
Method Article
Détection de Tilapia lac Virus à l’aide de RT-PCR classique et SYBR Green RT-qPCR
Dans cet article
Résumé
Ce protocole diagnostique Tilapia lac Virus (TiLV) dans les tissus de tilapia à l’aide de méthodes de RT-PCR. L’ensemble de la méthode est décrite de la dissection des tissus à l’extraction d’ARN totale, suivie de la synthèse de cDNA et détection de TiLV par PCR classique ou PCR quantitative à l’aide d’ADN double brin contraignant un colorant fluorescent obligatoire.
Résumé
Cette méthode vise à faciliter la détection rapide, sensible et spécifique du Virus de lac de Tilapia (TiLV) dans les tissus de tilapia. Ce protocole peut être utilisé dans le cadre de programmes de surveillance, les mesures de biosécurité dans les laboratoires de recherche fondamentale TiLV. L’étalon-or des diagnostics de virus implique généralement l’isolement du virus suivie des techniques complémentaires telles que de la réaction en chaîne polymérase de transcription inverse (RT-PCR) pour vérification supplémentaire. Cela peut être fastidieux, chronophage et nécessite généralement des échantillons de tissus fortement infectés par le virus. L’utilisation de RT-quantitative (q) PCR dans la détection des virus est avantageuse en raison de sa nature quantitative, haute sensibilité, spécificité, évolutivité et son temps rapide causera. Ici, l’ensemble de la méthode de PCR basée approches pour TiLV détection est décrite, de tilapia orgue sectionnement, total acide ribonucléique (ARN) extraction à l’aide d’une solution de thiocyanate-phénol-chloroforme de guanidium, quantification de RNA, suivie d’une PCR en deux étapes entraînant le protocole, la synthèse de l’acide désoxyribonucléique complémentaire (ADNc) et la détection de TiLV par PCR classique ou identification quantitative via qPCR SYBR green à l’aide de je le teindre. PCR classique exige des étapes post-PCR et informera simplement la présence du virus. Cette dernière approche permettra de quantification absolue de la TiLV vers le bas pour aussi peu que 2 exemplaires et est donc particulièrement utile pour le diagnostic de la TiLV en cas subcliniques. Une description détaillée des deux approches de la PCR, des résultats représentatifs de deux laboratoires et une étude approfondie des paramètres critiques de tous les deux ont été incluses pour garantir que les chercheurs et les diagnostiqueurs trouvent leur plus convenables et il y a lieu méthode de détection TiLV.
Introduction
L’approvisionnement en poissons habitant global a atteint un nouveau record de 20 kg en 2014 et cela était dû à une croissance vigoureuse en aquaculture. L’aquaculture reste l’une des croissances secteurs produisant les aliments pour animaux dans le monde entier et est le secteur uniquement animaux destinés à l’alimentation qui croît plus rapidement que la population humaine1. Tilapiinés cichilds constituent les deuxième plus importants poissons d’eau douce d’élevage dans le monde entier avec une production mondiale totale de 6,4 millions de tonnes (MT) et une valeur estimative de 9,8 milliards de dollars en 2015,2. Les producteurs de top dix de tilapia sont la Chine (1,78 MT), Indonésie (1,12 MT) et l’Égypte (0,88 MT), suivie du Bangladesh, Vietnam, Philippines, Brésil, Thaïlande, Colombie et en Ouganda2. Il est prévu que la production de tilapia global sera environ 7,3 MT d’ici 2030,3. Tilapia sont devenus telle une source alimentaire importante non seulement parce qu’elles représentent une source bon marchée de protéines4 mais aussi parce qu’ils sont faciles à reproduire dans un large éventail de l’eau et le climat conditions5,6au titre. Il y a quelques décennies, on a cru qu’il y avait quelques maladies commercialement significatifs du tilapia en danger mais ce n’est plus vrai. Une maladie virale émergente appelée virose tilapia lac (TiLVD) est la première épidémie de maladie critique jamais trouvé chez le tilapia et l’industrie tout entière est menacée. Cette maladie a des conséquences socio-économiques graves et est une menace directe sur la sécurité alimentaire de millions de personnes en Afrique7, Asie et Amérique du Sud. Au début de 2018, l’Organisation mondiale de la santé animale (OIE) a signalé que l’agent étiologique de cette maladie, la TiLV, avait été officiellement détecté sur trois continents, qui couvre huit pays8 et étant donné que cette carte d’information agent pathogène a été mis à jour il y a signalé plus de TiLV en Tanzanie, Ouganda9, Indonésie10, Taiwan11 et Pérou12. TiLV est un virus à ARN simple brin roman décrit est un virus analogue à orthomyxo car il contient une variété de caractéristiques qui rappelle d’autres orthomyoxoviruses comme la grippe ou infectieuse anémie du saumon (AIS) de virus13. Il a été identifié à la suite de pertes massives du tilapia d’élevage et sauvage dans le lac de Galilée, Israël14. Par la suite, les éclosions de maladies semblables dénommé mortalités d’été et le délai d’un mois du syndrome de mortalité associé à une infection TiLV ont été signalés dans le tilapia du Nil (Oreochromis niloticus) en Egypte15 et du Nil et le tilapia rouge hybride (Oreochromis spp.) en Thaïlande16, respectivement. Détection de virus animaux aquatiques est historiquement réalisée par la croissance et l’isolement du virus en culture cellulaire. Diverses lignées cellulaires ont été testées pour la propagation et l’isolement des TiLV, y compris, les cellules de E-11 dérivés du poisson-serpent poisson (Ophiocephalus striatus)17,18, OmB et TmB provenant Oreochromis mossambicus18et OnlB et OnlL originaires du Nile tilapia (o. niloticus)19. Alors que la culture de virus a l’avantage qu’il fournit du matériel pour d’autres expériences, il a l’inconvénient qu’il faut au moins 4-7 jours pour observer la formation de l’effet cytopathique (ECP) et c’est essentiel, différents virus piscine, qui sont plus aptes à REPLICATE peut être multiplié et produire CPE similaire.
Dans les dernières décennies, il y a eu abandon des méthodes de diagnostic traditionnels, souvent fastidieuses telles que culture cellulaire, sérologie et détection de l’antigène et remplacement avec acide nucléique plus rapide et plus sensible basé détection essais20, 21. Cela est évident par le fait que de nombreux essais de qPCR ont été élaborés comme des méthodes de diagnostic importants pour une multitude de maladies virales chez les animaux aquatiques, comme pour AIS22,23, virus de la septicémie hémorragique virale (SHV)24 ,25, bétanodavirus26,27 alphavirus salmonidés28, poisson iridovirus29, anguillidés herpèsvirus de type 1 (AngHV1)30et Lymphocystis maladie virus (LCDV)31 . Méthodes robustes pour la surveillance de diagnostic et les agents pathogènes sont urgemment requis pour réduire la propagation de la TiLV. Ces méthodes devraient permettre la détection précoce de l’infection avant les signes cliniques se développent et de détection de virus faibles charges. À ce jour, différents protocoles PCR dont RT-PCR14,32, imbriqués de RT-PCR18, semi imbriquées RT-PCR33et RT-qPCR32,,34 ont été développés pour la détection des TiLV dans les tissus des poissons. Une comparaison d’isolement RT-qPCR et virus dans des lignées de cellules sensibles pour la détection de la TiLV a révélé que la RT-qPCR était 1 000 fois plus sensible que l' isolement de virus32. Bien que chaque protocole PCR publiée a rapporté des sensibilités différentes pour la détection des TiLV, la plupart des tests sont très sensibles avec les limites de détection de copies virales à 7,5 copies33, 7 exemplaires18 ou 2 copies32 par réaction.
Le but de cet article des méthodes est d’expliquer, en détail, comment réaliser des tests de détection TiLV, commençant par prélèvement tissulaire de tilapia, à extraction totale de l’ARN, synthèse de cDNA et puis TiLV PCR spécifique basé dosages. Plus précisément, les protocoles complets de RT-PCR classique et également axée sur le vert SYBR RT-qPCR ont été décrits faire appel à un large éventail de scientifiques visant à détecter la TiLV. Le premier est moins sensible mais est généralement une option moins onéreuse de détection. Cette dernière nécessite des infrastructures plus élaborées comme une machine PCR quantitative et réactifs plus chers, mais il a l’avantage d’être quantitative, rapide et très sensible, ce qui signifie qu’il peut être utilisé pour la détection des TiLV dans infraclinique poissons infectés. La RT-PCR et la RT-qPCR protocoles ont été réalisées dans deux laboratoires différents avec des isolats géographiques distincts de TiLV et l’inclus résultats mettent en évidence la sensibilité et la reproductibilité des essais décrits ici.
Protocole
Le protocole d’utilisation des animaux pour la présente étude a été approuvé par le Comité d’éthique animale Université Kasetsart sous le numéro de permis ACKU 59-VET-016.
Remarque : Veuillez vous référer à la Table des matières pour des informations détaillées concernant les réactifs et le matériel proposé pour ce protocole.
1. prélèvement d’échantillons de tissus
- Euthanasier le poisson à l’aide d’un surdosage d’huile de clou de girofle (le volume dépend de la taille du poisson et de la concentration des produits, généralement plus de 3 mL/L). Plonger un quart des ciseaux pinces et mayo dans éthanol à 95 % (v/v) suivi par la combustion de l’appareil à l’aide d’une lampe à alcool pour stériliser le matériel.
Remarque : Le méthanesulfonate de tricaïne (MS-222) peut être utilisé au lieu de l’huile de clou de girofle. - Trouver le foie et couper un petit morceau (environ 20 à 100 mg) ou recueillir 200 µL de mucus à l’aide de la lamelle couvre-objet ou lame chirurgicale pour enlever le mucus d’antérieur postérieur du poisson tilapia et placer les échantillons dans un tube de microtubes de 1,5 mL.
- Échantillons de processus immédiatement, stockent dans un ARN stabilisant de solution ou déplacent à-80 ° C jusqu'à l’utilisation ultérieure.
Remarque : La plus grande tâche en travaillant avec de l’ARN est préparation de molécules d’ARN intacts et en les gardant intact tout au long des manipulations ultérieures. L’épine dorsale de RNA est naturellement plus sensible aux lésions de l’ADN. Extraction et l’isolement de l’ARN total des cellules des tissus nécessite technique de laboratoire prudent ; prendre toutes dispositions pour prévenir la contamination de la RNase en portant des gants, à l’aide de réactifs, équipements, vaisselle en plastique, verrerie, eau libre de RNase, espace de travail et à l’aide de bouts filtrants pour le pipetage.
2. Guanidium Thiocyanate - phénol - chloroforme Extraction d’ARN
- Ajouter 1 mL de solution de monophasique contenant du phénol et guanidine isothiocyanate dans un tube contenant l’échantillon de tissu de la section 1.
ATTENTION : Cette solution est très toxique et doit être manipulée avec soin dans une hotte à flux laminaire avec un équipement de protection et en portant les bon gants lunettes, de vêtements et de sécurité. - Broyer l’échantillon de tissu à l’aide d’un homogénéisateur de pilon de tissus jusqu'à homogène.
NOTE : Échantillons peuvent également être homogénéisés à l’aide d’un homogénéisateur de puissance combinée avec des perles en céramique. S’assurer que l’échantillon de tissu est complètement homogène avant de passer à l’étape suivante ou arrêter le protocole ici et stocker les échantillons totalement homogénéisés à-80 ° C jusqu'à l’utilisation ultérieure. - Ajouter 200 µL de chloroforme pour séparation de phase.
ATTENTION chloroforme est un potentiel stupéfiant et est extrêmement dangereux. Il doit être manipulé avec soin dans une hotte à flux laminaire avec un équipement de protection, ainsi qu’en portant les bon gants lunettes, de vêtements et de sécurité. Comme moins toxique alternatif, 1-Bromo-3-chloropropane peut également être utilisé.
NOTE : Échelle les volumes vers le haut ou vers le bas, le cas échéant. Par exemple, si un seul a été utilisé 500 µL de solution de monophasique contenant du phénol et guanidine isothiocyanate, ensuite seulement ajouter 100 µL de chloroforme à cette étape.- Mélanger les échantillons bien par inversion pendant 15 s.
- Incuber les échantillons pendant 3 min à température ambiante (RT).
- Centrifuger pendant 15 min à 12 000 × g et 4 ° C.
Remarque : Il faut une séparation nette entre dans une phase organique inférieure, une interphase blanc et une phase aqueuse supérieure contenant ARN. Cette phase supérieure est normalement incolore, mais selon le type et la quantité de tissu homogénéisé, il peut avoir un aspect rose clair. - Transférer la phase aqueuse supérieure (environ 500 µL) dans un tube de microcentrifuge frais sans déranger l’interphase.
Remarque : ne pas essayer de transférer la phase aqueuse ensemble, laisser une petite quantité pour éviter toute contamination potentielle de l’ARN contenant une phase aqueuse avec l’organique ou interphase. - Ajouter 1 volume d’alcool isopropylique 100 % à précipiter l’ARN.
- Éventuellement, si de très petites quantités de tissu ont été utilisées, puis ajoutez 1 µL (5-10 µg) de glycogène RNase-libre à chaque échantillon pour favoriser la précipitation efficace d’ARN. Cela facilitera l’identification de la pastille de RNA étape 2.8.
Remarque : Le glycogène agit comme un transporteur de l’ARN et évitera les petites quantités d’ARN de coller à la paroi du tube. - Mix tubes bien en le retournant plusieurs fois.
- Stocker les échantillons pendant 2 h à une nuit à-20 ° C.
- Éventuellement, si de très petites quantités de tissu ont été utilisées, puis ajoutez 1 µL (5-10 µg) de glycogène RNase-libre à chaque échantillon pour favoriser la précipitation efficace d’ARN. Cela facilitera l’identification de la pastille de RNA étape 2.8.
- Centrifuger les échantillons pendant 15 min à 12 000 x g et 4 ° C.
- Jeter le surnageant, en faisant attention de ne pas déloger la pastille de RNA au fond du tube de microcentrifuge.
- Ajouter 1 mL d’éthanol 75 % (v/v) et mélanger les échantillons d’ARN en renversant le tube plusieurs fois.
- Centrifuger pendant 15 minutes à 10 000 x g et 4 ° C.
Remarque : Le protocole peut être arrêté ici et les échantillons comprenant le culot de RNA dans 75 % d’éthanol peuvent être conservés à-20 ° C jusqu'à l’utilisation ultérieure. - Jeter le surnageant, être vigilant ne pas à déloger le culot de RNA au fond du tube de microcentrifuge.
- Éventuellement, répétez les étapes 2,9-2.11 à l’aide d’éthanol à 70 % (v/v). Bien laver le culot de RNA minimisera tout sel ou report de contaminants qui peut-être interférer sera sensibles applications en aval.
- Faire ressortir l’éthanol restant à l’aide d’une pipette et ensuite sécher le culot de RNA à température ambiante pendant pas plus de 5 à 10 min.
NOTE : trop secs granulés sera difficiles à remettre en suspension. - Ajouter 30-60 µL d’eau exempte de RNase, préchauffée à 55-60 ° C à solubiliser le culot de RNA.
- Placez l’ARN sur la glace pour une utilisation immédiate ou conserver à-80 ° C pour une utilisation ultérieure.
3. mesurer la Concentration d’ARN à l’aide d’un spectrophotomètre Micro-Volume
- Changer les paramètres du spectrophotomètre à l’ARN.
- Utiliser 1-2 µL d’eau exempte de RNase comme blanc.
- 1-2 µL de chaque échantillon de RNA permet d’évaluer la quantité de RNA.
- Enregistrer les lectures à 230 nm, 260 nm et 280 nm pour chaque échantillon.
- Diluer l’ARN à 200 ng/µL en utilisant l’eau exempte de RNase.
4. synthèse de l’ADN complémentaire (ADNc) utilisant l’ARN total
- Mélanger 1 µg d’ARN total de protocole 2, 2 µM oligo (dT), mélange de dNTPs 0,5 mM et amener le volume final à 10 µL d’eau exempte de nucléase. Pour ce faire, préparez un RT-master-mix selon le nombre de contrôles et d’échantillons à tester.
Remarque : Les commandes sont un échantillon de moins-la transcriptase inverse (-RT) dans laquelle l’enzyme RT est remplacé avec de l’eau exempte de nucléase (voir étape 4.3) et un aucun contrôle de modèle (NTC) dans lequel l’eau exempte de nucléase est ajouté au master-mix au lieu de descripteur d’ARN.- Mélanger les échantillons bien en pipettant également, suivie d’une centrifugation courte.
- Incuber les échantillons à 65 ° C pendant 5 min, suivie d’une incubation de 2 min sur la glace.
- Brièvement, centrifuger les échantillons pour recueillir tout le liquide dans le fond des tubes.
- Ajouter 1 x tampon de transcriptase inverse, 100 U de la transcriptase inverse et porter le volume final de chaque échantillon à 20 µL en utilisant de l’eau exempte de nucléase.
- Mélanger les échantillons bien en pipettant également, suivie d’une centrifugation courte.
- Incuber les échantillons à 42 ° C pendant 60 min, suivie à 85 ° C pendant 5 min.
- Diluer le cDNA synthétisé à une concentration désirée en ajoutant un volume approprié d’eau exempte de nucléase et mettre l’ADNc sur glace pour un usage immédiat ou conserver à-20 ° C pour une utilisation ultérieure.
5. TiLV PCR classique
- Utilisez l’ARNC, les échantillons et les contrôles, générées en protocole 4 de l’article en tant que modèles pour une réaction de PCR en utilisant l’une des paires d’amorces établie détaillés dans le tableau 1, ainsi que d’une ADN polymérase de choix.
Remarque : Un contrôle supplémentaire de la non-modèle (NTC) devrait être inclus ici en substituant des ADNc pour eau exempte de nucléase dans la réaction de PCR. Un contrôle positif, le cas échéant, doit également être inclus comprenant déjà vérifiée TiLV échantillons positifs ou la TiLV appropriée de fragments d’ADNc cloné dans à un plasmide. - Préparer un mélange-maître PCR conformément aux lignes directrices du système DNA polymérase dans l’utilisation et le nombre d’échantillons et des contrôles à tester. Ce mélange devrait inclure l’amorce vers l’avant, apprêt inverse, dNTPs, MgCl2 et le sélectionné l’ADN polymérase, ainsi que sa mémoire tampon.
- Conformément aux directives de la DNA polymérase sélectionné, allient la quantité suggérée de l’ADNc des échantillons et contrôle le volume désigné de master-mix.
NOTE : Préparer un 0,5 x excès de réaction est souvent bénéfique puisque certains de la master-mix est perdu pendant le pipetage. - Effectuer des PCR cyclisme conditions conformément aux lignes directrices du système utilisé l’ADN polymérase et utilisant une température de recuit appropriée pour les amorces en cours d’utilisation (tableau 1). Habituellement, un tel programme impliquera une dénaturation initiale à 95 ° C pendant 2 à 5 min, suivie de 30 à 40 cycles de dénaturation à 95 ° C pendant 30 s, recuit à la température recommandée pendant 30 s et l’allongement à 72 ° C pendant 30 s, suivie d’une élongation finale à 72 ° C pendant 5-10 min.
- Charger 5-15 µL de chaque réaction de PCR et une échelle d’ADN appropriée dans sur puits d’un gel d’agarose de 1 à 2 %, selon la taille prévue du produit PCR. Séparer l’amplification de l’ADN par électrophorèse sur gel et tacher le gel par le bromure d’éthidium (EthBr) pour faciliter la visualisation des bandes d’ADN de la taille attendue (tableau 1) dans une machine de documentation gel utilisant la lumière UV.
ATTENTION : Le bromure d’éthidium est toxique ; il doit être manipulé avec soin, en portant les vêtements de protection appropriés et des gants de sécurité.
| Segment de cible TiLV génome | Primer avant 5' - 3' | Inverser l’apprêt 5' - 3' | Taille de produit PCR (bp) | Tm ° C | Référence d’origine | ||||
| 1 | CCAAACGTTATCTCTTAATTACGCAC | GCAAATATTTCTCTCATTCGCCT | 1641 | 50 | Surachetpong et coll., 2017 | ||||
| 1 | CCTCATTCCTCGTTGTGTAAGT | AGGAGTTGCTGTTGGGTTATAG | 1000 | 62 | Cris et coll., 2018 | ||||
| 2 | ACTCTCTATTACCAAATACATTTACT | TTACCATATATATAGTGAAGGC | 1445 | 45 | Surachetpong et coll., 2017 | ||||
| 2 | GTCCAGGGCGGTATGTATTG | CTTACGGCTGACAAGTCTCTAAG | 834 | 62 | Cris et coll., 2018 | ||||
| 3 | GTTGGGCACAAGGCATCCTA | TATCACGTGCGTACTCGTTCAGT | 250 | 56 | Eyngor et coll., 2014 | ||||
| 3 | TATGCAGTACTTTCCCTGCC | TTGCTCTGAGCAAGAGTACC | 491 | 57 | Eyngor et coll., 2014 | ||||
| 3 | ACCCCTTAATCCTTAATAGACCGTTA | CCCATAATCCTCTATTAGAACGTCGT | 1352 | 50 | Surachetpong et coll., 2017 | ||||
| 3 | GTCGAGGCATTCCAGAAGTAAG | GAGCTAAGGGAACGGCTATTG | 834 | 62 | Cris et coll., 2018 | ||||
| 4 | AGCAGCAGCAGGAGAAAGAG | ACCGTCCTGTTTCTGAATGG | 358 | 60 | Nicholson et al., 2017 | ||||
| 4 | CCAAAGTTTACTCCTATTACCCAGA | GCAAATCTTTCTCCAATTACCGTCT | 1250 | 50 | Surachetpong et coll., 2017 | ||||
| 4 | GCCCAATGGTTCCCATATCT | GCCCAATGGTTCCCATATCT | 524 | 62 | Cris et coll., 2018 | ||||
| 5 | CCAAATGTTTCTCTTATCTCAGACTC | CTTTTTCTCAGTTTACCACTTTATG | 1087 | 57 | Surachetpong et coll., 2017 | ||||
| 5 | CAACTCTTAGCCTCCGGAATAC | CGTTCTGCACTGGGTTACA | 696 | 62 | Cris et coll., 2018 | ||||
| 6 | CCAAATTTTACCTCTCGCAT | TCAAGCACTTAAAACTGTACC | 1027 | 45 | Surachetpong et coll., 2017 | ||||
| 6 | CCCACACGACAGGACATATAG | GAGTTGGCTTAGGGTGATAAGA | 948 | 62 | Cris et coll., 2018 | ||||
| 7 | CTCTCTTTGCATTGCATACCGT | GACCAATTATCCCTGCTTTCA | 704 | 57 | Surachetpong et coll., 2017 | ||||
| 7 | TCCTTTAGGGATTGGCACTAAC | TTCCATCGACTGCTCCTAGA | 486 | 62 | Cris et coll., 2018 | ||||
| 8 | ACCTCATCTACACTAACATTTCCA | TCATCATTACACAAATGGAGTAGCT | 637 | 50 | Surachetpong et coll., 2017 | ||||
| 8 | CTTAAGGGCCATCCTGTCATC | TGGCTCAAATCCCAACACTAA | 476 | 62 | Cris et coll., 2018 | ||||
| 9 | TTGGTGATGTCACGATGGATA | AGTTCTATCGCCAGCCATGT | 351 | 60 | Nicholson et al., 2017 | ||||
| 9 | ACAAGTCCGATTACTTTTTCCGC | TCTTTCTCACGTCCTTAAAGTCA | 530 | 50 | Surachetpong et coll., 2017 | ||||
| 9 | GATATCCTCCACATGACCCTTC | GTACGTCACTTTGTGCCATTAC | 261 | 62 | Cris et coll., 2018 | ||||
| 10 | AACCCTACTAACACCAAATATAGCT | CTTTCCCTCTGACACCCTGT | 450 | 50 | Surachetpong et coll., 2017 | ||||
| 10 | TCCTCTCTGTCCCTTCTGTT | CAGGATGAGTGTGGCAGATTAT | 276 | 62 | Cris et coll., 2018 | ||||
Table 1. Paires d’amorces publiées pour l’amplification de l’ADNc TiLV, terminaison de PCR. L’apprêt mis en gras ont été utilisées pour produire les résultats représentatifs, illustrés à la Figure 3 a et 3 b.
6. TiLV Quantitative Polymerase Chain Reaction (qPCR)
- À l’aide d’un plasmide contenant la TiLV appropriée segment génomique 3 ARNC comme une norme, comme pTiLV32, préparer une série de dilutions 10 fois dupliqué ou dupliquée.
- Préparer un mélange-maître de qPCR pour tous les échantillons, les normes et les contrôles, en tenant compte du fait que les réactions doivent être effectuées en double ou triple utilisant 0,4 µL d’eau exempte de nucléase, 0,3 µL d’apprêt avant, 0,3 µL d’apprêt inverse et 5 µL de 2 x SYBR Green ADN polymérase master-mix par réaction.
- Utilisez les amorces à une concentration de 10 µM et les informations de l’apprêt et la norme pTiLV comme suit :
Apprêt en avant : TiLV-112F (5'-CTGAGCTAAAGAGGCAATATGGATT-3')
Inverser l’apprêt : TiLV-112R (5'-CGTGCGTACTCGTTCAGTATAAGTTCT-3')
PTiLV:10 standard pg/µL
Remarque : Si le nombre total d’échantillons et des contrôles est 10 et sera jouée en géométrie, cela équivaut à un master-mix de qPCR comprenant 12 µL exempte de nucléase eau, apprêt avant de 9 µL, 9 amorces inverse µL et 150 µL de SYBR Green ADN polymérase master-mix. Achetés dans le commerce 2 x universel SYBR Green ADN polymérase maître-mélanges contiennent tous les composants nécessaires pour la réaction de qPCR, nommément SYBR Green I teindre, hot-start Taq DNA polymerase, dNTPs, MgCl2 et colorants référence passive. Protéger le mélange maître SYBR Green de la lumière.
- Utilisez les amorces à une concentration de 10 µM et les informations de l’apprêt et la norme pTiLV comme suit :
- Distribuer 6 µL de qPCR master-mix dans qPCR bande tubes ou une plaque bien 96 compatibles avec la machine de qPCR en cours d’utilisation.
- Ajouter 4 µL de matrice d’ADNc, des contrôles ou des normes de TiLV dilués en série dans les tubes ou le puits de la plaque 96 puits.
- Fermer les tubes de qPCR ou sceller la plaque 96 puits avec un couvercle de plaque compatible pour la machine de qPCR en cours d’utilisation
- Doucement effleurer les tubes de qPCR pour mélanger la solution et la centrifuger les tubes de qPCR ou 96 plaque bien en utilisant une centrifugeuse pour recueillir tout le liquide dans le fond des vaisseaux.
- Placer les tubes ou la plaque dans le thermocycleur en temps réel.
- Programme du thermocycleur qPCR pour effectuer une dénaturation initiale à 95 ° C pendant 3 min, suivi de 40 cycles de 95 ° C pour 10 s et 60 ° C pendant 30 s pour apprêt recuit et l’élongation, se terminant par une fusion étape courbe de 65 ° C à 95 ° C avec un incrément de 0,5 ° C / 5 s.
- Sélectionnez SYBR comme colorant fluorophore, puis inconnu comme un type d’échantillon et insérer un nom dans une zone de nom d’échantillon.
- Ouvrez le couvercle de la machine de la RT-qPCR et placez une bande de qPCR dans les puits assignées, puis fermez le couvercle.
- Effectuer le test de RT-qPCR avec les conditions choisies. La machine commence à courir après que le couvercle a atteint la température désirée. Recueillir la fluorescence de l’échantillon après chaque étape de l’extension pour surveiller la progression de la réaction.
Remarque : La machine de qPCR et logiciels connexes automatiquement calculer tous les paramètres du test et afficher les courbes de l’amplification en temps réel, tandis que la courbe d’étalonnage et de la courbe de fusion seront générés à la fin du cycle de qPCR. - Effectuer des analyses de données et acquisition de s’assurer d’abord que la fonte courbes pour chaque échantillon et standard ont un pic d’uniform à la température prévue de l’amplicon.
- Évaluer les courbes de l’amplification des échantillons et des normes et la série le seuil dans une région étaient que le taux d’amplification de l’ADNc est le même dans tous les échantillons. Ceci est normalement effectuée automatiquement par le logiciel, mais doit être soigneusement contrôlé.
- Calculer le nombre de copies TiLV à l’aide de la courbe d’étalonnage.
Résultats
Suivant le protocole décrit dans la section 1, tilapia moribonde rouge hybride présentant des signes cliniques de l’infection (Figure 1 a) de la TiLV ont été euthanasiés par bain dans une concentration élevée d’huile de clou de girofle, qui agit comme un anesthésique. Ont signalé des symptômes cliniques sont variables, mais les symptômes communs semblent être la léthargie, l’érosion de la peau et décoloration, exophtalmie, détaché des échelles, plaies/lésion ouverte et un comportement anormal15,16,33, 35,36, certaines d'entre elles est visible dans la Figure 1 a. La paroi abdominale a été supprimée pour collecter les organes internes comme le foie, la rate ou le rein (Figure 1 b). Mucus ont été aussi prélevés à ce stade en grattant doucement la peau de l’antéro-postérieur du poisson à l’aide d’une lamelle couvre-objet ou lame chirurgicale37.

Figure 1 . Collection de dissection et échantillon de tilapia. A. tilapia hybride rouge TiLV infectés avec peau leisons, rougeur autour de la bouche et l’opercule, l’érosion de la peau et l’opacité de la cornée. B. sections le tilapia rouge hybride permettant de prélèvement tissulaire du foie (au point d’une flèche bleue), rate ou organes rein. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
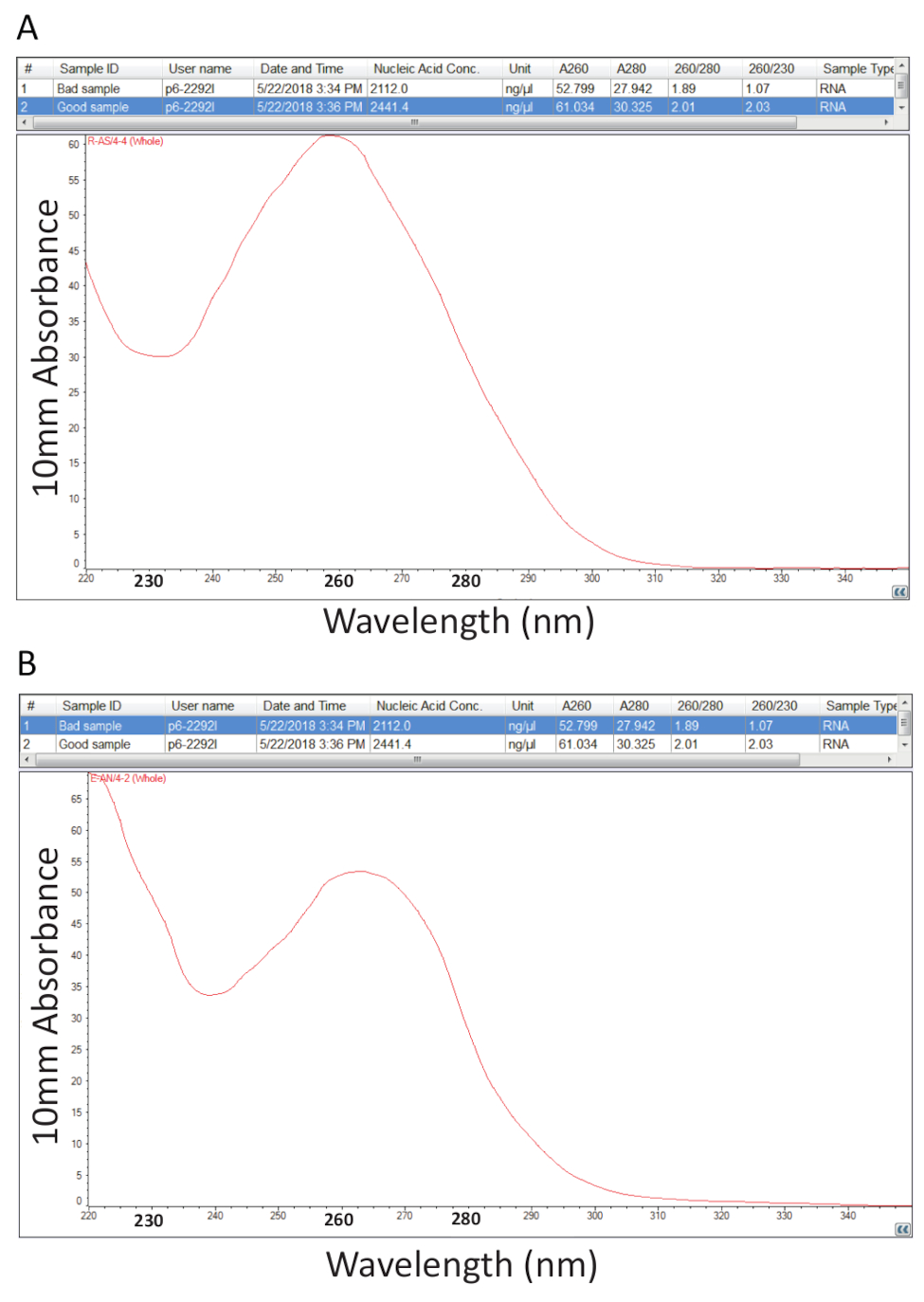
Par la suite, le protocole détaillé à l’article 2 pour l’extraction de Guanidium Thiocyanate-phénol-chloroforme de l’ARN total a été suivi et quantification de RNA énoncées à l’article 3 a été réalisée pour évaluer la pureté de l’échantillon par le calcul des ratios pureté et examen des profils spectraux (Figure 2). Figure 2 a montre un résultat représentatif d’une procédure d’extraction des RNA total succès, tandis que la Figure 2 b représente une mauvaise préparation d’ARN. Acides nucléiques ont maxima d’absorption à 260, tandis que les protéines ont les leurs à 280 nm. Le rapport entre les mesures à 260 nm et 280 nm indiquent la pureté de chaque échantillon et rapports de 1,9 à 2,1 indiquer RNA pur, comme c’est le cas pour l’exemple dans la Figure 2 a. Inférieur A260/280 de taux observé dans la Figure 2 b indique contamination possible protéine ou du phénol, reste de la procédure d’extraction de RNA. Absorbance à 230 nm peut être le résultat de la contamination de l’échantillon et le ratio de A260/230 nm est également calculé pour cette raison. Ce ratio devrait être de l’ordre de 2,0-2,2 pour les préparations d’ARN pures, comme en témoigne une valeur de 2,03 pour l’exemple dans la Figure 2 a, tandis que la Figure 2 b a un faible ratio A260/230 de 1,07 et le profil spectral montre un changement dans la fosse à 230 nm à 240 nm qui est révélateur de la guanidine résiduelle ou de phénol dans l’échantillon. Pour l’exemple illustré à la Figure 2 b, re-précipitant l’ARN pour éliminer la contamination peut améliorer la pureté de l’échantillon.

Figure 2 . Dosage spectrophotométrique d’ARN total extraite des tissus malades tilapia. A. les ratios de pureté et de profils spectraux d’une préparation réussie de RNA. B. comme A, sauf les représentant d’une procédure d’extraction pauvre RNA. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
Pour détecter TiLV par RT-PCR, échantillons purs tels que celui représenté dans la Figure 2 a ont été inversé transcrit (protocole 4) en ADNc et utilisé comme un modèle pour la PCR assay détaillées dans l’article 5 et de résultats représentatifs sont présentés dans la Figure 3 a. Amorces indiqués en gras dans le tableau 1 ont été utilisées pour amplifier un fragment de bp 491 TiLV segment génomique 314. Les amplicons ont été séparés par électrophorèse sur gel et colorées au bromure d’éthidium pour la visualisation. Figure 3 a montre les résultats d’une RT-PCR en deux étapes à l’aide de 4 échantillons de cDNA (S1-S4), provenant du foie de tilapia malade isolé en Thaïlande et dans chaque échantillon, une bande unique propre d’environ 500 bp peut être observée et échantillons 1-4 sont donc TiLV résultats positifs. Le même produit PCR a été obtenu sur l’échantillon de contrôle positif, composée de l’ADNc du segment TiLV 3 cloné dans un plasmide32 , alors que l’aucun contrôle de modèle (NTC) n’ont pas donné de produits PCR. Le test en Figure 3 b a été réalisé en utilisant les mêmes amorces comme dans la Figure 3 a , mais dans un laboratoire différent, en utilisant une approche de RT-PCR en une étape et avec 5 échantillons d’ARN dérivés les tissus du rein du tilapia originaires de l’Egyptian aquaculture 15. il a été déterminé à l’aide de ce test de détection que les échantillons 1, 3 et 5 sont TiLV positif tandis que les exemples 2 et 4 sont TiLV négatif car aucun produit de la PCR a été trouvé à la taille appropriée. Les témoins négatifs, dont deux au moins des contrôles de la transcriptase inverse et deux NTCs n’a pas généré des produits PCR. Un test de RT-PCR en une étape a également été effectué ciblant le gène ActinB tilapia. La taille de l’amplicon de 217 bp a été généré dans tous les échantillons (S1-S5) comme prévu38. Cet essai a été un contrôle de l’intégrité des échantillons d’ARN comme permettant un examen semi-quantitatif des échantillons positifs TiLV. Étant donné que le produit de Tilapia ActB généré est relativement égal, différences TiLV produit PCR spécifique généré peuvent être interprétés comme un reflet fidèle de la quantité de TiLV dans un échantillon de tissu donné.

Figure 3 . TiLV RT-PCR. A. cDNA produite à partir des tissus hépatiques du tilapia malade, prélevés de Thaïlande étaient écran pour TiLV infection en utilisant des amorces spécifiques pour le segment 3 (montré en gras dans le tableau 1) de TiLV à l’aide d’une RT-PCR 2-step. M = marqueur montré en paires de bases ; S1-S4 = échantillons 1-4 ; C1 = contrôle positif en utilisant pTiLV comme modèle PCR ; et C2 ne = aucun contrôle de modèle (NTC). B. une étape de RT-PCR en utilisant les mêmes amorces comme dans A et échantillons de tissus du rein du tilapia malade recueillis dans Egypte15. M = marqueur montré en paires de bases ; S1-S5 = échantillons 1-5. Les contrôles C1-C2 sont moins les contrôles de la transcriptase inverse et C3-C4 NTCs. panneau est une RT-PCR en une étape en utilisant des amorces dirigées contre la production de tilapia ActinB38 (voir le texte pour plus de détails) un produit de PCR de 217 de paires de bases. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
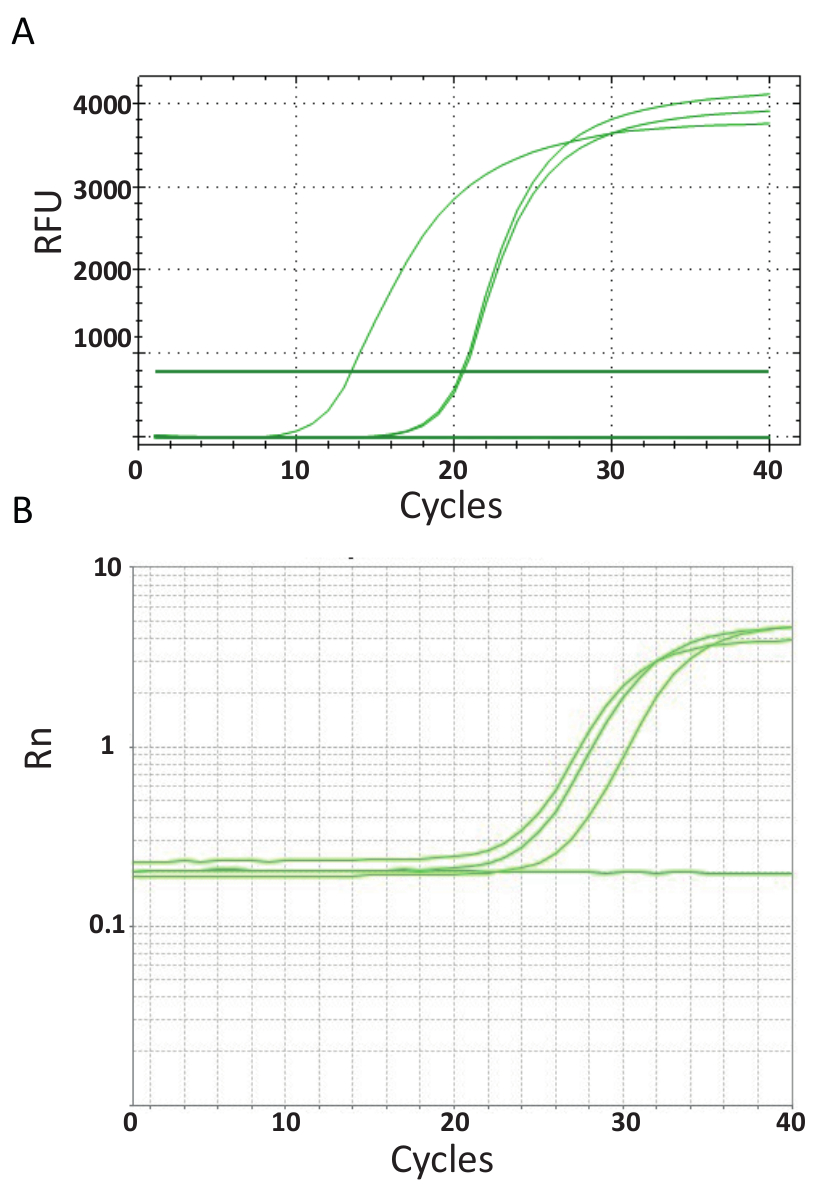
À la différence du point de terminaison PCRs représentées dans la Figure 3, qPCR dosages qui sont expliqués dans le protocole n° 6, mesurer la quantité de produit de PCR après chaque cycle de PCR. L’amplification de l’ADN cible est détectée à l’aide de molécules fluorescentes qui interagissent avec l’ADN produit de chaque ronde de réaction. Ici, SYBR Green je le teindre a été utilisé, qui s’intercale avec ADN bicaténaire. Le signal fluorescent est suivi au cours de la réaction et son intensité est liée à la quantité de produit formé de 39,40,41,42,43. TiLV qPCR essais ont été effectués comme indiqué dans le protocole n° 6 dans des laboratoires différents utilisant différents réactifs SYBR Green, machines de qPCR et des échantillons de différents pays. Les courbes d’amplification qui en résultent sont indiquées dans la Figure 4 a et 4 b. On peut constater que, pour chaque dosage, le cours de l’expérience a quatre phases : la phase linéaire au sol, début de la phase exponentiels, fin de la phase exponentielle et la phase de plateau. La phase linéaire au sol se produit pendant les premiers cycles lorsque duplication de l’ADN ne peut pas encore être identifiée en raison des quantités d’ADN produisant le ratio signal/contexte insuffisant. Fluorescence de la ligne de base est calculée au cours de cette phase. Par la suite, cible ADN commence à double concentration avec chaque cycle induisant le signal à devenir détectable au-dessus de fond et d’augmenter de façon exponentielle. L’efficacité de l’amplification (E) d’un test de qPCR bien optimisé est très élevée (près de 100 %) au début de la réaction et reste stable au cours de cette première phase exponentielle de l’amplification et c’est à ce stade que quantification est exécutée, lorsque efficacité de la réaction est toujours régulière. Dans les cycles plus tard, le signal commence à plateau, et l’intensité de la fluorescence est n’est plus en corrélation avec le nombre de copies modèle départ parce que les composantes de la réaction sont épuisés44. Saturation peut également se produire en raison de la concurrence des réactions re-recuites, les ratios de concentration changeant des composants, ou la quantité d’unités de l’enzyme de molécules de substrat d’ADN. Éventuellement, ces paramètres représentent les différences entre les courbes d’amplification pour les dosages, illustrés à la Figure 4 a et 4 b. Les contrôles inclus n’a pas généré ces courbes caractéristiques amplification.

Figure 4 . Amplification complote pour montrer l’accumulation du produit pendant la durée de la PCR en temps réel. A. courbes de l’Amplification des échantillons positifs TiLV provenant de Thaïlande, NTCs et le contrôle positif de plasmide utilisant un SYBR-Green j’ai test 2-step qPCR. Le graphique a été généré en traçant le nombre de la fluorescence relative (RFU) vs cycles. B. courbes d’Amplification des échantillons positifs TiLV dérivé de l’Egypte, comme dans la Figure 3 b et un NTC. La courbe d’amplification est la fluorescence du journaliste signal normalisé à la fluorescence du colorant ROX passif inclus dans le test (Rn) par rapport aux nombre de cycle. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
À la fin du cyclage thermique de qPCR sur les différentes machines dans chaque laboratoire, les données a été acquise et analysées. Figure 5 a et 5 b montrent des courbes de fusion représentatifs de l’analyses exécutées dans chaque laboratoire. Chaque machine de qPCR a été programmée pour effectuer une analyse des courbes de fusion à la fin. Ceci a été réalisé par progressivement augmenter la température et la surveillance de la fluorescence en fonction de la température. Lorsque la température est suffisamment élevée pour dénaturer l’ADN double brin, une forte baisse de fluorescence est enregistrée parce que la molécule de fluorophore est libérée. Le logiciel de chaque instrument de qPCR calculé la température de recuit (Tm) des données courbe fusion en traçant la température négative de vs dérivée première (Figure 5). On peut constater que dans la Figure 5 a et 5 b , que les produits formés dans les ensembles d’échantillons différents ont transition fusion homogène à la température prévue d’environ 80 ° C pour le dosage. Aucune autres pics à des températures plus basses ont été observés. En raison de leur petite taille, le Tm d’amorce-dimères est généralement inférieur à celui de l’objectif de la séquence d’ADN. Par conséquent, cette différence entre la Tmle rend facile à identifier les potentiels amorce-dimères ou autres produits d’amplification non spécifiques. Les contrôles n’ont pas généré des courbes de fusion comme TiLV échantillons positifs et normes et peuvent être considérées comme une ligne presque horizontale au bas des charts à la Figure 5 a et 5 b.

Figure 5 . Analyse de la courbe afin d’assurer la spécificité de dosage et de différents produits PCR peuvent être différenciés par leur fonte, faire fondre. A. analyse de la courbe des échantillons TiLV positifs provenant de Thaïlande, contrôle négatif et positif de plasmide contrôle de fonte. B. analyse de la courbe de TiLV échantillons positifs provenant d’Egypte, pTiLV normes et un NTC de fonte. Les graphiques en A et B les deux montrent le changement en fluorescence divisé par le changement de température complotèrent contre température pour produire une image claire de la dynamique de fusion. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
La plupart des machines de qPCR viennent avec un logiciel facilitant l’évaluation supplémentaire le qPCR exécuter et quantifiera les échantillons en générant une courbe d’étalonnage en plaçant automatiquement le cycle seuil (Ct) contre le logarithme du modèle des normes de la pTiLV Copiez le numéro tel qu’illustré à la Figure 6 a et 6 b pour les deux laboratoires indépendants. En bref, le Ct est l’unité utilisée pour l’évaluation des résultats de qPCR. La valeur de Ct désigne le nombre de cycles requis pour atteindre un niveau de signal de fluorescence seuil fixé. Plus le montant de départ modèle, moins il cycles est nécessaire pour atteindre un niveau de fluorescence détectable. En effet, des échantillons avec une forte sollicitation de la TiLV auront des valeurs det C plus faibles que les échantillons ayant une faible charge de TiLV comme chez les poissons avec une infection sub-clinique. Pour déterminer les valeurs det C, niveaux de fluorescence de fond sont tout d’abord déduites des données brutes. Ensuite, le logiciel associé au périphérique de qPCR sélectionne automatiquement un seuil de fluorescence, en cherchant les courbes de données pour chaque échantillon et comprenant un représentant det de C où l’échantillon a franchi le seuil. Cela se fait séparément pour chaque essai et chaque seuil doit être soigneusement évaluée, veiller à ce que le seuil a été fixé dans la partie logarithmique des courbes amplification et à un endroit où toutes les courbes sont parallèles. Ainsi, le spécifique Ct acquis est une valeur relative et c’est par rapport à la copie modèle numéro départ45, mais il est aussi spécifique pour la machine de qPCR et réactifs utilisés, l’efficacité de l’amplification par PCR et la sensibilité de détection. Ces paramètres contribuent à des différences observées à l’aide de la même épreuve dans la Figure 6.
Partir des courbes standards dans la Figure 6, les analyses de régression, y compris le calcul des pistes (m) de la courbe d’étalonnage et interceptions, amplification efficacités (100 x (101/m -1))46 et la linéarité de la réaction ont été effectuées. Analyse de la courbe d’étalonnage servaient aussi à confirmer la sensibilité (seuil de détection), de répétabilité et de reproductibilité du dosage. Théoriquement, la quantité d’ADN est doublée avec chaque PCR cycle, ce qui signifie que l’efficacité (E) est égale à 100 %. Cependant, dans la pratique une telle efficacité idéale est rarement atteint en raison des conditions PCR sous-optimal comme inhibition de l’ADN polymérase, contaminants, trop d' erreurs de cDNA et pipetage47. En général, amplification E varie de 90 à 110 % pour les tests de bons, dans la Figure 6 a une efficacité de 94,5 % a été calculée à l’aide des 8 échantillons dilués en série pTiLV, tandis que, l’efficacité du test dans l’essai illustré à la Figure 6 b à l’aide de pTiLV dilué en série 7 échantillons était de 101,2 %. Une efficacité de plus de 100 % est généralement due à la présence d’inhibiteurs de PCR dans le dosage. Analyse de régression linéaire de la courbe standard permet également le calcul du nombre de copies TiLV dans chaque échantillon de41,42,45, comme peut être observée pour les trois échantillons TiLV montrés en rouge en Figure 6 b qui est en accord avec les résultats pour les échantillons S1, S3 et S5 illustré à la Figure 3 b.

Figure 6 . RT-qPCR courbes standard. PCR en temps réel d’un facteur 10 dilutions successives du pTiLV, la norme utilisée dans les deux laboratoires. A. 8 échantillons dilués en série pTiLV ont été testés, tous de concentration connue et corrélés au nombre d’exemplaires TiLV / réaction. La courbe d’étalonnage a été générée en traçant journal copie numéro vs cycle seuil (Ct). La pente =-3.462, R2 = 0.9992 et l’efficacité est 94.47 %. B. comme en A, sauf pTiLV dilués en série 7 échantillons (vert) ont été testés et le graphique affiche le cycle seuil sur l’axe des y et le nombre de copies de TiLV (quantité) sur l’axe des abscisses. L’ordonnée à l’origine = 32.327, pente =-3.292, R2 = 0,98 et l’efficacité est 101,2 %. Pour les deux courbes de normes en A et B, la pente, l’ordonnée à l’origine et la valeurs des coefficients de corrélation (R2) sont utilisés pour comprendre la performance du test. Ce qui est important, valeur2 R devrait être proche de 1 car il s’agit d’une mesure de la linéarité de la courbe standard. La pente est utilisée pour mesurer l’efficacité PCR dans lequel une efficacité de 100 % correspond à une pente de-3.32, voir le texte principal de l’équation et obtenir des précisions. Une réaction de qPCR bon a généralement une efficacité entre 90-110 % de corrélation d’une pente entre-3.58 et-3.10. La courbe d’étalonnage est utilisée pour la quantification absolue des inconnus TiLV échantillons positifs et détermine le nombre exact de copies TiLV / réaction, comme c’est le cas pour les trois échantillons positifs TiLV couleur rouge b.
Discussion
TiLV a été pour la première fois en 2014 en Israël14 et depuis, il a été identifié dans plusieurs pays dont l’Egypte, la Colombie, Inde, Malaisie, Ouganda, Tanzanie et Thaïlande15,16,18, 35 , 48. prise de conscience mondiale, en particulier, chez le tilapia pays producteurs a placé plus d’attention sur le virus et les diverses restrictions et mesures de contrôle des autorités gouvernementales ont été appliquées pour tenter de prévenir la propagation de la TiLV. Ici, un protocole détaillé pour la détection de la TiLV dans les tissus de tilapia, portant sur le prélèvement d’échantillons, ARN, essais de synthèse, de PCR et de qPCR cDNA a été expliqué. Il y a différents aspects de ces méthodes qui justifient une discussion spécifique. TiLV a été identifié chez les poissons s’étendant sur une espèce de tilapia jusqu’ici, y compris le tilapia d’élevage hybrides (O. et diverses tailles9,12,14,15,49 niloticus x o. aureus)11,14, Nile tilapia (o. niloticus)9,10,14,15,16, 33 , 35 , 36 , 49 , 50 et rouge de tilapia (Oreochromis SP.)16,33,48,51, ainsi que dans sauvages Nile tilapia9,12, noir tilapia51, T. zilli14,15, S. galilaeus, o. aureus et T. simonis intermedia14 et très récemment TiLV a été identifié chez la carpe sauvage (Barbonymus schwanenfeldii)52. Des échantillons de tissus des organes internes (gill, rate, foie, cœur, rein) ou mucus37 peuvent être prélevés de tilapia moribond mais aussi sain quel que soit l’âge, la taille ou les espèces et traitées pour l’ARN. Au protocole d’extraction RNA total décrit utilise ici une solution de monophasique du thiocyanate de phénol et de guanidinium, qui est un agent chaotropique de dénaturation. Les tissus sont homogénéisés directement dans cette solution puis par l’ajout de chloroforme et centrifugation pour atteindre la phase de séparation dans lequel un ARN clair contenant une phase aqueuse supérieure, une interphase et une phase organique inférieure est généré. L’ARN est isolé dans la phase aqueuse par précipitation de l’isopropanol, lavée de l’ARN récupéré pour se débarrasser des contaminants. Isolement de l’ARN par cette méthodologie a été lancé par Piotr Chomczynski et Nicoletta Sacchi et parlait de guanidinium thiocyanate-phénol-chloroforme extraction53,54. Ce type de réactif utilisé pour l’extraction de l’ARN peut être acheté dans le commerce ou fait dans le laboratoire (voir la Table des matières pour plus d’informations). Ce protocole prend légèrement plu de méthodes fondées sur la colonne, telles que la purification à base de silice, mais en général, il est plus rentable et les rendements plus RNA.
Dans ce protocole, quantification ARN utilisant A260 valeurs a été exposée par lequel les valeurs de spectrophotométrie peuvent indiquer la qualité RNA (A260/A280 = 1,9 à 2,1). Tandis que cette méthode donnera une bonne indication de la pureté de l’échantillon, il ne peut absolument informer quant à la qualité de l’ARN extrait. Pour déterminer correctement si l’ARN est intacte ou partiellement dégradés, les échantillons peuvent être séparation par électrophorèse sur gel d’agarose dans lequel bavures du bromure d’éthidium teinté de 18 ans et bandes ARNr 28 s indiquent la dégradation de l’ARN. Vérification de la qualité de RNA peut-être inclure à l’aide d’un instrument lab-on-a-chip. En outre, il est également important de digérer l’ARN purifié à la DNase pour éliminer la contamination héberger l’ADN génomique, qui, selon les applications en aval peut conduire à des résultats erronés. Si l’hôte ADNg est encore contaminant l’échantillon RNA dans une large mesure, un traitement supplémentaire de la DNase i peut également être effectué à la fin de la procédure d’extraction de RNA (voir Table des matières).
Synthèse de l’ADN complémentaire peut influer grandement sur les résultats globaux de qPCR et constitue un aspect de la méthode qui peut introduire des variations. Le protocole d’ADNc préconisé ici comprend une installation de composant unique à l’aide d’oligo (dT) et donc seulement transcrit mRNA contenant la queue polyA. Il permet le contrôle utilisateur d’exactement quels composants à utiliser dans la réaction de transcription inverse et ce mode de cDNA de synthèse s’est avérée fructueuse pour TiLV détection32. Une alternative à ce set-up est un acheté dans le commerce-mélange réactionnel contenant tous les composants requis pour la réaction de transcription inverse et est très rapide et simple sans le Protocole habituel de plusieurs étape, pipetage et multi-température. C’est avantageux car il réduit la manutention et favorise l’uniformité dans l’ensemble de tous les échantillons. Ces mélanges en maître incluent souvent les oligo (décollement) et amorces aléatoires rendant applicables aux différents modèles de RNA et de générer des copies de l’ADNc représentant des séquences de toute la longueur de l’ARN dans une population (viral et tilapia héberger ARNm) et en théorie, toutes espèces d’ARN désiré peuvent alors être mesurée par conventionnels PCR ou qPCR d’un tel échantillon. Cette polyvalence est le principal avantage d’une approche de RT-PCR 2 étapes ; Il offre une piscine à long terme qui peut être utilisée à de nombreuses expériences différentes. Dans les résultats, une approche de RT-PCR en une étape a été représentée dans laquelle servaient les amorces spécifiques de séquence (tableau 1) et la RT et PCR ont été effectuées dans un tube (voir la liste de matériel). En général, des amorces spécifiques de séquence permettent une plus grande efficacité de RT de l’ARN de cible spécifique que l’utilisation d’amorçage aléatoire, mais la cible RNA est le seul qui peut être quantifié dans un tel échantillon de cDNA qui peut être le seul but de certains laboratoires (voir Table des matières pour des suggestions de produit synthèse de cDNA).
Alors que la RT-PCR classique semble être communément utilisé jusqu’ici dans la TiLV diagnostic9,13,14,15,16,17,18, 33 , 35 , 48 , 55. RT-qPCR s’est avéré être un outil plus puissant pour la détection et la quantification des rejets de petites quantités de TiLV dans les tissus de poissons ou de mucus32,37. En général, qPCR est largement utilisé dans les laboratoires de diagnostic de virologie clinique en raison de sa haute sensibilité, spécificité, bonne reproductibilité, une plage dynamique étendue et vitesse21. Si qPCR peut être initialement plus coûteux à mettre en oeuvre que la RT-PCR classique, il offre beaucoup d’avantages importants sur la PCR classique ; Il a un temps plus rapide retournement de l’échantillon de résultats et il ne nécessite aucune étape post-PCR. Ce dernier point signifie qu’il y a un risque minime de contamination de laboratoire et il peut s’adapter plus facilement à des situations de haut-débit comme en cas d’épidémies. En outre, il est intrinsèquement plus sensible que conventionnelle RT-PCR, ce qui est d’une importance vitale pour détecter une charge virale faible dans les infections subcliniques21. Cela nécessiterait une approche PCR imbriquée nécessitant une transcription inverse, deux davantage les réactions de PCR, analyse de gel d’agarose puis électrophorèse sur gel. Ces nombreuses étapes prennent beaucoup de temps et augmentent les chances d’erreurs ou de contamination. Néanmoins, grâce à sa haute sensibilité, RT-qPCR exige une conception expérimentale rigoureuse et une connaissance approfondie des techniques de quantification pour générer des résultats précis56,,57.
Le fluorophore de liaison ADN, SYBR Green I a été démontrée dans le présent protocole. C’est un colorant non spécifique des liaison ADN dsDNA et donc la spécificité du test se trouve entièrement dans le jeu d’amorces, ce qui peut générer des faux positifs,58. Par conséquent, alors que l’ADN double brin fonte analyse de la courbe réalisée à la fin de chaque PCR est un élément particulièrement important de la réaction PCR parce qu’elle confirme ce qu’un amplicon PCR du bon Tm est produit (Ceci devrait aussi être réalisé en gel électrophorèse lors de nouveaux tests sont en cours d’exécution). Le Tm d’un fragment d’ADN dépend d’une variété de caractéristiques telles que sa longueur, séquence, composition de GC, complémentarité des brins, la concentration ainsi que sur la mémoire tampon composants et exhausteurs de PCR. Les analyses de courbe fusion dans les résultats représentatifs, illustrés de deux laboratoires n’ont pas révélé la présence d’amorce-dimères ou autres produits PCR non désirés, mais si c’est observé avec d’autres échantillons et/ou des montages expérimentaux, puis l’essai doit être ré-optimisé. Des technologies plus avancées de qPCR ne nécessitent pas une telle mesure de courbe de fusion et en effet, depuis ce document a été rédigé, des méthodes une TaqMan basé TiLV RT-qPCR développée utilisant deux amorces et une sonde rendant hautement spécifiques TiLV34.
Sans aucun doute, les amorces conçues pour des dosages de RT-qPCR sont fondamentales pour la réussite du test et les amorces ici ont été conçus d’après les données génomiques d’accessibles TiLV à la fois32. Toutefois, les virus à ARN sont bien connus de présentent des taux de mutation élevé et souches possibles seront échapperont les tests diagnostiques actuels, comme a été observée pour l’AIS59. Il sera toujours difficile pour ces types de virus générer un universel pan-TiLV RT-qPCR analyse et ces tests seront seulement continuellement améliorées si plus TiLV données génomiques des lieux et des périodes de temps deviennent disponibles.
Enfin, il est essentiel pour courir en double ou triple si possible, les réactions dans les deux intra et inter qPCR dosages. Si les valeurs det C sont très élevés, alors que l’utilisation des répliques est particulièrement importante de s’assurer que la réaction de PCR est fiable et reproductible. En général, si répliquent des données provenant de réactions varie de plus de 0,5 cycles, les réactions doivent être répétés et si les valeurs det C varient constamment cycles > 0,5 en réplique, le dosage doit être ré-optimisé. L’utilisation d’un robot de pipetage intégrée qPCR aide énormément à cette question, mais c’est un outil de luxe. Comme pour toute expérience, les contrôles adéquats et appropriés d’inclusion sont de plus grande importance au développement de dosages moléculaires robustes, en particulier dans les laboratoires de diagnostic où ces tests doivent être accrédités. Contrôles devraient inclure positif (échantillon positif de TiLV, plasmide TiLV standard) et des échantillons témoins négatifs (NTC et -RT) ainsi que la détection des gènes domestiques tilapia endogène. Ces contrôles ne peuvent pas être sous-estimée et devraient être inclus dans chaque série de tests afin de bien comprendre la qualité de chaque étape du test et d’interpréter correctement les résultats.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous sommes reconnaissants à l’Institut de bactériologie vétérinaire, la faculté Vetsuisse, l’Université de Berne pour leur soutien. Ce travail a été financé par le Comité pour la Promotion de début carrière des chercheurs universitaires et d’égalité des sexes à la faculté Vetsuisse, Université de Berne en finançant des modèle de 120 % attribué à PN. WS et PR sont pris en charge par le Centre for Advanced Studies pour l’Agriculture et alimentation, Institut des hautes études, l’Université Kasetsart, Bangkok, Thaïlande sous la Promotion de l’enseignement supérieur recherche et National recherche Université Project of Thailand, Bureau de la Thaïlande de Commission, ministère de l’éducation, l’enseignement supérieur. Nous tenons à remercier le Dr. Kwanrawee Sirikanchana pour son récit et Piyawatchara Sikarin pour le montage de la vidéo.
matériels
| Name | Company | Catalog Number | Comments |
| Tissue collection | Step 1 | ||
| Tricaine methanesulfonate | Sigma-Aldrich | E10521 | An alternative to clove oil. Step 1.1 |
| RNAlater stabilization solution | Thermo Fisher Scientific | AM7020 | For storing tissues if they cannot be processed immediately Step 1.3 |
| RNA extraction | Step 2 | ||
| TRIreagent | Sigma-Aldrich | Step 2.1 | |
| TRIzol | Thermo Fisher Scientific (Invitrogen) | 15596026 | Step 2.1 |
| GENEzol | Geneaid | GZR100 | Step 2.1 |
| Trisure | Bioline | BIO-38032 | Step 2.1 |
| Homemade solution | - | - | 94.53 g/L (800 mM) guanidine thiocyanate 30.45 g/L (400 mM) ammonium thiocyanate 8.20 g/L (100 mM) sodium acetate 380 mL/L (38 % v/v) phenol 50 mL/L (5 % v/v) glycerol 1.0 g/L (0.1 % w/v) 8-quinolinol, pH 5.0 Store up to 2 years at 4oC Step 2.1 |
| MagNA Lyser Green Beads | Roche | 3358941001 | An alternative tissue homogenization method used in conjunction with tissue lysing machines detailed below Step 2.2 |
| Lysing Matrix D, 2 mL Tube | MP BIOMEDICALS | 116913050 | |
| Chloroform | Sigma-Aldrich | C2432 | Step 2.3 |
| Chloroform | RCI Labscan | AR1027E-G2.5L | Step 2.3 |
| 1-Bromo-3-chloropropane | Sigma-Aldrich | B9673 | A less toxic alternative to chloroform Step 2.3 |
| Isopropanol (GC) ≥ 99.8 % | Sigma-Aldrich | 59300 | Step 2.6 |
| Isopropanol (ACS, ISO Reag. Ph Eur) | Merck KGaA (EMSURE) | 1.09634.2500 | Step 2.6 |
| Glycogen, molecular biology grade (e.g., Sigma, cat. no. G1767) | Thermo Fisher Scientific (Thermo Scientific) | R0551 | Useful step if tissue starting material is small to maximise RNA precipitation optional |
| Ethanol (purity (GC) ≥ 99.9 % | Sigma-Aldrich (EMD Millipore) | 1.00983 | Step 2.9 |
| Ethanol (ACS, ISO Reag. Ph Eur) | Merck (EMSURE) | 1.00983.2500 | Step 2.9 |
| Nuclease-free water | Promega | P1193 | Step 2.13 |
| Nuclease-free water | Multicell | 809-115-CL | Step 2.13 |
| Ambion TURBO DNA-free kit | Thermo Fisher Scientific (Invitrogen) | AM1907 | Can be performed at the end of the RNA extraction protocol optional |
| cDNA synthesis | Step 4 | ||
| Viva cDNA Synthesis Kit | Vivantis | cDSK01 | Step 4.1 & 4.3 |
| ReverTra Ace qPCR RT MasterMix with gDNA remover | Toyobo | A1172K | An alternative option see discussion |
| ReverTra Ace qPCR RT Kit | Toyobo | FSQ-101 | An alternative option see discussion |
| AffinityScript Multiple Temperature Reverse Transcriptase | Agilent Technologies | 600107 | An alternative option |
| PCR | Step 5 | ||
| DNA polymerase systems: | Step 5.2 | ||
| - Platinum II Hot-Start Green PCR Master Mix (2X) | Thermo Fisher Scientific (Invitrogen) | 14001012a | Step 5.2 |
| - GoTaq Mastermix | Promega | M7122 | Step 5.2 |
| Separate PCR mixture components: | Step 5.2 | ||
| 10mM dNTP Mix | Vivantis | NP2409 | Step 5.2 |
| 25mM MgCl2 | Thermo Fisher Scientific | R0971 | Step 5.2 |
| 10X Taq Buffer with KCl | Thermo Fisher Scientific | 00348114 | Step 5.2 |
| Taq DNA polymerase | Vivantis | PL1202 | Step 5.2 |
| - Verso 1-step RT-PCR ReddyMix with ThermoPrime Taq | Thermo Fisher Scientific | AB1454 | One step RT-PCR exemplified in Figure 3B |
| Gel electrophoresis: | For visulation of PCR products from steps 5.1-5.4 | ||
| Ethidium Bromide solution (10 mg/mL) | Thermo Fisher Scientific | 17898 | Step 5.5 |
| Tris/Acetic/EDTA (TAE) buffer: | Step 5.5 | ||
| - Tris | Vivantis | PR0612-1KG | Step 5.5 |
| - Acetic acid (glacial) (ACS, ISO, Reag. Ph Eur) | Merck KGaA (EMSURE) | 1.00063.2500 | Step 5.5 |
| - Ethylenediaminetetraacetic acid (EDTA) | BIO-RAD | 161-0729 | Step 5.5 |
| Agarose | Vivantis | PC0701-100G | Step 5.5 |
| DNA ladders and markers | Vivantis | NL1405 | Step 5.5 |
| DNA gel loading dye (6X) | Thermo Fisher Scientific | R0611 | Step 5.5 |
| qPCR | Step 6 | ||
| PowerUP SYBR Green Master Mix | Thermo Fisher Scientific (Applied Biosystems) | A25779 | Exemplified in Figures 4-6B Step 6.2 |
| iTaq Universal SYBR Green Supermix | BIO-RAD | 1725120 | Exemplified in the video and in Figures 4-6A Step 6.2 |
| Equipment | |||
| Dounce tissue grinder pestle | Sigma-Aldrich | P1110 | Protocol 2 |
| MagNA Lyser Instrument | Roche | 3358976001 | An alternative tissue homogenizing option for protocol 2 which are used in conjunction with the lysing beads detailed above Step 2.2 |
| FastPrep-24 5G Homogenizer | MP BIOMEDICALS | 116005500 | |
| Refrigerated microcentrifuge | Eppendorf | Eppendorf 5427R | Protocol 2 Step 2.4, 2.7 & 2.10 |
| Refrigerated microcentrifuge | Eppendorf | Eppendorf 5418R | |
| Heat box | Labnet | AccuBlock Digital Dry Bath | Protocol 2 Step 2.13 |
| Microvolume spectrophotometer | Thermo Fisher Scientific (Applied Biosystems) | Nanodrop 2000 | Protocol 3 Step 3.1 - 3.4 |
| PCR machine | BIO-RAD | T100 Thermal Cycler | Protocol 5 Step 5.4 |
| Power supply | BIO-RAD | PowerPac HC | Protocol 5 Step 5.5 |
| Horizontal gel electrophoresis | BIO-RAD | Mini ReadySub-Cell GT Cell #1704487edu | Protocol 5 Step 5.5 |
| Mini microcentrifuge | Corning | LSE 6766 | Useful to quickly spin down PCR reaction tubes in protocols 4, 5 & 6 Step 6.5.1 |
| Microcentrifuge | LioFuge | LM-60 | Step 6.5.1 |
| qPCR machine and software | Thermo Fisher Scientific | 7500 Fast Real-Time PCR System with 7500 Software v2.0 | Protocol 6 Step 6.6-6.8 |
| qPCR machine and software | BIO-RAD | CFX96 Touch Real-Time PCR Detection System with CFX Manager software | |
| General Materials | |||
| Mayo scissors | Step 1.1-1.2 | ||
| Forceps | Step 1.1-1.2 | ||
| Pipette | Rainin | Pipette-Lite XLS | |
| Aerosol-barrier pipette tips | Sigma-Aldrich | Z333328, Z333336, Z333344 | |
| Nuclease-free 1.5-ml microcentrifuge tubes | Eppendorf |
Références
- FAO. . The State of World Fisheries and Aquaculture, 2014. Opportunities and Challenges. , (2014).
- FAO. . The State of World Fisheries and Aquaculture, 2016. Contributing to Food Security and Nutrition for all. , (2016).
- WorldBank. . FISH TO 2030: Prospects for Fisheries and Aquaculture. Agriculture and Environmental Services Discussion Paper 03. , (2013).
- Wing-Keong, N., Nicholas, R. A review of the nutrition and feeding management of farmed tilapia throughout the culture cycle. Reviews in Aquaculture. 5 (4), 220-254 (2013).
- Cleasby, N., et al. The socio-economic context for improving food security through land based aquaculture in Solomon Islands: A peri-urban case study. Marine Policy. 45, 89-97 (2014).
- Ponzoni Raul, W., et al. Genetic improvement of Nile tilapia (Oreochromis niloticus) with special reference to the work conducted by the WorldFish Center with the GIFT strain. Reviews in Aquaculture. 3 (1), 27-41 (2011).
- Hounmanou, Y. M. G., et al. Tilapia lake virus threatens tilapiines farming and food security: Socio-economic challenges and preventive measures in Sub-Saharan Africa. Aquaculture (Amsterdam, Netherlands). 493, 123-129 (2018).
- OIE. . Tilapia Lake Virus (TiLV) - a novel orthomyxo-like virus. OIE technical disease cards. , (2018).
- Mugimba, K. K., et al. Detection of tilapia lake virus (TiLV) infection by PCR in farmed and wild Nile tilapia (Oreochromis niloticus) from Lake Victoria. Journal of Fish Diseases. , (2018).
- Koesharyani, I., Gardenia, L., Widowati, Z., Khumaira, D. D., Rustianti, Studi kasus infeksi tilapia lake virus (tilv) pada ikan nila (Oreochromis niloticus). Jurnal Riset Akuakultur. 13 (1), 85-92 (2018).
- OIE. . Tilapia lake virus disease (TiLV), Chinese Taipei. Immediate Notification. , (2017).
- OIE. . Tilapia Lake Virus Disease (TiLV), Peru. Immediate Notification. , (2018).
- Bacharach, E., et al. Characterization of a Novel Orthomyxo-like Virus Causing Mass Die-Offs of Tilapia. MBio. 7 (2), e00431-e00416 (2016).
- Eyngor, M., et al. Identification of a novel RNA virus lethal to tilapia. Journal of Clinical Microbiology. 52 (12), 4137-4146 (2014).
- Nicholson, P., et al. Detection of Tilapia Lake Virus in Egyptian fish farms experiencing high mortalities in 2015. Journal of Fish Diseases. 40 (12), 1925-1928 (2017).
- Surachetpong, W., et al. Outbreaks of Tilapia Lake Virus Infection, Thailand, 2015-2016. Emerging Infectious Diseases. 23 (6), 1031-1033 (2017).
- Tattiyapong, P., Dachavichitlead, W., Surachetpong, W. Experimental infection of Tilapia Lake Virus (TiLV) in Nile tilapia (Oreochromis niloticus) and red tilapia (Oreochromis spp.). Veterinary Microbiology. 207, 170-177 (2017).
- Kembou Tsofack, J. E., et al. Detection of Tilapia Lake Virus in Clinical Samples by Culturing and Nested Reverse Transcription-PCR. Journal of Clinical Microbiology. 55 (3), 759-767 (2017).
- Thangaraj, R. S., et al. Derivation of two tilapia (Oreochromis niloticus) cell lines for efficient propagation of Tilapia Lake Virus (TiLV). Aquaculture (Amsterdam, Netherlands). 492, 206-214 (2018).
- Hanson, L. A., Rudis, M. R., Vasquez-Lee, M., Montgomery, R. D. A broadly applicable method to characterize large DNA viruses and adenoviruses based on the DNA polymerase gene. Virology Journal. 3, 28-28 (2006).
- Josko, D. Molecular virology in the clinical laboratory. Clinical Laboratory Science. 23 (4), 231-236 (2010).
- Munir, K., Kibenge, F. S. Detection of infectious salmon anaemia virus by real-time RT-PCR. Journal of Virological Methods. 117 (1), 37-47 (2004).
- Snow, M., et al. Developement, application and validation of a Taqman real-time RT-PCR assay for the detection of infectious salmon anaemia virus (ISAV) in Atlantic salmon (Salmo salar). Developments in Biologicals. 126, 133-145 (2006).
- Matejusova, I., McKay, P., McBeath, A. J., Collet, B., Snow, M. Development of a sensitive and controlled real-time RT-PCR assay for viral haemorrhagic septicaemia virus (VHSV) in marine salmonid aquaculture. Diseases of Aquatic Organisms. 80 (2), 137-144 (2008).
- Garver, K. A., et al. Development and validation of a reverse transcription quantitative PCR for universal detection of viral hemorrhagic septicemia virus. Diseases of Aquatic Organisms. 95 (2), 97-112 (2011).
- Dalla Valle, L., et al. Development of a sensitive and quantitative diagnostic assay for fish nervous necrosis virus based on two-target real-time PCR. Veterinary Microbiology. 110 (3-4), 167-179 (2005).
- Hodneland, K., Garcia, R., Balbuena, J. A., Zarza, C., Fouz, B. Real-time RT-PCR detection of betanodavirus in naturally and experimentally infected fish from Spain. Journal of Fish Diseases. 34 (3), 189-202 (2011).
- Hodneland, K., Endresen, C. Sensitive and specific detection of Salmonid alphavirus using real-time PCR (TaqMan). Journal of Virological Methods. 131 (2), 184-192 (2006).
- Wang, X. W., Ao, J. Q., Li, Q. G., Chen, X. H. Quantitative detection of a marine fish iridovirus isolated from large yellow croaker, Pseudosciaena crocea, using a molecular beacon. Journal of Virological Methods. 133 (1), 76-81 (2006).
- van Beurden, S. J., et al. Development and validation of a real-time PCR assay for the detection of anguillid herpesvirus 1. Journal of Fish Diseases. 39 (1), 95-104 (2016).
- Ciulli, S., et al. Development and application of a real-time PCR assay for the detection and quantitation of lymphocystis disease virus. Journal of Virological Methods. 213, 164-173 (2015).
- Tattiyapong, P., Sirikanchana, K., Surachetpong, W. Development and validation of a reverse transcription quantitative polymerase chain reaction for tilapia lake virus detection in clinical samples and experimentally challenged fish. Journal of Fish Diseases. 41 (2), 255-261 (2018).
- Dong, H. T., et al. Emergence of tilapia lake virus in Thailand and an alternative semi-nested RT-PCR for detection. Aquaculture (Amsterdam, Netherlands). 476, 111-118 (2017).
- Waiyamitra, P., et al. A TaqMan RT-qPCR assay for tilapia lake virus (TiLV) detection in tilapia. Aquaculture (Amsterdam, Netherlands). 497, 184-188 (2018).
- Behera, B. K., et al. Emergence of Tilapia Lake Virus associated with mortalities of farmed Nile Tilapia Oreochromis niloticus (Linnaeus 1758) in India. Aquaculture (Amsterdam, Netherlands). 484, 168-174 (2018).
- Ferguson, H. W., et al. Syncytial hepatitis of farmed tilapia, Oreochromis niloticus (L.): a case report. Journal of Fish Diseases. 37 (6), 583-589 (2014).
- Liamnimitr, P., Thammatorn, W., U-thoomporn, S., Tattiyapong, P., Surachetpong, W. Non-lethal sampling for Tilapia Lake Virus detection by RT-qPCR and cell culture. Aquaculture (Amsterdam, Netherlands). 486, 75-80 (2018).
- Yang, C. G., et al. Evaluation of reference genes for quantitative real-time RT-PCR analysis of gene expression in Nile tilapia (Oreochromis niloticus). Gene. 527 (1), 183-192 (2013).
- Bustin, S. A. Real-time, fluorescence-based quantitative PCR: a snapshot of current procedures and preferences. Expert Review of Molecular Diagnostics. 5 (4), 493-498 (2005).
- Fleige, S., Pfaffl, M. W. RNA integrity and the effect on the real-time qRT-PCR performance. Molecular Aspects of Medicine. 27 (2-3), 126-139 (2006).
- Kubista, M., et al. The real-time polymerase chain reaction. Molecular Aspects of Medicine. 27 (2-3), 95-125 (2006).
- Mackay, I. M., Arden, K. E., Nitsche, A. Real-time PCR in virology. Nucleic Acids Research. 30 (6), 1292-1305 (2002).
- Wong, M. L., Medrano, J. F. Real-time PCR for mRNA quantitation. Biotechniques. 39 (1), 75-85 (2005).
- Bustin, S. A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. Journal of Molecular Endocrinology. 25 (2), 169-193 (2000).
- Heid, C. A., Stevens, J., Livak, K. J., Williams, P. M. Real time quantitative PCR. Genome Research. 6 (10), 986-994 (1996).
- Rutledge, R. G., Côté, C. Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Research. 31 (16), e93-e93 (2003).
- Svec, D., Tichopad, A., Novosadova, V., Pfaffl, M. W., Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomolecular Detection and Quantification. 3, 9-16 (2015).
- Amal, M. N. A., et al. A case of natural co-infection of Tilapia Lake Virus and Aeromonas veronii in a Malaysian red hybrid tilapia (Oreochromis niloticus × O. mossambicus) farm experiencing high mortality. Aquaculture (Amsterdam, Netherlands). 485, 12-16 (2018).
- Fathi, M., et al. Identification of Tilapia Lake Virus in Egypt in Nile tilapia affected by ‘summer mortality’ syndrome. Aquaculture (Amsterdam, Netherlands). 473, 430-432 (2017).
- OIE. . Tilapia Lake Virus disease (TiLV), Philippines. Immediate Notification. , (2017).
- OIE. . Tilapia lake virus disease (TiLV), Malaysia. Immediate Notification. , (2017).
- Abdullah, A., et al. First detection of tilapia lake virus (TiLV) in wild river carp (Barbonymus schwanenfeldii) at Timah Tasoh Lake, Malaysia. Journal of Fish Diseases. 41 (9), 1459-1462 (2018).
- Chomczynski, P., Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 162 (1), 156-159 (1987).
- Chomczynski, P., Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nature Protocols. 1 (2), 581-585 (2006).
- Del-Pozo, J., et al. Syncytial Hepatitis of Tilapia ( Oreochromis niloticus L.) is Associated With Orthomyxovirus-Like Virions in Hepatocytes. Veterinary Pathology. 54 (1), 164-170 (2017).
- Bustin, S. A., et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry. 55 (4), 611-622 (2009).
- Purcell, M. K., Getchell, R. G., McClure, C. A., Garver, K. A. Quantitative polymerase chain reaction (PCR) for detection of aquatic animal pathogens in a diagnostic laboratory setting. Journal of Aquatic Animal Health. 23 (3), 148-161 (2011).
- Simpson, D. A., Feeney, S., Boyle, C., Stitt, A. W. Retinal VEGF mRNA measured by SYBR green I fluorescence: A versatile approach to quantitative PCR. Molecular Vision. 6, 178-183 (2000).
- Kibenge, M. J., et al. Discovery of variant infectious salmon anaemia virus (ISAV) of European genotype in British Columbia, Canada. Virology Journal. 13, 3 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.