Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
In vitro Chemische Kartierung von G-Quadruplex-DNA-Strukturen durch Bis-3-Chlorpiperidine
In diesem Artikel
Zusammenfassung
Bis-3-Chloropiperidine (B-CePs) sind nützliche chemische Sonden, um G-Quadruplex-Strukturen in DNA-Matrizen in vitro zu identifizieren und zu charakterisieren. Dieses Protokoll beschreibt das Verfahren zur Durchführung von Sondierungsreaktionen mit B-CePs und zur Auflösung von Reaktionsprodukten durch hochauflösende Polyacrylamid-Gelelektrophorese.
Zusammenfassung
G-Quadruplexe (G4s) sind biologisch relevante, nicht-kanonische DNA-Strukturen, die eine wichtige Rolle bei der Genexpression und bei Krankheiten spielen und wichtige therapeutische Ziele darstellen. Für die in vitro Charakterisierung von DNA innerhalb potenzieller G-Quadruplex-bildender Sequenzen (PQS) werden zugängliche Methoden benötigt. B-CePs sind eine Klasse von Alkylierungsmitteln, die sich als nützliche chemische Sonden für die Untersuchung der Struktur höherer Ordnung von Nukleinsäuren erwiesen haben. In dieser Arbeit wird ein neuer chemischer Mapping-Assay beschrieben, der die spezifische Reaktivität von B-CePs mit dem N7 von Guaninen ausnutzt, gefolgt von einer direkten Strangspaltung an den alkylierten Gs.
Um G4-Falten von ungefalteten DNA-Formen zu unterscheiden, verwenden wir B-CeP 1, um das Thrombin-bindende Aptamer (TBA) zu untersuchen, eine 15-mer-DNA, die in der Lage ist, die G4-Anordnung anzunehmen. Die Reaktion von B-CeP-antwortenden Guaninen mit B-CeP1 führt zu Produkten, die durch hochauflösende Polyacrylamid-Gelelektrophorese (PAGE) auf Einzelnukleotidebene aufgelöst werden können, indem einzelne Alkylierungsaddukte und DNA-Strangspaltung an den alkylierten Guaninen lokalisiert werden. Die Kartierung mit B-CePs ist ein einfaches und leistungsfähiges Werkzeug für die In-vitro-Charakterisierung von G-Quadruplex-bildenden DNA-Sequenzen, das die genaue Lokalisierung von Guaninen ermöglicht, die an der Bildung von G-Tetrades beteiligt sind.
Einleitung
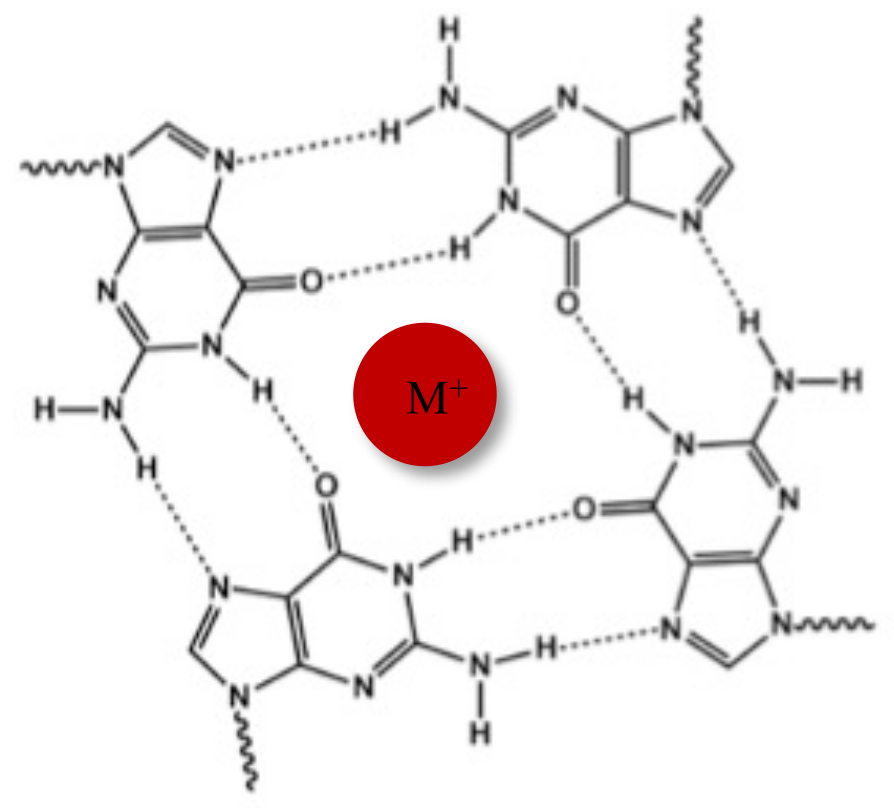
Neben der typischen Watson-Crick-Doppelhelix können Nukleinsäuren aufgrund ihrer Guanin-reichen Sequenzen verschiedene Sekundärstrukturen annehmen, wie z.B. die alternative G-Quadruplex (G4)-Form. Die G4-Struktur basiert auf der Bildung von planaren Tetrameren, sogenannten G-Tetraden, in denen vier Guanine durch Hoogsteen-Wasserstoffbrückenbindungen wechselwirken. G-Tetraden werden durch monovalente Kationen, die im Zentrum des Guaninkerns koordiniert sind, gestapelt und weiter stabilisiert (Abbildung 1)1.

Abbildung 1: Schematische Darstellung einer G-Quadruplex-Struktur. (A) Schematische Darstellung einer G-Tetrade. Das planare Array wird durch Hoogsteen-Basenpaarung und durch ein zentrales Kation (M+) stabilisiert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Sequenzen mit vier oder mehr Durchläufen von mindestens zwei aufeinanderfolgenden Guanin-Nukleotiden sind potentielle G-Quadruplex-bildende Sequenzen (PQS), die sich in G-Quadruplex-Strukturen falten können. PQS befinden sich in vielen verschiedenen zellulären Kontexten, wie z.B. an Telomeren, Genpromotoren, ribosomaler DNA und Rekombinationsstellen, und sind an der Regulation vieler biologischer Prozesse beteiligt2. Daher ist die Identifizierung und experimentelle Validierung von G4s im menschlichen Genom, die derzeit hauptsächlich mit computergestützten Werkzeugen durchgeführt wird, ein biologisch relevantes Thema3. Um computergestützte Vorhersagen zu unterstützen oder unvorhergesehene G4-Strukturen zu erkennen, wird hier eine auf chemischer Kartierung basierende Methode zur Identifizierung der G4-Bildung in einem DNA-Template gezeigt, die die präzise Identifizierung von Guaninen ermöglicht, die die G-Tetradenstruktur bilden.
Der beschriebene chemische Mapping-Assay nutzt die unterschiedliche Reaktivität von Bis-3-Chlorpyperidinen (B-CePs) mit Guaninen nach der Bildung von G4-Strukturen. Aufgrund ihrer hohen Reaktivität mit Nukleophilen 4,5,6,7,8,9 sind B-CePs Nukleinsäure-Alkylanzien mit der Fähigkeit, sehr effizient mit der N7-Position von Guanin-Nukleotiden10 zu reagieren. Auf die Alkylierung folgt die Depurinierung und die Strangspaltung in einzel- und doppelsträngigen DNA-Konstrukten. Im Gegensatz dazu sind Guanine, die an der Bildung der G-Tetraden in G4-Anordnungen beteiligt sind, unempfindlich gegen die B-CeP-Alkylierung, da die N7-Positionvon Guanin an den Hoogsteen-Wasserstoffbrückenbindungen beteiligt ist. Diese spezifische Reaktivität von B-CePs ermöglicht nicht nur den Nachweis von G4-Strukturen, sondern auch die Identifizierung der Guanine, die die Tetrade bilden, da sie aus ihrem relativen Schutz vor Alkylierung im Vergleich zu Guaninen in einzel- und doppelsträngiger DNA abgeleitet werden können.
Das chemische Kartierungsprotokoll wird hier unter Verwendung von B-CeP 1 (Abbildung 2A) als Sonde für die Charakterisierung von Thrombin-bindendem Aptamer (TBA) vorgestellt, einer 15-mer-DNA, die in der Lage ist, die G4-Anordnung in Gegenwart von Kaliumkationen anzunehmen11,12. Die G4-Anordnung von TBA (G4-TBA) wird direkt mit zwei Kontrollen verglichen, nämlich TBA in der einzelsträngigen Form (ssTBA) und TBA, die zu ihrer komplementären Sequenz getempert wurde, um das doppelsträngige Konstrukt (dsTBA) zu bilden (Tabelle 1). Die Produkte der Sondierungsreaktionen werden durch hochauflösende Polyacrylamid-Gelelektrophorese (PAGE) auf Einzelnukleotidebene aufgelöst, indem einzelne Alkylierungsaddukte und DNA-Strangspaltungen an den alkylierten Guaninen lokalisiert werden. Die Visualisierung auf dem Gel wird durch Konjugation des TBA-Oligonukleotids mit einem Fluorophor an seinem 3'-Ende ermöglicht (Tabelle 1). Dieses Protokoll zeigt, wie TBA in seinen verschiedenen Konformationen (G4 und Kontrollen) gefaltet wird und wie Sondierungsreaktionen mit B-CePs gefolgt von PAGE durchgeführt werden.
Protokoll
1. Nukleinsäure- und chemische Sondenvorbereitung
- Nukleinsäuren
HINWEIS: Das Oligonukleotid mit der Bezeichnung "TBA" ist die 15-mer-DNA-Sequenz 5'-GGT-TGG-TGT-GGT-TGG-3', die am 3'-Ende durch das Fluorophor 5-Carboxyfluorescein (FAM) markiert wird, um die Visualisierung auf dem Gel zu ermöglichen. Das unmarkierte Oligonukleotid "cTBA" ist seine DNA-komplementäre Sequenz 5'-CCA-ACC-ACA-CCA-ACC-3'. TBA und cTBA werden verwendet, um die drei verschiedenen Strukturen zu erhalten, wie in Tabelle 1 dargestellt.- Autoklavenspitzen und 0,5-ml-Röhrchen, um sterile Einwegartikel zu erhalten und Kontaminationen zu vermeiden.
- Bereiten Sie Stammlösungen vor, in denen jedes Oligonukleotid in Reinstwasser auf eine Endkonzentration von 100 μM gelöst wird. Bestimmen Sie die genaue Oligonukleotidkonzentration mit einem UV-Vis-Spektralphotometer (UV-Vis) unter Verwendung des vom Hersteller angegebenen Extinktionskoeffizienten bei 260 nm.
ANMERKUNG: Extinktionskoeffizienten: 164.300 M-1 cm-1 und 138.600 M-1 cm-1 wurden für TBA bzw. cTBA verwendet. - TBA- und cTBA-Stammlösungen bei -20 °C lagern (monatelang unter diesen Bedingungen).
- Verbindung B-CeP 1
HINWEIS: Die Verbindung B-CeP 1 wird wie zuvor berichtetsynthetisiert 6.- Bereiten Sie die B-CeP 1 Stammlösung bei ~10 mM vor. Wiegen Sie ~1 mg der lyophilisierten Verbindung mit einer Analysenwaage in einem Abzug und lösen Sie sie in 100% Dimethylsulfoxid (DMSO).
- Berechnen Sie die genaue Wirkstoffkonzentration auf der Grundlage der tatsächlich verwendeten Menge an Verbindung und DMSO (d = 1,1 g/cm3).
HINWEIS: Behandeln Sie die Verbindung immer mit Handschuhen (sowohl bei Gefriertrocknung als auch beim Auflösen in DMSO)13,14.
Tabelle 1: In diesem Protokoll verwendete Oligonukleotidstrukturen. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
2. Faltung von Nukleinsäurekonstrukten
- Vorbereitung von Puffern
- Es wird eine BPE-Pufferlösung (Biphosphat-Ethylendiamintetraessigsäure [EDTA], 5x: 2 mM NaH2PO 4, 6 mM Na2HPO4, 1 mMNa2EDTA, pH7,4) und eine Lösung von 500 mM KCl in Reinstwasser hergestellt. Filtern Sie die Lösungen durch Filter mit einer Porengröße von 0,22 μm.
HINWEIS: Die besten Ergebnisse erzielen Sie, wenn Sie frisch zubereitete Lösungen verwenden. BPE-Puffer kann bei 4 °C bis zu 15 Tage gelagert werden.
- Es wird eine BPE-Pufferlösung (Biphosphat-Ethylendiamintetraessigsäure [EDTA], 5x: 2 mM NaH2PO 4, 6 mM Na2HPO4, 1 mMNa2EDTA, pH7,4) und eine Lösung von 500 mM KCl in Reinstwasser hergestellt. Filtern Sie die Lösungen durch Filter mit einer Porengröße von 0,22 μm.
- Falten von G4-TBA-, ssTBA- und dsTBA-Proben im Heat-Refolding-Verfahren
HINWEIS: Kaliumkationen sind notwendig, um die G4-Struktur (G4-TBA) zu falten. Kaliumkationen dürfen nicht in die Faltungslösung der Kontrollen ssTBA und dsTBA gegeben werden.- Bereiten Sie 40 μl einer 4 μM-Lösung von G4-TBA in 1x BPE und 100 mM KCl vor. Denaturieren Sie die Oligonukleotid-G4-TBA-Lösung, indem Sie das Röhrchen 5 Minuten lang auf 95 °C erhitzen und langsam auf Raumtemperatur (RT) abkühlen, damit sich die TBA in G-Quadruplexe falten kann.
- Bereiten Sie 40 μl einer 4 μM Lösung von ssTBA in 1x BPE vor. Führen Sie die in Schritt 2.2.1 beschriebene Hitzerückfaltung durch, um TBA in seiner einzelsträngigen Form zu falten.
- Bereiten Sie 40 μl einer 4 μM-Lösung von dsTBA vor, indem Sie äquimolare Mengen von TBA und cTBA in 1x BPE mischen. Führen Sie das oben beschriebene Verfahren der Wärmerückfaltung (Schritt 2.2.1) durch, damit TBA zu seiner komplementären Sequenz cTBA glüht und die doppelsträngige Form von TBA bildet.
HINWEIS: Das endgültige Volumen jeder Faltlösung basiert auf der Anzahl der Proben für die Sondierungsreaktionen, wobei zu berücksichtigen ist, dass für jede Probe 5 μl 4 μM-Lösung benötigt werden. Bereiten Sie von jeder Lösung ein wenig überschüssiges Volumen vor, um Pipettierfehler zu vermeiden.
3. Sondierungsreaktionen
HINWEIS: Sondierungsreaktionen müssen unmittelbar nach dem Hitzerückfaltungsverfahren durchgeführt werden.
- Wenn die Faltlösungen von G4-TBA, ssTBA und dsTBA auf RT abgekühlt sind, wird eine Kurzschleuderzentrifugation (7.000 × g für 5-8 s bei RT) durchgeführt und die Sondierungsreaktionen gestartet.
- Bereiten Sie 21 leere 0,5-ml-Autoklavierröhrchen vor. Organisieren Sie sie in drei Sätzen mit jeweils sieben Röhrchen im Rack für Laborproben, wie in Tabelle 2 angegeben.
HINWEIS: Jeder Spaltensatz entspricht den drei verschiedenen TBA-Faltungsbedingungen G4-TBA, ssTBA und dsTBA. Jede Reihe entspricht drei verschiedenen Inkubationszeiten. Jede Zelle innerhalb der Säule entspricht der endgültigen B-CeP 1-Sondenkonzentration (Tabelle 2). Achten Sie darauf, die Röhrchen deutlich zu beschriften. - Geben Sie 3 μl Reinstwasser in jedes Röhrchen.
- Geben Sie 5 μl gefaltetes G4-TBA in jedes Röhrchen des ersten Satzes (Schritt 3.2). Geben Sie 5 μl gefaltetes ssTBA in jedes Röhrchen des zweiten Sets. Geben Sie 5 μl gefaltetes dsTBA in jedes Röhrchen des dritten Sets.
- Verdünnen Sie die B-CeP 1 Stammlösung auf 250 μM und 25 μM in Reinstwasser.
HINWEIS: Verdünnungen der chemischen B-CeP 1-Sonde müssen frisch zubereitet und sofort mit dem DNA-Substrat umgesetzt werden, um konkurrierende Reaktionen mit Wasser zu vermeiden. - Die Proben werden mit 2 μl der entsprechenden B-CeP 1-Verdünnung (25 μM und 250 μM) versetzt. Die Verbindung wird in den drei Kontrollproben (C) durch Reinstwasser ersetzt, um die unterschiedlich gefalteten TBAs in Abwesenheit der Verbindung zu analysieren. Alle Proben werden bei 37 °C inkubiert.
- Nach 1 h, 4 h und 15 h Inkubation wird die Reaktion gestoppt, indem die Röhrchen bis zum nächsten Schritt bei -20 °C platziert werden.
HINWEIS: Die Proben können unter diesen Bedingungen einige Tage gelagert werden. - Trocknen Sie die Proben in einer Vakuumzentrifuge.
HINWEIS: Die getrockneten Proben können wochenlang bei -20 °C gelagert werden, bevor mit der PAGE-Analyse fortgefahren wird.
Tabelle 2: Proben für die Sondierungsreaktionen (Strukturen, Sondenkonzentrationen und Inkubationszeit). Jeder Spaltensatz entspricht den drei verschiedenen TBA-Faltungsbedingungen (G4-TBA, ssTBA und dsTBA). Jede Reihe entspricht drei verschiedenen Inkubationszeiten (1, 4, 15 h). Jede Zelle innerhalb der Säule entspricht der endgültigen B-CeP 1-Sondenkonzentration (5 oder 50 μM). Die Kontrolle (C) für jeden Satz entspricht einer Probe der unterschiedlich gefalteten TBAs, die für die längere Zeit (15 h) in Abwesenheit von Verbindung inkubiert wurden. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
4. Hochauflösende SEITE
- Herstellung der denaturierenden Polyacrylamidlösung
HINWEIS: Bereiten Sie im Voraus 500 ml 20%ige denaturierende Polyacrylamid-Gellösung vor. Etwa 80 ml dieser Lösung werden für jedes Experiment verwendet. Verwenden Sie eine Braunglasflasche oder decken Sie eine Glasflasche mit Aluminiumfolie ab, um die Lösung bei RT aufzubewahren.
ACHTUNG: Polyacrylamid ist neurotoxisch. Tragen Sie bei allen Schritten der Gelvorbereitung und des Gießens Handschuhe und einen Laborkittel. Entsorgen Sie polymerisiertes Acrylamid in einem geeigneten Karton für kontaminierte Materialien.- 210 g Harnstoff werden in einem 1-Liter-Becherglas eingewogen. 250 ml 40%ige Acrylamid/Bisacrylamid-Lösung (19/1) und 50 ml 10x FSME (890 mM Tris-HCl, 890 mM Borat, 20 mM EDTA, pH 8) zugeben.
- Stellen Sie das Becherglas auf eine Rührplatte und mischen Sie die Lösung mit einem Rührstab. Decken Sie das Becherglas während des Mischens mit Aluminiumfolie ab, um Spritzer und Verunreinigungen zu vermeiden.
- Mischen Sie die Lösung, bis sich der Harnstoff vollständig aufgelöst hat und die Lösung klar ist.
HINWEIS: Dieser Schritt kann viele Stunden dauern. Um die Auflösung von Harnstoff zu fördern, fügen Sie ein kleines Aliquot Wasser hinzu, ohne das gewünschte Endvolumen zu überschreiten. - Entfernen Sie den Rührstab. Gießen Sie die Lösung in einen Zylinder und fügen Sie Wasser zu einem genauen Endvolumen von 500 ml hinzu.
- Aufbau der Gel-Apparatur
- Reinigen Sie zwei Platten (eine gekerbte und eine ungekerbte) mit 70%igem Ethanol, lassen Sie sie trocknen und behandeln Sie die Platten dann mit einer Dimethyldichlorsilanlösung.
HINWEIS: Die Silanisierung kann übersprungen werden, obwohl sie die Freisetzung des Gels von einer der Platten unterstützt, wenn das Gel-Sandwich auseinandergenommen wird.
VORSICHT: Fassen Sie die Silanisierungslösung mit Handschuhen an und führen Sie die Plattenbehandlung mit dieser Lösung in einem Abzug durch. - Platzieren Sie die 0,4-mm-Abstandshalter entlang der langen Kanten der längeren Platte, legen Sie die kurze Platte aufeinander und richten Sie die beiden Platten unten aus.
- Lege mehrere Lagen Papierklebeband entlang aller Kanten außer der Oberseite.
- Um Leckagen während des Gießens zu vermeiden, fügen Sie eine zusätzliche Schicht Klebeband auf die Unterseite des Gels auf.
- Befestigen Sie die Seiten des Glassandwiches mit sauberen Klammern gemäß den Anweisungen des Lieferanten (verschiedene Lieferanten verwenden leicht unterschiedliche Apparaturen, Sandwichklemmen und Dichtungen).
- Reinigen Sie zwei Platten (eine gekerbte und eine ungekerbte) mit 70%igem Ethanol, lassen Sie sie trocknen und behandeln Sie die Platten dann mit einer Dimethyldichlorsilanlösung.
- Gießen des Gels
HINWEIS: Gießen Sie das Gel bei RT (25 °C), da die Polyacrylamidpolymerisation temperaturempfindlich ist.- Unmittelbar vor Gebrauch werden 80 ml der zuvor hergestellten denaturierenden Polyacrylamidlösung (Schritt 4.1), 450 μl einer 10%igen m/V-Ammoniumpersulfatlösung (APS) und 45 μl Tetramethylethylendiamin (TEMED) in ein Becherglas gegeben.
- Mischen Sie die Lösung und gießen Sie sie mit einer 50-ml-Spritze schnell zwischen die Glasplatten. Führen Sie den Kamm mit der gewünschten Anzahl von Vertiefungen zwischen die Glasplatten ein, um Blasen zu vermeiden. Füge bei Bedarf eine Gellösung hinzu, um das Sandwich vollständig zu füllen. Setzen Sie vier Klammern auf den Kamm, um sie nach unten zu drücken und eine gleichmäßige Verteilung der Vertiefungen zu ermöglichen.
- Lassen Sie das Gel mindestens 45 Minuten lang polymerisieren.
- Das Gel laufen lassen
- Entfernen Sie nach der Polymerisation alle Klammern und Papierbandschichten. Entfernen Sie den Kamm langsam und spülen Sie die Vertiefungen gründlich mit destilliertem Wasser aus.
- Befolgen Sie die Anweisungen des jeweiligen Lieferanten, um das Gel-Sandwich korrekt in das vertikale Gelelektrophoresegerät zu legen.
- FSME-Laufpuffer (1x: 89 mM Tris-HCl, 89 mM Borat, 2 mM EDTA, pH 8) in deionisiertem Wasser vorbereiten und sowohl den oberen als auch den unteren Behälter mit dem Puffer füllen.
- Erwärmen Sie die Platten, indem Sie einen Vorlauf der Gelelektrophorese für mindestens 30 Minuten bei 50 W durchführen.
- Denaturierungsgel-Ladepuffer (DGLB: 1 M Tris-HCl, 80 % Formamid, 50 % Glycerin, 0,05 % Bromphenolblau) in Reinstwasser herstellen.
HINWEIS: GLB hilft dabei, die Bewegung der Oligonukleotidproben in das Gelsystem zu verfolgen und ermöglicht es, die Proben in die Vertiefungen des Gels zu laden. Das Vorhandensein des Denaturierungsmittels Formamid ermöglicht es DNA-Spezies, sich nach Größe zu trennen, selbst in einem nicht denaturierenden PAGE. - Die getrockneten Proben (Proben aus Schritt 3.8) werden in 5 μl DGLB resuspendiert.
- Reinigen Sie vor dem Laden der Proben die Vertiefungen mit einer kleinen Spritze und dem TBE-Puffer in der oberen Pufferkammer, um den Harnstoff aus den Vertiefungen zu entfernen.
HINWEIS: Wiederholen Sie diesen Schritt mehrmals, um die Vertiefungen genau zu reinigen und so schwer zu interpretierende Bänder zu vermeiden. - Laden Sie die Proben in die sauberen Vertiefungen und notieren Sie sich die Reihenfolge der Beladung.
- Lassen Sie die Gelelektrophorese 2 Stunden lang bei 50 W laufen, oder zumindest so lange, bis der Bromphenolblaufarbstoff 2/3 des Gels heruntergelaufen ist.
- Bildgebung des Gels
- Schalten Sie nach der Elektrophorese die Stromversorgung aus, entfernen Sie das Glassandwich und reinigen Sie die Gläser.
- Detektieren Sie die Fluoreszenz der FAM-markierten Oligonukleotidbanden durch Scannen mit einem Gel-Imager.
Ergebnisse
Abbildung 2 zeigt ein repräsentatives Ergebnis eines durchgeführten chemischen Mapping-Assays, wie im Protokoll beschrieben, mit B-CeP 1 auf dem TBA-Oligonukleotid, das in drei verschiedenen Strukturen gefaltet ist. Die G-Quadruplex-Anordnung von TBA (G4-TBA) wurde durch Faltung des Oligonukleotids in BPE und in Gegenwart des K+-Kations erhalten, während die einzelsträngige Form derselben TBA-Sequenz (ssTBA) in Abwesenheit von Kalium gefaltet wurde. Das doppelsträngige Konstr...
Diskussion
G-Quadruplexe sind Nukleinsäure-Sekundärstrukturen, die sich typischerweise innerhalb von Guanin-reichen DNA-Sequenzen falten und aufgrund ihrer Assoziation mit genetischer Kontrolle und Krankheiten wichtige Forschungsziele sind. Die chemische Kartierung durch B-CePs ist ein nützliches Protokoll für die Charakterisierung von DNA-G4s, das verwendet werden kann, um die Guaninbasen zu identifizieren, die an der Bildung von G-Tetrades unter physiologischen Salzbedingungen beteiligt sind.
Die i...
Offenlegungen
Die Autoren haben keine Interessenkonflikte offenzulegen.
Danksagungen
Diese Arbeit wurde vom Institut für Pharmazeutische und Pharmakologische Wissenschaften der Universität Padua (PRIDJ-BIRD2019) unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| Acrylamide/bis-acrylamide solution 40% | Applichem | A3658 | R45-46-20/21-25-36/38-43-48/23/ 24/25-62 |
| Ammonium per-sulfate (APS) | Sigma Aldrich | A7460 | |
| Analytical balance | Mettler Toledo | ||
| Autoclave | pbi international | ||
| Boric acid | Sigma Aldrich | B0252 | |
| Bromophenol blue Brilliant blue R | Sigma Aldrich | B0149 | |
| di-Sodium hydrogen phosphate dodecahydrate | Fluka | 71649 | |
| DMSO | Sigma Aldrich | 276855 | |
| DNA oligonucleotides | Integrated DNA Technologies | synthesis of custom sequences | |
| EDTA disodium | Sigma Aldrich | E5134 | |
| Formamide | Fluka | 40248 | H351-360D-373 |
| Gel imager | GE Healtcare | STORM B40 | |
| Glycerol | Sigma Aldrich | G5516 | |
| Micro tubes 0.5 mL | Sarstedt | 72.704 | |
| Potassium Chloride | Sigma Aldrich | P9541 | |
| Sequencing apparatus | Biometra | Model S2 | |
| Silanization solution I | Fluka | 85126 | H225, 314, 318, 336, 304, 400, 410 |
| Sodium phosphate monobasic | Carlo Erba | 480086 | |
| Speedvac concentrator | Thermo Scientific | Savant DNA 120 | |
| TEMED | Fluka | 87689 | R11-21/22-23-34 |
| Tris-HCl | MERCK | 1.08387.2500 | |
| Urea | Sigma Aldrich | 51456 | |
| UV-Vis spectrophotometer | Thermo Scientific | Nanodrop 1000 |
Referenzen
- Davis, J. T. G-quartets 40 years later: from 5'-GMP to molecular biology and supramolecular chemistry. Angewandte Chemie. 43 (6), 668-698 (2004).
- Varshney, D., Spiegel, J., Zyner, K., Tannahill, D., Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nature Reviews Molecular Cell Biology. 21 (8), 459-474 (2020).
- Chambers, V. S., et al. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nature Biotechnology. 33 (8), 877-881 (2015).
- Zuravka, I., Sosic, A., Gatto, B., Gottlich, R. Synthesis and evaluation of a bis-3-chloropiperidine derivative incorporating an anthraquinone pharmacophore. Bioorganic & Medicinal Chemistry Letters. 25 (20), 4606-4609 (2015).
- Zuravka, I., Roesmann, R., Sosic, A., Gottlich, R., Gatto, B. Bis-3-chloropiperidines containing bridging lysine linkers: Influence of side chain structure on DNA alkylating activity. Bioorganic & Medicinal Chemistry. 23 (6), 1241-1250 (2015).
- Zuravka, I., et al. Synthesis and DNA cleavage activity of bis-3-chloropiperidines as alkylating agents. ChemMedChem. 9 (9), 2178-2185 (2014).
- Sosic, A., Gottlich, R., Fabris, D., Gatto, B. B-CePs as cross-linking probes for the investigation of RNA higher-order structure. Nucleic Acids Research. 49 (12), 6660-6672 (2021).
- Sosic, A., et al. Bis-3-chloropiperidines targeting TAR RNA as a novel strategy to impair the HIV-1 nucleocapsid protein. Molecules. 26 (7), 1874 (2021).
- Sosic, A., et al. In vitro evaluation of bis-3-chloropiperidines as RNA modulators targeting TAR and TAR-protein interaction. International Journal of Molecular Sciences. 23 (2), 582 (2022).
- Sosic, A., et al. Direct and topoisomerase II mediated DNA damage by bis-3-chloropiperidines: The importance of being an earnest G. ChemMedChem. 12 (17), 1471-1479 (2017).
- Bock, L. C., Griffin, L. C., Latham, J. A., Vermaas, E. H., Toole, J. J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature. 355 (6360), 564-566 (1992).
- Paborsky, L. R., McCurdy, S. N., Griffin, L. C., Toole, J. J., Leung, L. L. The single-stranded DNA aptamer-binding site of human thrombin. The Journal of Biological Chemistry. 268 (28), 20808-20811 (1993).
- Carraro, C., et al. Behind the mirror: chirality tunes the reactivity and cytotoxicity of chloropiperidines as potential anticancer agents. ACS Medicinal Chemistry Letters. 10 (4), 552-557 (2019).
- Carraro, C., et al. Appended aromatic moieties in flexible bis-3-chloropiperidines confer tropism against pancreatic cancer cells. ChemMedChem. 16 (5), 860-868 (2021).
- Kypr, J., Kejnovska, I., Renciuk, D., Vorlickova, M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Research. 37 (6), 1713-1725 (2009).
- Onel, B., Wu, G., Sun, D., Lin, C., Yang, D. Electrophoretic mobility shift assay and dimethyl sulfate footprinting for characterization of G-quadruplexes and G-quadruplex-protein complexes. Methods in Molecular Biology. 2035, 201-222 (2019).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten