
Method Article
Rekonstitution membrangebundener minimaler Aktinkortices auf gestützten Lipiddoppelschichten
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt die Bildung von unterstützten Lipiddoppelschichten und die Zugabe von Zytoskelettfilamenten und Motorproteinen, um die Dynamik rekonstituierter, membrangebundener Zytoskelettnetzwerke mittels Fluoreszenzmikroskopie zu untersuchen.
Zusammenfassung
Die Oberfläche einer lebenden Zelle bietet eine vielseitige aktive Plattform für zahlreiche zelluläre Prozesse, die aus dem Zusammenspiel der Plasmamembran mit dem darunter liegenden Aktinkortex entstehen. In den letzten Jahrzehnten haben sich rekonstituierte, minimale Systeme, die auf unterstützten Lipiddoppelschichten in Kombination mit Aktinfilamentnetzwerken basieren, als sehr hilfreich erwiesen, um grundlegende Mechanismen und Konsequenzen membrangebundener Aktinnetzwerke zu entschlüsseln und die Funktionen einzelner membranassoziierter Proteine zu untersuchen. Hier beschreiben wir, wie solche aktiven Verbundsysteme in vitro rekonstruiert werden können, die aus flüssigkeitsgestützten Lipiddoppelschichten bestehen, die über membranassoziierte Aktin-bindende Proteine an dynamische Aktinfilamente und Myosinmotoren gekoppelt sind, die leicht über die Totalreflexionsfluoreszenzmikroskopie beobachtet werden können. Ein Open-Chamber-Design ermöglicht es, das System Schritt für Schritt zusammenzubauen und viele Parameter wie Linkerproteinkonzentration, Aktinkonzentration, Aktinfilamentlänge, Aktin/Myosin-Verhältnis sowie ATP-Spiegel systematisch zu steuern. Schließlich diskutieren wir, wie die Qualität des Systems kontrolliert werden kann, wie häufig auftretende Probleme erkannt und behoben werden können und einige Einschränkungen dieses Systems im Vergleich zur lebenden Zelloberfläche.
Einleitung
Die Plasmamembran einer lebenden tierischen Zelle interagiert ständig mit dem benachbarten Aktinzytoskelett, und zusammen bilden sie ein aktives Verbundmaterial, das eine Vielzahl zellulärer Funktionen erfüllt 1,2. Um Prozesse an dieser Lipidmembran-Aktin-Grenzfläche zu untersuchen, hat sich die Rekonstitution von Zytoskelettnetzwerken auf gestützten Lipiddoppelschichten (SLBs) als sehr hilfreich erwiesen. Dieser minimale Systemansatz ermöglicht die präzise Kontrolle der Komponenten des Zytoskelett-Netzwerks und der Lipidzusammensetzung. Im Vergleich zu den freistehenden Lipidmembranen riesiger unilamellärer Vesikel ermöglicht die planare Geometrie von SLBs den effizienten Einsatz modernster Mikroskopietechniken wie Super-Resolution3,4, Total Internal Reflection Fluorescence (TIRF)5,6,7 oder interferometrische Streuung 8 Untersuchung der räumlichen Organisation und Dynamik von Zytoskelettnetzwerken. TIRF bietet den höchsten Kontrast für fluoreszierend markierte Komponenten, da das Signal von ungebundenen markierten Molekülen in der Lösung, die zum Hintergrundsignal beitragen, minimal ist.
Hier beschreiben wir ein grundlegendes Protokoll für die Bildung von Actomyosin-Netzwerken, die an unterstützte Lipiddoppelschichten gebunden sind, die in der Praxis weit verbreitet sind, um die Physik aktiver, quasi-2D-Netzwerke 9,10,11 und ihre Wirkung auf die Membranorganisation 3,5,12,13,14,15,16 zu untersuchen (Abbildung 1 ). Dieser Ansatz ist nicht auf Aktin-basierte Netzwerke beschränkt, sondern kann auch leicht angepasst werden, um Mikrotubuli, Zwischenfilamente oder zusammengesetzte Netzwerke gemischter Natur zu erforschen und eine Vielzahl von Wechselwirkungen zwischen Lipidmembranproteinen und Zytoskelettkomponenten mit oberflächensensitiven Mikroskopiemethoden zu untersuchen.
Um dieses Protokoll fokussiert zu halten, haben wir eine detaillierte Beschreibung der Reinigung und Markierung von Aktin- und Myosinproteinen oder Details zur Abstimmung und Kontrolle der Kontraktilität und Organisation von Actomyosin-Netzwerken ausgeschlossen. Man sollte auf andere Protokolle verweisen, die neben diesem in der JoVE Methods Collection, In Vitro Reconstitution of Cytoskeleton Networks for Biomaterials, Biophysics and Active Matter Research17 veröffentlicht sind.

Abbildung 1: Schematische Darstellung des in vitro Aktin-Membran-aktiven Verbundsystems. Erstellt mit Biorender. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
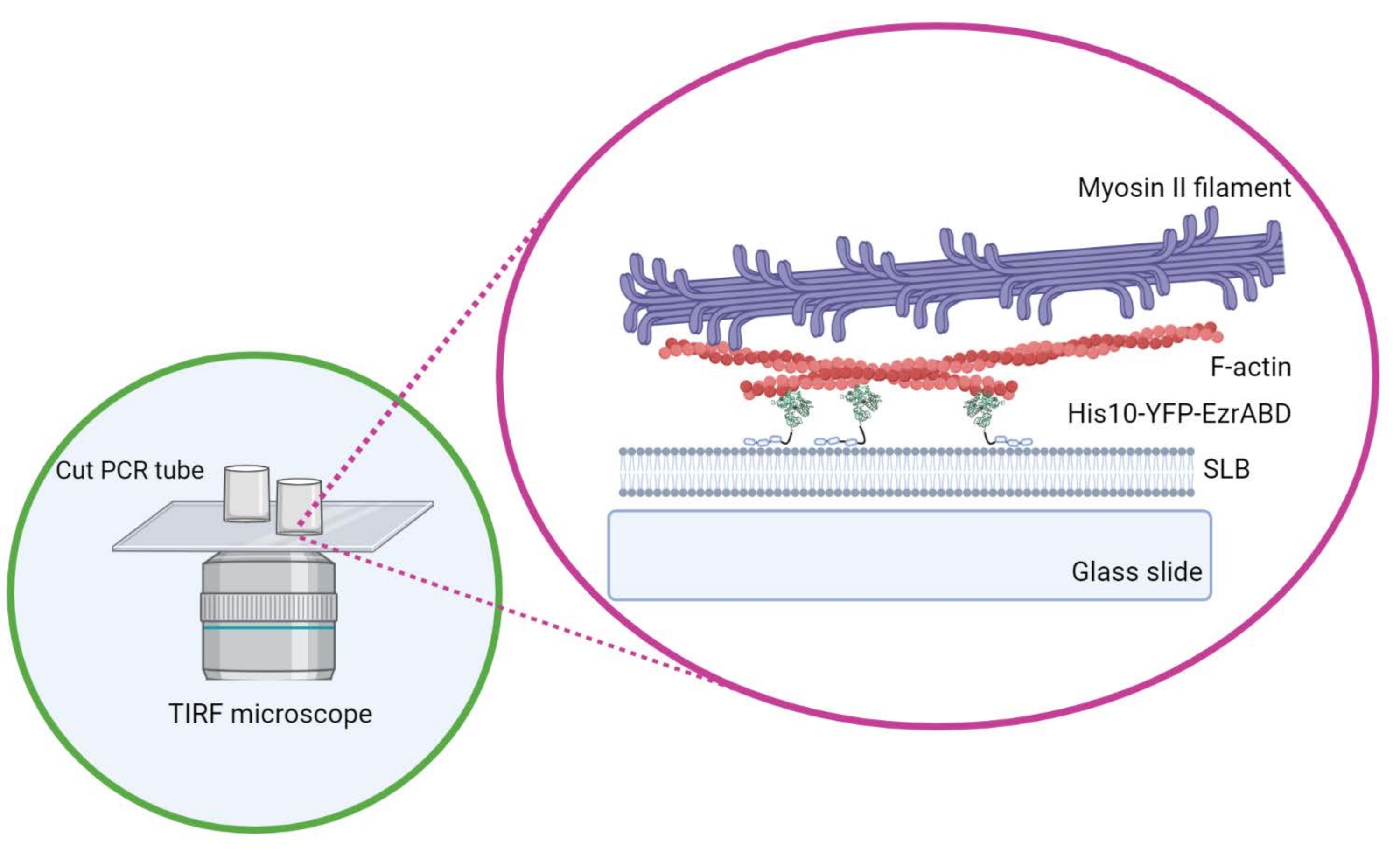{kind=link}
Protokoll
1. Reagenzien und Ausrüstung
- Bereiten Sie neue Puffer vor, wie in Tabelle 1 aufgeführt. Verwenden Sie hochreines, entionisiertes Wasser mit einem spezifischen Widerstand von 18,2 MΩ·cm bei 25 °C. Sterilisieren Sie alle Puffer, indem Sie sie unter Vakuum durch 0,22-μm-Filter leiten. Entgasen Sie die für die Säulenchromatographie verwendeten Puffer.
- Reinigen Sie das Aktin der Skelettmuskulatur wie zuvor beschrieben18,19. Fügen Sie 20% Glycerin zur endgültigen gereinigten G-Aktinlösung hinzu und machen Sie Aliquots von 500 μL (für Markierungs- oder Massenexperimente) und 10 μL (für Einzelexperimente) Volumen. Die Aliquots werden durch Eintauchen der Röhrchen in flüssigen Stickstoff für 30 s schockgefroren und anschließend bis zu 18 Monate bei −80 °C gelagert.
HINWEIS: Alternativ kann gereinigtes Aktin- oder Acetonpulver im Handel erworben werden. - Markieren Sie gereinigte Skelettmuskel-G-Aktin mit fluoreszierenden Maleimidfarbstoffen wie zuvor beschrieben5. Bestimmung der Konzentration und des Markierungsgrades des Proteins durch Spektrophotometrie unter Verwendung von korrigiertem A290nm für Aktin (εactin = 26.600 M-1 cm-1) und Aλmax des Farbstoffs. Aliquots von 10 μL herstellen und schockgefrieren, indem Sie die Röhrchen 30 s lang in flüssigen Stickstoff tauchen und bis zu 18 Monate bei −80 °C lagern.
HINWEIS: Die Markierung mit lysinkonjugierenden NHS-Estern erzeugt nicht-funktionelles Aktin und sollte vermieden werden. - Reinigen Sie die Skelettmuskulatur Myosin II, indem Sie das Protokoll20 befolgen. Führen Sie SDS-PAGE mit 10% Polyacrylamid-Gel gefolgt von Coomassie-Färbung aus, um den Reinheitsgrad des Proteins21 zu bestimmen. Lagern Sie gereinigte Skelettmuskulatur Myosin-II bei −20 °C in flüssiger Form in Myosin II Puffer mit 50% Glycerin.
HINWEIS: Das gespeicherte Myosin II kann bis zu 2 Jahre verwendet werden. - Markieren Sie gereinigtes Myosin-II mit fluoreszierenden Maleimidfarbstoffen wie zuvor beschrieben5. Vermeiden Sie die Kennzeichnung von Myosinmotoren mit NHS-Esterfarbstoffen. Die Konzentration und der Grad der Markierung werden durch Spektrophotometrie unter Verwendung von korrigiertem A280nm Myosin II und Aλmax des Farbstoffs bestimmt. Das recycelte Myosin II (dunkel oder etikettiert) bei 4 °C lagern und innerhalb von 6 Wochen aufbrauchen.
- Reinigung von Capping-Protein
- Erhalten Sie murines Capping-Protein, indem Sie einem früheren Protokoll22 folgen. Führen Sie SDS-PAGE mit 10% Polyacrylamid-Gel gefolgt von Coomassie-Färbung aus, um den Reinheitsgrad des Proteins zu bestimmen. Messen Sie die Konzentration mit A280nm Capping-Protein (εCP = 99.530 M-1 cm-1).
- Fügen Sie 20% Glycerin zur Proteinlösung hinzu und stellen Sie 5 μL Aliquots in 200 μL PCR-Röhrchen her. Tauchen Sie die Röhrchen in flüssigen Stickstoff und lagern Sie sie bis zu 2 Jahre bei −80 °C.
HINWEIS: Die Capping-Proteinaktivität wird durch Polymerisation fester Mengen an fluoreszierendem G-Aktin in Gegenwart unterschiedlicher Capping-Proteinmengen überprüft. Die Filamente werden dann unter einem Mikroskop abgebildet und ihre Längenverteilung quantifiziert. Je höher die relative Konzentration des Capping-Proteins ist, desto kürzer sind die Aktinfilamentverteilungen. Siehe Köster et al.5.
- Exprimieren Sie ein fluoreszierendes Membran-Aktin-Linkerprotein, z. B. verwenden Sie für dieses Protokoll 10xHis-YFP-EzrinABD (HYE), exprimieren Sie es in Bl21DE3* Escherichia Coli und reinigen Sie es wie zuvor beschrieben23. Bestimmen Sie die Konzentration des Proteins durch Spektrophotometrie.
- Lagern Sie das Protein in kleinen Aliquots in einem Gelfiltrationschromatographiepuffer (oder einem anderen geeigneten Puffer) mit 20% Glycerin bei −80 °C. Unter diesen Bedingungen ist das Protein über 2 Jahre stabil.
HINWEIS: Die Wahl des Aktin-Membran-Linkerproteins und des Fluoreszenzmarkers hängt von der Art der Frage ab, die man behandelt. In den letzten Jahren wurde eine breite Palette von Lipidbindungsstrategien entwickelt, darunter Histidin-markierte Proteine24, Biotin-Streptavidin 25 und einzelsträngige DNA26. - Herstellung von mehrschichtigen Vesikeln (MLVs)
HINWEIS: Der Workflow von MLVs zu unterstützten Lipiddoppelschichten ist in Abbildung 2 dargestellt.- 5-10 Durchstechflaschen aus gelbem Glas in ein 200-ml-Glasbecherglas geben. Füllen Sie das Becherglas mit 2% iger Reinigungslösung, gerade genug, um die Glasfläschchen unter Wasser zu tauchen. Beschallen Sie sie in einem Wasserbad für 30 min bei vollem Puls und 65 °C.
- Nehmen Sie die Durchstechflaschen aus der Lösung heraus und spülen Sie sie gründlich mit destilliertem Wasser ab. Die Durchstechflaschen in ein Glasbecherglas mit 2 N NaOH geben und 20 min lang beschallen. Während dieses Schritts ist keine Heizung erforderlich.
- Nehmen Sie die Durchstechflaschen aus der NaOH-Lösung heraus und spülen Sie sie gründlich mit destilliertem Wasser ab. Trocknen Sie die Durchstechflaschen in einem Heißluftofen bei 65 °C für 2 h oder länger.
- Bewahren Sie die gereinigten Durchstechflaschen bis zu 6 Wochen in einem sauberen Becherglas auf, das mit transparenter Folie verschlossen ist.
VORSICHT: Führen Sie die folgenden Schritte in einem chemischen Abzug durch. Behandeln Sie Chloroform und die Lipidlösungen mit gasdichten Hamilton-Glasspritzen, um eine Kontamination durch Kunststoff zu vermeiden. - Spülen Sie die Hamilton-Spritzen und einige braune Glasdurchstechflaschen mehrmals mit reinem Chloroform ab. Nehmen Sie das in Glasampullen gelagerte Lipidpulver aus dem Gefrierschrank -20 °C und fügen Sie ausreichende Mengen Chloroform hinzu, um die Lipidpulver auf Konzentrationen von 10-25 mg/ml aufzulösen.
- Die Lösung aus der Ampulle in eine frisch gereinigte Durchstechflasche aus Braunglas geben und entsprechend beschriften. Führen Sie diesen Schritt auf Eis durch, um die Verdunstung von Chloroform zu reduzieren.
- Stellen Sie eine DOPC-Stammlösung mit einer Konzentration von 10-25 mg/ml und DGS-NTA-Ni2+ mit einer Konzentration von 1-10 mg/ml her.
- Um eine funktionierende Lipidmischung herzustellen, nehmen Sie eine saubere Glasdurchstechflasche und spülen Sie sie 2x mit Chloroform ab. Geben Sie 300 μL reines Chloroform in die Durchstechflasche, um als Basis für eine bessere Mischung der Komponenten zu dienen. Dies hat keinen Einfluss auf die Endkonzentrationen der Lipide, da das gesamte Chloroform in den nächsten Schritten ausgetrocknet wird.
- Fügen Sie der Durchstechflasche gemessene Mengen an Stammlipidlösungen hinzu, um die gewünschten Arbeitslipidmischungen herzustellen. Die Ziellipidkonzentration im Lipidrehydratationspuffer beträgt 4 mM. Trocknen Sie das Lipidgemisch unter einem langsamen Strom vonN2-Gas in der chemischen Haube bei Raumtemperatur. Dieser Schritt kann bis zu 30 Minuten für jede Durchstechflasche dauern.
- Nachdem das gesamte Lösungsmittel ausgetrocknet ist, vakuum-austrocknen Sie den Lipidfilm für >2 h bei Raumtemperatur, um alle Spuren von Chloroform zu entfernen. Resuspendieren Sie die getrocknete Lipidmischung in einem Lipidrehydratationspuffer für eine endgültige Lipidkonzentration von 4 mM.
- Inkubieren Sie für 5-10 min, um eine Rehydratation der Lipide zu ermöglichen. Wirbeln Sie die Lipidlösung für etwa 30 s, um MLVs zu bilden.
- Machen Sie 0,5-1 ml Aliquots der MLVs in 1,5 mL Mikrozentrifugenröhrchen. Die Röhrchen in flüssigen Stickstoff tauchen, mit einer transparenten Folie verschließen und bei −20 °C lagern (bis zu 6 Wochen).
HINWEIS: Die Lipidkonzentrationen werden so gewählt, dass ausreichend große Volumina möglich sind, die ein zuverlässiges Pipettieren mit den Hamilton-Spritzen ermöglichen. Wenn die zur Herstellung der Brühe erforderlichen Volumina zu groß sind, um das getrocknete Lipidpulver aufzulösen, nehmen Sie mehrere Verdünnungen der Brühe vor, um eine reproduzierbare Mischung verschiedener Lipide zu gewährleisten.
- Herstellung von kleinen unilamellaren Vesikeln (SUVs)
- Nehmen Sie ein Aliquot MLVs aus dem Speicher von −20 °C heraus und tauen Sie es bei Raumtemperatur auf. Die Vesikel durch Eintauchen des Mikrozentrifugenröhrchens in flüssigen Stickstoff für 15-30 s schockieren und sofort in ein auf 45 °C eingestelltes Wasserbad geben, bis die Lösung vollständig aufgetaut ist (1-2 min). Wiederholen Sie den obigen Gefrier-Tau-Zyklus 10x-15x, bis die Lösung weniger trüb aussieht.
HINWEIS: Stellen Sie die Temperatur des Wasserbades höher ein als die Übergangstemperatur der aufzutauenden Lipidmischung, um eine gleichmäßige Lipidmischung zu ermöglichen. - Equilibrieren Sie einen spritzenbasierten Mini-Extruder, der mit einer 80-nm-Polycarbonat-Filtermembran mit SUV-Rehydratationspuffer ausgestattet ist. Stellen Sie sicher, dass das System keine Leckagen oder Blasen aufweist. Während das Extrusionsverfahren monodisperse SUVs mit minimaler Lipidschädigung ergibt, können Lipidmischungen mit negativer Ladung an der Polycarbonatmembran haften.
- Führen Sie die aufgetaute Lipidlösung vorsichtig durch den voräquilibrierten Extruder von einer Seite zur anderen und dann zurück. Wiederholen Sie den Zyklus 5x-10x, bis die Lipidlösung sichtbar klar wird, was auf die Bildung von SUVs mit ~100 nm Durchmesser hinweist.
- Zentrifugieren Sie die extrudierte Suspension (oder beschallen Sie die Lösung; siehe Hinweis unten) bei 15.000 x g für 60 min bei 4 °C, um die Lipidablagerungen abzuspritzen. Sammeln Sie die oberen 80% der Lösung, ohne das Pellet zu stören und ohne Blasen zu erzeugen. Der Überstand mit den SUVs wird in ein frisches Mikrozentrifugenröhrchen überführt und bis zu 6 Tage auf Eis gelagert.
HINWEIS: Eine Alternative zur Zentrifugation ist die Spitzenbeschallung, die wie folgt durchgeführt wird. Schalten Sie einen Mikrospitzen-Ultraschallgerät ein und stellen Sie die folgenden Einstellungen ein: Amplitude = 30% des Maximums, EINSCHALTZEIT = 10 s, AUS-Zeit = 60 s. Reinigen Sie die Spitze des Mikrobeschallers mit entionisiertem Wasser, gefolgt von 2 N NaOH, Chloroform und erneut entionisiertem Wasser. Tauchen Sie die Ultraschallspitze in jede dieser Lösungen und beschallen Sie sie für 1-2 Zyklen mit den obigen Einstellungen. Tauchen Sie die saubere Spitze in die gefrieraufgetaute Vesikellösung und beschallen Sie sie für 3-6 Zyklen auf Eis, bis die Lösung klar wird. - Überprüfen Sie nach der Zentrifugation auf Anzeichen eines hohen Lipidabbaus oder einer fehlgeschlagenen Lipidextrusion wie die Bildung eines dünnen weißlichen Films und/oder eines deutlich sichtbaren Pellets. Fahren Sie in diesen Fällen nicht fort und wiederholen Sie die SUV-Vorbereitungsschritte erneut.
HINWEIS: Die Haltbarkeit von SUVs kann für verschiedene Lipidgemische unterschiedlich sein. SUVs aus DOPC: DGS-NTA-Ni2+ sind für diese Experimente bis zu 6 Tage stabil. Tipps zur Behebung häufig auftretender Probleme finden Sie in Tabelle 2.
- Nehmen Sie ein Aliquot MLVs aus dem Speicher von −20 °C heraus und tauen Sie es bei Raumtemperatur auf. Die Vesikel durch Eintauchen des Mikrozentrifugenröhrchens in flüssigen Stickstoff für 15-30 s schockieren und sofort in ein auf 45 °C eingestelltes Wasserbad geben, bis die Lösung vollständig aufgetaut ist (1-2 min). Wiederholen Sie den obigen Gefrier-Tau-Zyklus 10x-15x, bis die Lösung weniger trüb aussieht.

Abbildung 2: Schematische Darstellung des Workflows von der Herstellung mehrschaliger Vesikel und kleiner unilamellärer Vesikel bis zur Bildung von unterstützten Lipiddoppelschichten. Erstellt mit Biorender. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
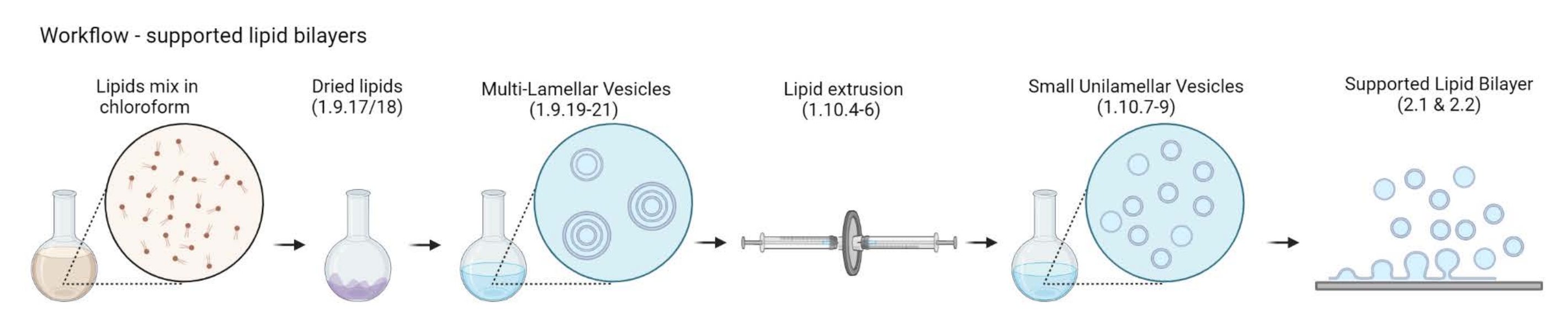{kind=link}
2. Rekonstitution membrangebundener Aktinnetzwerke
- Vorbereitung von Probenkammern
- Nehmen Sie 3-5 rechteckige Glasdeckgläser und legen Sie sie in ein Coplin-Glas. Schalten Sie den Badsonikator ein und stellen Sie die Temperatur auf 65 °C ein. Füllen Sie das Coplin-Glas mit 2% iger Reinigungslösung, um die Deckgläser vollständig einzutauchen, und legen Sie es für 30 Minuten im vollen Pulsmodus in den Sonicator.
- Verwenden Sie eine stumpfe PTFE-beschichtete Pinzette, um die Deckgläser nacheinander aus dem Glas zu nehmen. Spülen Sie sie gründlich mit destilliertem Wasser ab und stellen Sie sie in ein anderes Coplin-Glas, das mit 2 N NaOH gefüllt ist.
- Beschallen Sie die Deckgläser für 20 Minuten im vollen Pulsmodus. Entfernen Sie die Deckgläser nacheinander, spülen Sie sie gründlich mit destilliertem Wasser ab und stellen Sie sie in ein anderes Coplin-Glas, das mit destilliertem Wasser gefüllt ist.
HINWEIS: Optional die Deckgläser 20 min in destilliertem Wasser beschallen und dann erneut mit destilliertem Wasser abspülen. - Nehmen Sie unmittelbar vor Beginn des Versuchs das Glas mit den Deckgläsern in eine chemische Haube, die mit einerN2-Gaszufuhr ausgestattet ist.
- Optimieren Sie den Luftdruck desN2-Gasstroms durch Versuch und Irrtum, so dass er gerade ausreicht, um Wasser von der Deckglasoberfläche zu verdrängen, ohne sie zu brechen. Richten Sie den Gasstrom N2 parallel zur Deckglasebene aus, um die Möglichkeit eines Bruchs des Deckglases zu verringern.
- Verwenden Sie Handschuhe und Pinzetten, um die Deckgläser nacheinander aus dem Glas zu nehmen, um sie unter dem Strom N2 zu trocknen. Trocknen Sie beide Seiten jedes Deckglases und legen Sie sie auf ein sauberes Kunststoffgitter mit einem Deckel. Legen Sie die Box mit den Deckgläsern in einen Exsikkator, um den Kontakt mit Staubpartikeln in der Luft zu vermeiden.
HINWEIS: Die N 2-getrockneten Deckgläser können in einem Exsikkator gelagert werden, wo sie bis zu2 Tage hydrophil bleiben können. Diese Strategie kann nützlich sein, wenn viele Doppelschichten für das Experiment benötigt werden oder wenn das Experiment länger als 8 Stunden dauert. - Nehmen Sie autoklavierte PCR-Röhrchen und schneiden Sie ihre Lider und unteren konischen Hälften mit einer scharfen chirurgischen Klinge aus. Nehmen Sie die zylindrischen halbgeschnittenen Tuben nacheinander, tragen Sie UV-härtenden Klebstoff auf den glatten Rand jedes geschnittenen Röhrchens auf und legen Sie ihn umgekehrt auf ein frisch gereinigtes Deckglas, so dass der Rand flach auf dem Deckglas sitzt.
- Bewegen Sie den Zylinder nicht seitlich, sobald er auf dem Deckglas positioniert ist, um sicherzustellen, dass der Klebstoff nicht in den zentralen Raum der Kammer verschüttet wird. Rechteckige Deckgläser können bequem bis zu drei Reaktionskammern aufnehmen, und die runden können nur eine in der Mitte aufnehmen (Abbildung 1).
- Legen Sie die kammerführenden Deckgläser in einen UV-Ozonreiniger mitO2-Versorgung und Vakuum (oder verwenden Sie einen UV-Strahler). Schalten Sie das UV-Licht ein und leuchten Sie für 3-5 Minuten, damit der Klebstoff polymerisieren kann. Führen Sie eine längere Beleuchtung durch (10-15 min), um die Hydrophilie des Deckglases und damit die Qualität der Lipiddoppelschicht zu verbessern.
- Lagern Sie die trockenen, UV-beleuchteten Probenkammern bis zu 8 h in kleinen Kunststoffboxen (z. B. leere rechteckige Deckglasboxen), die in transparente Folie eingewickelt sind, um den Kontakt mit Staubpartikeln in der Luft zu reduzieren.
HINWEIS: Ein stetiger Strom vonO2 in Gegenwart von UV-Licht bildet Ozon- und Sauerstoffradikale, die organische Verunreinigungen von der Oberfläche des Deckglases entfernen können. Ein Vakuum verhindert das Austreten von giftigem Ozon, das während des Prozesses gebildet wird. - Nehmen Sie die Deckgläser heraus und testen Sie die Kammern auf Leckagen, indem Sie sie mit destilliertem Wasser auffüllen. Jede Kammer kann bis zu ~150 μL Probe aufnehmen. Entsorgen Sie die undichten Kammern.
HINWEIS: Eine weitere großartige und sichere Reinigungsoption ist der Plasmareiniger. Die Zeit- und Leistungseinstellungen hängen vom Modell ab, aber achten Sie darauf, die Glasobjektträger nicht mit Plasma zu überbehandeln, da dies zu einer Verringerung der Lipidbeweglichkeit führt. Die Oberflächenbehandlung kann die Beweglichkeit der Lipide27 beeinträchtigen, wie bei längerer Behandlung mit der Reinigungslösung (>45 min) oder NaOH (>30 min) beobachtet wurde.
- Herstellung von unterstützten Lipiddoppelschichten
- Waschen Sie jede Kammer mit SLB-Formationspuffer (oder 1x PBS), um Oberflächenverunreinigungen zu entfernen, wobei am Ende 100 μL Puffer übrig bleiben. Markieren Sie den Füllstand des Puffers bei 100 μL mit einem permanenten Marker, um Änderungen im Volumen reproduzierbar zu verfolgen.
- 2 μL 0,1 MCaCl2 in die Kammer geben. Dies verbessert die Adsorption der Vesikel an die Glasoberfläche und verstärkt im nächsten Schritt die Doppelschichtbildung. 8 μL der SUV-Lösung (ab Schritt 1.10) in jede Kammer geben und 15 min bei 25 °C inkubieren.
HINWEIS: Das Volumen der hinzuzufügenden SUV-Mischung kann geschätzt werden, indem die Gesamtzahl der Lipide (mit einer durchschnittlichen Fläche von 0,72 nm2) berechnet wird, die benötigt werden, um den exponierten hydrophilen Bereich des Bohrlochs vollständig mit zwei Lipidschichten abzudecken. - Die ungebundenen Vesikel mit Aktinmotilitätspuffer (1x KMEH) abwaschen. Entfernen Sie zunächst 50 μL des SLB-Formationspuffers, so dass nur 50 μL in der Probenkammer verbleiben. Zweitens fügen Sie 100 μL 1x KMEH in die Kammer hinzu. Mischen Sie vorsichtig und entfernen Sie dann 100 μL des Puffers, ohne den Boden zu berühren.
HINWEIS: Es ist wichtig, beim Waschen sanft zu sein. Stellen Sie sicher, dass die Pipettenspitze den Boden der Kammer nicht berührt. Halten Sie die Pipette geneigt, um den Pufferfluss an die Wand der Kammer und nicht direkt an die Doppelschicht zu lenken, da ein direkter Fluss die Doppelschicht stören kann. Achten Sie darauf, beim Pipettieren keine Luftblasen einzuführen, da Luft die Lipiddoppelschicht erreichen und Defekte verursachen kann. - Wiederholen Sie die Waschgänge 10x, indem Sie 100 μL 1x KMEH hinzufügen und 100 μL entfernen.
- 10 μL 1 mg/ml β-Casein in die Doppelschicht geben, vorsichtig mischen und 5-10 min inkubieren. β-Casein blockiert die Bereiche auf dem Deckglas, an denen sich die Doppelschicht nicht gebildet hat. Waschen Sie β-Casein 3x mit 1x KMEH ab, wie in Schritt 2.2.3 beschrieben. und bringen Sie den Puffer wieder auf die 100-μL-Marke.
- Zugabe von Membran-Aktin-Linker
- Nehmen Sie während der β-Casein-Inkubation (Schritt 2.2.5.) ein Aliquot Membran-Aktin-Linkerprotein aus −80 °C, tauen Sie es schnell bei 37 °C auf und halten Sie es dann auf Eis. Das Aliquot wird mit einem Proteinverdünnungspuffer auf eine Konzentration von 1 μM verdünnt.
- Fügen Sie das Linkerprotein in einer definierten Endkonzentration (typischerweise 5-20 nM) hinzu und mischen Sie vorsichtig. Um ein schnelles Gleichgewicht des Proteins in der Kammer zu gewährleisten, fügen Sie Volumina hinzu, die größer als 20 μL sind, indem Sie das Linkerprotein mit 1x KMEH vormischen.
- 40 min bei Raumtemperatur inkubieren. 3x mit 1x KMEH-Puffer waschen, um das ungebundene HSE-Protein zu entfernen (wie in Schritt 2.2.3.). Bringen Sie den Pufferstand in jeder Kammer wieder auf die 100-μL-Marke. Die Probe ist nun bereit für die Bildgebung.
- Qualitätsbeurteilung der Lipiddoppelschicht
HINWEIS: Dies ist ein optionaler Schritt, der nicht jedes Mal ausgeführt werden muss. Wir empfehlen, diese Bewertung jedes Mal durchzuführen, wenn frische SUVs aus gefrorenen MLV-Beständen hergestellt werden.- Schalten Sie das Mikroskop, die Anregungslaser und die Detektionskameras ein. Stellen Sie sicher, dass der Laser ausgerichtet ist, das Objektiv gereinigt ist und die Software bereit ist, Bilder aufzunehmen.
- Öl auf das 100-fache Objektiv geben, die Probe auf den Mikroskoptisch montieren und das Objektiv auf die Doppelschicht fokussieren. Stellen Sie sicher, dass die Laserposition so ist, dass sie eine vollständige interne Reflexion auf der Probe erfährt. Verwenden Sie einen 488 nm Anregungslaser, um die Fluoreszenzintensitätsverteilung des doppelschichtgebundenen 10xHis-YFP-EzrinABD zu überprüfen.
HINWEIS: Doppelschichten von guter Qualität zeigen eine großräumige, gleichmäßige Verteilung der Fluoreszenzintensität. Schlechte Doppelschichten zeigen intensive und fleckige fluoreszierende Flecken. - Um die Integrität der Doppelschicht zu bestimmen, führen Sie einen FRAP-Test durch.
- Wählen Sie einen Bereich von Interesse auf der Doppelschicht und zeichnen Sie einige Bilder des Sichtfelds unter Verwendung von Bildbedingungen auf, die ein Signal-Rausch-Verhältnis von 5:1 oder höher bieten. Pausieren Sie die Aufnahme und schließen Sie die Feldmembran des TIRF-Mikroskops, um einen konzentrierten Laserstrahl auf einen kleinen kreisförmigen Bereich der Doppelschicht zu fokussieren, um die Fluorophore lokal zu bleichen.
- Schalten Sie den Laser auf seine maximale Leistung ein, um den kleinen Bereich für 3-10 s zu photobleichen, und schalten Sie dann den Laser aus. Öffnen Sie die Feldblende wieder auf ihren ursprünglichen Radius, stellen Sie den Bildgebungszustand wieder auf die Einstellungen (vor dem Bleichmittel) ein und setzen Sie sofort die Aufzeichnung der Wiederherstellung des Fluoreszenzsignals im Sichtfeld fort.
- Überprüfen Sie, ob die Doppelschicht flüssig ist. Gute Doppelschichten mit normaler lateraler Diffusion erholen sich schnell, während sich schlechte Doppelschichten langsam oder gar nicht erholen (Abbildung 3). Wenn der Bilayer nicht wiederhergestellt wird, überprüfen Sie den Abschnitt zur Fehlerbehebung und starten Sie ihn neu. Speichern Sie die Bilder als 16-Bit-TIFF-Dateien. Für eine quantitative Schätzung des Diffusionskoeffizienten ist Schritt 3 zu prüfen. unter.
- Polymerisation von fluoreszierendem Aktin
HINWEIS: Um Zeit zu sparen, beginnen Sie mit der Polymerisation von Aktin während der Inkubationszeit des HSE-Proteins, das an die Doppelschicht bindet (Schritt 2.3.) oder während der Qualitätsbewertung der Doppelschicht (Schritt 2.4.).- Mischen Sie unmarkiertes und fluoreszierend markiertes G-Aktin in einem Molverhältnis von 10:1 und füllen Sie es mit G-Buffer auf, so dass die Konzentration von G-Aktin 20 μM beträgt. Die Konzentration, bei der Aktin schließlich polymerisiert wird, beträgt 1/4 dieses Wertes. 1/10 von 10x ME Puffer zu der Mischung für eine 1x Lösung geben und 2 min inkubieren. In diesem Schritt werden die an G-Aktin gebundenenCa2 +-Ionen durchMg2+ -Ionen ersetzt. Stellen Sie sicher, dass das Endvolumen ein Vielfaches von 10 μL aufweist.
- Fügen Sie die gewünschte Menge an Capping-Protein wie folgt hinzu. Eine Durchstechflasche mit verschließender Proteinbrühe schnell bei 37 °C auftauen und dann auf Eis halten. Mit G-Puffer so verdünnen, dass die Konzentration des Capping-Proteins nun doppelt so hoch ist wie die gewünschte Endkonzentration in der Polymerisationsmischung. Geben Sie der Aktinmischung aus Schritt 2.5.1 ein gleiches Volumen der verdünnten Verschließproteinlösung hinzu.
- Zum Schluss fügen Sie der Reaktionsmischung ein gleiches Volumen an frischem 2x-Zielpuffer hinzu. Das Endvolumen der Lösung sollte das Vierfache des Volumens der Aktinmischung am Ende von Schritt 2.5.2 betragen. Stellen Sie sicher, dass die Endkonzentration von KMEH 1x, von ATP 1 mM, von BSA 1 mg / ml und von G-Aktin 5 μM beträgt.
Im Dunkeln bei 25 °C für 45-60 min inkubieren, damit die Polymerisation stattfinden kann.
HINWEIS: Dies wird als Zielpufferstrategie bezeichnet, bei der ein Volumen Mg 2+ G-Aktin (Schritt 2.5.1.) mit einem Volumen Capping-Proteinmix (Schritt 2.5.2.) und zwei Volumen2x Zielpuffer (Schritt 2.5.3.) gemischt wird. Dies erleichtert es, die Menge an Aktin nach oben oder unten zu skalieren und die relative Konzentration des Capping-Proteins (oder eines anderen Aktinmodulators; Abbildung 4).
- Zugabe von fluoreszierenden Aktinfilamenten
- Schneiden Sie einige 200-μL-Spitzen mit einer scharfen Klinge oder einer Schere ab, um sie stumpf zu machen. Das erforderliche Volumen an 5 μM polymerisiertem Aktin (ab Schritt 2.5.3.) wird vorsichtig mit einer stumpfen Pipettenspitze (um ein Scheren von Aktinfilamenten zu verhindern) pipettiert und in ein sauberes autoklaviertes PCR-Röhrchen gegeben.
- Geben Sie 1x KMEH in das Röhrchen, um das Volumen >20 μL zu machen, und mischen Sie vorsichtig, um ein Scheren von F-Aktin zu vermeiden. Entfernen Sie aus der montierten Probenkammer ein gleiches Volumen des Puffers.
- Geben Sie die polymerisierte Aktinlösung in die Kammer und pipettieren Sie vorsichtig 3x auf und ab, ohne die Doppelschicht am Boden zu berühren. Dadurch können sich Aktinfilamente gleichmäßig auf der Doppelschicht verteilen. Montieren Sie die Probe auf dem TIRF-Mikroskop (siehe Schritt 2.4.1. und Schritt 2.4.2.).
- Man kann den Prozess der F-Aktin-Bindung an die Doppelschicht aufzeichnen. Inkubieren für 20-30 min. Nehmen Sie einige Bilder aus verschiedenen Sichtfeldern auf, nachdem F-Actin zusätzlich einen stabilen Zustand erreicht hat. Beobachten Sie Veränderungen in der räumlichen Organisation von 10xHis-YFP-EzrinABD vor (homogen) und nach Aktinorganisation.
HINWEIS: HYE wird in Abwesenheit von Aktin gleichmäßig über die Lipiddoppelschicht verteilt. Bei Zugabe von Aktinfilamenten kolokalisiert HYE mit F-Aktin. Das Ausmaß der Kolokalisation hängt von der Aktinbindungsaffinität des Linkerproteins ab; Je stärker die Affinität, desto höher die Kolokalisation und desto langsamer die laterale Beweglichkeit des Linkerproteins (Abbildung 5).
- Zugabe von Myosin II
- Nach 30 Minuten Aktininkubation montieren Sie die Probe wieder auf das Mikroskop (wenn es nicht montiert wurde). Überprüfen Sie das Signal in den Linkerprotein- und F-Aktin-Kanälen. Passen Sie die Bildgebungsbedingungen bei Bedarf an.
- Wählen Sie eine gute Region mit gleichmäßigem Linkerproteinsignal und gleichmäßig gestreuten Aktinfilamenten und ohne Artefakte für eine lange Zeitrafferaufnahme. Nehmen Sie 10-15 Bilder bei 0,1-0,2 Hz vor Myosin auf und pausieren Sie die Aufnahme. Pipettieren Sie das erforderliche Volumen an recyceltem Muskelmyosin-II aus der Durchstechflasche mit einer stumpfen Pipettenspitze (um das Scheren von Myosinfilamenten zu verhindern) und fügen Sie es einem sauberen autoklavierten PCR-Röhrchen hinzu.
- Sofort 1x KMEH in die Tube geben, um das Volumen >20 μL zu machen und vorsichtig zu mischen. Man kann auch ATP, ATP-Regenerationsmischung, Photostabilisierungsmittel usw. hinzufügen. während dieses Schritts. Entfernen Sie vorsichtig ein gleiches Volumen des Puffers aus der montierten Probenkammer, ohne sie zu stören.
- Geben Sie die Myosinlösung vorsichtig in die Probenkammer. Pipettieren Sie nicht nach oben und unten, da dies die oberflächengebundenen Filamente stört. Nehmen Sie sofort die Zeitrafferaufnahme wieder auf und beobachten Sie, wie sich das System vom Prä-Myosin-Zustand zu ATP-befeuerten kontraktilen Acto-Myosin-Flüssen und der Asternbildung zu einem ATP-erschöpften gestauten Zustand entwickelt (siehe repräsentative Ergebnisse).
- Nehmen Sie Hintergrundbilder für alle Kanäle mit einem reinen Puffersample auf. Speichern Sie alle Bilder als 16-Bit-.tiff-Dateien. In Tabelle 2 finden Sie Tipps zum Beheben häufiger Probleme.

Abbildung 3: Qualitätsbeurteilung der Doppelschichten mit schnellem FRAP-Assay. Unterstützte Lipiddoppelschichten (SLBs), die aus DOPC- und Ni-NTA-Lipiden (98:2 mol%) hergestellt werden, sind mit HYE (10xHis-YFP-markierter Membran-Aktin-Linker) beschichtet. Nachdem das ungebundene Protein ausgewaschen wurde, wird die fluoreszierende Doppelschicht unter einem TIRF-Mikroskop abgebildet. Ein kleiner Bereich auf der Doppelschicht wird mit hoher Laserleistung photogebleicht und die Erholung der Fluoreszenz aufgezeichnet. (A) Eine gute Doppelschicht erholt sich immer schnell, mit einem erwarteten Diffusionskoeffizienten von 1-1,5 μm2/s für die in diesem Fall verwendete Lipidzusammensetzung. (B) Schlechte Doppelschichten erholen sich sehr langsam oder erholen sich überhaupt nicht. (C) Repräsentative Bilder von schlechten Doppelschichten: (C-i) eine Doppelschicht mit Löchern, (C-ii) eine Doppelschicht mit großen, unbeweglichen Lipidflecken und (C-iii) eine Doppelschicht mit kleinen, unbeweglichen Punkten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 4: Schematische Darstellung der Polymerisation von Aktin mit der Zielpuffermethode. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
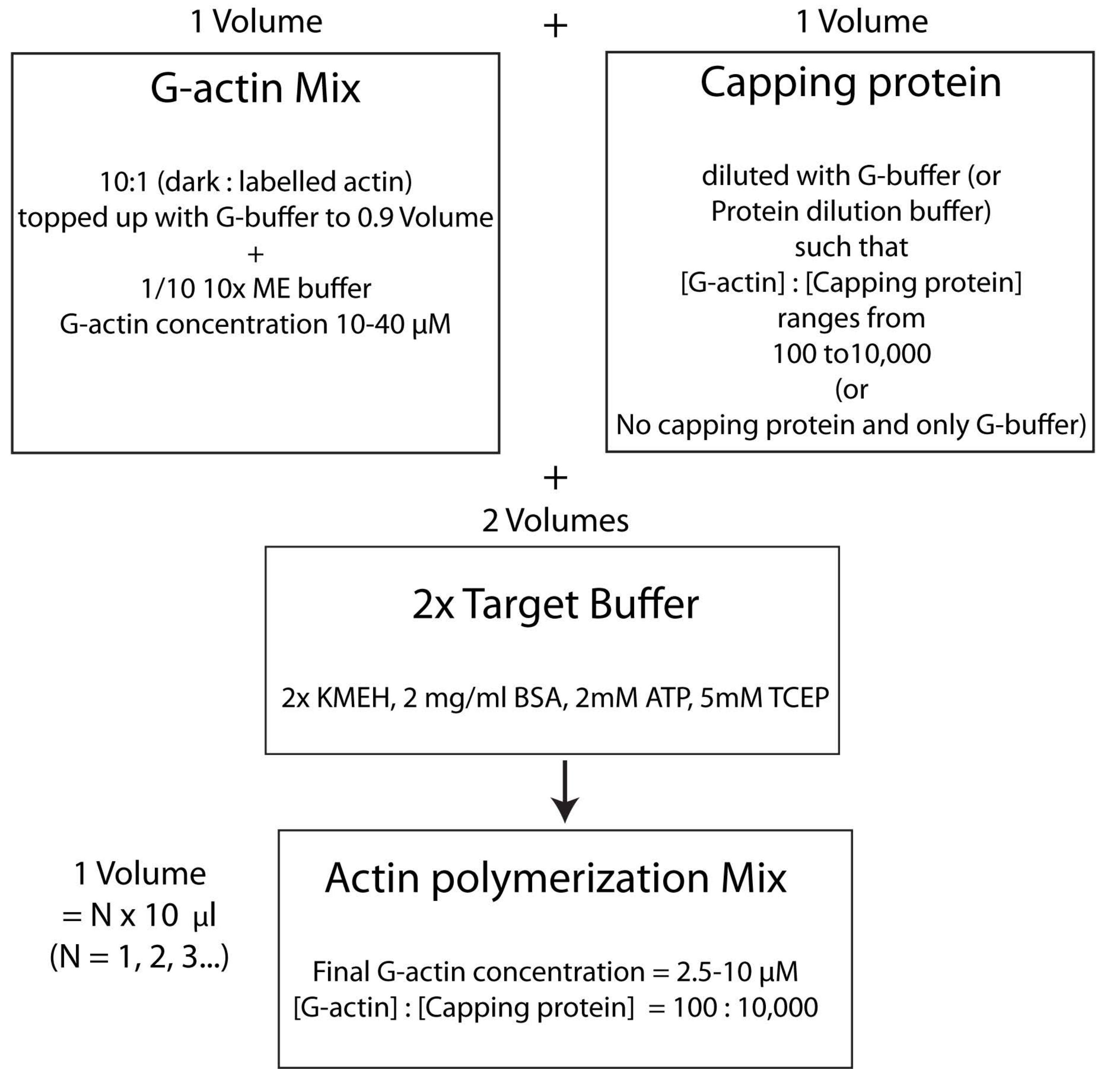{kind=link}

Abbildung 5: Räumliche Organisation von HYE bei Bindung an F-Aktin. TIRF-Schnappschüsse, die die räumliche Organisation von HYE vor und nach der Zugabe von Aktinfilamenten zeigen (markiert mit Atto-635-Malemid). Die HYE-Organisation ist vor der Zugabe von F-Aktin homogen und wird entlang von Aktinfilamenten kolokalisiert und coaligniert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
3. Datenanalyse
- Subtrahieren Sie mit der Fidschi-Software (https://imagej.net) den Hintergrund von den Linkerproteinbildern (ab Schritt 2.4.). Messen Sie die mittleren Intensitätswerte aus dem gebleichten Punkt und einem Referenzbereich.
- Normalisieren Sie die Zeitspuren vom gebleichten Fleck und dem Referenzbereich auf die Intensität ihrer jeweiligen Intensitätswerte vor dem Bleichmittel. Teilen Sie jeden Zeitpunkt in den normalisierten gebleichten Bereichswerten durch die entsprechenden Zeitpunkte in der normalisierten Zeitablaufverfolgung des Referenzbereichs. Korrigieren Sie die resultierende normalisierte Zeitspur für den Hintergrund und für systematische Intensitätsschwankungen während der Aufnahme (globales Photobleichen, Z-Drift usw.).
- Verwenden Sie eine anpassungsfreie, manuelle Methode28 , um den Diffusionskoeffizienten der zweischichtigen gebundenen Proteine zu schätzen. Kurz gesagt, die Halbzeit des Wiederherstellungsprofils, τ1/2, kann berechnet werden, indem man den Zeitpunkt betrachtet, zu dem das normalisierte Erholungsprofil die Hälfte seines stationären Zustands erreicht:

Hier ist F 0 die mittlere Intensität im gebleichten Bereich im ersten Bild nach dem Photobleaching, und F∞ ist der langfristige stationäre Wert der Erholung der Doppelschicht. - Schätzen Sie den effektiven Bleichradius, re, ein Parameter, der die Diffusion während des Photobleichens korrigiert, aus einem Linienscan des Postbleichfleckprofils29. Die halbe Breite mindestens halber Breite dieses Zeilenscans, der durch die Mitte des Bleichflecks verläuft, r 1/2, bezieht sich auf re wie folgt:

Das in Schritt 2.8.3 berechnete τ1/2, das in Schritt 2.8.4 berechnete r e und der ursprünglich eingestellte Bleichradius rn werden zur Berechnung des Diffusionskoeffizienten (D) nach folgender Formel verwendet:
- Verwenden Sie eine anpassungsfreie, manuelle Methode28 , um den Diffusionskoeffizienten der zweischichtigen gebundenen Proteine zu schätzen. Kurz gesagt, die Halbzeit des Wiederherstellungsprofils, τ1/2, kann berechnet werden, indem man den Zeitpunkt betrachtet, zu dem das normalisierte Erholungsprofil die Hälfte seines stationären Zustands erreicht:
- Bildanalyse von Actomyosin-Astern
- Subtrahieren Sie mit Fidschi den Hintergrund von allen aufgezeichneten Bildern in allen Kanälen. Korrigieren Sie die Bilder mithilfe der Flat-Field-Korrektur auf ungleichmäßige Beleuchtungs- oder Interferenzmuster.
HINWEIS: Man kann farbige Plastikobjektträger verwenden, die gute flache Proben sind, um solche Korrekturen durchzuführen. Für das Linkerprotein und die Aktinfilamente auf einer planaren Doppelschicht kann man auch die durchschnittliche Projektion mehrerer Prä-Myosin-Bilder verwenden, um kanalspezifische Beleuchtungskorrekturkarten zu erstellen.- Für den hier gezeigten HYE-Kanal nehmen Sie eine durchschnittliche Intensitätsprojektion mehrerer HYE-Bilder (aufgenommen aus verschiedenen Regionen der Lipiddoppelschicht vor Myosin zusätzlich). Wenden Sie einen geeigneten Gaußfilter (σ = 50 Pixel bis 80 Pixel) auf die durchschnittliche Projektion an (aus Prä-Myosin-Bildern oder einer beliebigen flachen Standardprobe).
- Konvertieren Sie das gefilterte Bild in ein 32-Bit-Bild. Teilen Sie alle Pixelwerte durch den Mittelwert des gesamten Bildes. Dadurch erhalten Sie eine normalisierte Korrekturkarte für den HYE-Kanal. Teilen Sie alle Bilder im HYE-Kanal mit dieser Karte für die Korrektur des flachen Feldes. Erstellen Sie Korrekturkarten für andere Kanäle mit der gleichen Strategie.
- Korrekt für Photobleiche mit einer exponentiellen oder einfachen Verhältnismethode (abhängig vom Intensitätszerfallsprofil) in Fidschi.
- Um eine zeitliche x-y-Fehlausrichtung (translatorische Bewegung) zu korrigieren, führen Sie alle photobleichkorrigierten Kanäle zu einem einzigen Hyperstack zusammen. Wenden Sie mit dem Hyperstack-Reg-Plugin in Fidschi eine Rigid Body oder Translation Transformation an.
- Schließlich teilen Sie den ausgerichteten Hyperstack in einzelne Kanäle auf und speichern Sie sie separat als 16-Bit-TIFF-Stacks zur weiteren Analyse.
- Subtrahieren Sie mit Fidschi den Hintergrund von allen aufgezeichneten Bildern in allen Kanälen. Korrigieren Sie die Bilder mithilfe der Flat-Field-Korrektur auf ungleichmäßige Beleuchtungs- oder Interferenzmuster.
Ergebnisse
Zur Darstellung wird hier ein typisches Postbleichprofil aus dem 1. Bild nach dem Photobleaching (Bild bei t = 0 s in Abbildung 3A) und dessen Anpassung an folgende Funktion28 (siehe Abbildung 6A) gezeigt:

Der Wert von r e (23,94 μm), der durch die Anpassung an diese Kurve berechnet wird, ist dem in Schritt 2.8.4 berechneten Wert von r e sehr ähnlich. (23,24 μm). Hier ist K ein Bleichtiefenparameter, der direkt von F0 geschätzt werden kann (beschrieben in Schritt 2.8.4.). In ähnlicher Weise zeigt Abbildung 6B das Wiederherstellungsprofil und seine Anpassung an die folgende Funktion28:

Wir finden, dass der angepasste Wert des Diffusionskoeffizienten 1,34 μm 2/s beträgt, ein Wert, der eng mit dem Wert von 1,39 μm 2/s übereinstimmt, der nach der Formel in Schritt2.8.4 berechnet wird. Hier steht MF für den mobilen Anteil der Lipiddoppelschicht, der den Anteil der gebleichten Population darstellt, der sich erholt. Die Mobilität lipidverankerter Moleküle hängt natürlich von der Lipidzusammensetzung und ihrem physikalischen Zustand (Flüssig- oder Gelphase) ab. Für unsere Experimente mit DOPC-basierten Lipidmembranen sollte die Mobilität >1 μm2/s betragen, und die mobile Fraktion sollte nicht kleiner als 0,9 sein, um eine gute Lipiddoppelschicht anzuzeigen. Wir empfehlen die Verwendung der manuellen anpassungsfreien Methode für einen schnellen Test der Qualität und Beweglichkeit der Doppelschicht. Die Anpassungsmethode kann bei der Automatisierung der Analyse für viele FRAP-Kurven nützlich sein. Wenn man ein anspruchsvolleres FRAP-Experiment durchführen möchte, um die Diffusion im System systematisch zu charakterisieren, empfehlen wir dem Leser diesen Review von Lorén et al.30, um mehr Details zu Anpassungsmodellen und möglichen Fallstricken im Versuchsdesign zu erhalten.

Abbildung 6: Quantifizierung des Diffusionskoeffizienten von Lipiddoppelschichten. (A) Linienprofil des ersten Bildes nach dem Photobleichen (t = 0 s in Abbildung 3A) und seine Anpassung an Gleichung 4 zur Berechnung des effektiven Bleichradius. (B) Das Wiederfindungsprofil des gebleichten Bereichs und seine Anpassung an Gleichung 5 zur Berechnung des Diffusionskoeffizienten und des mobilen Anteils. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Ein typisches Ergebnis der oben beschriebenen Experimente, das den dynamischen Aufbau und die Organisation eines Acto-Myosin-Netzwerks zeigt, das auf einer durch TIRF-Mikroskopie abgebildeten unterstützten Lipiddoppelschicht verknüpft ist, ist in Abbildung 7 und Supplementary Video S1 dargestellt.
Abbildung 7 zeigt eine Bildmontage des Linkerproteins F-Aktin und Myosin-II.

Abbildung 7: Kontraktile Actomyosinflüsse treiben die lokale Clusterbildung des Membran-Aktin-Linkerproteins HYE an. TIRF-Schnappschüsse von HYE (YFP-markiert), Aktinfilamenten (markiert mit Atto-635-Malemid) und Myosin-II-Filamenten (markiert mit Atto-565-Malemid) nach Zugabe von Myosin II zu einem SLB, das HYE und F-Aktin enthält. Die Zeit ist oben angegeben: 0 min ist unmittelbar bevor fluoreszierende Myofilamente im TIRF-Feld erscheinen. HYE und F-Aktin werden vor der Myosin-Addition homogen über die Lipiddoppelschicht verteilt (0 min). Die Myosinaktivität induziert kontraktile Actomyosin-Ströme, die im stationären Zustand (15 min) zu asterartigen Strukturen austreten und die lokale Clusterbildung der gekoppelten Membrankomponente (HYE) vorantreiben. Die unterste Zeile ist eine Verschmelzung von Aktin- (gelb) und Myosin-II-Bildern (Magenta), die die Organisation von Aktin und Myosin zu verschiedenen Zeitpunkten zeigen. Die Bilder, die bei der Erstellung dieser Montagen verwendet wurden, wurden in Fidschi für Hintergrundsignale, ungleichmäßige Intensitätsmuster und translatorische Bewegungen korrigiert. Maßstabsbalken = 10 μm. Weitere Informationen finden Sie unter Ergänzendes Video S1. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
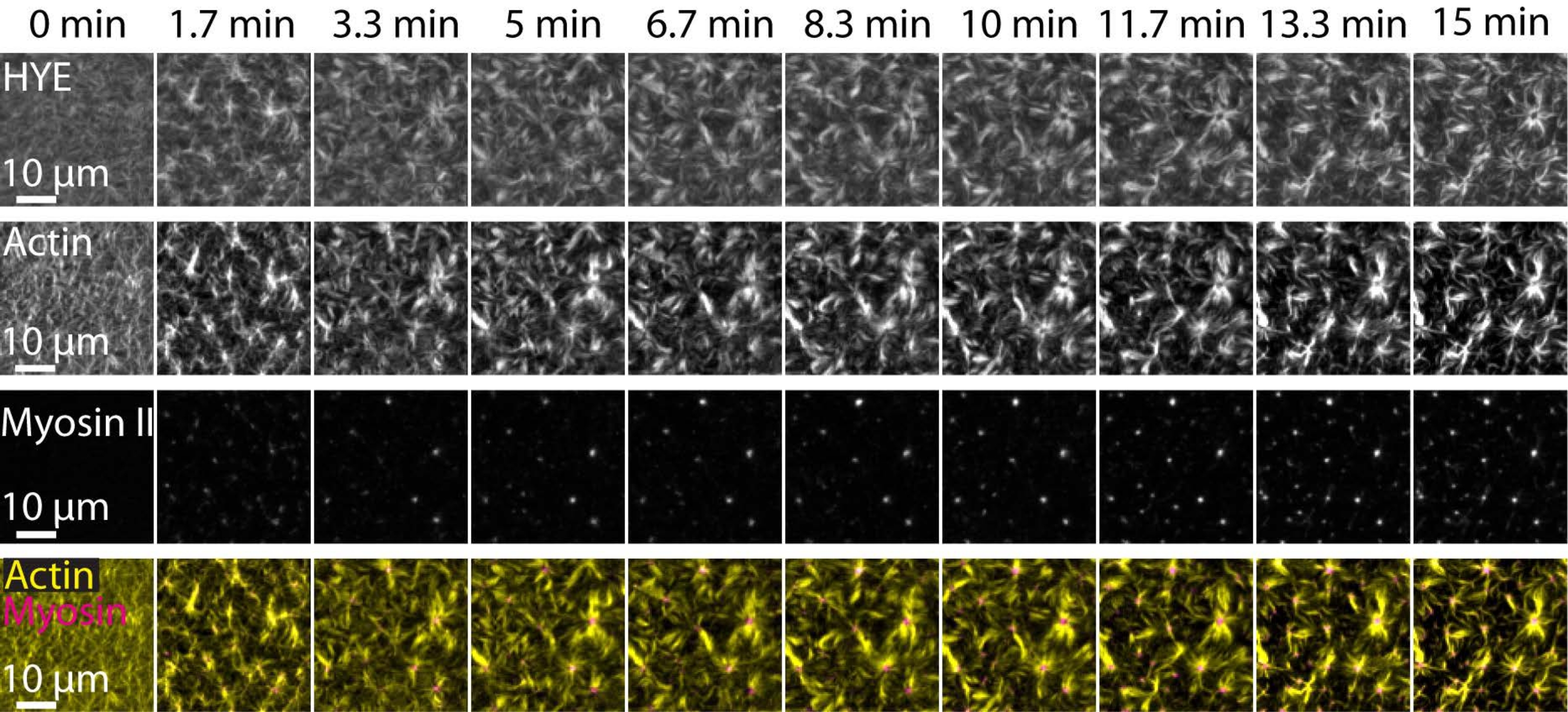{kind=link}
| Name des Puffers | Zusammensetzung | |
| Lipid-Rehydratationspuffer | 50 mM HEPES, 150 mM NaCl, 5% Saccharose, pH 7,5 | |
| SLB-Formationspuffer | 50 mM HEPES, 150 mM NaCl, pH 5-6 | |
| SLB-Speicherpuffer | 50 mM HEPES, 150 mM NaCl, pH 7,2 | |
| Protein-Verdünnungspuffer | 20 mM HEPES, 100 mM KCl, 1mM TCEP oder DTT, pH 7,2 | |
| 1X ME- oder Actin-Ionenaustauschpuffer | 50 mM MgCl2, 0,2 mM EGTA, 10 mM HEPES, pH 7,2 (bei 4°C lagern) | |
| 1X KMEH oder Aktin Polymerisationspuffer | 50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 50 mM HEPES, pH 7,2 | |
| 100 mM ATP-Vorrat | 100 mM ATP Dinatriumsalz, 50 mM Tris, 50 mM NaCl, 5 mM MgCl2, 2 mM EGTA, pH 7,5 (lagern bei -20°C) | |
| 2x Zielpuffer | 2x KMEH, 2 mg/ml BSA, 2mM ATP, 5mM TCEP (gelagert bei 4°C) | |
| G-Puffer | 2 mM Tris, 0,1 mM CaCl2, 0,2 mM ATP, 0,5 mM TCEP, 0,04 % NaN3, pH 8 (bei 4°C lagern) | |
| Myosin II Puffer | 500 mM KCl, 1 mM EDTA, 10-20 mM Hepes, pH 7,0 | |
| Gelfiltrations-Chromatographie-Puffer | 50 mM Tris-HCl, 150-300 mM NaCl, 5 mM TCEP, 0,1% Tween-20, pH 7,5 | |
| Capping Protein Speicherpuffer | 10 mM Tris· Cl, 50 mM NaCl, 1 mM TCEP, pH 7,5, 20% Glycerin | |
Tabelle 1: Liste der in diesem Protokoll verwendeten Pufferzusammensetzungen.
| Häufige Probleme und deren Fehlerbehebung | Problem | Verursachen | Mögliche Lösungen | ||||||
| 1 | Lipiddoppelschicht zeigt keine Diffusion | Die wahrscheinlichste Ursache für dieses Problem ist verschmutztes Deckglas, das auftreten kann, wenn die Reinigungslösung gealtert ist oder die Heizung während der Badbeschallung nicht stattgefunden hat. Diese Doppelschichten haben ein "vesikuläres" Aussehen, da die geplatzten Vesikel am Deckglas haften, aber nicht miteinander verschmelzen. Die Verwendung von MLVs, die älter als 6 Wochen oder SUVs älter als 6 Tage sind, oder die Zugabe geringer Mengen an SUVs kann ebenfalls zur Bildung vesikulärer Doppelschichten führen. | Verwenden Sie eine frische Reinigungslösung. Stellen Sie sicher, dass die Heizung eingeschaltet ist und die Temperatur zwischen 45-65 ° C liegt. Verwenden Sie frische Lipidmischungen. (Die Verwendung einer fluoreszierenden Lipidsonde im Vergleich zu einer fluoreszierenden Proteinsonde kann sich manchmal anders manifestieren. Wenn z.B. die Doppelschicht Subbeugungsdefekte aufweist und der Oberflächenpassivierungsschritt übersprungen wird (oder nicht funktioniert), zeigt die Lipidsonde eine gleichmäßige Intensitätsverteilung, aber die fluoreszierende Proteinsonde kann helle fluoreszierende Flecken aufweisen.) | ||||||
| 2 | Lipiddoppelschicht hat helle Flecken | Eine lange Inkubation von SUVs für die Doppelschichtbildung kann eine Lipiddoppelschicht erzeugen, die insgesamt diffundiert, aber gelegentlich helle Flecken aufweist. Diese Patches können mehrschichtige Doppelschichten sein, die große Mengen fluoreszierender Sonden anziehen können. | 15-20 min Inkubation mit SUVs ist ausreichend. Stellen Sie sicher, dass die Sonde nicht aggregiert: Ein schneller harter Spin des Linkerproteins (300 x g für 15 min bei 4 °C) kann die Aggregate entfernen | ||||||
| 3 | Lipiddoppelschicht hat dunkle Löcher | Dies geschieht, wenn die Doppelschicht aus alten SUVs hergestellt und für längere Stunden (> 4 Stunden nach der Bildung) abgebildet wird oder sich der pH-Wert der Lösung aufgrund längerer Bildgebung drastisch ändert (z. B. im hohen ATP-Zustand und in Gegenwart bestimmter Sauerstofffänger) oder wenn die Oberfläche mit Beta-Casein überpassiviert ist (Zugabe von zu viel Beta-Casein für mehr als 10-15 min und oder nicht auswaschen). | Verwenden Sie frische Lipide. Reduzieren Sie die Bildrate oder die effektive Laserbeleuchtungszeit. Verwenden Sie Puffer mit höherer Pufferkapazität. | ||||||
| 4 | Lipiddoppelschicht zeigt langsame Diffusion | Lipiddoppelschichten mit hohem Cholesterinanteil, lange gesättigte Lipide oder geladene Lipide diffundieren langsamer. | In solchen Fällen bereiten Sie Ihre Probe bei hoher Temperatur vor. Man kann auch eine einfache, getestete Lipidzusammensetzung als Kontrolle zusammen mit komplexen und ungetesteten Lipidzusammensetzungen verwenden. Stellen Sie sicher, dass das Glas sauber ist. | ||||||
| 5 | Aktin polymerisiert nicht | Zielpuffer ist alt, G-Aktin-Bestand ist zu alt, altes und neues G-Aktin wurden co-polymerisiert. | Stellen Sie sicher, dass das Ca 2+ vor der Polymerisation (mit ME-Puffer) durch Mg2+ ersetzt wird. Verwenden Sie frische ATP-Mg2+ Brühe. Verwenden Sie frisch recyceltes G-Actin. Stellen Sie sicher, dass die Konzentration von F-Aktin (in Bezug auf G-Aktin), die der Doppelschicht zugesetzt wird, höher als 0,2 μM ist. Für niedrigere Konzentrationen verwenden Sie Phalloidin-stabilisiertes F-Aktin. | ||||||
| 6 | Aktin bindet nicht an die Doppelschicht | Membran-Aktin-Linker wird nicht oder in sehr geringer Konzentration hinzugefügt - dies kann aus der Fluoreszenz des Linkerproteins abgeleitet werden. Wenn die Fluoreszenz anständig ist, hat der Membran-Aktin-Linker die Aktinbindungskapazität verloren. Auch wenn das Linkerprotein unspezifisch an die Glasoberfläche gebunden ist (wenn die Doppelschicht schlecht ist), rekrutiert es möglicherweise keine Aktinfilamente. | Stellen Sie sicher, dass die Doppelschicht diffundiert. Verwenden Sie frisches Linkerprotein | ||||||
| 7 | Fluoreszierendes F-Aktin-Signal ist schwach | Das Verhältnis von markiertem zu dunklem Aktin ist zu gering. Entweder ist das markierte Aktin oder das unmarkierte Aktin zu alt und sie copolymerisieren nicht miteinander. | Recyceln Sie Aktin erneut und versuchen Sie die Beschäftigung mit frisch recyceltem Aktin. Lichtschäden können F-Aktin zerstören oder depolymerisieren; Wenn möglich, verwenden Sie rote oder tiefrote Farbstoffe für Aktin (und Myosin). | ||||||
| 8 | Myosin zeigt keine Kontraktilität | Es kann beobachtet werden, dass nach Zugabe von ATP zum Myosin-infundierten System keine Kontraktilität des Acto-Myosins vorliegt. | Überprüfen Sie, ob die Myosinkonzentration oder der Reinheitsgrad gut ist. Verwenden Sie frisch recyceltes Myosin (innerhalb von 6 Wochen nach dem Recycling verwenden). Die Zugabe von frischem ATP zum Myosin-Mix kann helfen. Entgasungspuffer und Verwendung von Sauerstofffängern usw. kann Lichtschäden der Motoren reduzieren. Weitere Informationen finden sich in den Protokollen von Plastino et al. oder Stam et al. derselben Methodensammlung | ||||||
| 9 | Deckglas ist nicht hydrophil | Deckglas wird nicht richtig gereinigt. | Sauberes hydrophilllisches Deckglas ist entscheidend für die Bildung von Lipiddoppelschichten. Eine nützliche, visuelle Anzeige der Hydrophilie des Deckglases nach dem Reinigungsprotokoll ist die Beobachtung der Benetzung des Glases durch Wasser. Fügen Sie eine kleine Menge Wasser zu einem flach liegenden Deckglas hinzu. Das Wasser bleibt in Form eines runden Tröpfchens, wenn das Deckglas nicht richtig gereinigt wird. Das gleiche Wasservolumen breitet sich jedoch aus und bildet eine dünne Schicht auf einem behandelten hydrophilen Deckglas. Anhand dieses Benetzungsverhaltens des Wassers auf der Deckglasoberfläche kann festgestellt werden, ob die Reinigungsschritte mit der Reinigungslösung/NaOH funktioniert haben. | ||||||
Tabelle 2: Anleitung zur Fehlerbehebung mit einer Zusammenfassung häufiger Probleme und entsprechender Lösungen.
Ergänzendes Video S1: Kontraktile Actomyosinflüsse treiben lokales Clustering des Membran-Aktin-Linkerproteins HYE voran. TIRF-Zeitraffer von HYE (YFP-markiert), Aktinfilamenten (markiert mit Atto-635-Maleimid) und Myosin-II-Filamenten (markiert mit Atto-565-Malemid) nach Zugabe von Myosin II zu einem SLB, das HYE und F-Aktin enthält. Die Zeit ist oben angegeben: 0 min ist unmittelbar bevor fluoreszierende Myofilamente im TIRF-Feld erscheinen. Maßstabsbalken = 10 μm. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
Dieses Protokoll bietet eine vielseitige Plattform und einen Ausgangspunkt für Designexperimente zur Untersuchung der Membran-Kortex-Schnittstelle von Zellen. Kritische Schritte sind die Herstellung sauberer Objektträger, die Verwendung frischer Lipide für eine effiziente SUV-Bildung (beides beeinflusst die Qualität von SLBs) und die Verwendung von frisch recycelten Myosin-II-Proteinen für die dynamische Reorganisation von Aktinfilamenten. Bei der Abbildung von Dynamiken über einen langen Zeitraum ist es sehr wichtig, ein Sauerstofffängersystem zu integrieren (z. B. Protocatechusäure und Protocatechuate 3 4-Dioxygenase 5,31).
Das Open-Chamber-Design ermöglicht die sequentielle Zugabe von Komponenten zu einem bestehenden System, ohne Lipidflüsse zu induzieren. Dies kann ein wichtiger Vorteil gegenüber häufig verwendeten, geschlossenen Kammeransätzen sein oder mit verkapselten Proteinen in Liposomenarbeiten 36. Konträre Effekte wie proteininduzierte Membranverformungen können mit glasadsorbierten Lipiddoppelschichten nicht untersucht werden.
Die Lipiddoppelschichten können mit einer Vielzahl von Lipidzusammensetzungen gebildet werden. Es beginnt mit der Adsorption der Lipidvesikel an die hydrophile Glasoberfläche, gefolgt von entweder spontaner Vesikelruptur aufgrund von Oberflächen-Vesikel und direkten Vesikel-Vesikel-Wechselwirkungen oder die adsorbierten Vesikel erreichen eine kritische Abdeckung, nach der ein kleiner Teil der Vesikel reißt und aktive Kanten bildet, was schließlich zur Doppelschichtbildung führt32 . Neben Glas können verschiedene Substrate verwendet werden, um gestützte Lipiddoppelschichten zu bilden, wie z.B. Glimmer (z.B. für die Rasterkraftmikroskopie), weiche Substrate (z.B. Poly-di-methyl-siloxan), Polymerpolster33,34,35, die sich zwischen Löchern von Elektronenmikroskopiegittern 14 spannen. Tröpfchengrenzflächendoppelschichten sind eine weitere interessante Methode zur Erzeugung stabiler, freistehender Lipiddoppelschichten36. Der Einschluss von Acto-Myosin-Netzwerken in Vesikel oder Emulsionen ist eine sehr leistungsfähige Methode, um dieses minimale System in einer zellähnlichen Geometrie37,38 zu untersuchen, und die an anderer Stelle39 ausführlich beschrieben wird.
Offenlegungen
Die Autoren haben keine Interessenkonflikte zu erklären.
Danksagungen
Diese Arbeit wurde vom AXA Forschungsfonds und dem Warwick-Wellcome Quantitative Biomedicine Programme (Wellcome ISSF, RMRCB0058) für DVK, NCBS-TIFR für AB und ST und dem Wellcome-DBT Margdarshi Fellowship (IA/M/15/1/502018) für SM unterstützt. DVK dankt auch der Biophysikalischen Gesellschaft für die Ermöglichung der virtuellen Netzwerkveranstaltung "Herausforderungen beim Verständnis von Mehrkomponenten-Zytoskelettnetzwerken von der molekularen bis zur mesoskala", was zur Erstellung dieser Protokollsammlung beigetragen hat.
Materialien
| Name | Company | Catalog Number | Comments |
| 1,2 dipalmitoyl-sn-glycero-3-phosphoethanolamine-N- (lissamine rhodamine B sulfonyl) | Avanti Polar Lipids | 810158 | 16:0 RhoPE |
| 1,2-dioleoyl-sn-glycero-3- [(N-(5-amino-1 carboxypentyl) iminodiacetic acid) succinyl] (nickel salt) | Avanti Polar Lipids | 790404 | DGS-NTA-Ni2+ |
| 1,2-dioleoyl-sn-glycero-3-phosphocholine | Avanti Polar Lipids | 850375 | DOPC |
| 1,2-dipalmitoyl-sn-glycero-3-phosphocholine | Avanti Polar Lipids | 850355 | DPPC |
| Amber glass vials | ThermoFisher | B7990-2A | |
| ATP disodium salt | Sigma Aldrich | A26209 | |
| Attofluor cell chamber | ThermoFisher | A7816 | |
| Bath sonicator | GT Sonic | 1860QTS | |
| beta-casein | Sigma Aldrich | C6905 | |
| CaCl2 | ThermoFisher | 12135 | |
| chloroform | Sigma Aldrich | 650471 | alternatively from Electron Microscopy Sciences, 50980296 |
| Cover slips, #1, 25 mm diameter, Gold Seal | Harvard Apparatus | 64-0705B | |
| Cover slips, #1, 40x22 mm, Gold Seal | ThermoFisher | 48404-031 | |
| EDTA | ThermoFisher | G12635 | |
| EGTA | Himedia | MB130 | |
| Gas tight glass syringes, with removebla needle, blunt, volumes 10 µL, 100 µL, 500 µL | Hammilton | 1700 series | |
| Hellmanex III | Hellma Analytics | Z805939 | cleaning solution |
| HEPES | Himedia | RM380 | |
| KaH2PO4 | ThermoFisher | G13405 | |
| KCl | ThermoFisher | G13305 | |
| KOH | ThermoFisher | G26708 | |
| Lipid extruder | Avanti Polar Lipids | 61000-1EA | |
| MgCl2 | ThermoFisher | G15535 | |
| Microtip sonicator | Sonics | VC750 | 3 mm Tip diameter |
| Na2CO3 | ThermoFisher | G15955 | |
| NaCl | Himedia | GRM853 | |
| NaH2PO4 | ThermoFisher | G15825 | |
| NaOH | ThermoFisher | G27815 | |
| Nikon Ti Eclipse TIRF microscope | Nikon | With a TIRF unit connected through a polarization-conserving optical fibre to an Agilent monolithic laser combiner MLC400 with multiple laser lines with a 100X, 1.45 NA Nikon Oil Objective with two 512 x 512-pixel EMCCD cameras (Photometrics Evolve 512) with a 100X, 1.45 NA Nikon Oil Objective with two 512 x 512-pixel EMCCD cameras (Photometrics Evolve 512) | |
| NOA88 | Norland Products | 8801 | |
| PTFE Coated Tweezer Style #2A | Structure Probe | 0S2AT-XD | |
| Refrigerated microcentrifuge | Eppendorf | 5424R | |
| Sucrose | ThermoFisher | G15925 | |
| UV-illuminator | Novascan | PSD PRO-UV | needs vacuum and O2 supply |
Referenzen
- Köster, D. V., Mayor, S. Cortical actin and the plasma membrane: Inextricably intertwined. Current Opinion in Cell Biology. 38, 81-89 (2016).
- Rao, M., Mayor, S. Active organization of membrane constituents in living cells. Current Opinion in Cell Biology. 29, 126-132 (2014).
- Honigmann, A., et al. A lipid bound actin meshwork organizes liquid phase separation in model membranes. eLife. 3, 01671(2014).
- Das, A., et al. Stratification relieves constraints from steric hindrance in the generation of compact actomyosin asters at the membrane cortex. Science Advances. 6 (11), (2020).
- Köster, D. V., et al. Actomyosin dynamics drive local membrane component organization in an in vitro active composite layer. Proceedings of the National Academy of Sciences. 113 (12), 1645-1654 (2016).
- Vogel, S. K., Heinemann, F., Chwastek, G., Schwille, P. The design of MACs (minimal actin cortices). Cytoskeleton. 70 (11), 706-717 (2013).
- Murrell, M., Thoresen, T., Gardel, M. Reconstitution of contractile actomyosin arrays. Methods in Enzymology. 540, 265-282 (2014).
- Mosby, L. S., et al. Myosin II filament dynamics in actin networks revealed with interferometric scattering microscopy. Biophysical Journal. 118 (8), 1946-1957 (2020).
- Seara, D. S., et al. Entropy production rate is maximized in non-contractile actomyosin. Nature Communications. 9 (1), 4948(2018).
- Linsmeier, I., et al. Disordered actomyosin networks are sufficient to produce cooperative and telescopic contractility. Nature Communications. 7, 12615(2016).
- Vogel, S. K., Petrasek, Z., Heinemann, F., Schwille, P. Myosin motors fragment and compact membrane-bound actin filaments. eLife. 2, 00116(2013).
- Ditlev, J. A., et al. A composition-dependent molecular clutch between T cell signaling condensates and actin. eLife. 8, 42695(2019).
- Banjade, S., Rosen, M. K. Phase transitions of multivalent proteins can promote clustering of membrane receptors. eLife. 3, 04123(2014).
- Heinemann, F., Vogel, S. K., Schwille, P. Lateral membrane diffusion modulated by a minimal actin cortex. Biophysical Journal. 104 (7), 1465-1475 (2013).
- Vogel, S. K., Greiss, F., Khmelinskaia, A., Schwille, P. Control of lipid domain organization by a biomimetic contractile actomyosin cortex. eLife. 6, 24350(2017).
- Block, S. Brownian motion at lipid membranes: A comparison of hydrodynamic models describing and experiments quantifying diffusion within lipid bilayers. Biomolecules. 8 (2), 30(2018).
- JoVE, JoVE. JoVE Methods Collection. In vitro reconstitution of cytoskeleton networks for biomaterials, biophysics and active matter research. Journal of Visualized Experiments. , Cambridge, MA. (2022).
- Pardee, J. D., Spudich, J. A. Purification of muscle actin. Methods in Enzymology. 85, 164-181 (1982).
- Spudich, J. A., Watt, S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. Journal of Biological Chemistry. 246 (15), 4866-4871 (1971).
- Pollard, T. D. Myosin purification and characterization. Methods in Cell Biology. 24, 331-371 (1982).
- JoVE Science Education Database. Separating protein with SDS-PAGE. Journal of Visualized Experiments. , Cambridge, MA. (2022).
- Bieling, P., et al. WH2 and proline-rich domains of WASP-family proteins collaborate to accelerate actin filament elongation. The EMBO Journal. 37 (1), 102-121 (2018).
- Shrivastava, R., Köster, D., Kalme, S., Mayor, S., Neerathilingam, M. Tailor-made ezrin actin binding domain to probe its interaction with actin in-vitro. PLoS One. 10 (4), 0123428(2015).
- Nye, J. A., Groves, J. T. Kinetic control of histidine-tagged protein surface density on supported lipid bilayers. Langmuir. 24 (8), 4145-4149 (2008).
- Honigmann, A., et al. Scanning STED-FCS reveals spatiotemporal heterogeneity of lipid interaction in the plasma membrane of living cells. Nature Communications. 5, 5412(2014).
- Selden, N. S., et al. Chemically programmed cell adhesion with membrane-anchored oligonucleotides. Journal of the American Chemical Society. 134 (2), 765-768 (2012).
- Seu, K. J., et al. Effect of surface treatment on diffusion and domain formation in supported lipid bilayers. Biophysical Journal. 92 (7), 2445-2450 (2007).
- Kang, M., Day, C. A., Kenworthy, A. K., DiBenedetto, E. Simplified equation to extract diffusion coefficients from confocal FRAP data. Traffic. 13 (12), 1589-1600 (2012).
- Kang, M., Day, C. A., Kenworthy, A. K. A novel computational framework for D(t) from Fluorescence Recovery after Photobleaching data reveals various anomalous diffusion types in live cell membranes. Traffic. 20 (11), 867-880 (2019).
- Lorén, N., et al. Fluorescence recovery after photobleaching in material and life sciences: Putting theory into practice. Quarterly Reviews of Biophysics. 48 (3), 323-387 (2015).
- Alvarado, J., Sheinman, M., Sharma, A., MacKintosh, F. C., Koenderink, G. H. Molecular motors robustly drive active gels to a critically connected state. Nature Physics. 9 (7), 1-7 (2013).
- Andrecka, J., Spillane, K. M., Ortega-Arroyo, J., Kukura, P. Direct observation and control of Supported lipid bilayer formation with interferometric scattering microscopy. ACS Nano. 7 (12), 10662-10670 (2013).
- Lin, W. -C., et al. Supported membrane formation, characterization, functionalization, and patterning for application in biological science and technology. Current Protocols in Chemical Biology. 2 (4), 235-269 (2010).
- Richter, R. P., Bérat, R., Brisson, A. R. Formation of solid-supported lipid bilayers: An integrated view. Langmuir. 22 (8), 3497-3505 (2006).
- Sapuri-Butti, A. R., Butti, R. C., Parikh, A. N. Characterization of supported membranes on topographically patterned polymeric elastomers and their applications to microcontact printing. Langmuir. 23 (25), 12645-12654 (2007).
- Leptihn, S., et al. Constructing droplet interface bilayers from the contact of aqueous droplets in oil. Nature Protocols. 8 (6), 1048-1057 (2013).
- Bashirzadeh, Y., Moghimianavval, H., Liu, A. P. Encapsulated actomyosin patterns drive cell-like membrane shape changes. iScience. 25 (5), 104236(2022).
- Bashirzadeh, Y., Wubshet, N., Litschel, T., Schwille, P., Liu, A. P. Rapid encapsulation of reconstituted cytoskeleton inside giant unilamellar vesicles. Journal of Visualized Experiments. (177), e63332(2021).
- Murrell, M., Chen, S., Sun, Z. G. In vitro reconstitution of actin cytoskeleton inside giant unilamellar vesicles. Journal of Visualized Experiments. , (2022).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten