
Method Article
Rilevamento di Tilapia Lago Virus mediante RT-PCR convenzionale e SYBR Green RT-qPCR
In questo articolo
Riepilogo
Questo protocollo diagnosi Tilapia Lago Virus (TiLV) nei tessuti di tilapia utilizzando metodologie di RT-PCR. L'intero metodo è descritto da dissezione del tessuto per estrazione di RNA totale, seguita dalla sintesi del cDNA e rilevamento di TiLV da PCR convenzionale o PCR quantitativa utilizzando dsDNA vincolante una tintura fluorescente legante.
Abstract
Lo scopo di questo metodo è quello di facilitare l'individuazione veloce, sensibile e specifico di Tilapia Lago Virus (TiLV) nei tessuti di tilapia. Questo protocollo può essere usato come parte di programmi di sorveglianza, le misure di biosicurezza e nei laboratori di ricerca di base TiLV. Il gold standard di sistema diagnostico del virus in genere comporta l'isolamento del virus seguita da tecniche complementari quali la reazione a catena della polimerasi d'inversione-trascrizione (RT-PCR) per le necessarie verifiche. Questo può essere ingombrante, che richiede tempo e in genere richiede campioni di tessuto pesantemente infettati da virus. L'uso di RT-PCR (q) nella rilevazione di virus quantitativa è vantaggioso a causa della sua natura quantitativa, alta sensibilità, specificità, scalabilità e il suo rapido tempo di risultato. Qui, l'intero metodo di PCR approcci basati per TiLV rilevamento è descritto, da tilapia organo sezionamento, totale dell'acido ribonucleico (RNA) estrazione utilizzando una soluzione di tiocianato-fenolo-cloroformio di guanidium, la quantificazione di RNA, seguita da una PCR in due fasi protocollo che comportano, sintesi di acido desossiribonucleico complementare (cDNA) e rilevamento di TiLV da PCR convenzionale o identificazione quantitativa via qPCR mediante SYBR green tingere. PCR convenzionale richiede passaggi post-PCR e semplicemente vi informerà circa la presenza del virus. Quest'ultimo approccio permetterà per quantificazione assoluta di TiLV giù a appena 2 copie ed è quindi eccezionalmente utile per la diagnosi di TiLV nei casi sub-clinici. Una descrizione dettagliata dei due approcci PCR, risultati rappresentativi da due laboratori e una discussione approfondita dei parametri critici di entrambi sono stati inclusi per garantire che i ricercatori e diagnosti trovano la loro più adatto e applicabile Metodo di rilevamento TiLV.
Introduzione
La fornitura di pesce pro capite globale ha raggiunto un nuovo record di 20 kg nel 2014 e questo era a causa della crescita vigorosa in acquacoltura. L'acquacoltura rimane uno dei settori di produzione mangime più rapida crescita in tutto il mondo ed è il settore di produzione alimentare solo animale che sta crescendo più velocemente della popolazione umana1. Tilapiine cichilds costituiscono i secondo più importanti pesci d'acqua dolce coltivati in tutto il mondo con una produzione globale totale di 6,4 milioni di tonnellate (MT) e un valore stimato di 9,8 miliardi di dollari nel 20152. I dieci produttori principali di tilapia sono Cina (1,78 MT), Indonesia (1.12 MT) ed Egitto (0,88 MT), seguita dal Bangladesh, Vietnam, Filippine, Brasile, Thailandia, Colombia e Uganda2. Si prevede che la produzione globale di tilapia sarà circa 7,3 MT da 20303. Tilapia sono diventati tali una fonte di cibo globale importante non solo perché sono una fonte economica di proteina4 ma anche perché sono facili da allevare in capacità sotto una vasta gamma di acqua e clima condizioni5,6. A pochi decenni fa, si credeva che c'erano poche malattie commercialmente significativi minacciando agricoltura tilapia, ma questo non è più vero. Una malattia virale emergente chiamata malattia di tilapia Lago virus (TiLVD) è la prima epidemia di malattia critica mai trovati a tilapia e l'intero settore è a rischio. Questa malattia ha gravi conseguenze socio-economiche ed è una minaccia diretta sulla sicurezza alimentare per milioni di persone in Africa7, Asia e Sud America. All'inizio del 2018, l'organizzazione mondiale per la salute animale (OIE) ha riferito che l'agente eziologico di questa malattia, TiLV, era stato rilevato ufficialmente in tre continenti che copre otto paesi8 , poiché questa scheda di informazioni di agente patogeno era Aggiornato ci sono state altre segnalazioni di TiLV in Tanzania, Uganda9, Indonesia10, Taiwan11 e Perù12. TiLV è un romanzo single-stranded RNA virus descritto per essere un orthomyxo-come il virus perché contiene una varietà di caratteristiche che ricorda altri orthomyoxoviruses come l'Influenza o infettiva salmon anemia virus (ISAV)13. In primo luogo è stato identificato nel guaime delle massicce perdite di tilapia selvatico e di allevamento nel lago di Galilea, Israele14. Da allora in poi, simili epidemie definito come mortalità di estate e il mese di una sindrome di mortalità associata con l'infezione di TiLV sono stati segnalati in Nile tilapia (Oreochromis niloticus) in Egitto15 e Nilo e tilapia ibrido rosso (Oreochromis spp.) in Thailandia16, rispettivamente. Rilevamento di virus animali acquatiche avviene storicamente di crescita e l'isolamento del virus in coltura cellulare. Varie linee cellulari sono stati testati per la propagazione e l'isolamento di TiLV compreso, le cellule E-11 derivate da snakehead pesce (striatus Ophiocephalus)17,18, OmB e TmB provenienti da Oreochromis mossambicus18e OnlB e uniche provenienti da Nile tilapia (o. niloticus)19. Mentre la cultura del virus ha il vantaggio che fornisce materiale per ulteriori esperimenti, ha lo svantaggio che richiede almeno 4-7 giorni per osservare la formazione degli effetti citopatici (CPE) e fondamentalmente, diversi virus piscine, che sono più in forma di replicare possono essere propagate e produrre simili CPE.
Negli ultimi decenni, c'è stato un allontanamento di metodi diagnostici tradizionali, spesso richiede tempo come coltura cellulare, sierologia e rilevazione dell'antigene e sostituzione con più veloce e più sensibile dell'acido nucleico basato rilevamento test20, 21. Questo è evidente dal fatto che molti saggi qPCR sono stati sviluppati come metodi diagnostici importanti per una moltitudine di malattie virali degli animali acquatici, come ad esempio per ISAV22,23, virus emorragico virale di setticemia (VHSV)24 ,25, betanodavirus26,27 salmonidi alphavirus28, pesce iridovirus29, popolazione herpesvirus 1 (AngHV1)30e Lymphocystis malattia virus (LCDV)31 . Metodi avanzati per la diagnosi e patogeno sorveglianza sono urgenti per ridurre la diffusione di TiLV. Tali metodi dovrebbero consentire diagnosi precoce dell'infezione prima di sviluppano segni clinici e rilevamento dei virus bassi carichi. Ad oggi, compresi RT-PCR14,32, diversi protocolli di PCR nested RT-PCR18, semi-nested RT-PCR33e RT-qPCR32,34 sono stati sviluppati per la rilevazione di TiLV nei tessuti del pesce. Un confronto dell'isolamento RT-qPCR e virus in linee cellulari sensibili per la rilevazione di TiLV ha rivelato che RT-qPCR era 1.000 volte più sensibile di isolamento del virus32. Anche se ogni protocollo PCR pubblicato ha segnalato diverse sensibilità per il rilevamento di TiLV, la maggior parte delle analisi sono altamente sensibile con i limiti di rilevazione di copie virali 7,5 copie33, 7 copie18 o 2 copie32 a reazione.
Lo scopo di questo articolo di metodi è quello di spiegare, in dettaglio, come eseguire le analisi di rilevamento TiLV, partendo da collezione di tessuto di tilapia, all'estrazione di RNA totale, sintesi del cDNA e quindi TiLV specifico PCR basata saggi. In particolare, sono stati descritti protocolli completi di RT-PCR convenzionale sia SYBR green-basato RT-qPCR fare appello a una vasta gamma di scienziati, con l'obiettivo di rilevare TiLV. Il primo è meno sensibile ma è in genere una soluzione meno costosa di rilevamento. Quest'ultimo richiede infrastrutture più elaborate come una macchina PCR quantitativa e reagenti più costosi, ma ha i vantaggi di essere quantitativa, veloce e altamente sensibile, che significa che può essere utilizzato per il rilevamento di TiLV in Sub-clinicamente pesci infetti. La RT-PCR e RT-qPCR protocolli sono stati effettuati in due diversi laboratori di distinte geografiche isolate di TiLV ed i risultati inclusi evidenzia la sensibilità e riproducibilità dei saggi qui descritto.
Protocollo
Il protocollo di uso di animali per questo studio è stato approvato dal comitato di etica animale Università Kasetsart sotto numero di permesso ACKU 59-VET-016.
Nota: Consultare la Tabella materiali per informazioni estese riguardanti i reagenti ed apparecchiature suggerito per questo protocollo.
1. i tessuti campionario
- Eutanasia il pesce utilizzando un sovradosaggio di olio di chiodi di garofano (il volume dipende la dimensione del pesce e la concentrazione di prodotti, di solito più di 3 mL/L). Immergere un quarto delle forbici pinze e mayo in etanolo al 95% (v/v) seguita da bruciare l'apparecchiatura utilizzando un bruciatore ad alcool per sterilizzare le attrezzature.
Nota: Tricaina metansolfonato (MS-222) può essere utilizzato invece di olio di chiodi di garofano. - Trovare il fegato e tagliare un piccolo pezzo (circa 20-100 mg) o raccogliere 200 µ l di muco utilizzando vetro di copertura o lama chirurgica per rimuovere muco dall'anteriore al posteriore del pesce tilapia e collocare i campioni in una microcentrifuga da 1,5 mL.
- Esempi di processo immediatamente, memorizzare in un RNA soluzione di stabilizzazione o spostarli a-80 ° C fino all'utilizzo ulteriore.
Nota: Il compito più grande nel lavoro con RNA è preparazione di molecole di RNA intatte e mantenendoli intatto durante eventuali manipolazioni successive. La spina dorsale di RNA è congenitalmente più sensibile ai danni del DNA. Estrazione e l'isolamento di RNA totale dalle cellule del tessuto richiede tecnica di laboratorio attenti; adottare tutte le disposizioni per evitare la contaminazione RNasi indossando guanti, utilizzando acqua gratuita RNasi, reagenti, attrezzature, oggetti in plastica, bicchieri, spazio di lavoro e utilizzando puntali con filtro per il pipettaggio.
2. Guanidium tiocianato - fenolo - cloroformio estrazione di RNA
- Aggiungere 1 mL di soluzione monofasica contenente fenolo e guanidina isotiocianato in una provetta contenente il campione di tessuto dalla sezione 1.
Attenzione: Questa soluzione è molto tossica e deve essere maneggiata con cura in una cappa a flusso laminare con dispositivi di protezione e indossando guanti occhiali, Abbigliamento e sicurezza adeguati. - Macinare il campione di tessuto usando un omogeneizzatore del pestello di tessuto fino a quando non è omogenea.
Nota: I campioni possono anche omogeneizzati utilizzando un omogeneizzatore di potenza combinato con perline di ceramica. Garantire che il campione di tessuto è omogeneizzato completamente prima di procedere al passaggio successivo o arrestare il protocollo qui e conservare i campioni completamente omogeneizzati a-80 ° C fino all'utilizzo ulteriore. - Aggiungere 200 µ l di cloroformio per separazione di fase.
ATTENZIONE cloroformio è un potenziale narcotico ed è estremamente pericoloso. Esso deve essere maneggiato con cura in una cappa a flusso laminare con dispositivi di protezione e anche indossando guanti occhiali, Abbigliamento e sicurezza adeguati. Come meno tossico alternativo, 1-Bromo-3-cloropropano può anche essere usato.
Nota: Scala i volumi su o giù dove appropriato. Ad esempio, se solo 500 µ l di soluzione monofasica contenente fenolo e guanidina isotiocianato è stato usato, quindi solo aggiungere 100 µ l di cloroformio in questa fase.- Miscelare i campioni anche di inversione per 15 s.
- Incubare i campioni per 3 min a temperatura ambiente (TA).
- Centrifuga per 15 min a 12.000 × g e 4 ° C.
Nota: Dovrebbe esserci una netta separazione in una fase organica inferiore, un bianco interfase e una fase acquosa superiore contenente RNA. Questa fase superiore è normalmente incolore, ma a seconda del tipo e la quantità di tessuto omogeneizzato, può avere un aspetto rosa chiaro. - Trasferire lo strato acquoso superiore (circa 500 µ l) ad un tubo del microcentrifuge fresco senza disturbare l'interfase.
Nota: non tenta di trasferire l'intera fase acquosa, lasciare una piccola quantità per evitare qualsiasi potenziale contaminazione del RNA contenente fase acquosa con l'organico o interfase. - Aggiungere 1 volume di isopropanolo 100% a precipitare il RNA.
- Facoltativamente, se sono state utilizzate molto piccole quantità di tessuto, quindi aggiungere 1 µ l (5-10 µ g) di glicogeno RNAsi-libera per ogni campione per promuovere efficiente precipitazione di RNA. Questo aiuterà l'identificazione della pallina del RNA in passo 2.8.
Nota: Il glicogeno funge da vettore del RNA e impedirà piccole quantità di RNA da attaccare al lato del tubo. - Mix tubi bene capovolgendo più volte.
- Conservare i campioni per 2 h per una notte a-20 ° C.
- Facoltativamente, se sono state utilizzate molto piccole quantità di tessuto, quindi aggiungere 1 µ l (5-10 µ g) di glicogeno RNAsi-libera per ogni campione per promuovere efficiente precipitazione di RNA. Questo aiuterà l'identificazione della pallina del RNA in passo 2.8.
- Centrifugare i campioni per 15 min a 12.000 x g e a 4 ° C.
- Eliminare il supernatante, facendo attenzione a non spostare la pallina del RNA nella parte inferiore del tubo del microcentrifuge.
- Aggiungere 1 mL di etanolo al 75% (v/v) e miscelare i campioni di RNA capovolgendo la provetta diverse volte.
- Centrifugare per 15 min 10.000 x g e a 4 ° C.
Nota: Il protocollo può essere fermato qui e i campioni comprendente la pallina del RNA in 75% etanolo possono essere conservati a-20 ° C fino all'ulteriore utilizzo. - Eliminare il supernatante, essere vigile non per sloggiare il pellet di RNA nella parte inferiore del tubo del microcentrifuge.
- Facoltativamente, ripetere i passaggi da 2.9-2.11 con etanolo al 70% (v/v). Accuratamente la pallina del RNA di lavaggio riduce al minimo qualsiasi sale o riporto di contaminanti che potrebbe interferire saranno sensibili applicazioni a valle.
- Tirare fuori l'etanolo rimanente utilizzando una pipetta e poi asciugare all'aria il pellet di RNA a temperatura ambiente per non più di 5-10 min.
Nota: pellet eccessivamente secchi sarà difficile risospendere. - 30-60 µ l di acqua RNAsi-libera, pre-riscaldato a 55-60 ° C per solubilizzare la pallina del RNA.
- Posizionare il RNA su ghiaccio per uso immediato o conservare a-80 ° C per un uso successivo.
3. quantificare la concentrazione di RNA usando uno spettrofotometro del Micro-Volume
- Passare le impostazioni dello spettrofotometro a RNA.
- Utilizzare 1-2 µ l di acqua RNAsi-libera come un vuoto.
- Utilizzare 1-2 µ l di ciascun campione di RNA per valutare la quantità di RNA.
- Registrare le letture a 230 nm, 260 nm e 280 nm per ciascun campione.
- Diluire il RNA a 200 ng / µ l utilizzando acqua RNAsi-libera.
4. sintesi di DNA complementare (cDNA) mediante RNA totale
- Mescolare 1 µ g di RNA totale da protocollo 2, 2 µM oligo (dT), miscela di 0,5 mM dNTPs e portare il volume finale di 10 µ l con acqua priva di nucleasi. Per questo, preparare un RT-master-mix in base al numero di campioni e controlli per essere testato.
Nota: I controlli sono un campione della trascrittasi inversa-minus (-RT) in cui l'enzima RT viene sostituito con acqua priva di nucleasi (Vedi punto 4.3) e un controllo privo di templato (NTC) in cui acqua priva di nucleasi è aggiunto al master-mix anziché modello di RNA.- Miscelare i campioni ben pipettando seguita da una breve centrifugazione.
- Incubare i campioni a 65 ° C per 5 min seguita da un'incubazione di 2 min sul ghiaccio.
- Centrifugare brevemente i campioni per raccogliere tutto il liquido sul fondo delle provette.
- Aggiungere tampone 1x trascrittasi inversa, della trascrittasi inversa di 100 U e portare il volume finale di ogni campione 20 µ l utilizzando acqua priva di nucleasi.
- Miscelare i campioni ben pipettando seguita da una breve centrifugazione.
- Incubare i campioni a 42 ° C per 60 min seguita da 85 ° C per 5 min.
- Diluire il cDNA sintetizzato ad una concentrazione desiderata aggiungendo un volume adeguato di acqua priva di nucleasi e mettere il cDNA sul ghiaccio per un uso immediato o conservare a-20 ° C per un uso successivo.
5. TiLV PCR convenzionale
- Utilizzare il cDNA, campioni e controlli, generati in protocollo sezione 4 come modelli per una reazione di PCR utilizzando uno qualsiasi delle coppie di primer stabilito indicate nella tabella 1, insieme a una polimerasi del DNA di scelta.
Nota: Un controllo no-modello aggiuntivo (NTC) deve essere inclusi qui sostituendo cDNA per acqua priva di nucleasi alla reazione di PCR. Un controllo positivo, se disponibile, dovrebbe anche essere incluso comprendente precedentemente verificato TiLV campioni positivi o il frammento di cDNA TiLV appropriato clonati un plasmide. - Preparare un master-mix di PCR secondo le linee guida del sistema DNA polimerasi in uso e il numero di campioni e controlli per essere testato. Questo mix dovrebbe includere il primer forward, reverse primer, dNTP, MgCl2 e la polimerasi del DNA selezionata, insieme al suo buffer.
- Secondo le linee guida della polimerasi del DNA selezionata, è possibile combinare il volume designato di master-mix con quantità suggerita di campioni di cDNA e campioni di controllo.
Nota: Preparare un 0,5 x eccesso di reazione è spesso utile poiché alcuni dei master-mix viene persa durante il pipettaggio. - Eseguire PCR ciclismo condizioni secondo le linee guida del sistema utilizzato DNA polimerasi e utilizzando un'appropriata temperatura di ricottura per il primer in uso (tabella 1). Di solito, un tale programma coinvolgerà una denaturazione iniziale a 95 ° C per 2-5 min, seguiti da 30-40 cicli di denaturazione a 95 ° C per 30 s, ricottura la temperatura raccomandata per 30 s e l'allungamento a 72 ° C per 30 s, seguita da un allungamento finale a 72 ° C per 5-10 min.
- Carico: 5-15 µ l di ogni reazione di PCR e un appropriato DNA ladder in pozzetti di un gel di agarosio 1-2%, a seconda della dimensione prevista del prodotto PCR. Separare il DNA amplificato mediante elettroforesi su gel e macchia il gel di bromuro di etidio (EthBr) per facilitare la visualizzazione delle bande di DNA di dimensioni previste (tabella 1) in una macchina di documentazione del gel usando la luce UV.
Attenzione: EtBr è tossico; deve essere maneggiato con cura, indossando l'abbigliamento protettivo adeguato e guanti di sicurezza.
| Segmento target TiLV genoma | Forward primer 5' - 3' | Reverse primer 5' - 3' | Formato del prodotto PCR (bp) | Tm ° C | Riferimento originale | ||||
| 1 | CCAAACGTTATCTCTTAATTACGCAC | GCAAATATTTCTCTCATTCGCCT | 1641 | 50 | Surachetpong et al., 2017 | ||||
| 1 | CCTCATTCCTCGTTGTGTAAGT | AGGAGTTGCTGTTGGGTTATAG | 1000 | 62 | Mugimba et al., 2018 | ||||
| 2 | ACTCTCTATTACCAAATACATTTACT | TTACCATATATATAGTGAAGGC | 1445 | 45 | Surachetpong et al., 2017 | ||||
| 2 | GTCCAGGGCGGTATGTATTG | CTTACGGCTGACAAGTCTCTAAG | 834 | 62 | Mugimba et al., 2018 | ||||
| 3 | GTTGGGCACAAGGCATCCTA | TATCACGTGCGTACTCGTTCAGT | 250 | 56 | Eyngor et al., 2014 | ||||
| 3 | TATGCAGTACTTTCCCTGCC | TTGCTCTGAGCAAGAGTACC | 491 | 57 | Eyngor et al., 2014 | ||||
| 3 | ACCCCTTAATCCTTAATAGACCGTTA | CCCATAATCCTCTATTAGAACGTCGT | 1352 | 50 | Surachetpong et al., 2017 | ||||
| 3 | GTCGAGGCATTCCAGAAGTAAG | GAGCTAAGGGAACGGCTATTG | 834 | 62 | Mugimba et al., 2018 | ||||
| 4 | AGCAGCAGCAGGAGAAAGAG | ACCGTCCTGTTTCTGAATGG | 358 | 60 | Nicholson et al., 2017 | ||||
| 4 | CCAAAGTTTACTCCTATTACCCAGA | GCAAATCTTTCTCCAATTACCGTCT | 1250 | 50 | Surachetpong et al., 2017 | ||||
| 4 | GCCCAATGGTTCCCATATCT | GCCCAATGGTTCCCATATCT | 524 | 62 | Mugimba et al., 2018 | ||||
| 5 | CCAAATGTTTCTCTTATCTCAGACTC | CTTTTTCTCAGTTTACCACTTTATG | 1087 | 57 | Surachetpong et al., 2017 | ||||
| 5 | CAACTCTTAGCCTCCGGAATAC | CGTTCTGCACTGGGTTACA | 696 | 62 | Mugimba et al., 2018 | ||||
| 6 | CCAAATTTTACCTCTCGCAT | TCAAGCACTTAAAACTGTACC | 1027 | 45 | Surachetpong et al., 2017 | ||||
| 6 | CCCACACGACAGGACATATAG | GAGTTGGCTTAGGGTGATAAGA | 948 | 62 | Mugimba et al., 2018 | ||||
| 7 | CTCTCTTTGCATTGCATACCGT | GACCAATTATCCCTGCTTTCA | 704 | 57 | Surachetpong et al., 2017 | ||||
| 7 | TCCTTTAGGGATTGGCACTAAC | TTCCATCGACTGCTCCTAGA | 486 | 62 | Mugimba et al., 2018 | ||||
| 8 | ACCTCATCTACACTAACATTTCCA | TCATCATTACACAAATGGAGTAGCT | 637 | 50 | Surachetpong et al., 2017 | ||||
| 8 | CTTAAGGGCCATCCTGTCATC | TGGCTCAAATCCCAACACTAA | 476 | 62 | Mugimba et al., 2018 | ||||
| 9 | TTGGTGATGTCACGATGGATA | AGTTCTATCGCCAGCCATGT | 351 | 60 | Nicholson et al., 2017 | ||||
| 9 | ACAAGTCCGATTACTTTTTCCGC | TCTTTCTCACGTCCTTAAAGTCA | 530 | 50 | Surachetpong et al., 2017 | ||||
| 9 | GATATCCTCCACATGACCCTTC | GTACGTCACTTTGTGCCATTAC | 261 | 62 | Mugimba et al., 2018 | ||||
| 10 | AACCCTACTAACACCAAATATAGCT | CTTTCCCTCTGACACCCTGT | 450 | 50 | Surachetpong et al., 2017 | ||||
| 10 | TCCTCTCTGTCCCTTCTGTT | CAGGATGAGTGTGGCAGATTAT | 276 | 62 | Mugimba et al., 2018 | ||||
Tabella 1. Coppie di primer pubblicati per l'amplificazione del cDNA TiLV utilizzando endpoint PCR. Il primer insieme mostrato in grassetto sono stati utilizzati per generare i risultati rappresentativi illustrati nella Figura 3A e 3B.
6. TiLV reazione a catena della polimerasi quantitativa (qPCR)
- Utilizzando un plasmide contenente il TiLV appropriato segmento genomico 3 cDNA come standard, ad esempio pTiLV32, preparare una serie di 10 volte diluizione seriale duplicato o triplicato.
- Preparare un qPCR master-mix per tutti i campioni, controlli e standard, tenendo conto che le reazioni dovrebbero essere eseguite in duplicato o in triplice copia utilizzando 0,4 µ l di acqua priva di nucleasi, 0,3 µ l di primer forward, 0,3 µ l di primer reverse e 5 µ l di 2x SYBR Green DNA polimerasi master-mix per reazione.
- Utilizzare il primer ad una concentrazione di 10 µM e le informazioni sul primer e il pTiLV standard come segue:
Primer in avanti: TiLV-112F (5'-CTGAGCTAAAGAGGCAATATGGATT-3')
Reverse primer: TiLV-112R (5'-CGTGCGTACTCGTTCAGTATAAGTTCT-3')
Standard pTiLV:10 pg / µ l
Nota: Se il numero totale di campioni e controlli è 10 e sarà eseguito in triplici copie, questo equivale a un qPCR master-mix composto da acqua priva di nucleasi di 12 µ l, 9 µ l forward primer, primer inverso di 9 µ l e 150 µ l di SYBR Green DNA polimerasi master-mix. Commercialmente acquistato 2 x universal SYBR Green DNA polimerasi master-miscele contengono tutti i componenti necessari per la reazione di qPCR, vale a dire, il SYBR Green tingere, avviamento a caldo la Taq DNA polimerasi, dNTP, MgCl2 e passivo riferimento coloranti. Proteggere il mix master SYBR Green dalla luce.
- Utilizzare il primer ad una concentrazione di 10 µM e le informazioni sul primer e il pTiLV standard come segue:
- Pipettare 6 µ l di qPCR master-mix in qPCR tubi in striscia o un piatto ben 96 compatibile con la macchina di qPCR in uso.
- Aggiungere 4 µ l di diluito serialmente TiLV standard, controlli o modello di cDNA in provette o pozzetti della piastra ben 96.
- Chiudere le provette qPCR o sigillare la piastra ben 96 con una copertura di piastra compatibile per la macchina di qPCR in uso
- Spostare delicatamente a scatti la qPCR di provette per mescolare la soluzione e spin giù i tubi qPCR o piastra ben 96 utilizzando una centrifuga per raccogliere tutto il liquido sul fondo dei vasi.
- Posizionare i tubi o la piastra nel termociclatore in tempo reale.
- Programmare il termociclatore qPCR per eseguire una denaturazione iniziale a 95 ° C per 3 min, seguiti da 40 cicli di 95 ° C per 10 s e 60 ° C per 30 s per la ricottura di primer e allungamento, terminando con un passo di curva fusione di 65 ° C a 95 ° C con un incremento di 0,5 ° C / 5 s.
- Selezionare SYBR come tintura fluoroforo, quindi selezionare sconosciuto come tipo di campione e inserire un nome nella casella di nome campione.
- Aprire il coperchio della macchina RT-qPCR e posizionare la striscia di qPCR nei pozzetti assegnati, quindi chiudere il coperchio.
- Eseguire l'analisi di RT-qPCR con le condizioni selezionate. La macchina inizierà a funzionare dopo il coperchio ha raggiunto la temperatura desiderata. Raccogliere la fluorescenza di ciascun campione dopo ogni passaggio di estensione per monitorare l'avanzamento della reazione.
Nota: La macchina di qPCR e relativi software automaticamente calcolare tutti i parametri del test e visualizzare le curve di amplificazione in tempo reale, mentre la curva standard e la curva di fusione verrà generati alla fine del ciclo di qPCR. - Eseguire analisi dei dati e acquisizione garantendo prima che la fusione le curve per ogni campione e standard hanno un picco uniforme alla temperatura prevista per l'amplicone.
- Valutare le curve di amplificazione dei campioni e standard e impostare la soglia in una regione sono stati che il tasso di amplificazione dei cDNA è lo stesso in tutti i campioni. Questo normalmente viene eseguito automaticamente dal software, ma deve essere controllato attentamente.
- Calcolare il numero di copie di TiLV utilizzando la curva standard.
Risultati
Seguendo il protocollo descritto nella sezione 1, tilapia ibrido rosso moribondo visualizzati da segni clinici di infezione TiLV (Figura 1A) sono stati eutanasizzati di balneazione in un'alta concentrazione di olio di chiodi di garofano, che agisce come un anestetico. Segnalati i sintomi clinici sono variabili, ma i sintomi comuni sembrano essere letargia, erosione della pelle e lo scolorimento, esoftalmia, scale indipendente, ferite/lesione aperta e comportamento anormale15,16,33, 35,36, alcuni di questi può essere visto chiaramente nella Figura 1A. La parete addominale è stato rimosso per raccogliere gli organi interni come il fegato, la milza o la testa del rene (Figura 1B). Campioni di muco inoltre sono stati raccolti in questa fase raschiando delicatamente la pelle da anteriore a posteriore dei pesci usando un vetro di copertura o chirurgico lama37.

Figura 1 . Raccolta di dissezione e campione di tilapia. A. tilapia ibrido rosso TiLV-infettati con leisons della pelle, rossore intorno alla bocca e opercolo, l'erosione della pelle ed opacità corneale. B. Sectioned ibrido rosso tilapia per consentire per raccolta di tessuto dal fegato (al punto di freccia blu), milza o rene testa organi. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
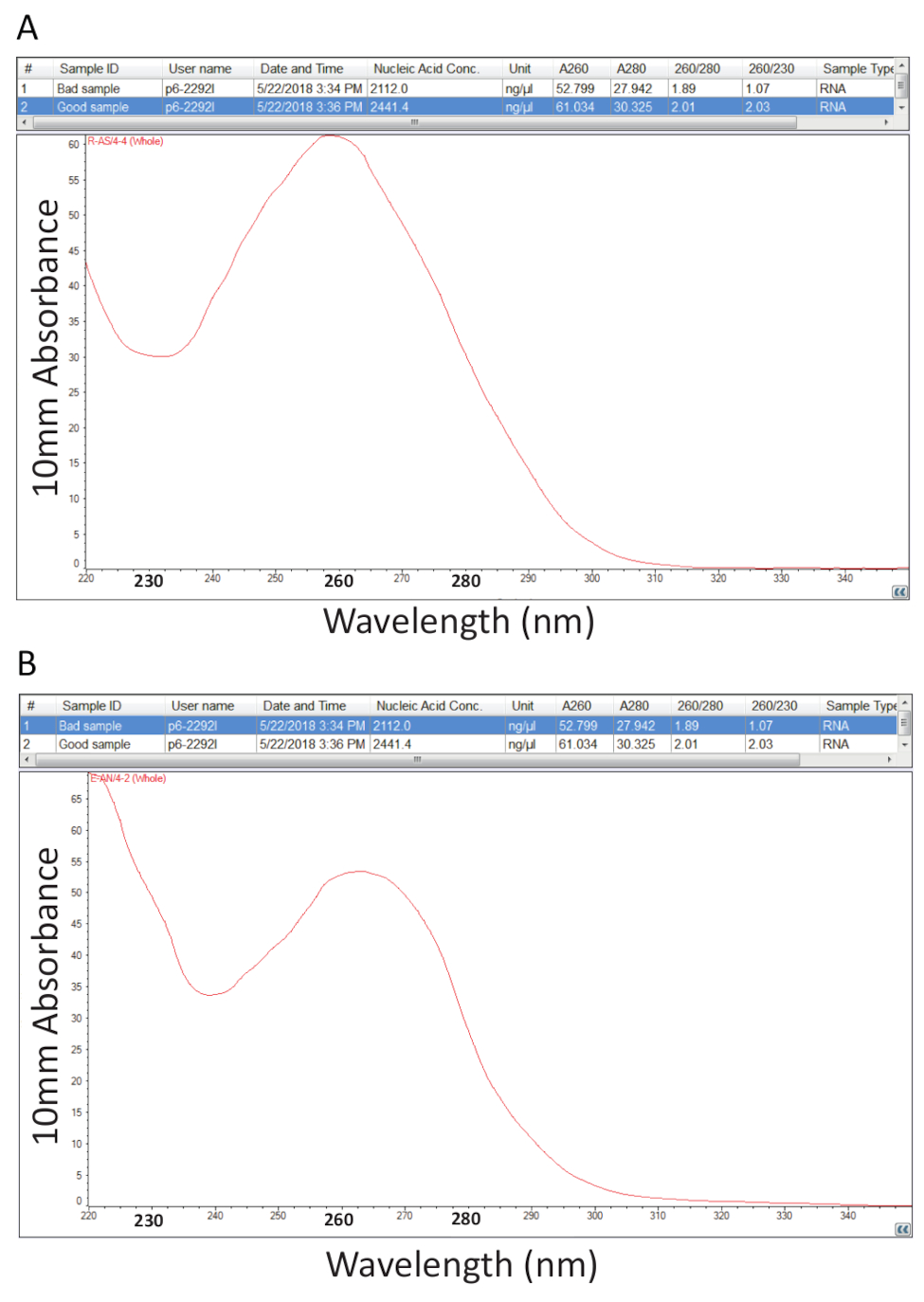
Da allora in poi, è stato seguito il protocollo dettagliato nella sezione 2 per l'estrazione di RNA totale Guanidium tiocianato-fenolo-cloroformio e quantificazione di RNA come descritto nella sezione 3 è stato effettuato per valutare la purezza del campione di calcolo dei rapporti di purezza e esame dei profili spettrali (Figura 2). La Figura 2A Mostra un risultato rappresentativo da una riuscita procedura di estrazione RNA totale, mentre Figura 2B rappresenta una scarsa preparazione di RNA. Gli acidi nucleici hanno maxima di assorbanza a 260, mentre proteine hanno la loro a 280 nm. Il rapporto delle misurazioni a 260 nm e 280 nm indicano la purezza di ogni campione e rapporti di 1,9 a 2,1 indicano RNA puro come è il caso per l'esempio in Figura 2A. Rapporti A260/280 più bassi osservati nella Figura 2B indicano eventuale contaminazione della proteina o fenolo rimasto dalla procedura di estrazione di RNA. Assorbanza a 230 nm può essere il risultato di contaminazione del campione e rapporto A260/230 nm viene calcolato anche per questo motivo. Questo rapporto dovrebbe essere nella gamma di 2.0-2.2 per le preparazioni di RNA puri come illustrato da un valore di 2.03 per l'esempio in Figura 2A, mentre Figura 2B ha un basso rapporto A260/230 di 1.07 e il profilo spettrale mostra uno spostamento nel trogolo a 230 nm verso 240 nm che è indicativo della guanidina residua o fenolo nel campione. Per l'esempio mostrato nella Figura 2B, ri-precipitare il RNA per rimuovere la contaminazione può migliorare la purezza del campione.

Figura 2 . Spettrofotometrica quantificazione di RNA totale estratto dai tessuti malati tilapia. A. rapporti di purezza e profili spettrali da una corretta preparazione di RNA. B. come A, ad eccezione del rappresentante di una procedura di estrazione di RNA povera. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Per rilevare TiLV mediante RT-PCR, campioni puri come quello rappresentato in Figura 2A erano inverso trascritto (protocollo n. 4) nel cDNA e utilizzato come un modello per la PCR analisi dettagliate nella sezione 5 e risultati rappresentativi sono mostrati in Figura 3A. Iniettori indicati grassetto nella tabella 1 sono stati utilizzati per amplificare un frammento di bp 491 di TiLV segmento genomico 314. I prodotti PCR sono stati separati mediante elettroforesi su gel e macchiati con EtBr per visualizzazione. Figura 3A Mostra i risultati di un two-step RT-PCR utilizzando 4 campioni di cDNA (S1-S4), derivati dal fegato di tilapia malato isolato in Thailandia e in ogni campione, una pulito singola banda di circa 500 bp può essere osservato e quindi, campioni 1-4 sono TiLV positivo. Lo stesso prodotto PCR è stato ottenuto dal campione di controllo positivo, costituito da cDNA del segmento TiLV 3 clonato in un plasmide32 mentre il nessun controllo del modello (NTC) non hanno reso i prodotti di PCR. L'analisi in Figura 3B è stata eseguita utilizzando i primers stessi come in Figura 3A , ma in un laboratorio differente, utilizzando un approccio di One-step RT-PCR e con 5 campioni di RNA derivati dai tessuti del rene testa di tilapia originari dell'acquacoltura egiziano 15. è stato determinato usando questa analisi di rilevamento che i campioni 1, 3 e 5 sono TiLV positivo mentre sono gli esempi 2 e 4 TiLV negativo poiché nessun prodotto di PCR è stato trovato alle dimensioni corrette. Controlli negativi, tra cui due meno controlli della trascrittasi inversa e due NTCs non ha generato alcun prodotti di PCR. Un test di One-step RT-PCR è stato anche effettuato mira il tilapia ActinB gene. La dimensione di amplicon di 217 bp è stata generata in ogni campione (S1-S5) come previsto38. Questo test è servito come un controllo dell'integrità dei campioni di RNA, nonché a consentire per un esame semi-quantitativa dei campioni positivi TiLV. Dato che il prodotto di Tilapia ActB generato è relativamente uguale, le differenze nella quantità di prodotto PCR specifico TiLV generato possono essere interpretate come un vero riflesso della quantità di TiLV in un campione di tessuto.

Figura 3 . TiLV RT-PCR. A. i campioni di cDNA prodotti dai tessuti al fegato di tilapia malato, raccolti da Thailandia erano schermo per TiLV infezione utilizzando primers specifici per segmento 3 (mostrato grassetto nella tabella 1) di TiLV usando un'analisi di RT-PCR 2-step. M = marcatore visualizzato in coppie di basi; S1-S4 = campioni 1-4; C1 = controllo positivo utilizzando pTiLV come un modello PCR; e C2 non = nessun controllo del modello (NTC). B. One-step RT-PCR utilizzando i primer stessi come in A e raccolti campioni da tessuti renali testa di tilapia malato da Egitto15. M = marcatore visualizzato in coppie di basi; S1-S5 = campioni 1-5. Controlli C1-C2 sono meno controlli della trascrittasi inversa e C3-C4 sono pannello NTCs. inferiore è un one-step RT-PCR utilizzando primers nei confronti di tilapia ActinB38 (per dettagli vedi testo) producendo un prodotto di PCR di 217 paia di basi. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
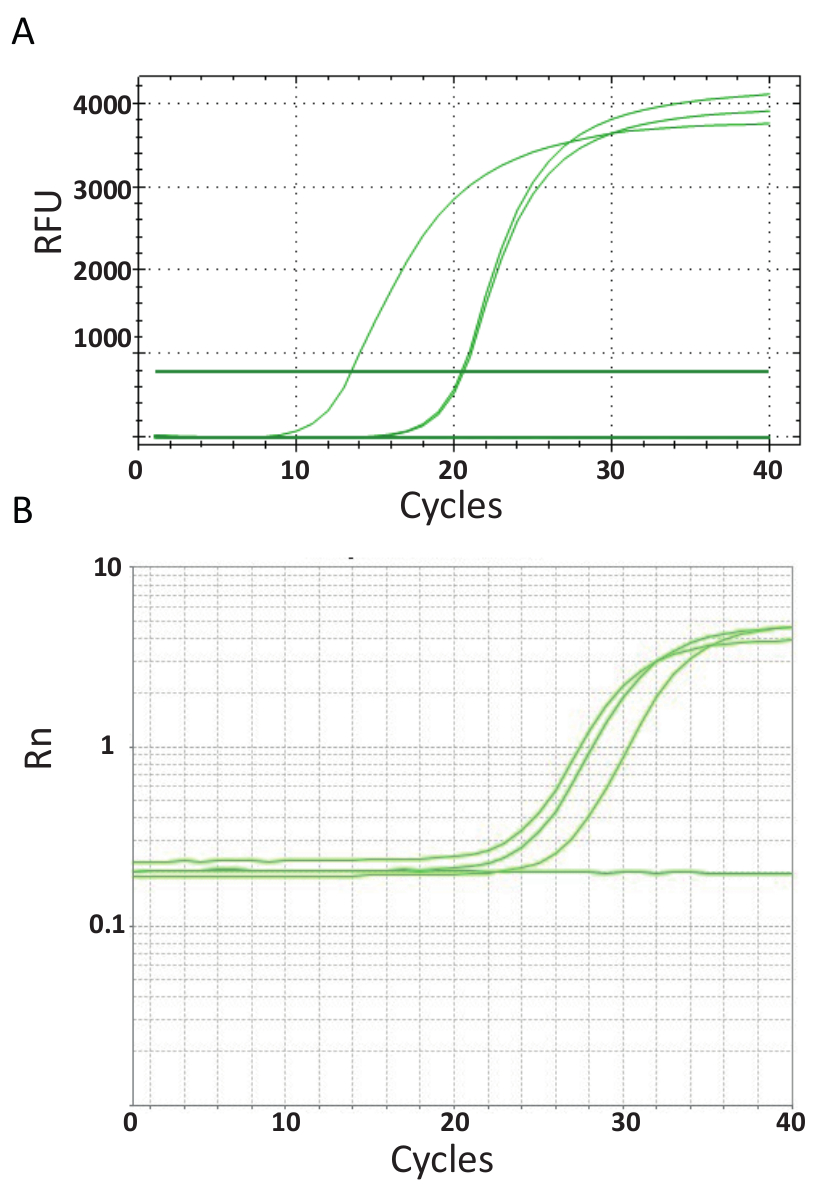
A differenza dell'endpoint PCRs rappresentato in Figura 3, saggi qPCR che sono spiegati nel protocollo n. 6, è possibile misurare la quantità di prodotto di PCR dopo ogni ciclo PCR. L'amplificazione del DNA bersaglio viene rilevata utilizzando molecole fluorescenti che interagiscono con il DNA generato da ogni turno di reazione. Qui, è stato utilizzato SYBR Green tingere, che intercala con DNA double-stranded. Il segnale fluorescente è seguito durante la reazione e la sua intensità si riferisce alla quantità di prodotto formato 39,40,41,42,43. TiLV qPCR analisi sono state effettuate come descritto nel protocollo n. 6 in laboratori diversi utilizzando diversi reagenti SYBR Green, qPCR macchine e campioni provenienti da diversi paesi. Le curve di amplificazione risultante sono illustrate in Figura 4A e 4B. Si può osservare che per ogni dosaggio, il corso dell'esperimento ha quattro fasi: la fase di terra lineare, fase esponenziale, fine della fase esponenziale e la fase di plateau. La fase di terra lineare si verifica durante i primi cicli dove la duplicazione del DNA ancora non può essere identificato a causa della quantità di DNA producendo rapporto segnale/rumore insufficiente. Fluorescenza della linea di base viene calcolato durante questa fase. Da allora in poi, il DNA bersaglio inizia a doppia concentrazione con ogni ciclo inducendo il segnale per diventare rilevabile sopra priorità bassa e aumentare in modo esponenziale. L'efficienza di amplificazione (E) di un dosaggio di qPCR ben ottimizzato è molto alta (vicino al 100%) all'inizio della reazione e rimane stabile durante questa prima fase esponenziale di amplificazione ed è a questo punto che quantificazione viene eseguita, quando efficienza di reazione è ancora stabile. Nei cicli successivi il segnale comincia a plateau, e l'intensità della fluorescenza non è non è più correlato e il numero di copia modello iniziale perché i componenti di reazione sono esausto44. Saturazione può verificarsi anche a causa della concorrenza da ri-ricottura reazioni, i mutevoli rapporti di concentrazione di componenti, o la quantità di enzima unità di molecole di DNA substrato. Eventualmente, tali parametri tenere conto delle differenze tra le curve di amplificazione per i dosaggi indicati in Figura 4A e 4B. I controlli inclusi non hanno generato queste curve di amplificazione caratteristico.

Figura 4 . Amplificazione trame mostrare l'accumulo di prodotto nel corso della durata del test PCR in tempo reale. A. curve di amplificazione di campioni positivi TiLV derivate da Thailandia, NTCs e controllo positivo plasmide utilizzando un SYBR-Green I qPCR 2-step test. Il grafico è stato generato tracciando il numero di cicli vs relativa fluorescenza (RFU). B. curve di amplificazione dei campioni positivi TiLV derivato dall'Egitto, come in Figura 3B e un NTC. La curva di amplificazione è la fluorescenza del reporter segnale normalizzato per la fluorescenza della tintura ROX passiva inclusa nel saggio (Rn) contro ciclo numero. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Alla fine del thermocycling qPCR su macchine diverse in ciascun laboratorio, i dati è stati acquistati e analizzati. Figura 5A e 5B mostrano curve di fusione rappresentante dalle analisi eseguite in ogni laboratorio. Ogni macchina di qPCR è stato programmato per eseguire un'analisi della curva di fusione alla fine. Questo è stato ottenuto in modo incrementale aumentando la temperatura e monitoraggio la fluorescenza in funzione della temperatura. Quando la temperatura è abbastanza alta per denaturare dsDNA, un forte calo in fluorescenza viene registrato perché la molecola di fluoroforo viene rilasciata. Il software di ogni strumento di qPCR calcola la temperatura di annealing (Tm) dai dati curva fusione tracciando il negativo primo derivato vs temperatura (Figura 5). Si vede che in Figura 5A e 5B che i prodotti formati nei set di campioni diversi hanno transizione uniforme di fusione alla temperatura prevista di circa 80 ° C per il dosaggio. Nessun altre cime a temperature più basse sono stati osservati. Grazie alle loro piccole dimensioni, i Tm di dimeri dell'iniettore è in genere inferiore a quello della destinazione sequenza del DNA. Pertanto, questa differenza fra i Tmlo rende facile identificare potenziali dimeri dell'iniettore o altri prodotti di amplificazione aspecifici. I controlli non genera curve di fusione come campioni positivi TiLV e standard e possono essere visto come una linea quasi orizzontale nella parte inferiore dei grafici nella Figura 5A e 5B.

Figura 5 . Sciogliere l'analisi delle curve per garantire la specificità dell'analisi e diversi prodotti di PCR possono essere differenziati per le loro caratteristiche di fusione. R. fondere analisi della curva di campioni TiLV positivi provenienti da Thailandia, controllo negativo e positivo plasmide controllo. B. analisi della curva di TiLV campioni positivi derivati da Egitto, pTiLV standard e un NTC di sciogliere. I grafici in A e B entrambi mostrare il cambiamento di fluorescenza diviso per il cambiamento di temperatura complottarono contro temperatura per produrre un'immagine chiara della fusione dinamica. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
La maggior parte delle qPCR macchine sono dotati di un software facilitare ulteriore valutazione la qPCR eseguire e quantificare i campioni generando una curva standard tracciando automaticamente il ciclo di soglia (Ct) contro il logaritmo del modello degli standard di pTiLV Copiare il numero come mostrato in Figura 6A e 6B per i due laboratori indipendenti. Brevemente, il Ct è l'unità utilizzata per la valutazione dei risultati qPCR. Il valore dit C denota il numero di cicli necessari per raggiungere un livello di segnale di fluorescenza soglia impostata. Maggiore sarà la quantità di avvio modelli, il meno cicli prende per raggiungere un livello rilevabile di fluorescenza. Infatti, i campioni con un carico elevato di TiLV avrà valori dit C inferiori rispetto i campioni con una bassa carica di TiLV come nei pesci con un'infezione sub-clinica. Per determinare i valori dit C, livelli di fluorescenza di fondo prima vengono dedotti dai dati grezzi. Successivamente, il software associato al dispositivo di qPCR selezionerà automaticamente una soglia di fluorescenza cercando le curve di dati per ogni campione e incorporando unat di C che rappresenta dove il campione varcato la soglia. Questa operazione viene eseguita separatamente per ogni dosaggio e ogni soglia deve essere valutato attentamente, assicurando che la soglia è stata impostata nella parte logaritmica delle curve di amplificazione e in un luogo dove tutte le curve sono parallele. Così, la specifica Ct acquisito è un valore relativo ed è relativo alla partenza modello copia numero45, ma è anche specifica per la macchina di qPCR e dei reagenti utilizzati, l'efficienza di amplificazione di PCR e la sensibilità di rilevazione. Questi parametri contribuiscono alle differenze osservate usando l'analisi stessa nella Figura 6.
Le curve standard in Figura 6, le analisi di regressione, tra cui il calcolo della curva standard piste (m) e intercetta, amplificazione efficienze (100 x (101/m -1))46 e linearità della reazione sono stati effettuati. Curva standard analisi inoltre sono state usate per confermare la sensibilità (limite di rilevazione), ripetibilità e la riproducibilità del test. Teoricamente, la quantità di DNA viene raddoppiata a ogni PCR ciclo, vale a dire che l'efficienza (E) è pari al 100%. Tuttavia, in pratica un livello di efficienza tale ideale raramente viene raggiunto a causa di sub-ottimale condizioni di PCR come, l'inibizione della DNA polimerasi, contaminanti, troppo cDNA e pipettaggio errori47. In genere, intervallo di amplificazione E da 90-110% per i dosaggi buoni, in Figura 6A un'efficienza del 94,5% è stata calcolata usando 8 campioni diluiti in serie pTiLV, mentre, l'efficienza del test nel test illustrato nella Figura 6B utilizzando 7 pTiLV diluito serialmente campioni è stato 101,2%. Un'efficienza di oltre il 100% è solitamente dovuto la presenza di inibitori della PCR nel dosaggio. Analisi di regressione lineare della trama standard permette anche per il calcolo del numero di copie di TiLV in ogni campione41,42,45, come può essere osservato per i tre campioni di TiLV mostrati in rosso in Figura 6B che è in linea con i risultati dei campioni S1, S3 e S5 illustrato nella Figura 3B.

Figura 6 . RT-qPCR curve standard. PCR in tempo reale di 10 volte diluizioni seriali del pTiLV, lo standard utilizzato in entrambi i laboratori. R. 8 pTiLV diluito serialmente campioni sono stati testati, tutti di concentrazione nota e correlati al numero di copie TiLV / reazione. La curva standard è stata generata tracciando registro copia numero vs ciclo soglia (Ct). La pendenza =-3.462, R2 = 0.9992 e l'efficienza è 94.47%. B. come in A, tranne pTiLV diluito in serie 7 sono stati testati campioni (verde) e il grafico Visualizza il ciclo di soglia sull'asse y e il numero di copie di TiLV (quantità) sull'asse x. L'intercetta y = 32.327, pendenza =-3.292, R2 = 0.98 e l'efficienza è 101,2%. Per entrambe le curve standard in A e B, la pendenza, intercetta y e i valori del coefficiente di correlazione (R2) vengono utilizzate per comprendere le prestazioni del dosaggio. D'importanza, R2 valore deve essere a 1, poiché è una misura della linearità della curva standard. La pendenza è utilizzata per misurare l'efficienza PCR in cui efficienza 100% corrisponde ad una pendenza di -3.32, vedere testo principale per l'equazione e ulteriori dettagli. Una reazione buona qPCR ha generalmente un'efficienza tra 90-110% correlazione ad una pendenza di tra-3.58 e-3.10. La curva standard è usata per quantificazione assoluta dei campioni positivi TiLV sconosciuti e determina il numero esatto di TiLV copie / reazione, come è il caso per i tre campioni positivi TiLV di colore rosso in B.
Discussione
TiLV in primo luogo è stato segnalato nel 2014 in Israele14 e da allora, è stato identificato in diversi paesi tra i quali Malesia, Colombia, Egitto, Tanzania, Uganda, India e Thailandia15,16,18, 35 , 48. consapevolezza globale, in particolare, nei paesi produttori di tilapia ha posto maggiore attenzione su virus e varie restrizioni e sono state attuate misure di controllo da parte delle autorità di governo tentando di impedire la diffusione di TiLV. Qui, un protocollo dettagliato per il rilevamento di TiLV in tessuto di tilapia, che copre la raccolta del campione, isolamento del RNA, saggi di sintesi, PCR e qPCR di cDNA è stato spiegato. Ci sono vari aspetti di questi metodi che giustificano la discussione specifica. TiLV è stato identificato nei pesci che coprono una varietà di formati9,12,14,15,49 e specie di tilapia finora, tra cui allevamento ibrido tilapia (O. niloticus x o. aureus)11,14, Nile tilapia (o. niloticus)9,10,14,15,16, 33 , 35 , 36 , 49 , 50 e red tilapia (Oreochromis SP.)16,33,48,51, come bene come in wild Nile tilapia9,12, nero Tilapia51, T. zilli14,15, S. galilaeus, aureus o. e T. simonis intermedia14 e molto recentemente TiLV è stata identificata in carpa selvatica (Barbonymus schwanenfeldii)52. Campioni di tessuto da organi interni (gill, milza, fegato, cuore, rene testa) o muco37 possono essere raccolti da sano così come moribonda tilapia indipendentemente dall'età, taglia o specie ed elaborati per l'isolamento di RNA. Il protocollo di estrazione di RNA totale delineato qui usa una soluzione monofasica di fenolo e guanidinio tiocianato, che è un agente denaturante caotropici. I tessuti sono omogeneizzati direttamente in questa soluzione, seguita dall'aggiunta di cloroformio e centrifugazione per realizzare la separazione di fase in cui viene generato un RNA chiaro contenente la fase acquosa superiore, un'interfase e una fase organica inferiore. il RNA è isolato dalla fase acquosa dalla precipitazione di isopropanolo, seguita da lavaggio del RNA recuperato per sbarazzarsi di contaminanti. Isolamento di RNA di questa metodologia è stata introdotta da Piotr Chomczynski e Nicoletta Sacchi ed è riferito a come guanidinio tiocianato-fenolo-cloroformio estrazione53,54. Questo tipo di reagente usato per l'estrazione di RNA possa essere acquistato o fatti in laboratorio in commercio (Vedi la Tabella materiali per maggiori informazioni). Questo protocollo richiede un po' più di metodi basati su colonna come la purificazione a base di silice, ma in generale, si è molto più conveniente e produce più RNA.
In questo protocollo, quantificazione di RNA usando valori di A260 è stato delineato per cui spettrofotometria valori possono indicare qualità di RNA (A260/A280 = 1,9-2,1). Mentre questo metodo vi darà una buona indicazione della purezza del campione, esso non può assolutamente informa circa la qualità del RNA estratto. Per determinare correttamente se il RNA è intatto o parzialmente degradato, i campioni possono essere separazione mediante elettroforesi su gel di agarosio in cui sbavature dell'EtBr macchiato 18S e 28S rRNA bande indicano degradazione del RNA. Ulteriore verifica della qualità di RNA può includere utilizzando uno strumento di lab-on-a-chip. Inoltre, è anche importante per digerire il RNA purificato con dnasi per rimuovere contaminanti ospito DNA genomico, che, a seconda delle applicazioni a valle, può portare a risultati falsi. Se host gDNA è ancora contaminare il campione di RNA in larga misura, un ulteriore trattamento di DNaseI può essere eseguito anche alla fine della procedura di estrazione RNA (Vedi Tabella materiali).
Sintesi del DNA complementari possono influenzare notevolmente i risultati complessivi di qPCR e costituisce un aspetto del metodo che possa introdurre variazione. Il protocollo di cDNA sostenuto qui è composto da un set-up singolo componente utilizzando oligo (dT) e così solo trascrive mRNA contenenti code di polyA. Permette il controllo di utente di esattamente i componenti da utilizzare in questa modalità di cDNA e la reazione di retrotrascrizione sintesi si sono rivelata vincente per TiLV rilevamento32. Un'alternativa a questo set-up è un commercialmente comprato master-mix contenente tutti i componenti necessari per la reazione di trascrizione inversa ed è molto veloce e semplice senza il consueto protocollo multi-step, pipettaggio e multitemperatura. Questo è vantaggioso perché riduce al minimo la manipolazione e promuove l'uniformità in tutti i campioni. Tali master-mescola spesso include sia oligo e casuale primer rendendolo applicabile a diversi modelli di RNA e generazione di copie di cDNA rappresentativo delle sequenze da tutta la lunghezza del RNAs in una popolazione (virale e tilapia ospitare mRNA) e in teoria, ogni specie di RNA desiderata quindi può essere misurata con PCR convenzionale o qPCR da tali un campione. Questa versatilità è il principale vantaggio di un approccio di 2-step RT-PCR; offre una piscina a lungo termine che può essere utilizzata per molti esperimenti diversi. Nei risultati, un approccio di One-step RT-PCR è stato rappresentato in cui sono stati utilizzati primers specifici di sequenza (tabella 1) e la RT e la PCR sono stati effettuati in un tubo (Vedi lista dei materiali). In generale, specifici primers sequenza consentono una maggiore efficienza di RT del RNA dell'obiettivo specifico rispetto all'utilizzo di adescamento casuale, ma l'obiettivo specifico del RNA è l'unico che può essere quantificata in un campione di cDNA che può essere l'unico scopo di alcuni laboratori (Vedi Tabella materiali per suggerimenti di prodotto di sintesi del cDNA).
Mentre la RT-PCR convenzionale sembra essere comunemente utilizzati finora a TiLV diagnosi9,13,14,15,16,17,18, 33 , 35 , 48 , 55. RT-qPCR ha dimostrato di essere uno strumento più potente per la rilevazione e la quantificazione delle piccole quantità di TiLV nei tessuti del pesce o muco32,37. In generale, qPCR è ampiamente usato nei laboratori diagnostici di virologia clinica grazie alla sua alta sensibilità, specificità, buona riproducibilità, ampia gamma dinamica e velocità21. Mentre qPCR può essere inizialmente più costoso da implementare di RT-PCR convenzionale, offrono molti vantaggi importanti sopra PCR convenzionale; ha un tempo più veloce svolta dall'esempio di risultati e non richiede alcuna procedura di post-PCR. Quest'ultimo punto significa che c'è il minimo rischio di contaminazione del laboratorio e può essere più facilmente adattato a situazioni di alto-rendimento come nel caso di epidemie. Inoltre, è intrinsecamente più sensibile di RT-PCR convenzionale, che è di vitale importanza per rilevare bassi carichi virali infezioni subcliniche21. Ciò richiederebbe un approccio PCR annidato che richiedono la trascrizione inversa, due ulteriori reazioni di PCR e quindi l'analisi di agarosio gel elettroforesi. Questi passaggi molti prendono un sacco di tempo e aumentano le possibilità di errori o di contaminazione. Tuttavia, dovuto la relativa alta sensibilità, RT-qPCR richiede meticolosa progettazione sperimentale e una conoscenza approfondita delle tecniche di quantificazione per generare risultati precisi56,57.
Il DNA associazione fluoroforo, SYBR Green attesta in questo protocollo. Si tratta di un colorante di associazione dsDNA DNA aspecifiche e così la specificità del test si trova interamente nel set di primers, che può generare falsi positivi58. Pertanto, mentre il dsDNA eseguita alla fine di ogni PCR analisi della curva di fusione è una parte particolarmente importante della reazione PCR perché conferma che solo un amplicone PCR del corretto Tm è prodotto (questo dovrebbe anche essere raggiunto da gel elettroforesi quando nuovi saggi sono in corso di attuazione). I Tm di un frammento di DNA è dipendente da una varietà di caratteristiche come la sua lunghezza, composizione GC, sequenza, strand complementarità, concentrazione nonché sulle buffer componenti ed esaltatori di PCR. Le analisi di curva fusione nei risultati rappresentativi mostrati da due laboratori non hanno rivelato la presenza di dimeri dell'iniettore o altri prodotti PCR non desiderati, ma se questo è osservato con altri campioni e/o set-up sperimentale, allora il dosaggio dovrebbe essere ri-ottimizzato. Tecnologie più avanzate di qPCR non richiedono tale fusione passo curva e infatti, poiché questo metodi paper è stato scritto, un TaqMan basato TiLV RT-qPCR è stato sviluppato utilizzando due primers ed una sonda che lo rende altamente specifico TiLV34.
Senza dubbio, il primer disegnati per le analisi di RT-qPCR sono fondamentali per il successo del test e i primer di qui sono stati progettati in base ai dati genomici TiLV pubblicamente disponibili presso il tempo32. Tuttavia, virus del RNA è ben noto a mostrare tassi di mutazione elevati e possibili ceppi sfuggiranno i test diagnostici, come è stato osservato per ISAV59. Sarà sempre difficile per tali tipi di virus generare una padella universale-TiLV RT-qPCR dosaggio e tali dosaggi saranno solo continuamente migliorate se resi disponibili più dati genomici TiLV da posizioni ampie e periodi di tempo.
Infine, è essenziale per eseguire duplicati o se possibile, in triplice copia reazioni in sia intra e inter saggi qPCR. Se i valori dit C sono molto alti, quindi l'uso delle ripetizioni è particolarmente importante per accertare che la reazione di PCR è affidabile e riproducibile. In generale, se i dati da replicano reazioni varia più di 0,5 cicli, le reazioni deve essere ripetuto e se i valori dit C variano costantemente cicli > 0,5 in replica, il dosaggio dovrebbe essere ri-ottimizzato. L'uso di un robot di pipettaggio qPCR integrato aiuta immensamente con questo problema, ma è uno strumento di lusso. Come con qualsiasi esperimento, i controlli di adeguata e appropriata di inclusione sono della massima importanza per lo sviluppo di saggi molecolari robusti, soprattutto nei laboratori diagnostici dove tali dosaggi devono essere accreditati. Controlli devono includere positivi (positivi TiLV campione, plasmide TiLV standard) ed esempi di controlli negativi (NTC e -RT), nonché l'individuazione di geni housekeeping tilapia endogeno. Tali controlli non possono essere sottovalutati e dovrebbero essere incluso in ogni analisi per comprendere correttamente la qualità di ogni passo del test e a interpretare correttamente i risultati.
Divulgazioni
Gli autori non hanno nulla a rivelare.
Riconoscimenti
Siamo grati per l'Istituto di batteriologia veterinaria, lezioni facoltà, Università di Berna per il loro sostegno. Questo lavoro è stato finanziato dal comitato accademico promozione di ricercatori all'inizio della carriera e della parità presso la facoltà di lezioni, Università di Berna dal 120% modello finanziamento assegnato a PN. WS e PR sono supportati da centro per gli studi avanzati per l'agricoltura e cibo, Institute for Advanced Studies, Kasetsart University, Bangkok, Thailandia sotto la promozione di ricerca di istruzione superiore e ricerca nazionale Università progetto di Thailandia, ufficio della Thailandia Commissione, Ministero dell'istruzione, dell'istruzione superiore. Vorremmo ringraziare il Dr. Kwanrawee Sirikanchana per la sua narrazione e Piyawatchara Sikarin per il video editing.
Materiali
| Name | Company | Catalog Number | Comments |
| Tissue collection | Step 1 | ||
| Tricaine methanesulfonate | Sigma-Aldrich | E10521 | An alternative to clove oil. Step 1.1 |
| RNAlater stabilization solution | Thermo Fisher Scientific | AM7020 | For storing tissues if they cannot be processed immediately Step 1.3 |
| RNA extraction | Step 2 | ||
| TRIreagent | Sigma-Aldrich | Step 2.1 | |
| TRIzol | Thermo Fisher Scientific (Invitrogen) | 15596026 | Step 2.1 |
| GENEzol | Geneaid | GZR100 | Step 2.1 |
| Trisure | Bioline | BIO-38032 | Step 2.1 |
| Homemade solution | - | - | 94.53 g/L (800 mM) guanidine thiocyanate 30.45 g/L (400 mM) ammonium thiocyanate 8.20 g/L (100 mM) sodium acetate 380 mL/L (38 % v/v) phenol 50 mL/L (5 % v/v) glycerol 1.0 g/L (0.1 % w/v) 8-quinolinol, pH 5.0 Store up to 2 years at 4oC Step 2.1 |
| MagNA Lyser Green Beads | Roche | 3358941001 | An alternative tissue homogenization method used in conjunction with tissue lysing machines detailed below Step 2.2 |
| Lysing Matrix D, 2 mL Tube | MP BIOMEDICALS | 116913050 | |
| Chloroform | Sigma-Aldrich | C2432 | Step 2.3 |
| Chloroform | RCI Labscan | AR1027E-G2.5L | Step 2.3 |
| 1-Bromo-3-chloropropane | Sigma-Aldrich | B9673 | A less toxic alternative to chloroform Step 2.3 |
| Isopropanol (GC) ≥ 99.8 % | Sigma-Aldrich | 59300 | Step 2.6 |
| Isopropanol (ACS, ISO Reag. Ph Eur) | Merck KGaA (EMSURE) | 1.09634.2500 | Step 2.6 |
| Glycogen, molecular biology grade (e.g., Sigma, cat. no. G1767) | Thermo Fisher Scientific (Thermo Scientific) | R0551 | Useful step if tissue starting material is small to maximise RNA precipitation optional |
| Ethanol (purity (GC) ≥ 99.9 % | Sigma-Aldrich (EMD Millipore) | 1.00983 | Step 2.9 |
| Ethanol (ACS, ISO Reag. Ph Eur) | Merck (EMSURE) | 1.00983.2500 | Step 2.9 |
| Nuclease-free water | Promega | P1193 | Step 2.13 |
| Nuclease-free water | Multicell | 809-115-CL | Step 2.13 |
| Ambion TURBO DNA-free kit | Thermo Fisher Scientific (Invitrogen) | AM1907 | Can be performed at the end of the RNA extraction protocol optional |
| cDNA synthesis | Step 4 | ||
| Viva cDNA Synthesis Kit | Vivantis | cDSK01 | Step 4.1 & 4.3 |
| ReverTra Ace qPCR RT MasterMix with gDNA remover | Toyobo | A1172K | An alternative option see discussion |
| ReverTra Ace qPCR RT Kit | Toyobo | FSQ-101 | An alternative option see discussion |
| AffinityScript Multiple Temperature Reverse Transcriptase | Agilent Technologies | 600107 | An alternative option |
| PCR | Step 5 | ||
| DNA polymerase systems: | Step 5.2 | ||
| - Platinum II Hot-Start Green PCR Master Mix (2X) | Thermo Fisher Scientific (Invitrogen) | 14001012a | Step 5.2 |
| - GoTaq Mastermix | Promega | M7122 | Step 5.2 |
| Separate PCR mixture components: | Step 5.2 | ||
| 10mM dNTP Mix | Vivantis | NP2409 | Step 5.2 |
| 25mM MgCl2 | Thermo Fisher Scientific | R0971 | Step 5.2 |
| 10X Taq Buffer with KCl | Thermo Fisher Scientific | 00348114 | Step 5.2 |
| Taq DNA polymerase | Vivantis | PL1202 | Step 5.2 |
| - Verso 1-step RT-PCR ReddyMix with ThermoPrime Taq | Thermo Fisher Scientific | AB1454 | One step RT-PCR exemplified in Figure 3B |
| Gel electrophoresis: | For visulation of PCR products from steps 5.1-5.4 | ||
| Ethidium Bromide solution (10 mg/mL) | Thermo Fisher Scientific | 17898 | Step 5.5 |
| Tris/Acetic/EDTA (TAE) buffer: | Step 5.5 | ||
| - Tris | Vivantis | PR0612-1KG | Step 5.5 |
| - Acetic acid (glacial) (ACS, ISO, Reag. Ph Eur) | Merck KGaA (EMSURE) | 1.00063.2500 | Step 5.5 |
| - Ethylenediaminetetraacetic acid (EDTA) | BIO-RAD | 161-0729 | Step 5.5 |
| Agarose | Vivantis | PC0701-100G | Step 5.5 |
| DNA ladders and markers | Vivantis | NL1405 | Step 5.5 |
| DNA gel loading dye (6X) | Thermo Fisher Scientific | R0611 | Step 5.5 |
| qPCR | Step 6 | ||
| PowerUP SYBR Green Master Mix | Thermo Fisher Scientific (Applied Biosystems) | A25779 | Exemplified in Figures 4-6B Step 6.2 |
| iTaq Universal SYBR Green Supermix | BIO-RAD | 1725120 | Exemplified in the video and in Figures 4-6A Step 6.2 |
| Equipment | |||
| Dounce tissue grinder pestle | Sigma-Aldrich | P1110 | Protocol 2 |
| MagNA Lyser Instrument | Roche | 3358976001 | An alternative tissue homogenizing option for protocol 2 which are used in conjunction with the lysing beads detailed above Step 2.2 |
| FastPrep-24 5G Homogenizer | MP BIOMEDICALS | 116005500 | |
| Refrigerated microcentrifuge | Eppendorf | Eppendorf 5427R | Protocol 2 Step 2.4, 2.7 & 2.10 |
| Refrigerated microcentrifuge | Eppendorf | Eppendorf 5418R | |
| Heat box | Labnet | AccuBlock Digital Dry Bath | Protocol 2 Step 2.13 |
| Microvolume spectrophotometer | Thermo Fisher Scientific (Applied Biosystems) | Nanodrop 2000 | Protocol 3 Step 3.1 - 3.4 |
| PCR machine | BIO-RAD | T100 Thermal Cycler | Protocol 5 Step 5.4 |
| Power supply | BIO-RAD | PowerPac HC | Protocol 5 Step 5.5 |
| Horizontal gel electrophoresis | BIO-RAD | Mini ReadySub-Cell GT Cell #1704487edu | Protocol 5 Step 5.5 |
| Mini microcentrifuge | Corning | LSE 6766 | Useful to quickly spin down PCR reaction tubes in protocols 4, 5 & 6 Step 6.5.1 |
| Microcentrifuge | LioFuge | LM-60 | Step 6.5.1 |
| qPCR machine and software | Thermo Fisher Scientific | 7500 Fast Real-Time PCR System with 7500 Software v2.0 | Protocol 6 Step 6.6-6.8 |
| qPCR machine and software | BIO-RAD | CFX96 Touch Real-Time PCR Detection System with CFX Manager software | |
| General Materials | |||
| Mayo scissors | Step 1.1-1.2 | ||
| Forceps | Step 1.1-1.2 | ||
| Pipette | Rainin | Pipette-Lite XLS | |
| Aerosol-barrier pipette tips | Sigma-Aldrich | Z333328, Z333336, Z333344 | |
| Nuclease-free 1.5-ml microcentrifuge tubes | Eppendorf |
Riferimenti
- FAO. . The State of World Fisheries and Aquaculture, 2014. Opportunities and Challenges. , (2014).
- FAO. . The State of World Fisheries and Aquaculture, 2016. Contributing to Food Security and Nutrition for all. , (2016).
- WorldBank. . FISH TO 2030: Prospects for Fisheries and Aquaculture. Agriculture and Environmental Services Discussion Paper 03. , (2013).
- Wing-Keong, N., Nicholas, R. A review of the nutrition and feeding management of farmed tilapia throughout the culture cycle. Reviews in Aquaculture. 5 (4), 220-254 (2013).
- Cleasby, N., et al. The socio-economic context for improving food security through land based aquaculture in Solomon Islands: A peri-urban case study. Marine Policy. 45, 89-97 (2014).
- Ponzoni Raul, W., et al. Genetic improvement of Nile tilapia (Oreochromis niloticus) with special reference to the work conducted by the WorldFish Center with the GIFT strain. Reviews in Aquaculture. 3 (1), 27-41 (2011).
- Hounmanou, Y. M. G., et al. Tilapia lake virus threatens tilapiines farming and food security: Socio-economic challenges and preventive measures in Sub-Saharan Africa. Aquaculture (Amsterdam, Netherlands). 493, 123-129 (2018).
- OIE. . Tilapia Lake Virus (TiLV) - a novel orthomyxo-like virus. OIE technical disease cards. , (2018).
- Mugimba, K. K., et al. Detection of tilapia lake virus (TiLV) infection by PCR in farmed and wild Nile tilapia (Oreochromis niloticus) from Lake Victoria. Journal of Fish Diseases. , (2018).
- Koesharyani, I., Gardenia, L., Widowati, Z., Khumaira, D. D., Rustianti, Studi kasus infeksi tilapia lake virus (tilv) pada ikan nila (Oreochromis niloticus). Jurnal Riset Akuakultur. 13 (1), 85-92 (2018).
- OIE. . Tilapia lake virus disease (TiLV), Chinese Taipei. Immediate Notification. , (2017).
- OIE. . Tilapia Lake Virus Disease (TiLV), Peru. Immediate Notification. , (2018).
- Bacharach, E., et al. Characterization of a Novel Orthomyxo-like Virus Causing Mass Die-Offs of Tilapia. MBio. 7 (2), e00431-e00416 (2016).
- Eyngor, M., et al. Identification of a novel RNA virus lethal to tilapia. Journal of Clinical Microbiology. 52 (12), 4137-4146 (2014).
- Nicholson, P., et al. Detection of Tilapia Lake Virus in Egyptian fish farms experiencing high mortalities in 2015. Journal of Fish Diseases. 40 (12), 1925-1928 (2017).
- Surachetpong, W., et al. Outbreaks of Tilapia Lake Virus Infection, Thailand, 2015-2016. Emerging Infectious Diseases. 23 (6), 1031-1033 (2017).
- Tattiyapong, P., Dachavichitlead, W., Surachetpong, W. Experimental infection of Tilapia Lake Virus (TiLV) in Nile tilapia (Oreochromis niloticus) and red tilapia (Oreochromis spp.). Veterinary Microbiology. 207, 170-177 (2017).
- Kembou Tsofack, J. E., et al. Detection of Tilapia Lake Virus in Clinical Samples by Culturing and Nested Reverse Transcription-PCR. Journal of Clinical Microbiology. 55 (3), 759-767 (2017).
- Thangaraj, R. S., et al. Derivation of two tilapia (Oreochromis niloticus) cell lines for efficient propagation of Tilapia Lake Virus (TiLV). Aquaculture (Amsterdam, Netherlands). 492, 206-214 (2018).
- Hanson, L. A., Rudis, M. R., Vasquez-Lee, M., Montgomery, R. D. A broadly applicable method to characterize large DNA viruses and adenoviruses based on the DNA polymerase gene. Virology Journal. 3, 28-28 (2006).
- Josko, D. Molecular virology in the clinical laboratory. Clinical Laboratory Science. 23 (4), 231-236 (2010).
- Munir, K., Kibenge, F. S. Detection of infectious salmon anaemia virus by real-time RT-PCR. Journal of Virological Methods. 117 (1), 37-47 (2004).
- Snow, M., et al. Developement, application and validation of a Taqman real-time RT-PCR assay for the detection of infectious salmon anaemia virus (ISAV) in Atlantic salmon (Salmo salar). Developments in Biologicals. 126, 133-145 (2006).
- Matejusova, I., McKay, P., McBeath, A. J., Collet, B., Snow, M. Development of a sensitive and controlled real-time RT-PCR assay for viral haemorrhagic septicaemia virus (VHSV) in marine salmonid aquaculture. Diseases of Aquatic Organisms. 80 (2), 137-144 (2008).
- Garver, K. A., et al. Development and validation of a reverse transcription quantitative PCR for universal detection of viral hemorrhagic septicemia virus. Diseases of Aquatic Organisms. 95 (2), 97-112 (2011).
- Dalla Valle, L., et al. Development of a sensitive and quantitative diagnostic assay for fish nervous necrosis virus based on two-target real-time PCR. Veterinary Microbiology. 110 (3-4), 167-179 (2005).
- Hodneland, K., Garcia, R., Balbuena, J. A., Zarza, C., Fouz, B. Real-time RT-PCR detection of betanodavirus in naturally and experimentally infected fish from Spain. Journal of Fish Diseases. 34 (3), 189-202 (2011).
- Hodneland, K., Endresen, C. Sensitive and specific detection of Salmonid alphavirus using real-time PCR (TaqMan). Journal of Virological Methods. 131 (2), 184-192 (2006).
- Wang, X. W., Ao, J. Q., Li, Q. G., Chen, X. H. Quantitative detection of a marine fish iridovirus isolated from large yellow croaker, Pseudosciaena crocea, using a molecular beacon. Journal of Virological Methods. 133 (1), 76-81 (2006).
- van Beurden, S. J., et al. Development and validation of a real-time PCR assay for the detection of anguillid herpesvirus 1. Journal of Fish Diseases. 39 (1), 95-104 (2016).
- Ciulli, S., et al. Development and application of a real-time PCR assay for the detection and quantitation of lymphocystis disease virus. Journal of Virological Methods. 213, 164-173 (2015).
- Tattiyapong, P., Sirikanchana, K., Surachetpong, W. Development and validation of a reverse transcription quantitative polymerase chain reaction for tilapia lake virus detection in clinical samples and experimentally challenged fish. Journal of Fish Diseases. 41 (2), 255-261 (2018).
- Dong, H. T., et al. Emergence of tilapia lake virus in Thailand and an alternative semi-nested RT-PCR for detection. Aquaculture (Amsterdam, Netherlands). 476, 111-118 (2017).
- Waiyamitra, P., et al. A TaqMan RT-qPCR assay for tilapia lake virus (TiLV) detection in tilapia. Aquaculture (Amsterdam, Netherlands). 497, 184-188 (2018).
- Behera, B. K., et al. Emergence of Tilapia Lake Virus associated with mortalities of farmed Nile Tilapia Oreochromis niloticus (Linnaeus 1758) in India. Aquaculture (Amsterdam, Netherlands). 484, 168-174 (2018).
- Ferguson, H. W., et al. Syncytial hepatitis of farmed tilapia, Oreochromis niloticus (L.): a case report. Journal of Fish Diseases. 37 (6), 583-589 (2014).
- Liamnimitr, P., Thammatorn, W., U-thoomporn, S., Tattiyapong, P., Surachetpong, W. Non-lethal sampling for Tilapia Lake Virus detection by RT-qPCR and cell culture. Aquaculture (Amsterdam, Netherlands). 486, 75-80 (2018).
- Yang, C. G., et al. Evaluation of reference genes for quantitative real-time RT-PCR analysis of gene expression in Nile tilapia (Oreochromis niloticus). Gene. 527 (1), 183-192 (2013).
- Bustin, S. A. Real-time, fluorescence-based quantitative PCR: a snapshot of current procedures and preferences. Expert Review of Molecular Diagnostics. 5 (4), 493-498 (2005).
- Fleige, S., Pfaffl, M. W. RNA integrity and the effect on the real-time qRT-PCR performance. Molecular Aspects of Medicine. 27 (2-3), 126-139 (2006).
- Kubista, M., et al. The real-time polymerase chain reaction. Molecular Aspects of Medicine. 27 (2-3), 95-125 (2006).
- Mackay, I. M., Arden, K. E., Nitsche, A. Real-time PCR in virology. Nucleic Acids Research. 30 (6), 1292-1305 (2002).
- Wong, M. L., Medrano, J. F. Real-time PCR for mRNA quantitation. Biotechniques. 39 (1), 75-85 (2005).
- Bustin, S. A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. Journal of Molecular Endocrinology. 25 (2), 169-193 (2000).
- Heid, C. A., Stevens, J., Livak, K. J., Williams, P. M. Real time quantitative PCR. Genome Research. 6 (10), 986-994 (1996).
- Rutledge, R. G., Côté, C. Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Research. 31 (16), e93-e93 (2003).
- Svec, D., Tichopad, A., Novosadova, V., Pfaffl, M. W., Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomolecular Detection and Quantification. 3, 9-16 (2015).
- Amal, M. N. A., et al. A case of natural co-infection of Tilapia Lake Virus and Aeromonas veronii in a Malaysian red hybrid tilapia (Oreochromis niloticus × O. mossambicus) farm experiencing high mortality. Aquaculture (Amsterdam, Netherlands). 485, 12-16 (2018).
- Fathi, M., et al. Identification of Tilapia Lake Virus in Egypt in Nile tilapia affected by ‘summer mortality’ syndrome. Aquaculture (Amsterdam, Netherlands). 473, 430-432 (2017).
- OIE. . Tilapia Lake Virus disease (TiLV), Philippines. Immediate Notification. , (2017).
- OIE. . Tilapia lake virus disease (TiLV), Malaysia. Immediate Notification. , (2017).
- Abdullah, A., et al. First detection of tilapia lake virus (TiLV) in wild river carp (Barbonymus schwanenfeldii) at Timah Tasoh Lake, Malaysia. Journal of Fish Diseases. 41 (9), 1459-1462 (2018).
- Chomczynski, P., Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 162 (1), 156-159 (1987).
- Chomczynski, P., Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nature Protocols. 1 (2), 581-585 (2006).
- Del-Pozo, J., et al. Syncytial Hepatitis of Tilapia ( Oreochromis niloticus L.) is Associated With Orthomyxovirus-Like Virions in Hepatocytes. Veterinary Pathology. 54 (1), 164-170 (2017).
- Bustin, S. A., et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry. 55 (4), 611-622 (2009).
- Purcell, M. K., Getchell, R. G., McClure, C. A., Garver, K. A. Quantitative polymerase chain reaction (PCR) for detection of aquatic animal pathogens in a diagnostic laboratory setting. Journal of Aquatic Animal Health. 23 (3), 148-161 (2011).
- Simpson, D. A., Feeney, S., Boyle, C., Stitt, A. W. Retinal VEGF mRNA measured by SYBR green I fluorescence: A versatile approach to quantitative PCR. Molecular Vision. 6, 178-183 (2000).
- Kibenge, M. J., et al. Discovery of variant infectious salmon anaemia virus (ISAV) of European genotype in British Columbia, Canada. Virology Journal. 13, 3 (2016).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati