
Method Article
Detección de Tilapia lago Virus usando RT-PCR convencional y SYBR Green RT-qPCR
En este artículo
Resumen
Este protocolo diagnóstico de Virus de lago de Tilapia (TiLV) en tejidos de tilapia utilizando metodologías de RT-PCR. El método completo se describe de la disección del tejido para extracción de RNA total, seguida de la síntesis de cDNA y detección de TiLV por PCR convencional o PCR cuantitativa utilizando dsDNA vinculante un tinte fluorescente vinculante.
Resumen
El objetivo de este método es facilitar la detección rápida, sensible y específica de Virus de lago de Tilapia (TiLV) en los tejidos de la tilapia. Este protocolo se puede utilizar como parte de programas de vigilancia, las medidas de bioseguridad en laboratorios de investigación básica TiLV. El patrón oro del diagnóstico de virus implica típicamente el aislamiento del virus seguido por técnicas complementarias como la reacción en cadena de polimerasa de transcripción inversa (RT-PCR) para la verificación de más. Esto puede ser engorroso, lento y requiere típicamente pesadamente infectadas con el virus de muestras de tejido. El uso de RT-cuantitativo (q) PCR en la detección de virus es ventajoso debido a su naturaleza cuantitativa, alta sensibilidad, especificidad, escalabilidad y su rápido tiempo de respuesta como resultado. Aquí, el método completo de PCR basado en enfoques para TiLV detección se describe, de tilapia órgano seccionamiento total ácido ribonucleico (ARN) extracción con una solución de tiocianato-fenol-cloroformo guanidínicos, cuantificación de RNA, seguido de una PCR de dos pasos Protocolo que entrañan, síntesis de ácido desoxirribonucleico complementario (cDNA) y detección de TiLV por PCR convencional o identificación cuantitativa mediante qPCR utilizando verde de SYBR tinte. PCR convencional requiere pasos de post-PCR y simplemente le informará sobre la presencia del virus. Este último enfoque permitirá una cuantificación absoluta de TiLV hasta tan poco como 2 copias y por lo tanto es excepcionalmente útil para la diagnosis de TiLV en casos subclínica. Una descripción detallada de los dos métodos PCR, resultados representativos de dos laboratorios y una discusión cuidadosa de los parámetros críticos de ambos han sido incluidas para que investigadores y diagnosticadores encuentran su más adecuada y aplicable método de detección TiLV.
Introducción
El suministro de pescado per cápita global alcanzó un nuevo récord de 20 kg en 2014 y esto era debido a un crecimiento vigoroso en la acuicultura. La acuicultura sigue siendo uno de los mayor crecimiento pienso producir sectores en todo el mundo y es el sector de producción de alimentos sólo animales que está creciendo más rápido que la población humana1. Cichilds tilapina comprenden el segundo pez de agua dulce cultivado en todo el mundo con una producción mundial total de 6,4 millones de toneladas (MT) y un valor estimado de 9,8 billones de dólares en 20152. El top 10 los productores de tilapia son China (1.78 MT), Indonesia (1.12 MT) y Egipto (0.88 MT), seguido por Bangladesh, Vietnam, Filipinas, Brasil, Tailandia, Colombia y Uganda2. Se espera que la producción mundial de tilapia será alrededor de 7.3 MT por 20303. Tilapia se han convertido en una fuente de alimento mundial importante, no sólo porque son una fuente barata de proteína4 sino también porque son fáciles de criar en capacidad bajo una amplia gama de agua y el clima condiciones5,6. Hace unas décadas se creía que había pocas enfermedades comercialmente importantes que amenazan el cultivo de tilapia, pero esto no es cierto. Una enfermedad viral llamada enfermedad de virus de lago de tilapia (TiLVD) es la primera epidemia de la enfermedad siempre crítico encontrado en tilapia y toda la industria está en riesgo. Esta enfermedad tiene graves consecuencias socio-económicas y es una amenaza directa sobre la seguridad alimentaria de millones de personas en África7, Asia y América del sur. En el comienzo del 2018, la Organización Mundial de Sanidad Animal (OIE) informó que el agente etiológico de esta enfermedad, TiLV, había sido detectado oficialmente en tres continentes, cubriendo ocho países8 y desde la ficha de este patógeno actualizado se ha informado más de TiLV en Tanzania, Uganda9, Indonesia10, Taiwán11 y Perú12. TiLV es un virus de ARN monocatenario novela se describe para ser un ortomixo-como virus ya que contiene una variedad de características que recuerdan a otros orthomyoxoviruses como gripe o infecciosa salmon anemia virus (ISAV)13. Primero fue identificado en las secuelas de pérdidas masivas de tilapias silvestres y cultivados en el lago de Galilea, Israel14. Después de eso, los brotes de enfermedades similares que se refiere como mortalidades de verano y un mes síndrome de mortalidad asociado a infección TiLV informaron en tilapia del Nilo (Oreochromis niloticus) en Egipto15 y Nilo y la tilapia híbrida roja (Oreochromis spp.) en Tailandia16, respectivamente. Detección de los virus animales acuáticos históricamente se realiza por aislamiento del virus en cultivo celular y crecimiento. Varias líneas celulares han sido probadas para la propagación y el aislamiento de TiLV incluyendo, E-11 en células de snakehead peces (striatus de Ophiocephalus)17,18, OmB y TmB procedentes de Oreochromis mossambicus18y OnlB y OnlL originarios del Nilo tilapia (o. niloticus)19. Mientras que el cultivo de virus tiene la ventaja que proporciona material para otros experimentos, tiene la desventaja que requiere al menos 4-7 días para observar la formación de efectos citopáticos (CPE) y crucial, diversos virus piscine, más apto para repetición puede ser propagado y producir CPE similar.
En las últimas décadas, ha habido un movimiento lejos de tradicionales, a menudo lentos métodos diagnósticos como el cultivo celular, serología y detección del antígeno y reemplazo con ácido nucleico más rápido y más sensible en la detección de pruebas20, 21. Esto es evidente por el hecho de que muchos ensayos de qPCR se han desarrollado como importantes métodos de diagnóstico para un sinfín de enfermedades virales en animales acuáticos, como vais22,23, virus hemorrágico viral del septicaemia (VHSV)24 ,25betanodavirus26,27 Alfavirus28, pescado iridovirus29, migración herpesvirus 1 (AngHV1)30y Lymphocystis enfermedad virus (LCDV)31 . Métodos robustos para vigilancia diagnóstico y patógeno son urgentemente necesarios para reducir la propagación de TiLV. Estos métodos deberían permitir la detección temprana de la infección antes de desarrollan signos clínicos y la detección de virus bajo cargas. Hasta la fecha, distintos protocolos PCR como RT-PCR14,32, RT-PCR anidada18, RT-PCR semi-anidada33y RT-qPCR32,34 han sido desarrollados para la detección de TiLV en tejidos de peces. Una comparación de RT-qPCR y aislamiento viral en líneas celulares susceptibles de detección TiLV reveló que RT-qPCR fue 1.000 veces más sensible que el aislamiento de virus32. Aunque cada protocolo PCR publicado ha divulgado diferentes sensibilidades para la detección de TiLV, la mayoría de ensayos son muy sensibles con los límites de detección de copias virales en 7.5 copias33, 7 copias18 o 2 copias32 por reacción.
El objetivo de este artículo de métodos es explicar, detalladamente, cómo llevar a cabo ensayos de detección TiLV, comenzando con la colección de tejidos de tilapia, a la extracción de RNA total, síntesis de cDNA y luego TiLV PCR específico basado en ensayos. Específicamente, se han descrito protocolos integrales de RT-PCR convencional y también basados en verde SYBR RT-qPCR para apelar a una amplia gama de científicos con el objetivo de detectar TiLV. El primero es menos sensible pero suele ser una opción más barata de detección. Este último requiere de infraestructura más elaborada como una máquina PCR cuantitativa y reactivos más costosos, pero tiene las ventajas de ser cuantitativa, rápida y muy sensible, lo que significa que puede ser utilizado para la detección de TiLV en sub-clínicamente peces infectados. La RT-PCR y RT-qPCR protocolos fueron realizados en dos laboratorios diferentes con distintas cepas geográficas de TiLV y el resaltado de resultados incluye la sensibilidad y reproducibilidad de los ensayos aquí descritos.
Protocolo
El protocolo de uso de animales para este estudio fue aprobado por el Comité de ética de Kasetsart University Animal bajo el permiso número ACKU 59-veterinario-016.
Nota: Consulte la Tabla de materiales para más información sobre los reactivos y equipos sugeridos para este protocolo.
1. tejido muestra colección
- Eutanasia a los peces con una sobredosis de aceite de clavo (el volumen depende del tamaño del pescado y la concentración de productos, generalmente más de 3 mL/L). Sumerja una cuarta parte de las tijeras pinzas y mayo en etanol al 95% (v/v) seguido de quema el equipo utilizando un mechero de alcohol para esterilizar el equipo.
Nota: Tricaine methanesulfonate (MS-222) se puede utilizar en lugar de aceite de clavo de olor. - Encuentra el hígado y cortar un pedazo pequeño (aproximadamente de 20-100 mg) o recoger 200 μL de moco con cubierta de vidrio o lámina quirúrgica para eliminar el moco de anterior a posterior de los pescados de la tilapia y coloque las muestras en un tubo de microcentrífuga de 1,5 mL.
- Process muestras inmediatamente, almacenan en un RNA estabilizar solución o moverlos a-80 ° C hasta su uso posterior.
Nota: La tarea más importante en el trabajo con RNA está preparando las moléculas de ARN intactas y mantenerlos indemnes a lo largo de cualquier manejos posteriores. El RNA es naturalmente más sensibles al daño de ADN. Extracción y aislamiento de ARN total de las células del tejido requiere técnica de laboratorio cuidadoso; tomar todas las disposiciones para prevenir la contaminación de la Rnasa por guantes, usar agua libre de ARNasa, reactivos, equipos, artículos de plástico, vidrio, espacio de trabajo y mediante el uso de extremidades de filtro para el pipeteo.
2. guanidínicos tiocianato - fenol - cloroformo extracción de ARN
- Añadir 1 mL de solución de monofásicos que contienen fenol con isotiocianato de guanidina en un tubo que contiene la muestra de tejido de la sección 1.
PRECAUCIÓN: Esta solución es muy tóxica y debe manipularse con cuidado en una campana de flujo laminar con equipo de protección y con el uso de los adecuados guantes protectores, gafas, ropa y seguridad. - Moler la muestra de tejido mediante un homogeneizador de tejidos Maja hasta homogéneo.
Nota: Las muestras pueden también ser homogeneizadas utilizando un homogeneizador de poder junto con granos de cerámica. Asegurar que la muestra de tejido es totalmente homogeneizada antes de proceder al siguiente paso o parar el protocolo aquí y almacenar las muestras totalmente homogeneizadas a-80 ° C hasta su uso posterior. - Añadir 200 μL de cloroformo para separación de fases.
Cloroformo de precaución es un potencial narcótico y es extremadamente peligroso. Debe manejarse con cuidado en una campana de flujo laminar con equipo de protección, así como con el uso de los adecuados guantes protectores, gafas, ropa y seguridad. Como un menos tóxicos alternativos, 1-Bromo-3-cloropropano puede también ser utilizado.
Nota: La escala de los volúmenes hacia arriba o hacia abajo en su caso. Por ejemplo, si sólo se utilizó 500 μl de solución monofásicos que contienen fenol con isotiocianato de guanidina, entonces sólo añadir 100 μl de cloroformo en este paso.- Muestras de mezclar bien por inversión durante 15 s.
- Incubar las muestras durante 3 min a temperatura ambiente (RT).
- Centrifugar durante 15 min a 12.000 x g a 4 ° C.
Nota: Debe haber una clara separación en una fase orgánica inferior, un blanco interfase y una fase acuosa superior que contiene RNA. Esta fase superior es normalmente incolora pero dependiendo del tipo y cantidad de tejido homogeneizado, puede tener una apariencia rosa luz. - Transferir la capa acuosa superior (aproximadamente 500 μl) a un tubo de microcentrífuga fresco sin alterar la interfase.
Nota: no intente transferir la fase acuosa toda, dejar una pequeña cantidad para evitar cualquier posible contaminación del RNA que contiene la fase acuosa con el orgánico o interfase. - Agregar 1 volumen de isopropanol 100% para precipitar el ARN.
- Opcionalmente, si se utilizan cantidades muy pequeñas de tejido, luego añadir 1 μl (5-10 μg) de glucógeno libre de Rnasa a cada muestra para promover la precipitación de RNA eficiente. Esto para facilitar la identificación de la pelotilla de RNA paso 2.8.
Nota: El glucógeno actúa como portador del ARN y evitará que pequeñas cantidades de ARN se adhiera a la pared del tubo. - Mezcla los tubos por inversión varias veces.
- Almacenar las muestras por 2 h para toda la noche a-20 ° C.
- Opcionalmente, si se utilizan cantidades muy pequeñas de tejido, luego añadir 1 μl (5-10 μg) de glucógeno libre de Rnasa a cada muestra para promover la precipitación de RNA eficiente. Esto para facilitar la identificación de la pelotilla de RNA paso 2.8.
- Centrifugar las muestras durante 15 min a 12.000 g y 4 ° C.
- Eliminar el sobrenadante, teniendo cuidado de no desalojar el pellet de RNA en el fondo del tubo de microcentrífuga.
- Añadir 1 mL de etanol al 75% (v/v) y mezclar las muestras de RNA invirtiendo el tubo varias veces.
- Centrifugar 15 min a 10.000 x g a 4 ° C.
Nota: El protocolo se puede detener aquí y las muestras que el pellet de RNA en etanol al 75% pueden ser almacenadas a-20 ° C hasta su uso posterior. - Eliminar el sobrenadante, que atenta no para desalojar el pellet de RNA en el fondo del tubo de microcentrífuga.
- Opcionalmente, repita los pasos 2.9-2.11 utilizando etanol al 70% (v/v). Cuidadosamente lavar el pellet de RNA minimizará cualquier sal o remanente de contaminantes que pueda interferir voluntad sensibles aplicaciones posteriores.
- Extraer el etanol restante con una pipeta y luego secar el pellet de RNA a temperatura de ambiente durante no más de 5 a 10 min.
Nota: pellets secados demasiado será difíciles que vuelva a suspender. - Añadir 30-60 μL de agua libre de ARNasa, previamente calentado a 55-60 ° C para solubilizar el precipitado de RNA.
- Coloque el ARN en hielo para su uso inmediato o tienda a-80 ° C para su uso posterior.
3. cuantificar la concentración de RNA utilizando un espectrofotómetro Micro volumen
- Cambiar la configuración del espectrofotómetro a RNA.
- Utilice 1-2 μl de agua libre de ARNasa como un espacio en blanco.
- Usar 1-2 μl de cada muestra de RNA para evaluar la cantidad de RNA.
- Registrar las lecturas a 230 nm, 260 nm y 280 nm para cada muestra.
- Diluir el RNA a 200 ng/μl con agua libre de ARNasa.
4. síntesis de ADN complementario (cDNA) utilizando RNA total
- 1 μg de ARN total de protocolo 2, 2 μm oligo (dT), mezcla de dNTPs de 0,5 mM de la mezcla y llevar el volumen final de 10 μl con agua libre de nucleasa. Para esto, prepare un RT-master-mix según el número de muestras y controles a probar.
Nota: Los controles son una muestra de la transcriptasa reversa de menos (-RT) en donde la enzima RT es reemplazada con agua libre de nucleasas (ver paso 4.3) y una sin plantilla control (NTC) en agua libre de nucleasa se agrega a la mezcla maestra en vez de plantilla del RNA.- Mezclar las muestras bien mediante pipeteo seguido por una breve centrifugación.
- Incubar las muestras a 65 ° C por 5 min seguido de una incubación de 2 minutos en hielo.
- Brevemente centrifugar las muestras a recoger todo el líquido en la parte inferior de los tubos.
- Añadir 1 Tampón de transcriptasa reversa x 100 U de la transcriptasa reversa y el volumen final de cada muestra a 20 μl con agua libre de nucleasa.
- Mezclar las muestras bien mediante pipeteo seguido por una breve centrifugación.
- Incubar las muestras a 42 ° C durante 60 min seguido de 85 ° C durante 5 minutos.
- Diluir el cDNA sintetizado a una concentración deseada mediante la adición de un volumen adecuado de agua libre de nucleasas y coloque el cDNA en hielo para su uso inmediato o almacenar a-20 ° C para su uso posterior.
5. TiLV PCR convencional
- Utilice el cDNA, muestras y controles, generados en protocolo sección 4 como plantillas para una reacción de polimerización en cadena usando cualquiera de los pares establecido cartilla detallados en la tabla 1, junto con una ADN polimerasa de elección.
Nota: Un control adicional de la plantilla de no (NTC) se debe incluir aquí mediante la sustitución de cDNA para agua libre de nucleasas en la reacción de PCR. Un control positivo, si está disponible, también debería incluirse que comprende previamente verificado TiLV muestras positivas o el correspondiente fragmento de cDNA TiLV clonada en a un plásmido. - Preparar una mezcla maestra de PCR según las directrices del sistema de polimerasa de la DNA en el uso y el número de muestras y controles a probar. Esta mezcla debe incluir el primer avance, primer revés, dNTPs, MgCl2 y polimerasa de la DNA seleccionada, junto con su búfer.
- Según las directrices de la polimerasa de ADN seleccionada, combinar el volumen señalado de mezcla maestra con sugerido cantidad de cDNA muestras y muestras de control.
Nota: Preparar un 0,5 x exceso de reacción es a menudo beneficioso puesto que algunos master-Mix se pierde durante el pipeteo. - Realizar PCR ciclismo condiciones según las directrices del sistema utilizado ADN polimerasa y usando una apropiada temperatura de recocido de las cartillas en uso (tabla 1). Generalmente, dicho programa implica una desnaturalización inicial a 95 ° C para 2-5 min, seguido de 30-40 ciclos de desnaturalización a 95 ° C por 30 s, recocido a la temperatura recomendada de 30 s y elongación a 72 ° C por 30 s, seguido por una elongación final a 72 ° C durante 5-10 min.
- Carga de 5-15 μl de cada reacción de PCR y una escalera apropiada de DNA en a los pocillos de un gel de agarosa al 1-2%, dependiendo del tamaño de producto PCR esperado. Separar el ADN amplificado por electroforesis del gel y el gel de la mancha por bromuro de etidio (EthBr) para facilitar la visualización de las bandas de ADN del tamaño esperado (tabla 1) en una máquina de documentación de gel con luz UV.
PRECAUCIÓN: EtBr es tóxica; deben manipularse con cuidado usando la ropa protectora adecuada y guantes de seguridad.
| Segmento de destino TiLV genoma | Primer avance 5' - 3' | Primer revés 5' - 3' | Tamaño del producto PCR (bp) | Tm ° C | Referencia original | ||||
| 1 | CCAAACGTTATCTCTTAATTACGCAC | GCAAATATTTCTCTCATTCGCCT | 1641 | 50 | Surachetpong et al., 2017 | ||||
| 1 | CCTCATTCCTCGTTGTGTAAGT | AGGAGTTGCTGTTGGGTTATAG | 1000 | 62 | Mugimba et al., 2018 | ||||
| 2 | ACTCTCTATTACCAAATACATTTACT | TTACCATATATATAGTGAAGGC | 1445 | 45 | Surachetpong et al., 2017 | ||||
| 2 | GTCCAGGGCGGTATGTATTG | CTTACGGCTGACAAGTCTCTAAG | 834 | 62 | Mugimba et al., 2018 | ||||
| 3 | GTTGGGCACAAGGCATCCTA | TATCACGTGCGTACTCGTTCAGT | 250 | 56 | Eyngor et al., 2014 | ||||
| 3 | TATGCAGTACTTTCCCTGCC | TTGCTCTGAGCAAGAGTACC | 491 | 57 | Eyngor et al., 2014 | ||||
| 3 | ACCCCTTAATCCTTAATAGACCGTTA | CCCATAATCCTCTATTAGAACGTCGT | 1352 | 50 | Surachetpong et al., 2017 | ||||
| 3 | GTCGAGGCATTCCAGAAGTAAG | GAGCTAAGGGAACGGCTATTG | 834 | 62 | Mugimba et al., 2018 | ||||
| 4 | AGCAGCAGCAGGAGAAAGAG | ACCGTCCTGTTTCTGAATGG | 358 | 60 | Nicholson et al., 2017 | ||||
| 4 | CCAAAGTTTACTCCTATTACCCAGA | GCAAATCTTTCTCCAATTACCGTCT | 1250 | 50 | Surachetpong et al., 2017 | ||||
| 4 | GCCCAATGGTTCCCATATCT | GCCCAATGGTTCCCATATCT | 524 | 62 | Mugimba et al., 2018 | ||||
| 5 | CCAAATGTTTCTCTTATCTCAGACTC | CTTTTTCTCAGTTTACCACTTTATG | 1087 | 57 | Surachetpong et al., 2017 | ||||
| 5 | CAACTCTTAGCCTCCGGAATAC | CGTTCTGCACTGGGTTACA | 696 | 62 | Mugimba et al., 2018 | ||||
| 6 | CCAAATTTTACCTCTCGCAT | TCAAGCACTTAAAACTGTACC | 1027 | 45 | Surachetpong et al., 2017 | ||||
| 6 | CCCACACGACAGGACATATAG | GAGTTGGCTTAGGGTGATAAGA | 948 | 62 | Mugimba et al., 2018 | ||||
| 7 | CTCTCTTTGCATTGCATACCGT | GACCAATTATCCCTGCTTTCA | 30° | 57 | Surachetpong et al., 2017 | ||||
| 7 | TCCTTTAGGGATTGGCACTAAC | TTCCATCGACTGCTCCTAGA | 486 | 62 | Mugimba et al., 2018 | ||||
| 8 | ACCTCATCTACACTAACATTTCCA | TCATCATTACACAAATGGAGTAGCT | 637 | 50 | Surachetpong et al., 2017 | ||||
| 8 | CTTAAGGGCCATCCTGTCATC | TGGCTCAAATCCCAACACTAA | 476 | 62 | Mugimba et al., 2018 | ||||
| 9 | TTGGTGATGTCACGATGGATA | AGTTCTATCGCCAGCCATGT | 351 | 60 | Nicholson et al., 2017 | ||||
| 9 | ACAAGTCCGATTACTTTTTCCGC | TCTTTCTCACGTCCTTAAAGTCA | 530 | 50 | Surachetpong et al., 2017 | ||||
| 9 | GATATCCTCCACATGACCCTTC | GTACGTCACTTTGTGCCATTAC | 261 | 62 | Mugimba et al., 2018 | ||||
| 10 | AACCCTACTAACACCAAATATAGCT | CTTTCCCTCTGACACCCTGT | 450 | 50 | Surachetpong et al., 2017 | ||||
| 10 | TCCTCTCTGTCCCTTCTGTT | CAGGATGAGTGTGGCAGATTAT | 276 | 62 | Mugimba et al., 2018 | ||||
Tabla 1. Publicado primer pares para la amplificación del cDNA TiLV mediante endpoint PCR. El primer set se muestra en negrita se utiliza para generar los resultados representativos se muestra en la Figura 3A y 3B.
6. TiLV reacción cuantitativa en cadena de polimerasa (qPCR)
- Usando un plásmido que contiene el apropiado TiLV cDNA genomic segmento 3 como un estándar, tal como pTiLV32, preparar una serie de dilución seriada 10 veces duplicados o triplicados.
- Preparar una mezcla maestra de qPCR para todas las muestras, estándares y controles, teniendo en cuenta que las reacciones se deben realizar en duplicar o triplicar utilizando 0,4 μL de agua libre de nucleasa, 0,3 μl de cebador forward, 0,3 μl de cebador reverso y 5 μl de 2 x SYBR Green Master-mix de polimerasa de la DNA por reacción.
- Utilizar los iniciadores en una concentración de 10 μm y la información de la cartilla y el pTiLV estándar como sigue:
Adelante la cartilla: TiLV-112F (5'-CTGAGCTAAAGAGGCAATATGGATT-3')
Revertir la cartilla: TiLV-112R (5'-CGTGCGTACTCGTTCAGTATAAGTTCT-3')
PTiLV:10 estándar pg/μl
Nota: Si el número total de muestras y controles es 10 y se realizará en triplicado, esto equivale a una mezcla maestra de qPCR que comprende 12 μl de agua libre de nucleasa, primer avance de 9 μl, cartilla reversa de 9 μl y 150 μL de mezcla de maestra de SYBR Green ADN polimerasa. Adquiridos comercialmente 2 x universal SYBR Green ADN polimerasa master-mezclas contienen todos los componentes necesarios para la reacción de la qPCR, a saber, el SYBR Green tinte, hot-start Taq ADN polimerasa, dNTPs, MgCl2 y tintes de referencia pasiva. Proteja la mezcla maestra SYBR Green de la luz.
- Utilizar los iniciadores en una concentración de 10 μm y la información de la cartilla y el pTiLV estándar como sigue:
- Añada 6 μl de qPCR master-mix en qPCR tira tubos o una placa bien 96 compatible con la máquina de qPCR en uso.
- Añadir 4 μL de cDNA plantilla, controles o normas TiLV diluidas en serie en los tubos o pocillos de la placa bien 96.
- Cerrar los tubos de qPCR o sellar la placa bien 96 con una cubierta de placa compatible para la máquina de qPCR en uso
- Suavemente mueva los qPCR de tubos para mezclar la solución y el giro de los tubos de qPCR o 96 placa bien utilizando una centrífuga para recoger todo el líquido en el fondo de los vasos.
- Coloque la placa o tubos en el termociclador en tiempo real.
- Programar el termociclador de qPCR para llevar a cabo una desnaturalización inicial a 95 ° C por 3 min, seguido de 40 ciclos de 95 ° C para 10 s y 60 ° C para 30 s para recocido de la cartilla y alargamiento, terminando con un paso de curva fusión de 65 ° C a 95 ° C con incrementos de 0.5 ° C / 5 s.
- Seleccione SYBR como fluoróforo tinte, entonces desconocido como un tipo de muestra e Introduzca un nombre en un cuadro de nombre de la muestra.
- Abra la tapa de la máquina de RT-qPCR y coloque la tira de qPCR en los pozos asignados, luego cierre la tapa.
- Realizar el ensayo de RT-qPCR con las condiciones seleccionadas. La máquina comenzará a funcionar después de que la tapa ha alcanzado la temperatura deseada. Recoger la fluorescencia de cada muestra después de cada paso de la extensión para supervisar el progreso de la reacción.
Nota: La máquina de qPCR y software relacionado automáticamente calcula todos los parámetros del análisis y mostrar las curvas de amplificación en tiempo real, mientras que la curva estándar y curva de fusión se generará al final del ciclo de la qPCR. - Realizar análisis de datos y adquisición primera asegurando que la fusión de curvas para cada muestra y estándar tienen un pico de uniforme a la temperatura esperada para el amplicon.
- Evaluar las curvas de amplificación de las muestras y estándares y conjunto el umbral en una región que la tasa de amplificación de los cDNAs es el mismo en todas las muestras. Esto normalmente es realizado automáticamente por el software pero se debe controlar.
- Calcular el número de copias TiLV usando la curva estándar.
Resultados
Siguiendo el protocolo descrito en la sección 1, tilapia híbrida roja moribundo mostrando signos clínicos de infección TiLV (figura 1A) fueron sacrificados por bañarse en una alta concentración de aceite de clavo, que actúa como un anestésico. Síntomas clínicos son variables, pero los síntomas parecen ser letargo, erosión de la piel y decoloración, exoftalmia, escalas separadas, heridas abierta, lesión y comportamiento anormal15,16,33, 35,36, algunas de ellas pueden verse claramente en la figura 1A. La pared abdominal fue quitada para recoger órganos internos como hígado, bazo o riñón principal (figura 1B). También se tomaron muestras de moco en esta etapa raspando suavemente la piel de la parte anterior a la posterior de los pescados usando un cristal o lámina quirúrgica37.

Figura 1 . Tilapia disección muestra colección. A. tilapia híbrida roja TiLV infectada con piel leisons, enrojecimiento alrededor de la boca y opérculo, erosión de la piel y opacidad de la córnea. B. Sectioned híbrida roja tilapia para permitir la colección de tejido del hígado (en el punto de flecha azul), bazo u órganos riñón cabeza. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
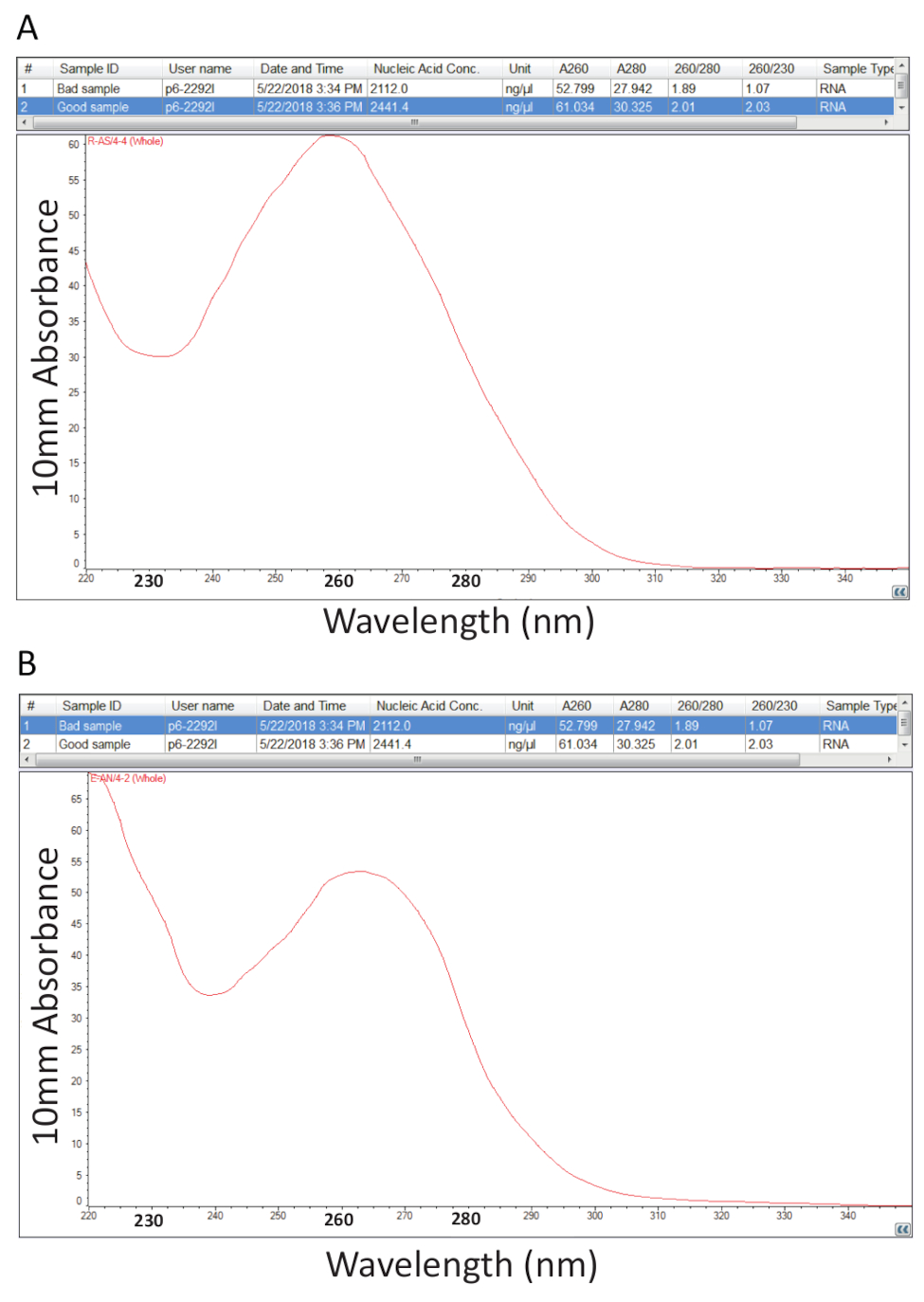
Después, se siguió el protocolo detallado en la sección 2 para la extracción de ARN total Guanidium tiocianato-fenol-cloroformo y cuantificación de RNA como se indica en la sección 3 se realizó para evaluar la pureza de la muestra mediante el cálculo de los ratios de pureza y examen de los perfiles espectrales (figura 2). Figura 2A muestra un resultado representativo de un procedimiento de extracción total acertado RNA, mientras que la figura 2B representa una mala preparación de RNA. Los ácidos nucleicos tienen máximos de absorbancia a 260 mientras que las proteínas tienen ellos a 280 nm. El cociente de las mediciones a 260 nm y 280 nm indican la pureza de cada muestra y ratios de 1.9 a 2.1 indican RNA puro como es el caso de la muestra en la figura 2A. Menores relaciones de A260/280 observados en la figura 2B indican posible contaminación de proteínas o fenol restante del procedimiento de extracción de RNA. Absorbancia a 230 nm puede ser el resultado de la contaminación de la muestra y la proporción de A260/230 nm se calcula también por este motivo. Esta relación debe ser en el rango de 2.0-2.2 para preparaciones de RNA puras tal como se ilustra por un valor de 2,03 para la muestra en la figura 2A, mientras que figura 2B tiene una baja relación A260/230 de 1.07 y el perfil espectral muestra un cambio en el canal a 230 nm a 240 nm que es indicativo de guanidina residual o de fenol en la muestra. Para la muestra que se muestra en la figura 2B, vuelva a precipitar el ARN para eliminar la contaminación puede mejorar la pureza de la muestra.

Figura 2 . Cuantificación espectrofotométrica de ARN total extraído de los tejidos enfermos de la tilapia. A. proporciones de pureza y perfiles espectrales de una exitosa preparación de RNA. B. como A, excepto el representante de un mal procedimiento de extracción de RNA. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Para detectar TiLV por RT-PCR, puras muestras como el representado en la figura 2A se reversa transcrito (Protocolo 4) en cDNA y utilizan como una plantilla para la polimerización en cadena del análisis detallado en sección 5 y resultados representativos se muestran en la Figura 3A. Cartillas se muestra en negrita en la tabla 1 se utiliza para amplificar un fragmento de bp 491 TiLV segmento genomic 314. Los productos PCR fueron separados por electroforesis en gel y manchados con EtBr para visualización. Figura 3A muestra los resultados de una RT-PCR de dos pasos con 4 muestras de cDNA (S1-S4), derivadas del hígado de la tilapia enferma aislada en Tailandia y en cada muestra, una sola limpia banda de aproximadamente 500 bp puede ser observado y por lo tanto, las muestras 1-4 son TiLV positiva. El mismo producto PCR se obtuvo de la muestra control positivo, formado por TiLV segmento 3 del cDNA clonado en un plásmido de32 , mientras que el no control de la plantilla (NTC) no dió los productos PCR. El ensayo en la figura 3B se realizó usando los mismos cebadores como en la Figura 3A , pero en un laboratorio diferente, utilizando un método de RT-PCR de un solo paso y con 5 muestras de RNA procedentes de los tejidos del riñón principal de tilapia originaria de acuicultura egipcia 15. se determinó usando este análisis de detección que las muestras 1, 3 y 5 son TiLV positivo mientras que las muestras 2 y 4 son TiLV negativo ya que ningún producto PCR se encontró en el tamaño correcto. Los controles negativos, incluyendo dos menos controles de transcriptasa inversa y dos NTCs no generaron los productos PCR. También se realizó un ensayo de RT-PCR de un paso dirigido a gene de ActinB de tilapia. El tamaño del amplicón de 217 bp se generó en todas las muestras (S1-S5) como esperado38. Este ensayo sirve como un control de la integridad de las muestras de RNA así como permitiendo un examen semi-cuantitativo de las muestras positivas TiLV. Dado que el producto generado de ActB de Tilapia es relativamente igual, las diferencias en la cantidad de TiLV específica producto PCR generado pueden interpretarse como un fiel reflejo de la cantidad de TiLV en una muestra de tejido dado.

Figura 3 . TiLV RT-PCR. A. las muestras de cDNA producidas a partir de tejidos del hígado de la tilapia enfermo, recogidos de Tailandia fueron pantalla TiLV infección utilizando iniciadores específicos para el segmento 3 (se muestra en negrita en la tabla 1) de TiLV utilizando un ensayo de RT-PCR de 2 pasos. M = marcador se muestra en pares de bases; S1-S4 = muestras 1-4; C1 = control positivo utilizando pTiLV como una plantilla de PCR; y C2 no = control de plantilla (NTC). B. RT-PCR de un solo paso usando los mismos cebadores como A y recogieron muestras de tejidos de riñón cabeza de tilapia enfermo de Egipto15. M = marcador se muestra en pares de bases; S1-S5 = muestras 1-5. Menos controles de transcriptasa reversa controles C1-C2 y C3-C4 son panel inferior NTCs. es una RT-PCR de un solo paso usando las cartillas contra tilapia ActinB38 (Ver texto para detalles) produciendo un producto PCR de 217 pares de bases. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
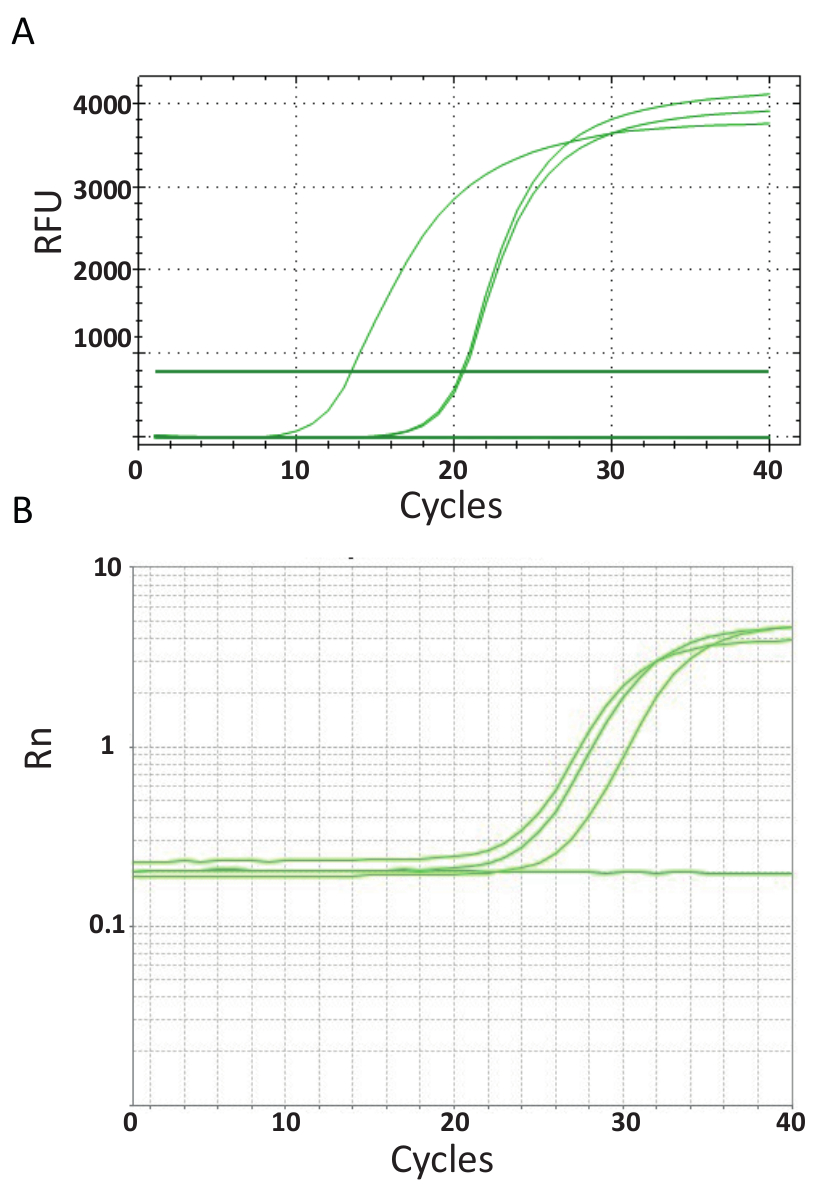
A diferencia de lo endpoint PCRs representados en la figura 3, ensayos de qPCR explicados en Protocolo 6, medir la cantidad de producto de PCR después de cada ciclo PCR. La amplificación del ADN diana se detecta uso de moléculas fluorescentes que interactúan con el DNA generado por cada ronda de reacción. Aquí, SYBR Green tinte fue utilizado, que intercala con ADN de doble hebra. La señal fluorescente es seguida durante la reacción y su intensidad se refiere a la cantidad de producto formado 39,40,41,42,43. Ensayos de qPCR TiLV se llevaron a cabo como se describe en el protocolo 6 en diferentes laboratorios utilizando diferentes reactivos de SYBR Green, máquinas de qPCR y muestras de diferentes países. Las curvas de amplificación resultantes se muestran en la Figura 4A y 4B. Se puede observar que para cada ensayo, el curso del experimento tiene cuatro fases: la fase lineal de la tierra, fase exponencial temprana, última fase exponencial y la fase de meseta. La fase lineal de la tierra se produce durante los primeros ciclos donde la duplicación de ADN todavía no puede ser identificada debido a cantidades de ADN produciendo cociente de señal/fondo insuficiente. Fluorescencia basal se calcula durante esta fase. Después de eso, ADN diana empieza a doble concentración con cada ciclo induciendo la señal detectable sobre el fondo y aumentar exponencialmente. La eficacia de la amplificación (E) de un análisis de qPCR bien optimizado es muy alta (casi 100%) en el inicio de la reacción y permanece estable durante la fase exponencial temprana de la amplificación y es en este punto que la cuantificación se lleva a cabo, cuando eficiencia de reacción es todavía constante. En ciclos posteriores la señal comienza a la meseta, y la intensidad de la fluorescencia no está correlacionada con el número inicial de copia de plantilla debido a los componentes de la reacción son agotados44. Saturación puede también ocurrir debido a la competencia de reacciones a recocido, los cocientes de concentración cambiante de los componentes, o la cantidad de unidades de enzima a las moléculas de sustrato de ADN. Posiblemente, estos parámetros representan las diferencias entre las curvas de amplificación para los ensayos que se muestra en la Figura 4A y 4B. Los controles incluyen no generaron las curvas de amplificación característica.

Figura 4 . Amplificación de parcelas muestran la acumulación de producto durante la duración del ensayo PCR en tiempo real. A. curvas de amplificación de las muestras positivas TiLV derivados de Tailandia, NTCs y positivo plásmido control mediante un SYBR-Green I ensayo qPCR de 2 pasos. La tabla fue generada mediante la representación de número de fluorescencia relativa (RFU) vs ciclo. B. curvas de amplificación de las muestras positivas TiLV derivan de Egipto, como en la figura 3B y una NTC. La curva de amplificación es la fluorescencia de la señal de reportero normalizada a la fluorescencia del tinte ROX pasivo incluido en el análisis (Rn) versus número de ciclo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Al final de la qPCR termociclaje rápido en máquinas diferentes en cada laboratorio, los datos se adquiridos y analizados. Figura 5A y 5B muestran curvas de fusión representativas de los ensayos realizados en cada laboratorio. Cada máquina de qPCR fue programado para realizar un análisis de la curva fusión al final. Esto se logró progresivamente aumentando la temperatura y monitoreo de la fluorescencia en función de la temperatura. Cuando la temperatura es lo suficientemente alta como para desnaturalizar dsDNA, una gota grande en fluorescencia se registra porque se libera la molécula de fluoróforo. El software de cada instrumento de qPCR calcula la temperatura de recocido (Tm) de los datos de la curva de fusión mediante el trazado de la temperatura vs derivados primera negativa (figura 5). Puede verse que en la figura 5A y 5B que los productos forman en los grupos diferentes de la muestra tienen transición fusión uniforme a la temperatura esperada de aproximadamente 80 ° C para el análisis. No hay otros picos a temperaturas más bajas se observaron. Debido a su pequeño tamaño, Tm de dímeros de primer es típicamente más bajo que el de la blanco de secuencia de ADN. Por lo tanto, esta diferencia entre la Tmfacilita identificar potenciales de dímeros de primer o de otros productos de la amplificación no específica. Los controles no se generaron curvas de fusión como muestras positivas TiLV y estándares y pueden ser vistos como una línea casi horizontal en la parte inferior de las cartas en la figura 5A y 5B.

Figura 5 . Fundir el análisis de la curva para asegurar la especificidad del ensayo y diversos productos PCR se pueden distinguir por sus características de fusión. A. análisis de la curva de muestras TiLV positivo proveniente de Tailandia, control negativo y control positivo plásmido del derretimiento. B. Análisis de la curva de TiLV muestras positivas derivadas de Egipto, pTiLV normas y una NTC del derretimiento. Las cartas en A y B ambos demuestran el cambio en fluorescencia dividida por el cambio de temperatura conspiraron contra temperatura para producir una imagen clara de la fusión dinámica. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Mayoría de qPCR las máquinas viene con un software facilitando la evaluación adicional del qPCR ejecutar y cuantificará las muestras mediante la generación de una curva estándar trazando automáticamente el ciclo umbral (Ct) contra el logaritmo de la plantilla de la pTiLV las normas Copie el número como se muestra en la figura 6A y 6B para los dos laboratorios independientes. Brevemente, el Ct es la unidad utilizada para evaluar los resultados de la qPCR. El valor det de C indica el número de ciclos necesario para alcanzar un nivel de señal de fluorescencia umbral fijado. Cuanto mayor sea la cantidad de a partir de plantilla, menos ciclos de se toma para alcanzar un nivel de fluorescencia detectable. En efecto, las muestras con una alta carga de TiLV tendrá valores det C más bajos que las muestras con una carga baja de TiLV como en los peces con una infección subclínica. Para determinar los valores det de C, los niveles de fluorescencia de fondo primero se deducen datos sin procesar. A continuación, el software asociado con el dispositivo de qPCR seleccionará automáticamente un umbral de fluorescencia buscando las curvas de datos para cada muestra e incorporando unt de C que representa donde la muestra cruzó el umbral. Esto se hace por separado para cada ensayo y cada umbral debe ser evaluado cuidadosamente, asegurando que el umbral se ha fijado en la parte logarítmica de las curvas de amplificación y en un lugar donde todas las curvas son paralelas. Así, la específica Ct adquirido es un valor relativo y es en relación con el de número de copia45de plantilla que se inicio, pero también es específico para la máquina de qPCR y reactivos utilizados, la eficacia de la amplificación por PCR y la sensibilidad de detección. Estos parámetros contribuyen a las diferencias observadas usando el mismo análisis en la figura 6.
De las curvas estándar en la figura 6, análisis de regresión, incluyendo cálculo de pendientes de la curva estándar (m) e intercepta, amplificación eficiencias (100 x 10 (1/m -1))46 y linealidad de la reacción se realizaron. Análisis de la curva estándar también fueron utilizados para confirmar la sensibilidad (límite de detección), repetibilidad y reproducibilidad del análisis. Teóricamente, la cantidad de ADN se duplica con cada PCR ciclo, lo que significa que la eficiencia (E) es igual a 100%. Sin embargo, en la práctica rara vez se alcanza una eficacia tal ideal debido a las condiciones PCR óptimas como inhibición de la ADN polimerasa, contaminantes, demasiado errores de cDNA y pipeteo47. Por lo general, amplificación E rango de 90-110% para los ensayos de buena, en la figura 6A , eficiencia del 94,5% se calculó en 8 muestras de pTiLV diluidos en serie, mientras que la eficiencia del ensayo en el ensayo que se muestra en la Figura 6B con pTiLV diluido en serie 7 muestras fue de 101,2%. Una eficacia de más del 100% suele ser debido a la presencia de inhibidores de la PCR en el análisis. Análisis de regresión lineal de la trama estándar también permite el cálculo del número de copias TiLV en cada muestra41,42,45, como se puede observar para las tres muestras TiLV que se muestra en rojo en Figura 6B que está en consonancia con los resultados de las muestras S1, S3 y S5 se muestra en la figura 3B.

Figura 6 . Curvas estándar de RT-qPCR. PCR en tiempo real de diluciones seriadas 10 veces de pTiLV, el estándar utilizado en ambos laboratorios. A. 8 muestras de pTiLV diluidos en serie fueron probadas, de concentración conocida y correlacionadas con el número de copias TiLV / reacción. La curva estándar se generó mediante la representación de log copia número vs ciclo umbral (Ct). La pendiente =-3.462, R2 = 0.9992 y la eficacia es 94.47%. B. al igual que en A, excepto pTiLV diluidos en serie 7 (verde) las muestras fueron probadas y la tabla muestra el umbral de ciclo en el eje y y el número de copia de TiLV (cantidad) en el eje x. El y-intercepto = 32.327, pendiente =-3.292, R2 = 0.98 y el rendimiento es de 101.2%. Por ambas curvas estándares en A y B, la pendiente, la intersección y valores del coeficiente de correlación (R2) son utilizados para entender el funcionamiento del ensayo. Lo importante es R2 valor debe ser cercano a 1 ya que es una medida de la linealidad de la curva estándar. La pendiente se utiliza para medir la eficiencia de la PCR en la que 100% de eficiencia corresponde a una pendiente de-3.32, véase el texto principal para la ecuación y otros datos. Una reacción buena qPCR generalmente tiene una eficiencia entre 90-110% correlacionando a una pendiente de entre-3.58 y-3.10. La curva estándar se utiliza para la cuantificación absoluta de desconocido TiLV muestras positivas y determina el número exacto de TiLV copias / reacción, como es el caso de las tres muestras positivas TiLV color rojo en la B.
Discusión
TiLV primero fue divulgado en 2014 en Israel14 y desde entonces, ha sido identificado en varios países incluyendo Egipto, Colombia, India, Malasia, Uganda, Tanzania y Tailandia15,16,18, 35 , 48. conciencia global, en particular, en países productores de tilapia ha puesto más atención en el virus y diversas restricciones y se han implementado medidas de control de las autoridades del gobierno tratar de evitar la propagación de TiLV. Aquí, un protocolo detallado para la detección de TiLV en tejido de tilapia, que cubre la recogida de muestras, aislamiento de RNA, análisis síntesis, PCR y qPCR de cDNA se ha explicado. Hay varios aspectos a estos métodos que merecen una discusión específica. TiLV ha sido identificado en los peces que abarca una variedad tamaños9,12,14,15,49 y especies de tilapia, incluyendo cultivo híbrido de tilapia (O. niloticus x o. aureus)11,14, Nilo tilapia (o. niloticus)9,10,14,15,16, 33 , 35 , 36 , 49 , 50 y rojo de la tilapia (Oreochromis SP.)16,33,48,51, como así como en la salvaje Nilo tilapia9,12, negro tilapia51, T. zilli14,15, S. galilaeus, o. aureus y T. simonis intermedia14 y muy recientemente TiLV fue identificado en carpa silvestre (Barbonymus schwanenfeldii)52. Muestras de tejido de órganos internos (gill, bazo, hígado, corazón, riñón principal) o moco37 pueden ser recogidas de tilapia sano como moribundo sin importar la edad, tamaño o especie y procesadas para el aislamiento de RNA. El protocolo de extracción de RNA total se indica aquí utiliza una solución monofásica de fenol y Guanidinio tiocianato, que es un agente de desnaturalización caotrópico. Los tejidos son homogeneizados directamente en esta solución seguida por la adición de cloroformo y centrifugación para lograr la separación de la fase en la que se genera un claro RNA que contiene la fase acuosa superior, una interfase y una fase orgánica inferior. El ARN está aislado de la fase acuosa por la precipitación del isopropanol, seguida de lavado del ARN recuperado para deshacerse de los contaminantes. Aislamiento del ARN de esta metodología fue pionero Piotr Chomczynski y Nicoletta Sacchi y fue referido como Guanidinio tiocianato-fenol-cloroformo extracción53,54. Este tipo de reactivo utilizado para la extracción de RNA puede ser comprado comercialmente o hecho en el laboratorio (véase la Tabla de materiales para más información). Este protocolo tiene un poco más de los métodos basados en la columna como la purificación basados en sílice, pero en general, es más rentable y da más RNA.
En este protocolo de cuantificación de RNA utilizando valores de A260 fue delineado por el que los valores de la espectrofotometría pueden indicar calidad de RNA (A260/A280 = 1.9-2.1). Mientras que este método dará una buena indicación de la pureza de la muestra, absolutamente no puede informar sobre la calidad del ARN extraído. Determinar correctamente si el ARN es intactas o parcialmente degradadas, las muestras pueden ser separación por electroforesis del gel de agarosa que mancharse de la EtBr manchado de 18 años y bandas de 28S rRNA indican degradación del RNA. Para verificación de calidad de RNA puede incluyen el uso de un instrumento de laboratorio en un chip. Además, también es importante para digerir el ARN purificado con DNasa I para eliminar contaminantes acoge la DNA genomic, que dependiendo de las aplicaciones posteriores puede conducir a resultados falsos. Si host gDNA todavía está contaminando la muestra de RNA en gran parte, también se puede realizar un tratamiento adicional de la DNaseI al final del procedimiento de extracción de RNA (véase Tabla de materiales).
La síntesis de ADN complementaria puede afectar en gran medida los resultados globales de la qPCR y es un aspecto del método que puede presentar variación. El protocolo de cDNA abogado aquí dispone de una configuración de un solo componente con oligo (dT) y así sólo transcribe mRNAs que contienen colas polyA. Permite el control de usuario de exactamente qué componentes a utilizar en la reacción de transcripción reversa y este modo de cDNA síntesis ha demostrado su eficacia para la detección de TiLV32. Una alternativa a esta configuración es una mezcla maestra compradas comercialmente que contiene todos los componentes necesarios para la reacción de transcripción reversa y es muy rápida y sencilla sin el protocolo habitual de multi-step, pipeteo y multi-temperatura. Esto es ventajoso porque reduce al mínimo la manipulación y promueve la uniformidad en todas las muestras. Tal maestro-se mezcla a menudo incluye oligo(dT) y cartillas al azar lo que es aplicable a diferentes plantillas del RNA y generar cDNA representante copias de secuencias de toda la longitud de los RNAs en una población (viral y anfitrión mRNA) y en teoría, puede medirse entonces cada especie deseada de RNA por PCR convencional o por qPCR de tal muestra. Esta versatilidad es la principal ventaja de un enfoque de RT-PCR de 2 pasos; ofrece una piscina a largo plazo que puede ser utilizada para muchos diversos experimentos. En los resultados, se ha representado un método de RT-PCR de un paso en donde se utilizaron primers específicos de secuencia (tabla 1) y la RT y PCR fueron realizadas en un tubo (véase lista de materiales). En general, primers específicos de secuencia permiten una mayor eficiencia de RT de la RNA específicos que el uso de cebado al azar, pero el objetivo específico de RNA es el único que puede cuantificarse en tal una muestra de cDNA que puede ser el único objetivo de ciertos laboratorios (ver Tabla de materiales para sugerencias de producto de síntesis de cDNA).
Mientras que RT-PCR convencional parece ser utilizados hasta ahora en el TiLV diagnóstico9,13,14,15,16,17,18, 33 , 35 , 48 , 55. RT-qPCR ha demostrado ser una herramienta más poderosa para la detección y cuantificación de pequeñas cantidades de TiLV en tejidos de peces o moco32,37. En general, qPCR es ampliamente utilizado en los laboratorios de diagnóstico de virología clínica debido a su alta sensibilidad, especificidad, buena reproducibilidad, amplio rango dinámico y velocidad21. QPCR puede ser inicialmente más caro de implementar que RT-PCR convencional, ofrecen muchas ventajas importantes sobre PCR convencional; tiene un más rápido tiempo de vuelta de la muestra resultados y no requiere ninguna medida de post-PCR. Este último punto significa que existe un riesgo mínimo de contaminación del laboratorio y se puede adaptar más fácilmente a situaciones de alto rendimiento como en el caso de brotes. Por otra parte, es inherentemente más sensible que los RT-PCR convencional, que es de vital importancia para detectar cargas virales bajas en infecciones sub clínicas21. Esto requeriría un enfoque PCR anidado requiere transcripción inversa, dos más reacciones de PCR y análisis de agarosa electroforesis del gel. Estos pasos muchos toman mucho tiempo y aumentan las posibilidades de errores o de contaminación. Sin embargo, debido a su alta sensibilidad, RT-qPCR exige diseño experimental meticuloso y un conocimiento profundo de las técnicas de cuantificación para generar resultados precisos56,57.
El ADN vinculante fluoróforo, SYBR Green se ha demostrado en el presente Protocolo. Es un tinte de unión de ADN inespecífico dsDNA y así la especificidad del ensayo se encuentra enteramente en el sistema de cartillas, que pueden generar falsos positivos58. Por lo tanto, mientras que el dsDNA fusión realizado al final de cada PCR Análisis de la curva es una parte especialmente importante de la reacción de PCR porque confirma que sólo un amplicon PCR de T correctom se produce (esto se debe también alcanzar por gel electroforesis, cuando se llevan a cabo nuevos ensayos). Tm de un fragmento de ADN depende de una variedad de características tales como su longitud, composición de GC, secuencia, filamento de la complementariedad, así como en la concentración del almacenador intermediario componentes y potenciadores de la polimerización en cadena. Los análisis de la curva fusión los resultados representativos que se muestra de dos laboratorios no reveló la presencia de dímeros de primer o de otros productos PCR no deseados pero si esto se observa con otras muestras y preparaciones para experimentos, el análisis debe ser volver a optimizado. Tecnologías más avanzadas de la qPCR no requieren tal fusión paso de curva y de hecho, desde este papel fue escrito, los métodos un TaqMan basado TiLV RT-qPCR se ha desarrollado utilizando dos iniciadores y sonda haciéndolo altamente TiLV específicas34.
Sin duda, los cebadores diseñados para los ensayos de RT-qPCR son fundamentales para el éxito del ensayo y aquí los cebadores fueron diseñados en base a los disposición del público datos genómicos TiLV en tiempo32. Sin embargo, los virus ARN son bien sabidos para exhibir tasas de mutación alta y posibles tensiones escaparán las pruebas diagnósticas actuales, como fue observado por ISAV59. Siempre va a ser difícil para tales tipos de virus generar un RT de pan-TiLV universal-ensayo qPCR y estos ensayos sólo continuamente mejorará si disponga de más datos genómicos TiLV de gran alcance lugares y periodos de tiempo.
Finalmente, es esencial ejecutar duplicadas o triplicadas si es posible, las reacciones en ambos intra y inter ensayos de qPCR. Si los valores det de C son muy altos, el uso de repeticiones es especialmente importante comprobar que la reacción de PCR es confiable y reproducible. En general, si replican datos de reacciones varía más de 0,5 ciclos, las reacciones debe repetirse y si los valores det de C varían constantemente ciclos de > 0,5 en replica, el ensayo debe volver a optimizar. El uso de un robot de pipeteado integrada qPCR ayuda enormemente con este tema, pero es una herramienta de lujo. Como con cualquier experimento, los controles adecuados y apropiados de inclusión son de suma importancia para el desarrollo de ensayos moleculares robustos, especialmente en laboratorios de diagnóstico donde tales ensayos tienen que estar acreditados. Deben incluir controles positivos (muestras positivas TiLV, TiLV plásmido estándar) y controles negativos (NTC y -RT) muestras así como la detección de genes housekeeping de tilapia endógena. Estos controles no se puede subestimar y se deben incluir en cada ensayo para entender adecuadamente la calidad de cada paso del análisis y a interpretar adecuadamente los resultados.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Agradecemos al Instituto de bacteriología veterinaria, Facultad Vetsuisse, Universidad de Berna por su apoyo. Este trabajo fue financiado por el Comité de promoción académica de investigadores de carrera temprana y equidad de género en la Facultad Vetsuisse, Universidad de Berna por 120% modelo financiación otorgada a PN. WS y PR son apoyados por el centro de estudios avanzados para la agricultura y alimentación, Instituto de estudios avanzados, Universidad de Kasetsart, Bangkok, Tailandia en la promoción de la investigación universitaria y nacional investigación Universidad proyecto de Tailandia, oficina de la educación superior Comisión, Ministerio de Educación de Tailandia. Nos gustaría agradecer al Dr. Kwanrawee Sirikanchana para su narración y Piyawatchara Sikarin para editar el video.
Materiales
| Name | Company | Catalog Number | Comments |
| Tissue collection | Step 1 | ||
| Tricaine methanesulfonate | Sigma-Aldrich | E10521 | An alternative to clove oil. Step 1.1 |
| RNAlater stabilization solution | Thermo Fisher Scientific | AM7020 | For storing tissues if they cannot be processed immediately Step 1.3 |
| RNA extraction | Step 2 | ||
| TRIreagent | Sigma-Aldrich | Step 2.1 | |
| TRIzol | Thermo Fisher Scientific (Invitrogen) | 15596026 | Step 2.1 |
| GENEzol | Geneaid | GZR100 | Step 2.1 |
| Trisure | Bioline | BIO-38032 | Step 2.1 |
| Homemade solution | - | - | 94.53 g/L (800 mM) guanidine thiocyanate 30.45 g/L (400 mM) ammonium thiocyanate 8.20 g/L (100 mM) sodium acetate 380 mL/L (38 % v/v) phenol 50 mL/L (5 % v/v) glycerol 1.0 g/L (0.1 % w/v) 8-quinolinol, pH 5.0 Store up to 2 years at 4oC Step 2.1 |
| MagNA Lyser Green Beads | Roche | 3358941001 | An alternative tissue homogenization method used in conjunction with tissue lysing machines detailed below Step 2.2 |
| Lysing Matrix D, 2 mL Tube | MP BIOMEDICALS | 116913050 | |
| Chloroform | Sigma-Aldrich | C2432 | Step 2.3 |
| Chloroform | RCI Labscan | AR1027E-G2.5L | Step 2.3 |
| 1-Bromo-3-chloropropane | Sigma-Aldrich | B9673 | A less toxic alternative to chloroform Step 2.3 |
| Isopropanol (GC) ≥ 99.8 % | Sigma-Aldrich | 59300 | Step 2.6 |
| Isopropanol (ACS, ISO Reag. Ph Eur) | Merck KGaA (EMSURE) | 1.09634.2500 | Step 2.6 |
| Glycogen, molecular biology grade (e.g., Sigma, cat. no. G1767) | Thermo Fisher Scientific (Thermo Scientific) | R0551 | Useful step if tissue starting material is small to maximise RNA precipitation optional |
| Ethanol (purity (GC) ≥ 99.9 % | Sigma-Aldrich (EMD Millipore) | 1.00983 | Step 2.9 |
| Ethanol (ACS, ISO Reag. Ph Eur) | Merck (EMSURE) | 1.00983.2500 | Step 2.9 |
| Nuclease-free water | Promega | P1193 | Step 2.13 |
| Nuclease-free water | Multicell | 809-115-CL | Step 2.13 |
| Ambion TURBO DNA-free kit | Thermo Fisher Scientific (Invitrogen) | AM1907 | Can be performed at the end of the RNA extraction protocol optional |
| cDNA synthesis | Step 4 | ||
| Viva cDNA Synthesis Kit | Vivantis | cDSK01 | Step 4.1 & 4.3 |
| ReverTra Ace qPCR RT MasterMix with gDNA remover | Toyobo | A1172K | An alternative option see discussion |
| ReverTra Ace qPCR RT Kit | Toyobo | FSQ-101 | An alternative option see discussion |
| AffinityScript Multiple Temperature Reverse Transcriptase | Agilent Technologies | 600107 | An alternative option |
| PCR | Step 5 | ||
| DNA polymerase systems: | Step 5.2 | ||
| - Platinum II Hot-Start Green PCR Master Mix (2X) | Thermo Fisher Scientific (Invitrogen) | 14001012a | Step 5.2 |
| - GoTaq Mastermix | Promega | M7122 | Step 5.2 |
| Separate PCR mixture components: | Step 5.2 | ||
| 10mM dNTP Mix | Vivantis | NP2409 | Step 5.2 |
| 25mM MgCl2 | Thermo Fisher Scientific | R0971 | Step 5.2 |
| 10X Taq Buffer with KCl | Thermo Fisher Scientific | 00348114 | Step 5.2 |
| Taq DNA polymerase | Vivantis | PL1202 | Step 5.2 |
| - Verso 1-step RT-PCR ReddyMix with ThermoPrime Taq | Thermo Fisher Scientific | AB1454 | One step RT-PCR exemplified in Figure 3B |
| Gel electrophoresis: | For visulation of PCR products from steps 5.1-5.4 | ||
| Ethidium Bromide solution (10 mg/mL) | Thermo Fisher Scientific | 17898 | Step 5.5 |
| Tris/Acetic/EDTA (TAE) buffer: | Step 5.5 | ||
| - Tris | Vivantis | PR0612-1KG | Step 5.5 |
| - Acetic acid (glacial) (ACS, ISO, Reag. Ph Eur) | Merck KGaA (EMSURE) | 1.00063.2500 | Step 5.5 |
| - Ethylenediaminetetraacetic acid (EDTA) | BIO-RAD | 161-0729 | Step 5.5 |
| Agarose | Vivantis | PC0701-100G | Step 5.5 |
| DNA ladders and markers | Vivantis | NL1405 | Step 5.5 |
| DNA gel loading dye (6X) | Thermo Fisher Scientific | R0611 | Step 5.5 |
| qPCR | Step 6 | ||
| PowerUP SYBR Green Master Mix | Thermo Fisher Scientific (Applied Biosystems) | A25779 | Exemplified in Figures 4-6B Step 6.2 |
| iTaq Universal SYBR Green Supermix | BIO-RAD | 1725120 | Exemplified in the video and in Figures 4-6A Step 6.2 |
| Equipment | |||
| Dounce tissue grinder pestle | Sigma-Aldrich | P1110 | Protocol 2 |
| MagNA Lyser Instrument | Roche | 3358976001 | An alternative tissue homogenizing option for protocol 2 which are used in conjunction with the lysing beads detailed above Step 2.2 |
| FastPrep-24 5G Homogenizer | MP BIOMEDICALS | 116005500 | |
| Refrigerated microcentrifuge | Eppendorf | Eppendorf 5427R | Protocol 2 Step 2.4, 2.7 & 2.10 |
| Refrigerated microcentrifuge | Eppendorf | Eppendorf 5418R | |
| Heat box | Labnet | AccuBlock Digital Dry Bath | Protocol 2 Step 2.13 |
| Microvolume spectrophotometer | Thermo Fisher Scientific (Applied Biosystems) | Nanodrop 2000 | Protocol 3 Step 3.1 - 3.4 |
| PCR machine | BIO-RAD | T100 Thermal Cycler | Protocol 5 Step 5.4 |
| Power supply | BIO-RAD | PowerPac HC | Protocol 5 Step 5.5 |
| Horizontal gel electrophoresis | BIO-RAD | Mini ReadySub-Cell GT Cell #1704487edu | Protocol 5 Step 5.5 |
| Mini microcentrifuge | Corning | LSE 6766 | Useful to quickly spin down PCR reaction tubes in protocols 4, 5 & 6 Step 6.5.1 |
| Microcentrifuge | LioFuge | LM-60 | Step 6.5.1 |
| qPCR machine and software | Thermo Fisher Scientific | 7500 Fast Real-Time PCR System with 7500 Software v2.0 | Protocol 6 Step 6.6-6.8 |
| qPCR machine and software | BIO-RAD | CFX96 Touch Real-Time PCR Detection System with CFX Manager software | |
| General Materials | |||
| Mayo scissors | Step 1.1-1.2 | ||
| Forceps | Step 1.1-1.2 | ||
| Pipette | Rainin | Pipette-Lite XLS | |
| Aerosol-barrier pipette tips | Sigma-Aldrich | Z333328, Z333336, Z333344 | |
| Nuclease-free 1.5-ml microcentrifuge tubes | Eppendorf |
Referencias
- FAO. . The State of World Fisheries and Aquaculture, 2014. Opportunities and Challenges. , (2014).
- FAO. . The State of World Fisheries and Aquaculture, 2016. Contributing to Food Security and Nutrition for all. , (2016).
- WorldBank. . FISH TO 2030: Prospects for Fisheries and Aquaculture. Agriculture and Environmental Services Discussion Paper 03. , (2013).
- Wing-Keong, N., Nicholas, R. A review of the nutrition and feeding management of farmed tilapia throughout the culture cycle. Reviews in Aquaculture. 5 (4), 220-254 (2013).
- Cleasby, N., et al. The socio-economic context for improving food security through land based aquaculture in Solomon Islands: A peri-urban case study. Marine Policy. 45, 89-97 (2014).
- Ponzoni Raul, W., et al. Genetic improvement of Nile tilapia (Oreochromis niloticus) with special reference to the work conducted by the WorldFish Center with the GIFT strain. Reviews in Aquaculture. 3 (1), 27-41 (2011).
- Hounmanou, Y. M. G., et al. Tilapia lake virus threatens tilapiines farming and food security: Socio-economic challenges and preventive measures in Sub-Saharan Africa. Aquaculture (Amsterdam, Netherlands). 493, 123-129 (2018).
- OIE. . Tilapia Lake Virus (TiLV) - a novel orthomyxo-like virus. OIE technical disease cards. , (2018).
- Mugimba, K. K., et al. Detection of tilapia lake virus (TiLV) infection by PCR in farmed and wild Nile tilapia (Oreochromis niloticus) from Lake Victoria. Journal of Fish Diseases. , (2018).
- Koesharyani, I., Gardenia, L., Widowati, Z., Khumaira, D. D., Rustianti, Studi kasus infeksi tilapia lake virus (tilv) pada ikan nila (Oreochromis niloticus). Jurnal Riset Akuakultur. 13 (1), 85-92 (2018).
- OIE. . Tilapia lake virus disease (TiLV), Chinese Taipei. Immediate Notification. , (2017).
- OIE. . Tilapia Lake Virus Disease (TiLV), Peru. Immediate Notification. , (2018).
- Bacharach, E., et al. Characterization of a Novel Orthomyxo-like Virus Causing Mass Die-Offs of Tilapia. MBio. 7 (2), e00431-e00416 (2016).
- Eyngor, M., et al. Identification of a novel RNA virus lethal to tilapia. Journal of Clinical Microbiology. 52 (12), 4137-4146 (2014).
- Nicholson, P., et al. Detection of Tilapia Lake Virus in Egyptian fish farms experiencing high mortalities in 2015. Journal of Fish Diseases. 40 (12), 1925-1928 (2017).
- Surachetpong, W., et al. Outbreaks of Tilapia Lake Virus Infection, Thailand, 2015-2016. Emerging Infectious Diseases. 23 (6), 1031-1033 (2017).
- Tattiyapong, P., Dachavichitlead, W., Surachetpong, W. Experimental infection of Tilapia Lake Virus (TiLV) in Nile tilapia (Oreochromis niloticus) and red tilapia (Oreochromis spp.). Veterinary Microbiology. 207, 170-177 (2017).
- Kembou Tsofack, J. E., et al. Detection of Tilapia Lake Virus in Clinical Samples by Culturing and Nested Reverse Transcription-PCR. Journal of Clinical Microbiology. 55 (3), 759-767 (2017).
- Thangaraj, R. S., et al. Derivation of two tilapia (Oreochromis niloticus) cell lines for efficient propagation of Tilapia Lake Virus (TiLV). Aquaculture (Amsterdam, Netherlands). 492, 206-214 (2018).
- Hanson, L. A., Rudis, M. R., Vasquez-Lee, M., Montgomery, R. D. A broadly applicable method to characterize large DNA viruses and adenoviruses based on the DNA polymerase gene. Virology Journal. 3, 28-28 (2006).
- Josko, D. Molecular virology in the clinical laboratory. Clinical Laboratory Science. 23 (4), 231-236 (2010).
- Munir, K., Kibenge, F. S. Detection of infectious salmon anaemia virus by real-time RT-PCR. Journal of Virological Methods. 117 (1), 37-47 (2004).
- Snow, M., et al. Developement, application and validation of a Taqman real-time RT-PCR assay for the detection of infectious salmon anaemia virus (ISAV) in Atlantic salmon (Salmo salar). Developments in Biologicals. 126, 133-145 (2006).
- Matejusova, I., McKay, P., McBeath, A. J., Collet, B., Snow, M. Development of a sensitive and controlled real-time RT-PCR assay for viral haemorrhagic septicaemia virus (VHSV) in marine salmonid aquaculture. Diseases of Aquatic Organisms. 80 (2), 137-144 (2008).
- Garver, K. A., et al. Development and validation of a reverse transcription quantitative PCR for universal detection of viral hemorrhagic septicemia virus. Diseases of Aquatic Organisms. 95 (2), 97-112 (2011).
- Dalla Valle, L., et al. Development of a sensitive and quantitative diagnostic assay for fish nervous necrosis virus based on two-target real-time PCR. Veterinary Microbiology. 110 (3-4), 167-179 (2005).
- Hodneland, K., Garcia, R., Balbuena, J. A., Zarza, C., Fouz, B. Real-time RT-PCR detection of betanodavirus in naturally and experimentally infected fish from Spain. Journal of Fish Diseases. 34 (3), 189-202 (2011).
- Hodneland, K., Endresen, C. Sensitive and specific detection of Salmonid alphavirus using real-time PCR (TaqMan). Journal of Virological Methods. 131 (2), 184-192 (2006).
- Wang, X. W., Ao, J. Q., Li, Q. G., Chen, X. H. Quantitative detection of a marine fish iridovirus isolated from large yellow croaker, Pseudosciaena crocea, using a molecular beacon. Journal of Virological Methods. 133 (1), 76-81 (2006).
- van Beurden, S. J., et al. Development and validation of a real-time PCR assay for the detection of anguillid herpesvirus 1. Journal of Fish Diseases. 39 (1), 95-104 (2016).
- Ciulli, S., et al. Development and application of a real-time PCR assay for the detection and quantitation of lymphocystis disease virus. Journal of Virological Methods. 213, 164-173 (2015).
- Tattiyapong, P., Sirikanchana, K., Surachetpong, W. Development and validation of a reverse transcription quantitative polymerase chain reaction for tilapia lake virus detection in clinical samples and experimentally challenged fish. Journal of Fish Diseases. 41 (2), 255-261 (2018).
- Dong, H. T., et al. Emergence of tilapia lake virus in Thailand and an alternative semi-nested RT-PCR for detection. Aquaculture (Amsterdam, Netherlands). 476, 111-118 (2017).
- Waiyamitra, P., et al. A TaqMan RT-qPCR assay for tilapia lake virus (TiLV) detection in tilapia. Aquaculture (Amsterdam, Netherlands). 497, 184-188 (2018).
- Behera, B. K., et al. Emergence of Tilapia Lake Virus associated with mortalities of farmed Nile Tilapia Oreochromis niloticus (Linnaeus 1758) in India. Aquaculture (Amsterdam, Netherlands). 484, 168-174 (2018).
- Ferguson, H. W., et al. Syncytial hepatitis of farmed tilapia, Oreochromis niloticus (L.): a case report. Journal of Fish Diseases. 37 (6), 583-589 (2014).
- Liamnimitr, P., Thammatorn, W., U-thoomporn, S., Tattiyapong, P., Surachetpong, W. Non-lethal sampling for Tilapia Lake Virus detection by RT-qPCR and cell culture. Aquaculture (Amsterdam, Netherlands). 486, 75-80 (2018).
- Yang, C. G., et al. Evaluation of reference genes for quantitative real-time RT-PCR analysis of gene expression in Nile tilapia (Oreochromis niloticus). Gene. 527 (1), 183-192 (2013).
- Bustin, S. A. Real-time, fluorescence-based quantitative PCR: a snapshot of current procedures and preferences. Expert Review of Molecular Diagnostics. 5 (4), 493-498 (2005).
- Fleige, S., Pfaffl, M. W. RNA integrity and the effect on the real-time qRT-PCR performance. Molecular Aspects of Medicine. 27 (2-3), 126-139 (2006).
- Kubista, M., et al. The real-time polymerase chain reaction. Molecular Aspects of Medicine. 27 (2-3), 95-125 (2006).
- Mackay, I. M., Arden, K. E., Nitsche, A. Real-time PCR in virology. Nucleic Acids Research. 30 (6), 1292-1305 (2002).
- Wong, M. L., Medrano, J. F. Real-time PCR for mRNA quantitation. Biotechniques. 39 (1), 75-85 (2005).
- Bustin, S. A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. Journal of Molecular Endocrinology. 25 (2), 169-193 (2000).
- Heid, C. A., Stevens, J., Livak, K. J., Williams, P. M. Real time quantitative PCR. Genome Research. 6 (10), 986-994 (1996).
- Rutledge, R. G., Côté, C. Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Research. 31 (16), e93-e93 (2003).
- Svec, D., Tichopad, A., Novosadova, V., Pfaffl, M. W., Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomolecular Detection and Quantification. 3, 9-16 (2015).
- Amal, M. N. A., et al. A case of natural co-infection of Tilapia Lake Virus and Aeromonas veronii in a Malaysian red hybrid tilapia (Oreochromis niloticus × O. mossambicus) farm experiencing high mortality. Aquaculture (Amsterdam, Netherlands). 485, 12-16 (2018).
- Fathi, M., et al. Identification of Tilapia Lake Virus in Egypt in Nile tilapia affected by ‘summer mortality’ syndrome. Aquaculture (Amsterdam, Netherlands). 473, 430-432 (2017).
- OIE. . Tilapia Lake Virus disease (TiLV), Philippines. Immediate Notification. , (2017).
- OIE. . Tilapia lake virus disease (TiLV), Malaysia. Immediate Notification. , (2017).
- Abdullah, A., et al. First detection of tilapia lake virus (TiLV) in wild river carp (Barbonymus schwanenfeldii) at Timah Tasoh Lake, Malaysia. Journal of Fish Diseases. 41 (9), 1459-1462 (2018).
- Chomczynski, P., Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 162 (1), 156-159 (1987).
- Chomczynski, P., Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nature Protocols. 1 (2), 581-585 (2006).
- Del-Pozo, J., et al. Syncytial Hepatitis of Tilapia ( Oreochromis niloticus L.) is Associated With Orthomyxovirus-Like Virions in Hepatocytes. Veterinary Pathology. 54 (1), 164-170 (2017).
- Bustin, S. A., et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry. 55 (4), 611-622 (2009).
- Purcell, M. K., Getchell, R. G., McClure, C. A., Garver, K. A. Quantitative polymerase chain reaction (PCR) for detection of aquatic animal pathogens in a diagnostic laboratory setting. Journal of Aquatic Animal Health. 23 (3), 148-161 (2011).
- Simpson, D. A., Feeney, S., Boyle, C., Stitt, A. W. Retinal VEGF mRNA measured by SYBR green I fluorescence: A versatile approach to quantitative PCR. Molecular Vision. 6, 178-183 (2000).
- Kibenge, M. J., et al. Discovery of variant infectious salmon anaemia virus (ISAV) of European genotype in British Columbia, Canada. Virology Journal. 13, 3 (2016).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados