
Method Article
Ricostituzione di cortecce di actina minima legate alla membrana su doppi strati lipidici supportati
In questo articolo
Riepilogo
Questo protocollo descrive la formazione di doppi strati lipidici supportati e l'aggiunta di filamenti citoscheletrici e proteine motorie per studiare la dinamica delle reti citoscheletriche ricostituite e legate alla membrana utilizzando la microscopia a fluorescenza.
Abstract
La superficie di una cellula vivente fornisce una piattaforma attiva versatile per numerosi processi cellulari, che derivano dall'interazione della membrana plasmatica con la corteccia di actina sottostante. Negli ultimi decenni, sistemi minimali ricostituiti basati su doppi strati lipidici supportati in combinazione con reti di filamenti di actina si sono dimostrati molto strumentali nel districare i meccanismi di base e le conseguenze delle reti di actina legate alla membrana, nonché nello studio delle funzioni delle singole proteine associate alla membrana. Qui, descriviamo come ricostituire tali sistemi compositi attivi in vitro che consistono in doppi strati lipidici supportati da fluidi accoppiati tramite proteine leganti l'actina associate alla membrana a filamenti di actina dinamici e motori di miosina che possono essere facilmente osservati tramite microscopia a fluorescenza a riflessione interna totale. Un design a camera aperta consente di assemblare il sistema in modo graduale e di controllare sistematicamente molti parametri come la concentrazione della proteina linker, la concentrazione di actina, la lunghezza del filamento di actina, il rapporto actina / miosina e i livelli di ATP. Infine, discutiamo come controllare la qualità del sistema, come rilevare e risolvere i problemi comuni e alcune limitazioni di questo sistema rispetto alla superficie della cellula vivente.
Introduzione
La membrana plasmatica di una cellula animale vivente interagisce costantemente con il citoscheletro di actina adiacente e insieme formano un materiale composito attivo che soddisfa una moltitudine di funzioni cellulari 1,2. Per studiare i processi a questa interfaccia membrana lipidica-actina, la ricostituzione delle reti citoscheletriche in cima ai doppi strati lipidici supportati (SLB) si è dimostrata molto utile. Questo approccio sistemico minimale consente il controllo preciso dei componenti della rete del citoscheletro e della composizione lipidica. Rispetto alle membrane lipidiche autoportanti delle vescicole unilamellari giganti, la geometria planare delle SLB consente un uso efficiente di tecniche di microscopia all'avanguardia come la super-risoluzione3,4, la fluorescenza a riflessione interna totale (TIRF)5,6,7 o lo scattering interferometrico 8 studiare l'organizzazione spaziale e la dinamica delle reti citoscheletriche. TIRF fornisce il più alto contrasto per i componenti marcati con fluorescenza, poiché il segnale delle molecole marcate non legate nella soluzione che contribuiscono al segnale di fondo è minimo.
Qui, descriviamo un protocollo di base per la formazione di reti di actomiosina legate a doppi strati lipidici supportati, che sono ampiamente utilizzati sul campo per studiare la fisica delle reti attive quasi-2D 9,10,11 e il loro effetto sull'organizzazione della membrana 3,5,12,13,14,15,16 (Figura 1 ). Questo approccio non è limitato alle reti basate sull'actina, ma può anche essere adattato facilmente per esplorare microtubuli, filamenti intermedi o reti composite di natura mista e per studiare una varietà di interazioni tra proteine della membrana lipidica e componenti citoscheletrici utilizzando metodi di microscopia sensibili alla superficie.
Per mantenere questo protocollo focalizzato, abbiamo escluso una descrizione dettagliata della purificazione e dell'etichettatura delle proteine di actina e miosina o dettagli su come sintonizzare e controllare la contrattilità e l'organizzazione delle reti di actomiosina. Si dovrebbe fare riferimento ad altri protocolli che sono pubblicati insieme a questo nella JoVE Methods Collection, In Vitro Reconstitution of Cytoskeleton Networks for Biomaterials, Biophysics and Active Matter Research17.

Figura 1: Schema del sistema composito attivo in vitro di actina-membrana. Creato con Biorender. Fare clic qui per visualizzare una versione ingrandita di questa figura.
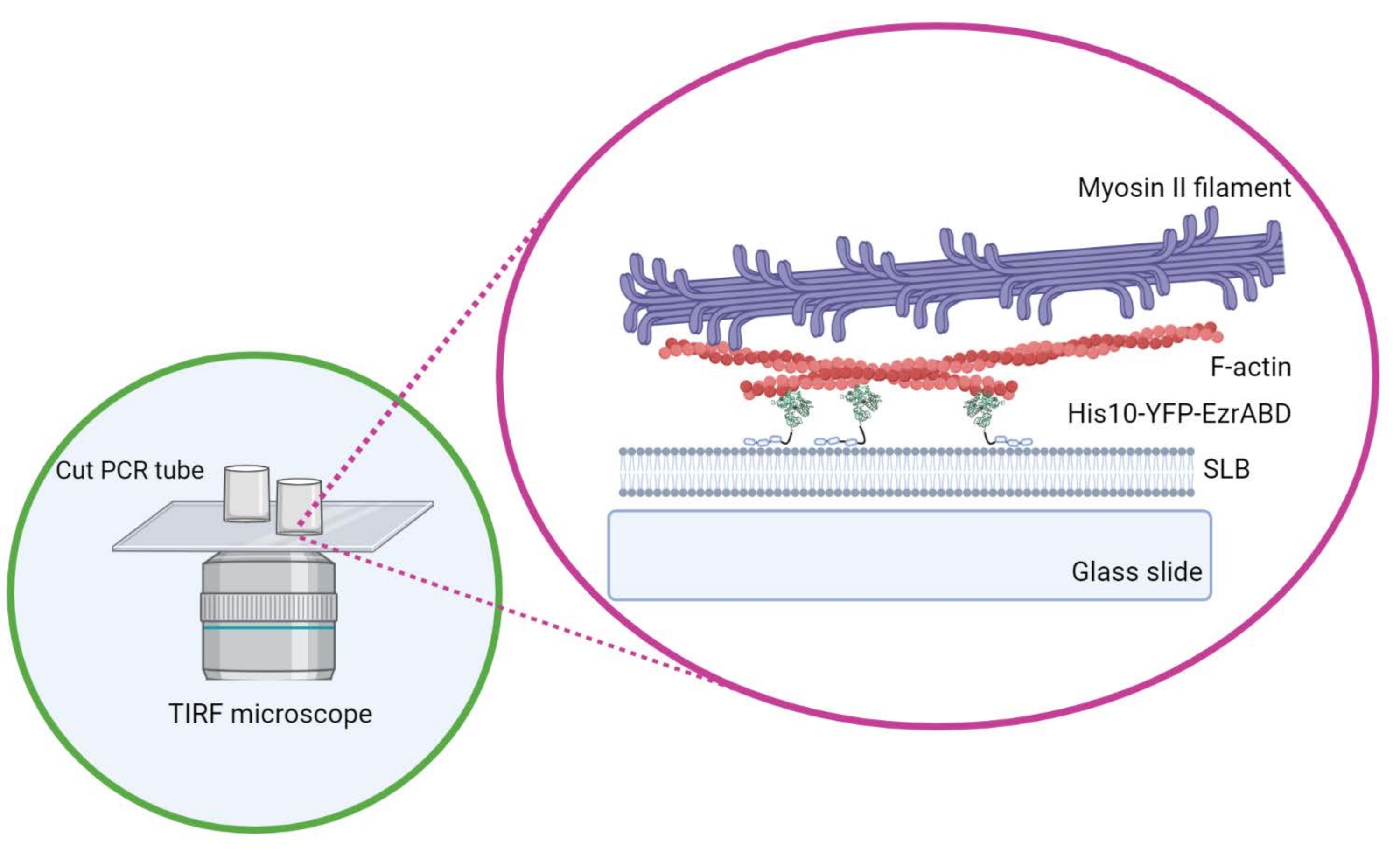{kind=link}
Protocollo
1. Reagenti e attrezzature
- Preparare nuovi buffer come indicato nella tabella 1. Utilizzare acqua ultrapura deionizzata con una resistività di 18,2 MΩ·cm a 25 °C. Sterilizzare tutti i tamponi facendoli passare attraverso filtri da 0,22 μm sotto vuoto. Degasare i buffer utilizzati per la cromatografia su colonna.
- Purificare l'actina del muscolo scheletrico come descritto in precedenza18,19. Aggiungere il 20% di glicerolo alla soluzione finale purificata di G-actina e fare aliquote di 500 μL (per l'etichettatura o esperimenti di massa) e 10 μL (per singoli esperimenti) volume. Flash congelare le aliquote immergendo i tubi in azoto liquido per 30 s e poi conservarli a -80 °C per un massimo di 18 mesi.
NOTA: In alternativa, l'actina purificata o la polvere di acetone possono essere acquistate in commercio. - Etichettare il muscolo scheletrico purificato G-actina con qualsiasi colorante maleimmide fluorescente come descritto in precedenza5. Determinare la concentrazione e il grado di marcatura della proteina mediante spettrofotometria utilizzando A290nm corretto per actina (εactina = 26.600 M-1 cm-1) e A λmax del colorante. Produrre aliquote da 10 μL e congelare immergendo i tubi in azoto liquido per 30 s e conservarli a -80 °C per un massimo di 18 mesi.
NOTA: L'etichettatura con esteri NHS coniuganti lisina creerà actina non funzionale e dovrebbe essere evitata. - Purificare la miosina muscolare scheletrica II seguendo il protocollo20. Eseguire SDS-PAGE utilizzando gel di poliacrilammide al 10% seguito da colorazione Coomassie per determinare il livello di purezza della proteina21. Conservare il muscolo scheletrico purificato miosina-II a -20 °C in forma liquida in tampone miosina II con glicerolo al 50%.
NOTA: La miosina II conservata può essere utilizzata per un massimo di 2 anni. - Etichettare la miosina-II purificata con qualsiasi colorante fluorescente maleimmide come descritto in precedenza5. Evitare di etichettare i motori della miosina con coloranti esteri del NHS. Determinare la concentrazione e il grado di marcatura mediante spettrofotometria utilizzando A280nm corretti di miosina II e Aλmax del colorante. Conservare la miosina II riciclata (scura o etichettata) a 4 °C e utilizzarla entro 6 settimane.
- Purificazione della proteina di tappatura
- Ottenere la proteina di copertura murina seguendo un protocollo precedente22. Eseguire SDS-PAGE utilizzando gel di poliacrilammide al 10% seguito da colorazione Coomassie per determinare il livello di purezza della proteina. Misurare la concentrazione utilizzando A280nm di proteina capping (εCP = 99.530 M-1 cm-1).
- Aggiungere il 20% di glicerolo alla soluzione proteica e fare 5 μL di aliquote in provette da 200 μL di PCR. Immergere i tubi in azoto liquido e conservarli a -80 °C per un massimo di 2 anni.
NOTA: L'attività della proteina capping viene controllata polimerizzando quantità fisse di G-actina fluorescente in presenza di diverse quantità di proteine capping. I filamenti vengono quindi ripresi al microscopio e la loro distribuzione della lunghezza viene quantificata. Maggiore è la concentrazione relativa della proteina di capping, più brevi sono le distribuzioni dei filamenti di actina. Vedi Köster et al.5.
- Esprimere una proteina fluorescente linker di membrana-actina, ad esempio, per questo protocollo utilizzare 10xHis-YFP-EzrinABD (HYE), esprimerla in Bl21DE3* Escherichia Coli, e purificare come descritto in precedenza23. Determinare la concentrazione della proteina mediante spettrofotometria.
- Conservare la proteina in piccole aliquote in tampone cromatografico a filtrazione su gel (o qualsiasi altro tampone appropriato) con glicerolo al 20% a -80 °C. In queste condizioni, la proteina è stabile per oltre 2 anni.
NOTA: La scelta della proteina linker della membrana di actina e del marcatore fluorescente dipende dal tipo di domanda che si sta affrontando. Negli ultimi anni è stata sviluppata una vasta gamma di strategie di collegamento lipidico, tra cui le proteine24 marcate con istidina, la streptavidina25 e il DNA26 a singolo filamento. - Preparazione di vescicole multilamellari (MLV)
NOTA: il flusso di lavoro dai MLV ai doppi strati lipidici supportati è illustrato nella Figura 2.- Posizionare 5-10 flaconcini di vetro ambrato in un becher di vetro da 200 ml. Riempire il becher con una soluzione detergente al 2%, quanto basta per immergere le fiale di vetro. Sonicarli a bagnomaria per 30 minuti a pieno ritmo e 65 °C.
- Estrarre i flaconcini dalla soluzione e sciacquarli abbondantemente con acqua distillata. Mettere i flaconcini in un becher di vetro contenente 2 N NaOH e sonicare per 20 minuti. Non è necessario alcun riscaldamento durante questa fase.
- Estrarre i flaconcini dalla soluzione di NaOH e risciacquare abbondantemente con acqua distillata. Asciugare i flaconcini all'interno di un forno ad aria calda impostato a 65 °C per 2 ore o più.
- Conservare i flaconcini puliti in un becher pulito sigillato con pellicola trasparente per un massimo di 6 settimane.
ATTENZIONE: Eseguire i seguenti passaggi all'interno di una cappa aspirante chimica. Maneggiare il cloroformio e le soluzioni lipidiche con siringhe di vetro Hamilton a tenuta di gas per evitare la contaminazione da plastica. - Risciacquare più volte le siringhe di Hamilton e alcuni flaconcini di vetro ambrato con cloroformio puro. Prelevare la polvere lipidica conservata in fiale di vetro dal congelatore a -20 °C e aggiungere volumi adeguati di cloroformio per sciogliere le polveri lipidiche a concentrazioni di 10-25 mg/ml.
- Trasferire la soluzione dalla fiala a un flaconcino di vetro ambrato appena pulito ed etichettarlo in modo appropriato. Eseguire questo passaggio sul ghiaccio per ridurre l'evaporazione del cloroformio.
- Preparare una soluzione madre DOPC con una concentrazione di 10-25 mg/ml e DGS-NTA-Ni2+ con una concentrazione di 1-10 mg/ml.
- Per fare una miscela lipidica funzionante, prendere una fiala di vetro pulita e sciacquarla 2x con cloroformio. Aggiungere 300 μL di cloroformio puro al flaconcino per servire come base per una migliore miscelazione dei componenti. Ciò non influirà sulle concentrazioni finali dei lipidi poiché tutto il cloroformio verrà essiccato nelle fasi successive.
- Aggiungere i volumi misurati di soluzioni lipidiche madre al flaconcino per ottenere le miscele lipidiche di lavoro desiderate. La concentrazione lipidica target nel tampone di reidratazione lipidica è di 4 mM. Asciugare la miscela lipidica sotto un flusso lento di gas N2 all'interno della cappa chimica a temperatura ambiente. Questo passaggio può richiedere fino a 30 minuti per ogni flaconcino.
- Dopo che tutto il solvente si è asciugato, essiccare sottovuoto il film lipidico per >2 ore a temperatura ambiente per rimuovere eventuali tracce di cloroformio. Risospendere la miscela lipidica essiccata nel tampone di reidratazione lipidica per una concentrazione lipidica finale di 4 mM.
- Incubare per 5-10 minuti per consentire la reidratazione dei lipidi. Vortice la soluzione lipidica per circa 30 s per formare MLV.
- Produrre 0,5-1 mL di aliquote dei MLV in provette da microcentrifuga da 1,5 ml. Immergere i tubi in azoto liquido, sigillare con una pellicola trasparente e conservare a -20 °C (per un massimo di 6 settimane).
NOTA: le concentrazioni di stock lipidici sono scelte per consentire volumi sufficientemente grandi che consentano un pipettaggio affidabile utilizzando le siringhe di Hamilton. Se i volumi necessari per produrre il brodo sono troppo grandi per sciogliere la polvere lipidica essiccata, effettuare più diluizioni del brodo per garantire una miscelazione riproducibile di vari lipidi.
- Preparazione di piccole vescicole unilamellari (SUV)
- Prelevare un'aliquota di MLV dal deposito a -20°C e scongelarla a temperatura ambiente. Flash congelare le vescicole immergendo il tubo della microcentrifuga in azoto liquido per 15-30 s e metterlo immediatamente in bagnomaria impostato a 45 °C fino a quando la soluzione non si è completamente scongelata (1-2 min). Ripetere il ciclo di congelamento-scongelamento sopra riportato 10x-15x fino a quando la soluzione appare meno torbida.
NOTA: Impostare la temperatura del bagno d'acqua più alta della temperatura di transizione della miscela lipidica da scongelare per consentire una miscelazione lipidica uniforme. - Equilibrare un mini estrusore a siringa dotato di una membrana filtrante in policarbonato da 80 nm con tampone di reidratazione SUV. Assicurarsi che non vi siano perdite o bolle nel sistema. Mentre il metodo di estrusione produce SUV monodispersi con danni lipidici minimi, le miscele lipidiche con carica negativa possono aderire alla membrana in policarbonato.
- Passare delicatamente la soluzione lipidica scongelata attraverso l'estrusore pre-equilibrato da un lato all'altro e poi indietro. Ripetere il ciclo 5x-10x fino a quando la soluzione lipidica diventa visibilmente chiara, indicando la formazione di SUV con ~ 100 nm di diametro.
- Centrifugare la sospensione estrusa (o sonicare la soluzione; vedere la nota sotto) a 15.000 x g per 60 minuti a 4 °C per pellettizzare i detriti lipidici. Raccogliere l'80% superiore della soluzione senza disturbare il pellet e senza creare bolle. Trasferire il surnatante contenente i SUV in un tubo di microcentrifuga fresco e conservare sul ghiaccio per un massimo di 6 giorni.
NOTA: Un'alternativa alla centrifugazione è la sonicazione della punta eseguita come segue. Accendere un sonicatore microtip e impostare le seguenti impostazioni: Ampiezza = 30% del massimo, Tempo di accensione = 10 s, Tempo di spegnimento = 60 s. Pulire la punta del microsonicatore con acqua deionizzata seguita da 2 N NaOH, cloroformio e di nuovo acqua deionizzata. Immergere la punta del sonicatore in ciascuna di queste soluzioni e sonicare per 1-2 cicli utilizzando le impostazioni di cui sopra. Immergere la punta pulita nella soluzione di vescicole congelata e sonicare per 3-6 cicli sul ghiaccio fino a quando la soluzione diventa limpida. - Dopo la centrifugazione, verificare la presenza di segni di elevata degradazione lipidica o di una mancata estrusione lipidica come la formazione di un sottile film biancastro e / o di un pellet chiaramente visibile. In questi casi, non procedere e ripetere nuovamente i passaggi di preparazione del SUV.
NOTA: La durata di conservazione dei SUV può differire per diverse miscele lipidiche. I SUV realizzati in DOPC: DGS-NTA-Ni2+ sono stabili fino a 6 giorni ai fini di questi esperimenti. Suggerimenti per risolvere i problemi comuni sono disponibili nella Tabella 2.
- Prelevare un'aliquota di MLV dal deposito a -20°C e scongelarla a temperatura ambiente. Flash congelare le vescicole immergendo il tubo della microcentrifuga in azoto liquido per 15-30 s e metterlo immediatamente in bagnomaria impostato a 45 °C fino a quando la soluzione non si è completamente scongelata (1-2 min). Ripetere il ciclo di congelamento-scongelamento sopra riportato 10x-15x fino a quando la soluzione appare meno torbida.

Figura 2: Schema che mostra il flusso di lavoro dalla preparazione di vescicole multilamellari e piccole vescicole unilamellari alla formazione di doppi strati lipidici supportati. Creato con Biorender. Fare clic qui per visualizzare una versione ingrandita di questa figura.
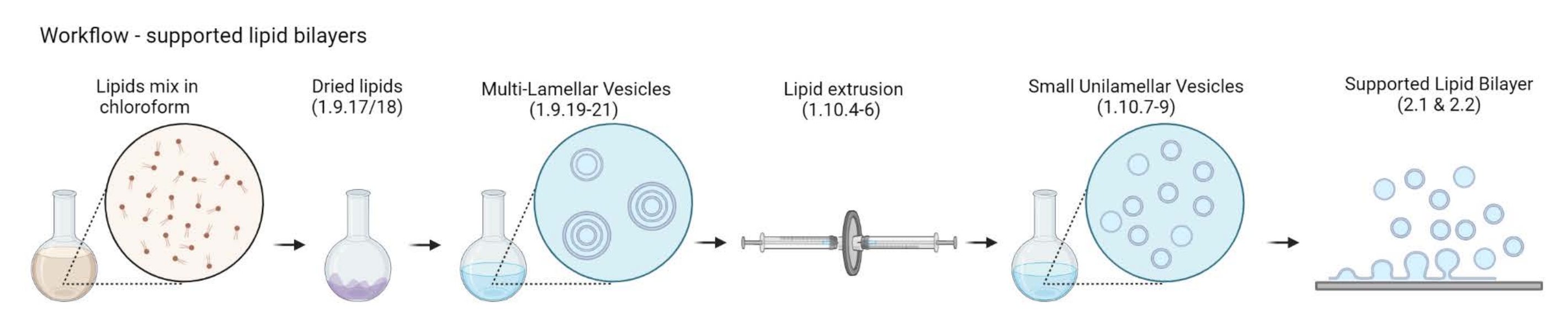{kind=link}
2. Ricostituzione di reti di actina legate a membrana
- Preparazione delle camere di campionamento
- Prendi 3-5 coprivetrini rettangolari e mettili all'interno di un barattolo Coplin. Accendere il sonicatore da bagno e impostare la temperatura su 65 °C. Riempire il barattolo Coplin con una soluzione detergente al 2% per immergere completamente i vetrini e posizionarlo nel sonicatore per 30 minuti in modalità a impulsi completi.
- Utilizzare una pinza smussata rivestita in PTFE per rimuovere i coprivetrini uno per uno dal barattolo. Risciacquarli accuratamente con acqua distillata e metterli in un altro barattolo Coplin riempito con 2 N NaOH.
- Sonica i coperchi per 20 minuti in modalità full pulse. Rimuovere i coperchi uno per uno, sciacquare abbondantemente con acqua distillata e metterli in un altro barattolo Coplin pieno di acqua distillata.
NOTA: Facoltativamente, sonicare i coperchi in acqua distillata per 20 minuti e quindi risciacquarli nuovamente con acqua distillata. - Immediatamente prima di iniziare l'esperimento, prendere il barattolo contenente i vetrini di copertura in una cappa chimica dotata di una fornitura di gas N2 .
- Ottimizzare la pressione dell'aria del flusso di gas N2 per tentativi ed errori in modo che sia sufficiente per spostare l'acqua dalla superficie del coperchio senza romperla. Allineare il flusso di gas N2 parallelamente al piano di copertura per ridurre la possibilità di rompere il coperchio.
- Utilizzare guanti e pinze per rimuovere i coprislip uno per uno dal barattolo per asciugarli sotto il flusso N2 . Asciugare entrambi i lati di ogni copricopertina e posizionarli su una griglia di plastica pulita con un coperchio. Posizionare la scatola con i vetrini di copertura in un essiccatore per evitare il contatto con particelle di polvere nell'aria.
NOTA: I coprivetrini essiccati N 2 possono essere conservati in un essiccatore dove possono rimanere idrofili per un massimo di2 giorni. Questa strategia potrebbe essere utile quando sono necessari molti doppi strati per l'esperimento o se l'esperimento richiede più di 8 ore. - Prendi i tubi PCR autoclavati e taglia i loro coperchi e le metà coniche inferiori con una lama chirurgica affilata. Prendi i tubi cilindrici tagliati a metà uno per uno, applica l'adesivo polimerizzabile UV sul bordo liscio di ciascun tubo tagliato e posizionalo invertito su un coprislip appena pulito in modo che il bordo si trovi piatto sul vetrino.
- Non spostare il cilindro lateralmente una volta posizionato sul coprivetrino per assicurarsi che la colla non si riversi nello spazio centrale della camera. Le copertine rettangolari possono ospitare comodamente fino a tre camere di reazione e quelle rotonde possono ospitarne solo una al centro (Figura 1).
- Inserire i coperchi dei cuscinetti della camera all'interno di un pulitore ad ozono UV con alimentazione O2 e aspirare (o utilizzare un illuminatore UV). Accendere la luce UV e illuminare per 3-5 minuti per consentire all'adesivo di polimerizzare. Eseguire un'illuminazione più lunga (10-15 min) per migliorare l'idrofilia del vetro di copertura e, quindi, la qualità del doppio strato lipidico.
- Conservare le camere di campionamento asciutte illuminate ai raggi UV per un massimo di 8 ore all'interno di piccole scatole di plastica (come scatole rettangolari vuote) avvolte in pellicola trasparente per ridurre il contatto con le particelle di polvere nell'aria.
NOTA: Un flusso costante di O2 in presenza di luce UV forma ozono e radicali dell'ossigeno che possono rimuovere le impurità organiche dalla superficie del vetrino. Un vuoto impedirà la fuoriuscita di ozono tossico che si forma durante il processo. - Estrarre i vetrini di copertura e verificare la presenza di perdite nelle camere riempiendole con acqua distillata. Ogni camera può contenere fino a ~ 150 μL di campione. Scartare le camere che perdono.
NOTA: Un'altra ottima e sicura opzione di pulizia è il detergente al plasma. Le impostazioni di tempo e potenza dipendono dal modello, ma assicurati di non trattare eccessivamente i vetrini con il plasma in quanto ciò comporterà una riduzione della mobilità lipidica. Il trattamento superficiale può influenzare la mobilità dei lipidi27, come è stato osservato con il trattamento prolungato con la soluzione detergente (>45 min) o NaOH (>30 min).
- Preparazione di doppi strati lipidici supportati
- Lavare ogni camera con tampone di formazione SLB (o 1x PBS) per rimuovere eventuali contaminanti superficiali, lasciando 100 μL di tampone alla fine. Contrassegnare il livello del tampone a 100 μL con un marcatore permanente per tracciare in modo riproducibile le variazioni del volume.
- Aggiungere 2 μL di 0,1 M CaCl2 alla camera. Ciò migliora l'adsorbimento delle vescicole sulla superficie del vetro, migliorando la formazione del doppio strato nella fase successiva. Aggiungere 8 μL della soluzione SUV (dal punto 1.10.) a ciascuna camera e incubare per 15 minuti a 25 °C.
NOTA: Il volume della miscela SUV da aggiungere può essere stimato calcolando il numero totale di lipidi (con una superficie media di 0,72 nm2) necessari per coprire completamente l'area idrofila esposta del pozzo con due strati lipidici. - Lavare via le vescicole non legate con tampone di motilità di actina (1x KMEH). In primo luogo, rimuovere 50 μL del tampone di formazione SLB, lasciando solo 50 μL nella camera del campione. In secondo luogo, aggiungere 100 μL di 1x KMEH alla camera. Mescolare delicatamente e quindi rimuovere 100 μL del tampone senza toccare il fondo.
NOTA: È importante essere delicati durante il lavaggio. Assicurarsi che la punta della pipetta non tocchi il fondo della camera. Tenere la pipetta inclinata in modo da dirigere il flusso del tampone verso la parete della camera e non direttamente sul doppio strato, poiché un flusso diretto potrebbe interrompere il doppio strato. Fare attenzione a non introdurre bolle d'aria durante il pipettaggio poiché l'aria potrebbe raggiungere il doppio strato lipidico e causare difetti in esso. - Ripetere i lavaggi 10x aggiungendo 100 μL di 1x KMEH e rimuovendo 100 μL.
- Aggiungere 10 μL di 1 mg/mL di β-caseina al doppio strato, mescolare delicatamente e incubare per 5-10 minuti. β-caseina blocca le regioni sul vetrino in cui il doppio strato non si è formato. Lavare via β-caseina 3x con 1x KMEH come descritto al punto 2.2.3. e riportare il livello del tampone al livello di 100 μL.
- Aggiunta di linker membrana-actina
- Durante l'incubazione della β-caseina (fase 2.2.5), estrarre un'aliquota della proteina linker actina di membrana da -80 °C, scongelarla rapidamente a 37 °C e tenerla sul ghiaccio. Diluire l'aliquota con tampone di diluizione proteica fino ad una concentrazione di 1 μM.
- Aggiungere la proteina linker ad una concentrazione finale definita (tipicamente 5-20 nM) e mescolare delicatamente. Per garantire un rapido equilibrio della proteina nella camera, aggiungere volumi superiori a 20 μL premiscelando la proteina linker con 1x KMEH.
- Incubare per 40 minuti a temperatura ambiente. Lavare 3 volte con 1x tampone KMEH per rimuovere la proteina HSE non legata (come al punto 2.2.3.). Riportare il livello del tampone in ciascuna camera al segno di 100 μL. Il campione è ora pronto per l'imaging.
- Valutazione della qualità del doppio strato lipidico
NOTA: questo è un passaggio facoltativo che non deve essere eseguito ogni volta. Raccomandiamo di eseguire questa valutazione ogni volta che i SUV freschi vengono prodotti da stock MLV congelati.- Accendi il microscopio, i laser di eccitazione e le telecamere di rilevamento. Assicurati che il laser sia allineato, che l'obiettivo sia pulito e che il software sia pronto per acquisire immagini.
- Mettere l'olio sull'obiettivo 100x, montare il campione sul palco del microscopio e focalizzare l'obiettivo sul doppio strato. Assicurarsi che la posizione del laser sia tale da subire una riflessione interna totale sul campione. Utilizzare un laser di eccitazione a 488 nm per verificare la distribuzione dell'intensità di fluorescenza del 10xHis-YFP-EzrinABD legato a doppio strato.
NOTA: I doppi strati di buona qualità mostrano una distribuzione uniforme su larga scala dell'intensità della fluorescenza. I doppi strati difettosi mostrano macchie fluorescenti intense e a chiazze. - Per determinare l'integrità del doppio strato, eseguire un test FRAP.
- Selezionare una regione di interesse sul bilayer e registrare alcune immagini del campo visivo utilizzando condizioni di imaging che forniscono un rapporto segnale/rumore di 5:1 o superiore. Mettere in pausa la registrazione e chiudere il diaframma di campo del microscopio TIRF per focalizzare un raggio laser concentrato su una piccola regione circolare del doppio strato per sbiancare localmente i fluorofori.
- Accendere il laser alla sua massima potenza per fotosbiancare la piccola regione per 3-10 s e quindi spegnere il laser. Riaprire il diaframma di campo al suo raggio originale, regolare nuovamente le impostazioni di imaging (pre-candeggina) e riprendere immediatamente per registrare il recupero del segnale fluorescente nel campo visivo.
- Controlla se il doppio strato è fluido. I doppi strati buoni con diffusione laterale normale recuperano velocemente, mentre i doppi strati cattivi recuperano lentamente o non recuperano affatto (Figura 3). Se il bilayer non viene ripristinato, controllare la sezione di risoluzione dei problemi e riavviare. Salvare le immagini come file TIFF a 16 bit. Per una stima quantitativa del coefficiente di diffusione, controllare il punto 3. sotto.
- Polimerizzazione dell'actina fluorescente
NOTA: Per risparmiare tempo, iniziare a polimerizzare l'actina durante il tempo di incubazione del legame della proteina HSE al doppio strato (fase 2.3.) o durante la valutazione della qualità del doppio strato (fase 2.4.).- Mescolare G-actina non etichettata e marcata con fluorescenza in un rapporto molare 10: 1 e rabboccarla con G-Buffer in modo che la concentrazione di G-actina sia di 20 μM. La concentrazione alla quale l'actina viene infine polimerizzata sarà 1/4 di questo valore. Aggiungere 1/10 di tampone ME 10x alla miscela per una soluzione 1x e incubare per 2 minuti. Questo passaggio sostituisce gli ioni Ca 2+ legati alla G-actina con ioni Mg2+. Assicurarsi che il volume finale sia in multipli di 10 μL.
- Aggiungere la quantità desiderata di proteine di tappatura come segue. Scongelare rapidamente un flaconcino di proteine tappando rapidamente a 37 °C e poi tenerlo in ghiaccio. Diluire con G-buffer in modo tale che la concentrazione della proteina di tappatura sia ora il doppio della concentrazione finale desiderata nella miscela di polimerizzazione. Aggiungere un volume uguale della soluzione proteica di tappatura diluita alla miscela di actina del punto 2.5.1.
- Infine, aggiungere un volume uguale di nuovo buffer target 2x al mix di reazione. Il volume finale della soluzione deve essere quattro volte il volume della miscela di actina alla fine della fase 2.5.2. Assicurarsi che la concentrazione finale di KMEH sia 1x, di ATP di 1 mM, di BSA sia di 1 mg/ml e di G-actina sia di 5 μM.
Incubare al buio a 25 °C per 45-60 minuti per consentire la polimerizzazione.
NOTA: Questa è chiamata strategia tampone target, in cui un volume di Mg 2+ G-actina (fase 2.5.1.) viene miscelato con un volume di mix proteico di capping (fase 2.5.2.) e due volumi di tampone target2x (punto 2.5.3.). Ciò rende più facile aumentare o diminuire la quantità di actina e alterare la concentrazione relativa della proteina capping (o di qualsiasi altro modulatore dell'actina; Figura 4).
- Aggiunta di filamenti fluorescenti di actina
- Tagliare alcune punte da 200 μL con una lama affilata o delle forbici per renderle smussate. Pipettare delicatamente il volume richiesto di actina polimerizzata da 5 μM (dal punto 2.5.3.) con una punta di pipetta smussata (per evitare il taglio dei filamenti di actina) e aggiungerla a una provetta PCR autoclavata pulita.
- Aggiungere 1x KMEH al tubo per ottenere il volume >20 μL e mescolare delicatamente per evitare il taglio di F-actina. Dalla camera di campionamento montata, rimuovere un volume uguale del buffer.
- Aggiungere la soluzione polimerizzata di actina nella camera e pipettare delicatamente su e giù 3 volte senza toccare il doppio strato sul fondo. Ciò consente ai filamenti di actina di distribuirsi uniformemente sul doppio strato. Montare il campione sul microscopio TIRF (vedere punto 2.4.1. e punto 2.4.2.).
- Si può registrare il processo di legame F-actina al doppio strato. Incubare per 20-30 min. Registra alcune immagini da diversi campi visivi dopo che l'aggiunta di F-actin ha raggiunto uno stato stazionario. Osservare il cambiamento nell'organizzazione spaziale di 10xHis-YFP-EzrinABD prima (omogeneo) e dopo l'organizzazione dell'actina.
NOTA: HYE è uniformemente distribuito sul doppio strato lipidico in assenza di actina. Dopo l'aggiunta di filamenti di actina, HYE colocalizza con F-actina. L'estensione della colocalizzazione dipende dall'affinità legante l'actina della proteina linker; più forte è l'affinità, maggiore è la colocalizzazione e più lenta è la mobilità laterale della proteina linker (Figura 5).
- Aggiunta di miosina II
- Dopo 30 minuti di incubazione dell'actina, rimontare il campione sul microscopio (se non è stato montato). Controllare il segnale nella proteina linker e nei canali F-actina. Regolare le condizioni di imaging, se necessario.
- Seleziona una buona regione con segnale proteico linker uniforme e filamenti di actina uniformemente sparsi e senza artefatti per una registrazione time-lapse lunga. Registra 10-15 fotogrammi a 0,1-0,2 Hz prima dell'aggiunta di miosina e metti in pausa la registrazione. Pipettare il volume richiesto di miosina-II muscolare riciclata dal flaconcino di riserva con una punta di pipetta smussata (per evitare il taglio dei filamenti di miosina) e aggiungere a un tubo PCR autoclavato pulito.
- Aggiungere immediatamente 1x KMEH al tubo per ottenere il volume >20 μL e mescolare delicatamente. Si può anche aggiungere ATP, miscela rigenerante ATP, agenti fotostabilizzanti, ecc. durante questo passaggio. Rimuovere con cautela un volume uguale del buffer dalla camera di campionamento montata senza disturbarla.
- Aggiungere delicatamente la soluzione di miosina alla camera del campione. Non pipettare su e giù in quanto disturberebbe i filamenti legati alla superficie. Riprendere immediatamente la registrazione time-lapse e osservare il sistema mentre si evolve dallo stato pre-miosina ai flussi contrattili di acto-miosina alimentati da ATP e alla formazione di aster a uno stato di blocco dell'ATP (vedere i risultati rappresentativi).
- Scatta immagini di sfondo per tutti i canali utilizzando un campione solo buffer. Salva tutte le immagini come file .tiff a 16 bit. Vedere la Tabella 2 per suggerimenti per risolvere i problemi comuni.

Figura 3: Valutazione della qualità dei doppi strati con test FRAP rapido. I doppi strati lipidici supportati (SLB) preparati da lipidi DOPC e Ni-NTA (98:2 mol%) sono rivestiti con HYE (linker membrana-actina 10xHis-YFP-tagged). Dopo che la proteina non legata è stata lavata via, il doppio strato fluorescente viene ripreso al microscopio TIRF. Una piccola regione sul doppio strato viene fotosbiancata con elevata potenza laser e viene registrato il recupero della fluorescenza. (A) Un buon doppio strato recupera sempre velocemente, con un coefficiente di diffusione atteso di 1-1,5 μm2/s per la composizione lipidica utilizzata in questo caso. (B) I doppi strati cattivi recuperano molto lentamente o non si riprendono affatto. (C) Immagini rappresentative di doppi strati difettosi: (C-i) un doppio strato con fori, (C-ii) un doppio strato con grandi chiazze lipidiche immobili e (C-iii) un doppio strato con piccoli punti immobili. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 4: Schema che mostra come polimerizzare l'actina usando il metodo del buffer target. Fare clic qui per visualizzare una versione ingrandita di questa figura.
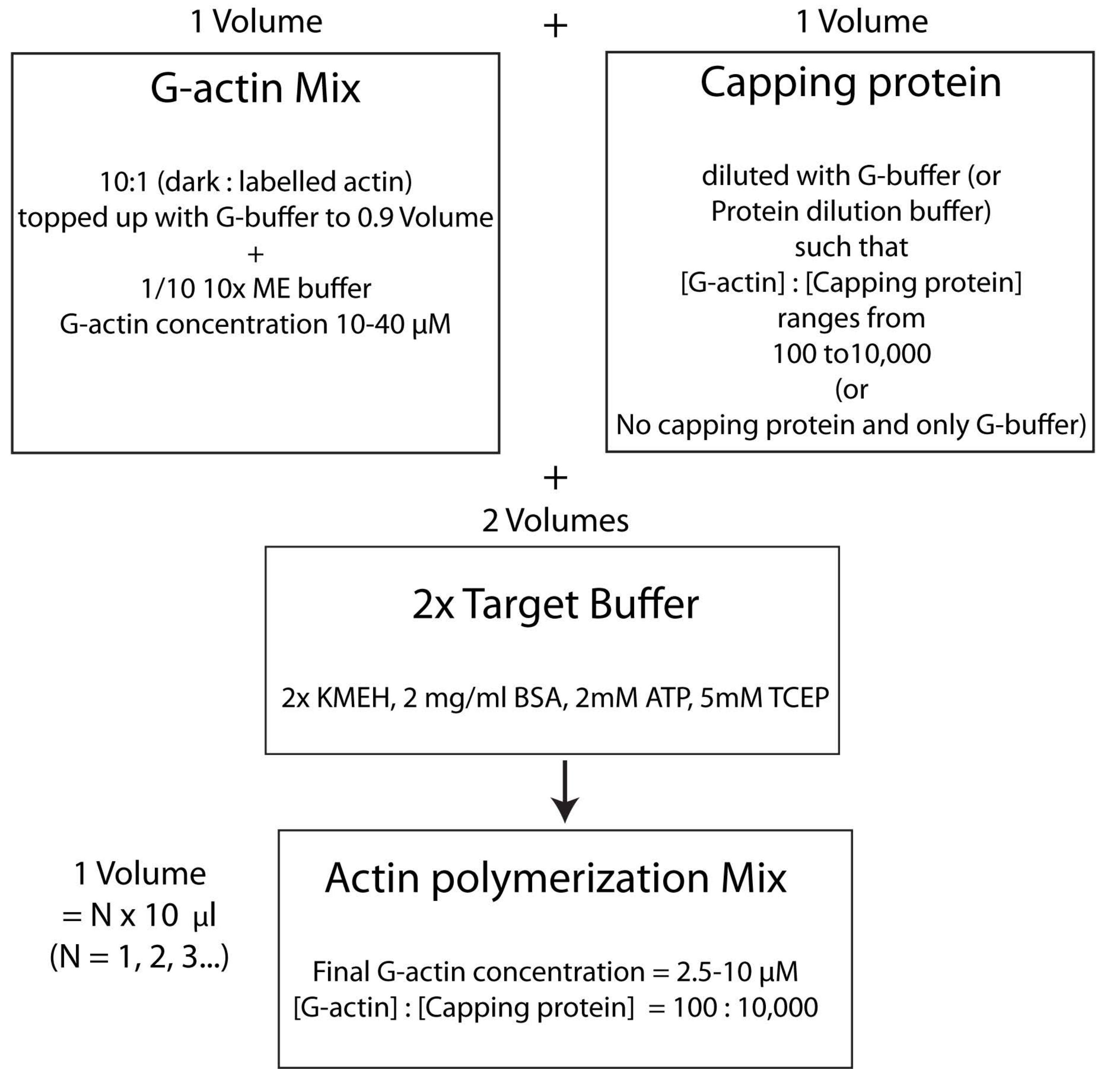{kind=link}

Figura 5: Organizzazione spaziale di HYE dopo il legame con F-actina. Istantanee TIRF che mostrano l'organizzazione spaziale di HYE prima e dopo l'aggiunta di filamenti di actina (etichettati con Atto-635 maleimide). L'organizzazione HYE è omogenea prima dell'aggiunta di F-actina e diventa colocalizzata e coallineata lungo i filamenti di actina. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
3. Analisi dei dati
- Utilizzando il software Fiji (https://imagej.net), sottrarre lo sfondo dalle immagini della proteina linker (dal punto 2.4.). Misurare i valori di intensità media dal punto sbiancato e da una regione di riferimento.
- Normalizzare le tracce temporali dal punto sbiancato e dalla regione di riferimento all'intensità dei rispettivi valori di intensità pre-candeggina. Dividere ogni punto temporale nei valori delle regioni sbiancate normalizzate per i rispettivi punti temporali nella traccia temporale della regione di riferimento normalizzata. Correggere la traccia temporale normalizzata risultante per lo sfondo e per eventuali fluttuazioni sistematiche di intensità durante l'acquisizione (photobleaching globale, z-drift, ecc.).
- Utilizzare un metodo manuale28 privo di calzature per stimare il coefficiente di diffusione delle proteine legate a doppio strato. In breve, il tempo dimezzato del profilo di recupero, τ1/2, può essere calcolato osservando il momento in cui il profilo di recupero normalizzato raggiunge la metà del suo stato stazionario:

Qui, F 0 è l'intensità media nella regione sbiancata nel primo fotogramma dopo il fotosbiancamento e F∞ è il valore a lungo termine dello stato stazionario del recupero del doppio strato. - Stimare il raggio effettivo della candeggina, re, un parametro che corregge la diffusione durante il fotosbiancamento, da una scansione a linea del profilo spot post candeggina29. La mezza larghezza a metà minimo di questa scansione lineare che passa attraverso il centro della macchia di candeggina, r 1/2, si riferisce a re come segue:

Il τ1/2 calcolato al punto 2.8.3., r e calcolato al punto 2.8.4., e il raggio di candeggina originariamente impostato, rn, sono utilizzati per calcolare il coefficiente di diffusione (D) utilizzando la seguente formula:
- Utilizzare un metodo manuale28 privo di calzature per stimare il coefficiente di diffusione delle proteine legate a doppio strato. In breve, il tempo dimezzato del profilo di recupero, τ1/2, può essere calcolato osservando il momento in cui il profilo di recupero normalizzato raggiunge la metà del suo stato stazionario:
- Analisi delle immagini degli astri di actomiosina
- Utilizzando Fiji, sottrarre lo sfondo da tutte le immagini registrate in tutti i canali. Correggete le immagini per qualsiasi illuminazione non uniforme o modello di interferenza utilizzando la correzione del campo piatto.
NOTA: Si possono usare diapositive di plastica colorata, che sono buoni campioni piatti per fare tali correzioni. Per la proteina linker e i filamenti di actina su un doppio strato planare, si può anche utilizzare la proiezione media di più immagini pre-miosina per creare mappe di correzione dell'illuminazione specifiche del canale.- Per il canale HYE, mostrato qui, prendere una proiezione di intensità media di più immagini HYE (registrate da diverse regioni del doppio strato lipidico prima dell'aggiunta di miosina). Applicare un filtro gaussiano appropriato (σ = da 50 pixel a 80 pixel) alla proiezione media (da immagini pre-miosina o da qualsiasi campione piatto standard).
- Convertire l'immagine filtrata in un'immagine a 32 bit. Dividere tutti i valori dei pixel per la media dell'intera immagine. Questo darà una mappa di correzione normalizzata per il canale HYE. Dividi tutte le immagini nel canale HYE con questa mappa per la correzione del campo piatto. Crea mappe di correzione per altri canali utilizzando la stessa strategia.
- Corretto per il fotosbiancamento utilizzando un metodo di rapporto esponenziale o semplice (a seconda del profilo di decadimento dell'intensità) nelle Figi.
- Per correggere qualsiasi disallineamento temporale x-y (movimento traslazionale), unire tutti i canali corretti per la fotocandeggina in un unico Hyperstack. Utilizzando il plugin Hyperstack-Reg nelle Fiji, applicare una trasformazione Corpo rigido o Traduzione.
- Infine, suddividere l'Hyperstack allineato in singoli canali e salvarli separatamente come stack TIFF a 16 bit per ulteriori analisi.
- Utilizzando Fiji, sottrarre lo sfondo da tutte le immagini registrate in tutti i canali. Correggete le immagini per qualsiasi illuminazione non uniforme o modello di interferenza utilizzando la correzione del campo piatto.
Risultati
Per la rappresentazione, qui viene mostrato un tipico profilo postbleach della 1a immagine dopo il photobleaching (immagine a t = 0 s nella Figura 3A) e il suo adattamento alla seguente funzione28 (vedi Figura 6A):

Il valore di r e (23,94 μm) calcolato dall'adattamento a questa curva è molto simile all'r e calcolato al punto 2.8.4. (23,24 μm). Qui, K è un parametro di profondità della candeggina che può essere stimato direttamente da F0 (descritto nel passaggio 2.8.4.). Analogamente, la Figura 6B mostra il profilo di recupero e il relativo adattamento alla seguente funzione28:

Troviamo che il valore adattato del coefficiente di diffusione è 1,34 μm 2/s, un valore che concorda strettamente con il valore di 1,39 μm 2/s che viene calcolato dalla formula nel passo2.8.4. Qui, MF sta per la frazione mobile del doppio strato lipidico che rappresenta la frazione della popolazione sbiancata che recupera. La mobilità delle molecole ancorate ai lipidi dipende, naturalmente, dalla composizione lipidica e dal suo stato fisico (fase liquida o gel). Per i nostri esperimenti che utilizzano membrane lipidiche basate su DOPC, la mobilità dovrebbe essere >1 μm2 / s e la frazione mobile non dovrebbe essere inferiore a 0,9 per indicare un buon doppio strato lipidico. Si consiglia l'uso del metodo manuale senza montaggio per un rapido test della qualità e della mobilità del doppio strato. Il metodo di adattamento può essere utile per automatizzare l'analisi per molte curve FRAP. Inoltre, se si desidera eseguire un esperimento FRAP più sofisticato per caratterizzare sistematicamente la diffusione nel sistema, raccomandiamo al lettore di questa recensione di Lorén et al.30 per maggiori dettagli sui modelli di adattamento e sulle potenziali insidie nella progettazione sperimentale.

Figura 6: Quantificazione del coefficiente di diffusione dei doppi strati lipidici. (A) Profilo della prima immagine dopo il fotosbiancamento (t = 0 s nella figura 3A) e sua corrispondenza con l'equazione 4 per calcolare il raggio effettivo di candeggina. (B) Il profilo di recupero della regione sbiancata e la sua idoneità all'equazione 5 per calcolare il coefficiente di diffusione e la frazione mobile. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
Un risultato tipico degli esperimenti sopra descritti che mostra l'assemblaggio dinamico e l'organizzazione di una rete di acto-miosina collegata su un doppio strato lipidico supportato ripreso dalla microscopia TIRF è rappresentato nella Figura 7 e nel Video supplementare S1.
La Figura 7 mostra un montaggio di immagini della proteina linker, F-actina e miosina-II.

Figura 7: I flussi contrattili di actomiosina guidano il clustering locale della proteina linker membrana-actina HYE. Istantanee TIRF di HYE (marcato YFP), filamenti di actina (marcati con Atto-635 maleimide) e filamenti di miosina II (marcati con Atto-565 maleimmide) dopo aggiunta di miosina II a un SLB contenente HYE e F-actina. Il tempo è indicato in alto: 0 min è immediatamente prima che i miofilamenti fluorescenti inizino a comparire nel campo TIRF. HYE e F-actina sono distribuiti omogeneamente sul doppio strato lipidico prima dell'aggiunta di miosina (0 min). L'attività della miosina induce flussi contrattili di actomosisina, che emergono in strutture simili ad aster allo stato stazionario (15 min), guidando il raggruppamento locale della componente accoppiata della membrana (HYE). La riga più bassa è una fusione di immagini di actina (gialla) e miosina II (magenta) che mostrano l'organizzazione di actina e miosina in diversi punti temporali. Le immagini utilizzate per realizzare questi montaggi sono state corrette nelle Figi per il segnale di fondo, i modelli di intensità non uniformi e il movimento traslazionale. Barra della scala = 10 μm. Per informazioni dettagliate, vedere Video supplementare S1. Fare clic qui per visualizzare una versione ingrandita di questa figura.
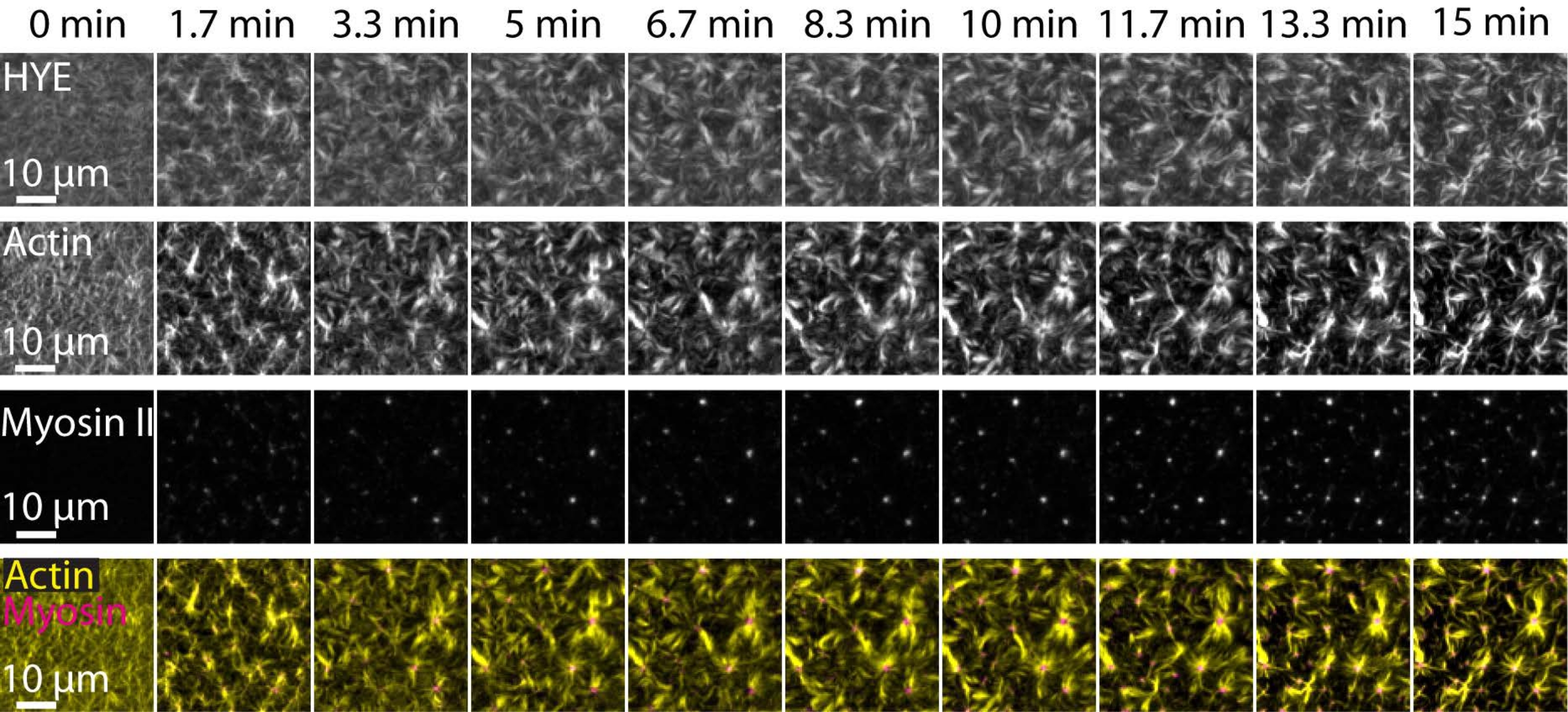{kind=link}
| Nome buffer | Composizione | |
| Tampone di reidratazione lipidica | 50 mM HEPES, 150 mM NaCl, 5% saccarosio, pH 7,5 | |
| Tampone di formazione SLB | 50 mM HEPES, 150 mM NaCl, pH 5-6 | |
| Buffer di archiviazione SLB | 50 mM HEPES, 150 mM NaCl, pH 7,2 | |
| Tampone di diluizione delle proteine | 20 mM HEPES, 100 mM KCl, 1mM TCEP o DTT, pH 7,2 | |
| 1X ME o tampone a scambio ionico actina | 50 mM MgCl2, 0,2 mM EGTA, 10 mM HEPES, pH 7,2 (conservare a 4°C) | |
| 1X tampone di polimerizzazione KMEH o actina | 50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 50 mM HEPES, pH 7,2 | |
| Stock ATP da 100 mM | 100 mM di sale disodico ATP, 50 mM Tris, 50 mM NaCl, 5 mM MgCl2, 2 mM EGTA, pH 7,5 (conservare a -20°C) | |
| 2x buffer di destinazione | 2x KMEH, 2 mg/ml BSA, 2mM ATP, 5mM TCEP (conservato a 4°C) | |
| G-buffer | 2 mM Tris, 0,1 mM CaCl2, 0,2 mM ATP, 0,5 mM TCEP, 0,04 % NaN3, pH 8 (conservare a 4°C) | |
| Tampone di miosina II | 500 mM KCl, 1 mM EDTA, 10-20 mM Hepes, pH 7,0 | |
| Tampone cromatografico con filtrazione su gel | 50 mM Tris-HCl, 150-300 mM NaCl, 5 mM TCEP, 0,1% Tween-20, pH 7,5 | |
| Tamponamento delle proteine Tampone di stoccaggio | 10 mM Tris· Cl, 50 mM NaCl, 1 mM TCEP, pH 7,5, 20% glicerolo | |
Tabella 1: Elenco delle composizioni di buffer utilizzate in questo protocollo.
| Problemi comuni e relativa risoluzione dei problemi | Problema | Causa | Possibili soluzioni | ||||||
| 1 | Il doppio strato lipidico non mostra diffusione | La causa più probabile di questo problema è il vetro di copertura sporco che può verificarsi quando la soluzione detergente è invecchiata o il riscaldamento non ha avuto luogo durante la sonicazione del bagno. Questi strati bistrati hanno un aspetto "vescicolare" perché le vescicole scoppiate si attaccano al vetro di copertura ma non si fondono tra loro. L'utilizzo di MLV più vecchi di 6 settimane o SUV più vecchi di 6 giorni, o l'aggiunta di basse quantità di SUV può anche portare alla formazione di un doppio strato vescicolare. | Utilizzare una soluzione detergente fresca. Assicurarsi che il riscaldatore sia acceso e che la temperatura sia compresa tra 45-65°C. Utilizzare miscele lipidiche fresche. (L'uso di una sonda lipidica fluorescente rispetto a una sonda proteica fluorescente a volte può manifestarsi in modo diverso. Ad esempio, se il doppio strato ha difetti di subdiffrazione e la fase di passivazione superficiale viene saltata (o non funziona), la sonda lipidica mostrerà una distribuzione uniforme dell'intensità, ma la sonda proteica fluorescente potrebbe mostrare punti fluorescenti luminosi.) | ||||||
| 2 | Il doppio strato lipidico ha chiazze luminose | La lunga incubazione di SUV per la formazione di due strati può creare un doppio strato lipidico che è complessivamente diffuso, ma con occasionali chiazze luminose. Questi cerotti possono essere doppi strati multistrato che possono attirare grandi quantità di sonda fluorescente. | 15-20 minuti di incubazione con SUV sono sufficienti. Assicurarsi che la sonda non si stia aggregando: una rapida rotazione dura della proteina linker (300 x g per 15 minuti a 4 °C) può rimuovere gli aggregati | ||||||
| 3 | Il doppio strato lipidico ha buchi scuri | Ciò accade quando il doppio strato è costituito da vecchi SUV e ripreso per ore prolungate (> 4 ore dopo la formazione), o il pH della soluzione cambia drasticamente a causa di immagini prolungate (ad esempio, nello stato ATP elevato e in presenza di alcuni scavenger di ossigeno), o quando la superficie è eccessivamente passivata con beta-caseina (aggiungendo troppa beta-caseina per più di 10-15 minuti e o non lavandola via). | Utilizzare lipidi freschi. Riduci la frequenza dei fotogrammi dell'immagine o il tempo effettivo di illuminazione laser. Utilizzare buffer con capacità di buffering superiore. | ||||||
| 4 | Il doppio strato lipidico mostra una diffusione lenta | I doppi strati lipidici con alta percentuale di colesterolo, lipidi saturi lunghi o lipidi carichi si diffondono più lentamente. | In questi casi, preparare il campione ad alta temperatura. Si può anche usare una composizione lipidica semplice e testata come controllo insieme a composizioni lipidiche complesse e non testate. Assicurati che il vetro sia pulito. | ||||||
| 5 | L'actina non polimerizza | Il buffer target è vecchio, lo stock di G-actina è troppo vecchio, la vecchia e la nuova G-actina sono state copolimerizzate. | Assicurarsi che il Ca 2+ sia sostituito da Mg2+ prima della polimerizzazione (utilizzando il tampone ME). Utilizzare brodo fresco di ATP-Mg2+. Utilizzare G-actina appena riciclata. Assicurarsi che la concentrazione di F-actina (in termini di G-actina) aggiunta al doppio strato sia superiore a 0,2 μM. Per concentrazioni più basse, utilizzare F-actina stabilizzata con falloidina. | ||||||
| 6 | L'actina non si lega al doppio strato | Il linker membrana-actina non viene aggiunto o aggiunto a concentrazioni molto basse, questo può essere dedotto dalla fluorescenza della proteina linker. Se la fluorescenza è decente, il linker membrana-actina ha perso la capacità di legare l'actina. Inoltre, se la proteina linker è legata in modo non specifico alla superficie del vetro (quando il doppio strato è cattivo), potrebbe non reclutare filamenti di actina. | Assicurarsi che il doppio strato si stia diffondendo. Utilizzare proteine linker fresche | ||||||
| 7 | Il segnale fluorescente F-actina è debole | Il rapporto tra actina marcata e actina scura è troppo basso. L'actina marcata o l'actina non etichettata sono troppo vecchie e non sono copolimerizzanti tra loro. | Riciclare di nuovo l'actina e riprovare la ploymerizzazione con actina appena riciclata. Il fotodanneggiamento può distruggere o depolimerizzare la F-actina; Se possibile, utilizzare coloranti rossi o rossi per l'actina (e la miosina). | ||||||
| 8 | La miosina non mostra contrattilità | si può osservare che dopo aver aggiunto ATP al sistema infuso di miosina, non vi è contrattilità dell'acto-miosina. | Controlla se la concentrazione di miosina o il livello di purezza sono buoni. Utilizzare miosina appena riciclata (utilizzare entro 6 settimane dopo il riciclaggio). L'aggiunta di ATP fresco al mix di miosina può aiutare. Tamponi di degasaggio e utilizzo di spazzini di ossigeno, ecc. può ridurre il fotodanneggiamento dei motori. Ulteriori informazioni possono essere trovate nei protocolli di Plastino et al. o Stam et al. della stessa raccolta di metodi | ||||||
| 9 | Il vetro di copertura non è idrofilo | Il vetro di copertura non viene pulito correttamente. | Il vetro di copertura idrofilo pulito è fondamentale per la formazione del doppio strato lipidico. Un'utile lettura visiva dell'idrofilia del vetro di copertura dopo il protocollo di pulizia è osservare la bagnatura del vetro da parte dell'acqua. Aggiungere un piccolo volume d'acqua a un coprislip piatto. L'acqua rimarrà sotto forma di una goccia rotonda se il coprislip non viene pulito correttamente. Tuttavia, lo stesso volume d'acqua si diffonderà e formerà uno strato sottile, su un coverglass idrofilo trattato. Questo comportamento bagnante dell'acqua sulla superficie del vetro di copertura può essere utilizzato per accertare se le fasi di pulizia con la soluzione detergente/NaOH hanno funzionato. | ||||||
Tabella 2: Guida alla risoluzione dei problemi che riepiloga i problemi comuni e le soluzioni corrispondenti.
Video supplementare S1: I flussi contrattili di actomiosina guidano il clustering locale della proteina linker membrana-actina HYE. Timelapse TIRF di HYE (marcato YFP), filamenti di actina (marcati con Atto-635 maleimide) e filamenti di miosina II (marcati con Atto-565 maleimmide) dopo aggiunta di miosina II a un SLB contenente HYE e F-actina. Il tempo è indicato in alto: 0 min è immediatamente prima che i miofilamenti fluorescenti inizino a comparire nel campo TIRF. Barra di scala = 10 μm. Clicca qui per scaricare questo file.
Discussione
Questo protocollo presenta una piattaforma versatile e un punto di partenza per progettare esperimenti per studiare l'interfaccia membrana-corteccia delle cellule. I passaggi critici sono la preparazione di vetrini puliti, utilizzando lipidi freschi per una formazione efficiente di SUV (entrambi influenzano la qualità degli SLB) e l'uso di proteine miosina II appena riciclate per la riorganizzazione dinamica dei filamenti di actina. Quando si visualizzano dinamiche per un lungo periodo, è molto importante incorporare un sistema scavenger di ossigeno (ad esempio, acido protocatechuico e protocatechuato 3 4-diossigenasi 5,31).
Il design a camera aperta consente l'aggiunta sequenziale di componenti a un sistema esistente senza indurre flussi lipidici. Questo può essere un vantaggio importante rispetto agli approcci a camera chiusa comunemente usati o al lavoro che utilizza proteine incapsulate all'interno dei liposomi36. Effetti contrari come la deformazione della membrana indotta da proteine non possono essere studiati con doppi strati lipidici adsorbiti di vetro.
I doppi strati lipidici possono essere formati con una vasta gamma di composizioni lipidiche. Inizia con l'adsorbimento delle vescicole lipidiche sulla superficie del vetro idrofilo, seguito dalla rottura spontanea della vescicola dovuta alle interazioni superficie-vescicola e vescicola-vescicola diretta o dalle vescicole adsorbite che raggiungono una copertura critica dopo di che una piccola frazione di vescicole si rompe, formando bordi attivi, che alla fine porta alla formazione del doppio strato32 . Oltre al vetro, vari substrati possono essere utilizzati per formare doppi strati lipidici supportati, come mica (ad esempio, per microscopia a forza atomica), substrati morbidi (ad esempio, poli-di-metil-silossano), cuscini polimerici33,34,35, che si estendono tra i fori delle griglie di microscopia elettronica 14. I doppi strati dell'interfaccia a goccia sono un altro metodo interessante per creare doppi strati lipidici stabili e indipendenti36. L'inclusione di reti di acto-miosina in vescicole o emulsioni è un metodo molto potente per studiare questo sistema minimo in una geometria simile a una cella37,38, e che è descritto in dettaglio altrove 39.
Divulgazioni
Gli autori non hanno conflitti di interesse da dichiarare.
Riconoscimenti
Questo lavoro è stato sostenuto dal fondo di ricerca AXA e dal Warwick-Wellcome Quantitative Biomedicine Programme (Wellcome ISSF, RMRCB0058) per DVK, NCBS-TIFR per AB e ST e dalla borsa di studio Wellcome-DBT Margdarshi (IA/M/15/1/502018) per SM. DVK desidera anche ringraziare la Biophysical Society per aver reso possibile l'evento di networking virtuale "Challenges in understanding multi-component cytoskeletal networks from the molecular to the meso-scale", che ha contribuito alla creazione di questa raccolta di protocolli.
Materiali
| Name | Company | Catalog Number | Comments |
| 1,2 dipalmitoyl-sn-glycero-3-phosphoethanolamine-N- (lissamine rhodamine B sulfonyl) | Avanti Polar Lipids | 810158 | 16:0 RhoPE |
| 1,2-dioleoyl-sn-glycero-3- [(N-(5-amino-1 carboxypentyl) iminodiacetic acid) succinyl] (nickel salt) | Avanti Polar Lipids | 790404 | DGS-NTA-Ni2+ |
| 1,2-dioleoyl-sn-glycero-3-phosphocholine | Avanti Polar Lipids | 850375 | DOPC |
| 1,2-dipalmitoyl-sn-glycero-3-phosphocholine | Avanti Polar Lipids | 850355 | DPPC |
| Amber glass vials | ThermoFisher | B7990-2A | |
| ATP disodium salt | Sigma Aldrich | A26209 | |
| Attofluor cell chamber | ThermoFisher | A7816 | |
| Bath sonicator | GT Sonic | 1860QTS | |
| beta-casein | Sigma Aldrich | C6905 | |
| CaCl2 | ThermoFisher | 12135 | |
| chloroform | Sigma Aldrich | 650471 | alternatively from Electron Microscopy Sciences, 50980296 |
| Cover slips, #1, 25 mm diameter, Gold Seal | Harvard Apparatus | 64-0705B | |
| Cover slips, #1, 40x22 mm, Gold Seal | ThermoFisher | 48404-031 | |
| EDTA | ThermoFisher | G12635 | |
| EGTA | Himedia | MB130 | |
| Gas tight glass syringes, with removebla needle, blunt, volumes 10 µL, 100 µL, 500 µL | Hammilton | 1700 series | |
| Hellmanex III | Hellma Analytics | Z805939 | cleaning solution |
| HEPES | Himedia | RM380 | |
| KaH2PO4 | ThermoFisher | G13405 | |
| KCl | ThermoFisher | G13305 | |
| KOH | ThermoFisher | G26708 | |
| Lipid extruder | Avanti Polar Lipids | 61000-1EA | |
| MgCl2 | ThermoFisher | G15535 | |
| Microtip sonicator | Sonics | VC750 | 3 mm Tip diameter |
| Na2CO3 | ThermoFisher | G15955 | |
| NaCl | Himedia | GRM853 | |
| NaH2PO4 | ThermoFisher | G15825 | |
| NaOH | ThermoFisher | G27815 | |
| Nikon Ti Eclipse TIRF microscope | Nikon | With a TIRF unit connected through a polarization-conserving optical fibre to an Agilent monolithic laser combiner MLC400 with multiple laser lines with a 100X, 1.45 NA Nikon Oil Objective with two 512 x 512-pixel EMCCD cameras (Photometrics Evolve 512) with a 100X, 1.45 NA Nikon Oil Objective with two 512 x 512-pixel EMCCD cameras (Photometrics Evolve 512) | |
| NOA88 | Norland Products | 8801 | |
| PTFE Coated Tweezer Style #2A | Structure Probe | 0S2AT-XD | |
| Refrigerated microcentrifuge | Eppendorf | 5424R | |
| Sucrose | ThermoFisher | G15925 | |
| UV-illuminator | Novascan | PSD PRO-UV | needs vacuum and O2 supply |
Riferimenti
- Köster, D. V., Mayor, S. Cortical actin and the plasma membrane: Inextricably intertwined. Current Opinion in Cell Biology. 38, 81-89 (2016).
- Rao, M., Mayor, S. Active organization of membrane constituents in living cells. Current Opinion in Cell Biology. 29, 126-132 (2014).
- Honigmann, A., et al. A lipid bound actin meshwork organizes liquid phase separation in model membranes. eLife. 3, 01671(2014).
- Das, A., et al. Stratification relieves constraints from steric hindrance in the generation of compact actomyosin asters at the membrane cortex. Science Advances. 6 (11), (2020).
- Köster, D. V., et al. Actomyosin dynamics drive local membrane component organization in an in vitro active composite layer. Proceedings of the National Academy of Sciences. 113 (12), 1645-1654 (2016).
- Vogel, S. K., Heinemann, F., Chwastek, G., Schwille, P. The design of MACs (minimal actin cortices). Cytoskeleton. 70 (11), 706-717 (2013).
- Murrell, M., Thoresen, T., Gardel, M. Reconstitution of contractile actomyosin arrays. Methods in Enzymology. 540, 265-282 (2014).
- Mosby, L. S., et al. Myosin II filament dynamics in actin networks revealed with interferometric scattering microscopy. Biophysical Journal. 118 (8), 1946-1957 (2020).
- Seara, D. S., et al. Entropy production rate is maximized in non-contractile actomyosin. Nature Communications. 9 (1), 4948(2018).
- Linsmeier, I., et al. Disordered actomyosin networks are sufficient to produce cooperative and telescopic contractility. Nature Communications. 7, 12615(2016).
- Vogel, S. K., Petrasek, Z., Heinemann, F., Schwille, P. Myosin motors fragment and compact membrane-bound actin filaments. eLife. 2, 00116(2013).
- Ditlev, J. A., et al. A composition-dependent molecular clutch between T cell signaling condensates and actin. eLife. 8, 42695(2019).
- Banjade, S., Rosen, M. K. Phase transitions of multivalent proteins can promote clustering of membrane receptors. eLife. 3, 04123(2014).
- Heinemann, F., Vogel, S. K., Schwille, P. Lateral membrane diffusion modulated by a minimal actin cortex. Biophysical Journal. 104 (7), 1465-1475 (2013).
- Vogel, S. K., Greiss, F., Khmelinskaia, A., Schwille, P. Control of lipid domain organization by a biomimetic contractile actomyosin cortex. eLife. 6, 24350(2017).
- Block, S. Brownian motion at lipid membranes: A comparison of hydrodynamic models describing and experiments quantifying diffusion within lipid bilayers. Biomolecules. 8 (2), 30(2018).
- JoVE, JoVE. JoVE Methods Collection. In vitro reconstitution of cytoskeleton networks for biomaterials, biophysics and active matter research. Journal of Visualized Experiments. , Cambridge, MA. (2022).
- Pardee, J. D., Spudich, J. A. Purification of muscle actin. Methods in Enzymology. 85, 164-181 (1982).
- Spudich, J. A., Watt, S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. Journal of Biological Chemistry. 246 (15), 4866-4871 (1971).
- Pollard, T. D. Myosin purification and characterization. Methods in Cell Biology. 24, 331-371 (1982).
- JoVE Science Education Database. Separating protein with SDS-PAGE. Journal of Visualized Experiments. , Cambridge, MA. (2022).
- Bieling, P., et al. WH2 and proline-rich domains of WASP-family proteins collaborate to accelerate actin filament elongation. The EMBO Journal. 37 (1), 102-121 (2018).
- Shrivastava, R., Köster, D., Kalme, S., Mayor, S., Neerathilingam, M. Tailor-made ezrin actin binding domain to probe its interaction with actin in-vitro. PLoS One. 10 (4), 0123428(2015).
- Nye, J. A., Groves, J. T. Kinetic control of histidine-tagged protein surface density on supported lipid bilayers. Langmuir. 24 (8), 4145-4149 (2008).
- Honigmann, A., et al. Scanning STED-FCS reveals spatiotemporal heterogeneity of lipid interaction in the plasma membrane of living cells. Nature Communications. 5, 5412(2014).
- Selden, N. S., et al. Chemically programmed cell adhesion with membrane-anchored oligonucleotides. Journal of the American Chemical Society. 134 (2), 765-768 (2012).
- Seu, K. J., et al. Effect of surface treatment on diffusion and domain formation in supported lipid bilayers. Biophysical Journal. 92 (7), 2445-2450 (2007).
- Kang, M., Day, C. A., Kenworthy, A. K., DiBenedetto, E. Simplified equation to extract diffusion coefficients from confocal FRAP data. Traffic. 13 (12), 1589-1600 (2012).
- Kang, M., Day, C. A., Kenworthy, A. K. A novel computational framework for D(t) from Fluorescence Recovery after Photobleaching data reveals various anomalous diffusion types in live cell membranes. Traffic. 20 (11), 867-880 (2019).
- Lorén, N., et al. Fluorescence recovery after photobleaching in material and life sciences: Putting theory into practice. Quarterly Reviews of Biophysics. 48 (3), 323-387 (2015).
- Alvarado, J., Sheinman, M., Sharma, A., MacKintosh, F. C., Koenderink, G. H. Molecular motors robustly drive active gels to a critically connected state. Nature Physics. 9 (7), 1-7 (2013).
- Andrecka, J., Spillane, K. M., Ortega-Arroyo, J., Kukura, P. Direct observation and control of Supported lipid bilayer formation with interferometric scattering microscopy. ACS Nano. 7 (12), 10662-10670 (2013).
- Lin, W. -C., et al. Supported membrane formation, characterization, functionalization, and patterning for application in biological science and technology. Current Protocols in Chemical Biology. 2 (4), 235-269 (2010).
- Richter, R. P., Bérat, R., Brisson, A. R. Formation of solid-supported lipid bilayers: An integrated view. Langmuir. 22 (8), 3497-3505 (2006).
- Sapuri-Butti, A. R., Butti, R. C., Parikh, A. N. Characterization of supported membranes on topographically patterned polymeric elastomers and their applications to microcontact printing. Langmuir. 23 (25), 12645-12654 (2007).
- Leptihn, S., et al. Constructing droplet interface bilayers from the contact of aqueous droplets in oil. Nature Protocols. 8 (6), 1048-1057 (2013).
- Bashirzadeh, Y., Moghimianavval, H., Liu, A. P. Encapsulated actomyosin patterns drive cell-like membrane shape changes. iScience. 25 (5), 104236(2022).
- Bashirzadeh, Y., Wubshet, N., Litschel, T., Schwille, P., Liu, A. P. Rapid encapsulation of reconstituted cytoskeleton inside giant unilamellar vesicles. Journal of Visualized Experiments. (177), e63332(2021).
- Murrell, M., Chen, S., Sun, Z. G. In vitro reconstitution of actin cytoskeleton inside giant unilamellar vesicles. Journal of Visualized Experiments. , (2022).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati