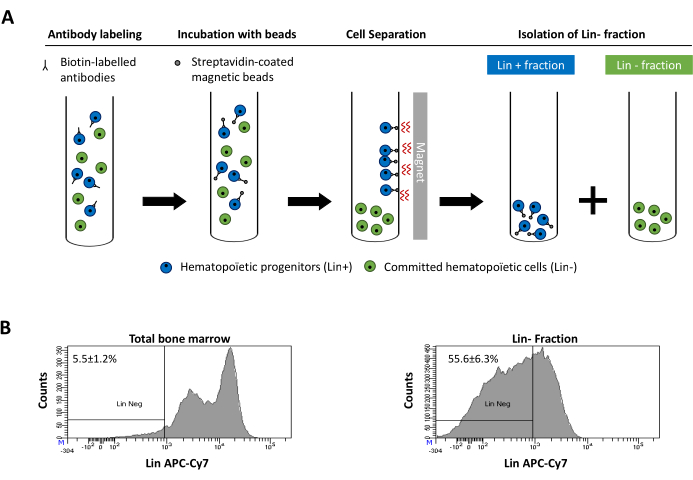
Figure 2: Magnetic depletion of lineage committed (Lin) cells. (A) Schematic representation of the magnetic depletion protocol. First, unsorted bone marrow cells are labeled with the biotin-conjugated rat anti-mouse antibody cocktail. Cells are then incubated with anti-rat Ig coated magnetic beads and subsequently subjected to the magnetic depletion using a strong magnet. The magnet will retain the labeled magnetic Lin+ fraction against the tube walls, while the unlabeled non-magnetic Lin- negative fraction will be collected in a new tube. (B) Lineage committed cells can be identified using fluorescent conjugated streptavidin. Typical analysis of the lineage expression in cells prior to magnetic depletion (total bone marrow) and after magnetic depletion (Lin- Fraction) N = 21. Please click here to view a larger version of this figure.
{kind=link}
to:
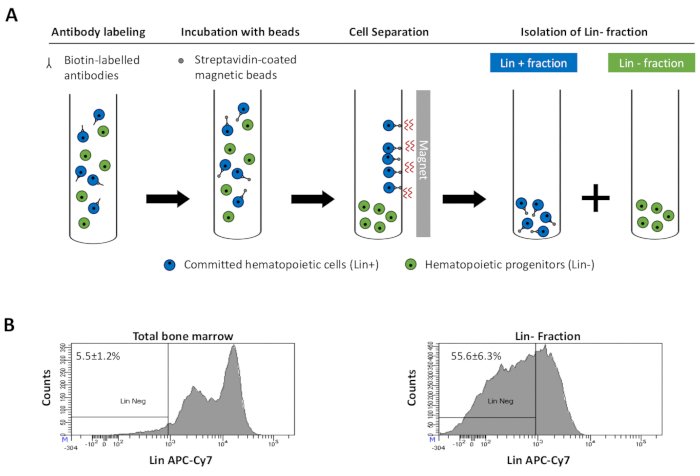
Figure 2: Magnetic depletion of lineage committed (Lin) cells. (A) Schematic representation of the magnetic depletion protocol. First, unsorted bone marrow cells are labeled with the biotin-conjugated rat anti-mouse antibody cocktail. Cells are then incubated with anti-rat Ig coated magnetic beads and subsequently subjected to the magnetic depletion using a strong magnet. The magnet will retain the labeled magnetic Lin+ fraction against the tube walls, while the unlabeled non-magnetic Lin- negative fraction will be collected in a new tube. (B) Lineage committed cells can be identified using fluorescent conjugated streptavidin. Typical analysis of the lineage expression in cells prior to magnetic depletion (total bone marrow) and after magnetic depletion (Lin- Fraction) N = 21. Please click here to view a larger version of this figure.
{kind=link}
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten