
Method Article
Erkennung von Tilapia See Virus mit konventionellen RT-PCR und SYBR Green RT-qPCR
In diesem Artikel
Zusammenfassung
Dieses Protokoll Diagnosen Tilapia See Virus (TiLV) in Tilapia Gewebe mit RT-PCR-Methoden. Die gesamte Methode ist von Gewebe Dissektion nach insgesamt RNA-Extraktion, gefolgt von cDNA Synthese und Erkennung von TiLV durch konventionelle PCR oder quantitative PCR mit DsDNA verbindlich einen Farbstoff Fluoreszenz Bindung beschrieben.
Zusammenfassung
Das Ziel dieser Methode ist es, den schnellen, empfindlichen und spezifischen Nachweis von Tilapia See Virus (TiLV) in Tilapia Gewebe zu erleichtern. Dieses Protokoll kann im Rahmen der Überwachungsprogramme, Biosicherheitsmaßnahmen und in TiLV Grundlagenforschung Labors verwendet werden. Der Goldstandard der Virus-Diagnostik umfasst in der Regel gefolgt von ergänzenden Techniken wie Reverse Transkription Polymerase-Kettenreaktion (RT-PCR) zur weiteren Überprüfung die Virusisolierung. Dies kann umständlich, zeitaufwendig und erfordert in der Regel Gewebeproben, die stark mit Virus infiziert. Die Verwendung von RT-quantitative (Q) PCR bei der Erkennung von Viren ist vorteilhaft wegen seiner quantitativen Natur, hohe Sensitivität, Spezifität, Skalierbarkeit und seiner schnellen Zeit zu führen. Hier basiert die gesamte Methode der PCR Ansätze für TiLV Erkennung beschrieben, von Tilapia Orgel strukturierendes, total Ribonukleinsäure (RNA) Extraktion mit einem Guanidium-Thiocyanat-Phenol-Chloroform-Lösung, RNA-Quantifizierung, gefolgt von einer zweistufigen PCR wird Protokoll mit, ergänzende Desoxyribonukleinsäure (DNA) Synthese und Erkennung von TiLV durch konventionelle PCR oder quantitative Identifikation über qPCR mit SYBR grün färben ich. Konventionelle PCR Post-PCR-Schritten und wird einfach über das Vorhandensein des Virus zu informieren. Der zweite Ansatz für absolute Quantifizierung von TiLV bis auf weniger als 2 Kopien ermöglicht und somit eignet sich besonders für TiLV Diagnose in Sub-klinische Fälle. Eine detaillierte Beschreibung der beiden PCR-Ansätze, repräsentative Ergebnisse aus zwei Labors und eine gründliche Diskussion der kritischen Parameter beider wurden aufgenommen, um sicherzustellen, dass Forscher und Diagnostikern ihre geeignet und anwendbar finden Methode der TiLV-Erkennung.
Einleitung
Die global-Kopf-Versorgung mit Fisch erreicht einen neuen Rekordwert von 20 kg im Jahr 2014 und das war aufgrund des kräftigen Wachstums in der Aquakultur. Aquakultur bleibt eine der am schnellsten wachsenden tierische Lebensmittel produzierenden Sektoren weltweit und ist der nur tierische Lebensmittel produzierenden Sektor, der schneller als die menschliche Bevölkerung1 wächst. Tilapiine Cichilds umfassen die zweitwichtigste Süßwasserfische weltweit mit einer weltweiten Gesamtproduktion von 6,4 Millionen Tonnen (MT) und einen geschätzten Wert von 9,8 Milliarden US-Dollar im Jahr 20152bewirtschaftet. Die Top-Ten-Produzenten von Tilapia sind China (1,78 MT), Indonesien (1.12 MT) und Ägypten (0,88 MT), gefolgt von Bangladesch, Vietnam, den Philippinen, Brasilien, Thailand, Kolumbien und Uganda2. Es wird erwartet, dass globale Tilapia Produktion rund 7,3 MT 20303. Tilapia sind solche eine wichtige globale Nahrungsquelle geworden, nicht nur weil sie eine preiswerte Quelle für Protein4 sondern auch weil sie leicht zu züchten in Kapazität unter einer Vielzahl von Wasser und Klima Bedingungen5,6. Vor ein paar Jahrzehnten glaubte man, dass gab es einige kommerziell bedeutenden Krankheiten bedrohen Tilapia Landwirtschaft aber das ist nicht mehr wahr. Eine aufstrebende Viruserkrankung namens Tilapia See Viruskrankheit (TiLVD) ist die erste jemals kritische Krankheit-Epidemie in Tilapia gefunden und die gesamte Branche ist gefährdet. Diese Krankheit hat gravierende sozio-ökonomischen Folgen und ist eine direkte Bedrohung auf die Ernährungssicherheit für Millionen von Menschen in Afrika7, Asien und Südamerika. Zum Jahresbeginn 2018 berichtet der Weltorganisation für Tiergesundheit (OIE), dass die ätiologische Agenten dieser Krankheit, TiLV, offiziell entdeckt worden waren, auf drei Kontinenten über acht Länder8 und da war dieser Erreger-Informationskarte Es aktualisiert wurden weitere Berichte des TiLV in Tansania, Uganda9, Indonesien10, Taiwan11 und Peru12. TiLV ist eine neuartige einsträngige RNA-Virus beschrieben, um eine Orthomyxo-ähnliche Viren sein, denn es eine Vielzahl von Eigenschaften erinnert an andere Orthomyoxoviruses wie Grippe oder infektiösen Lachsanämie Virus (ISAV)13 enthält. Es wurde zuerst in der Nachmahd von massiven Verlusten von wilden und gezüchteten Tilapia in den See von Galiläa, Israel14gekennzeichnet. Danach genannten ähnliche Ausbrüche von Krankheiten als Sommer-Sterblichkeit und die einmonatige Sterblichkeit-Syndrom mit TiLV Infektion assoziiert wurden im Nil Tilapia (Oreochromis Niloticus) in Ägypten15 und Nil und roten Hybrid Tilapia gemeldet (Oreochromis spp.) in Thailand16beziehungsweise. Erkennung von aquatischen tierischen Viren erfolgt historisch durch Wachstum und Isolierung von Viren in Zellkultur. Verschiedene Zelllinien wurden für die Ausbreitung und die Isolierung von TiLV einschließlich e-11 Zellen aus Snakehead Fisch (Ophiocephalus Striatus)17,18, OmB und TmB aus Oreochromis getestet Mossambicus18, und OnlB und OnlL aus Nil Tilapia (O. Niloticus)19. Während Virus Kultur den Vorteil hat, dass es Material für weitere Experimente bietet, hat es den Nachteil, dass es mindestens 4 bis 7 Tage erfordert, die Bildung von zytopathischen Effekte (CPE) zu beobachten und natürlich verschiedene piscine Viren, fitter sind replizieren kann weitergegeben werden und produzieren ähnliche CPE.
In den letzten Jahrzehnten gab es eine Abkehr von der traditionellen, oft zeitaufwändige diagnostische Methoden wie Zellkultur, Serologie und Antigennachweis und Ersatz mit schneller und empfindlicher Nukleinsäure-basierte Erkennung Tests20, 21. Dies wird deutlich durch die Tatsache, die viele qPCR-Assays für ISAV22,23, virale hämorrhagische Blutvergiftung Virus (VHSV)24 als wichtige diagnostische Methoden für eine Vielzahl von Viruserkrankungen in Wassertiere, wie z. B. entwickelt wurden ,25, Betanodavirus26,27 Lachsfische Alphavirus28, Fisch Iridovirus29, Anguillid Herpesvirus 1 (AngHV1)30und Lymphocystis Krankheit-Virus (LCDV)31 . Robuste Methoden für Diagnose und Erreger Überwachung sind dringend erforderlich, um die Verbreitung der TiLV zu verringern. Solche Methoden sollen frühzeitige Erkennung der Infektion, bevor klinische Symptome entwickeln und Erkennung von niedrigen Virus Lasten. Heute, verschiedene PCR-Protokolle einschließlich der RT-PCR14,32, nested RT-PCR18, halb-verschachtelte RT-PCR33und RT-qPCR32,34 für den Nachweis von TiLV entstanden im Gewebe der Fische. Ein Vergleich der RT-qPCR und Virus Isolation in anfälligen Zelllinien für die Erkennung der TiLV ergab, dass RT-qPCR 1.000 Mal empfindlicher als die Virus-Isolation-32. Obwohl jedes veröffentlichte PCR-Protokoll unterschiedliche Empfindlichkeit für den Nachweis von TiLV gemeldet hat, sind die meisten Tests sehr empfindlich mit der Nachweisgrenzen der viralen Kopien auf 7,5 Kopien33, 7 Kopien18 oder 2 Kopien32 pro Reaktion.
Diese Methoden Artikel soll erklären, im Detail, wie TiLV-Erkennung-Assays, beginnend mit Tilapia Gewebe Sammlung, total RNA-Extraktion, cDNA Synthese durchführen und dann TiLV spezifische PCR Assays basiert. Insbesondere wurden umfassende Protokolle von konventionellen RT-PCR und SYBR Green-basierte RT-qPCR Appell an eine Vielzahl von Wissenschaftlern mit dem Ziel, TiLV zu erkennen beschrieben. Erstere ist weniger empfindlich, aber ist in der Regel eine billigere Option Erkennung. Letzteres erfordert komplexere Infrastruktur wie eine quantitative PCR-Maschine und teurer Reagenzien, aber es hat die Vorteile der quantitativen, schnell und sehr empfindlich, was bedeutet, dass es für den Nachweis von TiLV im Sub klinisch verwendet werden kann infizierte Fische. Die RT-PCR und RT-qPCR Protokolle wurden in zwei verschiedenen Labors mit unterschiedlichen geographischen Isolaten von TiLV und die enthaltenen Ergebnisse Highlight die Empfindlichkeit und Reproduzierbarkeit der hier beschriebenen Tests durchgeführt.
Protokoll
Der Tiernutzung Protokoll für diese Studie wurde von der Ethikkommission unter Genehmigung Nummer ACKU 59-Tierarzt-016 Kasetsart Universität Tier genehmigt.
Hinweis: Lesen Sie bitte die Tabelle der Materialien für erweiterte Informationen über die Reagenzien und Geräte für dieses Protokoll vorgeschlagen.
(1) Gewebe Musterkollektion
- Einschläfern Sie den Fisch mit einer Überdosis von Nelkenöl (das Volumen hängt von der Größe der Fische und Konzentration der Produkte, in der Regel mehr als 3 mL/L). Tauchen Sie ein Viertel der Zange und Mayo Schere zu 95 % (V/V) Ethanol gefolgt von brennen die Ausrüstung mit einer Alkohol-Brenner um die Ausrüstung zu sterilisieren.
Hinweis: Tricaine Methanesulfonate (MS-222) kann anstelle von Nelkenöl verwendet werden. - Finden Sie die Leber und schneiden Sie ein kleines Stück (ca. 20-100 mg) oder sammeln Sie 200 µL des Schleims mit Deckglas oder chirurgischen Klinge entfernen Schleim aus anterior Posterior der Tilapia-Fisch und die Proben in einem 1,5 mL Microcentrifuge Schlauch zu.
- Prozesse-Proben sofort, speichern in eine RNA, die Lösung zu stabilisieren oder verschieben Sie sie bis-80 ° C bis zur weiteren Verwendung.
Hinweis: Die größte Aufgabe im Umgang mit RNA bereitet intakt RNA-Moleküle und unbeschädigt in jeder nachfolgenden Handhabungen zu halten. Das RNA-Rückgrat ist von Natur aus empfindlicher als DNA beschädigen. Extraktion und Isolierung von Gesamt-RNS von Gewebezellen erfordert sorgfältige Labortechnik; nehmen Sie alle Bestimmungen, RNase Kontamination durch das Tragen von Handschuhen, mit RNase freies Wasser, Reagenzien, Geräte, Kunststoff-Geschirr, Glaswaren, Arbeitsraum und mithilfe von Filter-Tips für Pipettieren zu verhindern.
2. Guanidium Thiocyanat - Phenol - Chloroform Extraktion von RNA
- Fügen Sie 1 mL der monophasischen Lösung mit Phenol und Guanidin erfolgt in einem Röhrchen mit Gewebeprobe von Abschnitt 1.
Achtung: Diese Lösung ist sehr giftig und muss in einer Laminar-Flow-Kapuze mit Schutzausrüstung und durch das Tragen der richtigen Schutzhandschuhe Brillen, Kleidung und Sicherheit mit großer Vorsicht behandelt werden. - Schleifen Sie die Gewebeprobe mit einem Gewebe Stößel Homogenisator bis homogene.
Hinweis: Proben können auch mit einem macht-Homogenisator kombiniert mit keramischen Perlen homogenisiert werden. Sicherzustellen Sie, dass die Gewebeprobe vollständig, bevor Sie mit dem nächsten Schritt fortfahren homogenisiert ist oder stoppen Sie das Protokoll hier und speichern Sie voll homogenisierten Proben bei-80 ° C bis zur weiteren Verwendung zu. - Fügen Sie 200 µL Chloroform zur Phasentrennung.
Vorsicht Chloroform ist ein potenzieller Betäubungsmittel und ist extrem gefährlich. Es sollte in einer Laminar-Flow-Kapuze mit Schutzausrüstung sowie durch das Tragen der richtigen Schutzhandschuhe Brillen, Kleidung und Sicherheit mit großer Vorsicht behandelt werden. Als eine weniger giftige alternative, 1-Bromo-3-Chloropropane kann auch verwendet werden.
Hinweis: Skaliert die Volumina nach oben oder unten gegebenenfalls. Zum Beispiel, wenn nur 500 µL monophasisch Lösung mit Phenol und Guanidin Herstellung verwendet wurde, dann nur fügen Sie 100 µL Chloroform bei diesem Schritt hinzu.- Mischen von Proben gut durch Umkehrung für 15 s.
- Inkubieren Sie Proben für 3 min bei Raumtemperatur (RT).
- Zentrifuge für 15 min bei 12.000 × g und 4 ° C.
Hinweis: Sollte es eine klare Trennung in eine niedrigere organische Phase, eine weiße Interphase und eine obere wässrige Phase, die RNA enthalten sein. Diese obere Phase ist normalerweise farblos, aber abhängig von der Art und Menge der homogenisierte Gewebe, kann es eine leichte rosa Aussehen haben. - Übertragen Sie die obere wässrige Schicht (ca. 500 µL) zu einem frischen Microcentrifuge Schlauch ohne zu stören die Interphase.
Hinweis: versuchen Sie nicht die gesamte wässrige Phase übertragen, lassen Sie eine kleine Menge auf eine mögliche Kontaminierung der RNA mit wässrigen Phase mit dem organischen zu verhindern oder interphase. - Fügen Sie 1 Volumen von 100 % Isopropanol, die RNA auszufällen.
- Optional, wenn sehr geringe Mengen von Gewebe verwendet wurden, dann fügen Sie 1 µL (5-10 µg hinzu) RNase-freie Glykogen zu jeder Probe, effiziente RNA Niederschlag zu fördern. Dies wird die Kennung des Pellet-RNA im Schritt 2.8 Hilfe.
Hinweis: Das Glykogen fungiert als Träger der RNA und verhindern, dass kleine Mengen an RNA festhalten an der Seite des Rohres. - Mix-Röhren auch durch Umkehrung mehrmals.
- Speichern Sie die Proben für 2 h bei-20 ° c über Nacht
- Optional, wenn sehr geringe Mengen von Gewebe verwendet wurden, dann fügen Sie 1 µL (5-10 µg hinzu) RNase-freie Glykogen zu jeder Probe, effiziente RNA Niederschlag zu fördern. Dies wird die Kennung des Pellet-RNA im Schritt 2.8 Hilfe.
- Zentrifuge Proben für 15 min bei 12.000 x g und 4 ° C.
- Verwerfen Sie den Überstand, ohne dabei die RNA-Pellet am unteren Microcentrifuge Schlauch zu verdrängen.
- 1 mL 75 % Ethanol (V/V) und mischen Sie RNA-Proben durch das Rohr mehrmals umdrehen.
- Zentrifuge für 15 min bei 10.000 x g und 4 ° C.
Hinweis: Das Protokoll kann hier gestoppt werden und die Proben aus den RNA-Pellet in 75 % Ethanol können bei-20 ° C bis zur weiteren Verwendung gespeichert werden. - Verwerfen Sie den Überstand zu wachen nicht, die RNA-Pellet am unteren Microcentrifuge Schlauch zu verdrängen.
- Gegebenenfalls wiederholen Sie die Schritte 2.9-2.11 mit 70 % igem Ethanol (V/V). Gründlich waschen der RNA-Pellet wird minimiert, kein Salz oder Verunreinigung Verschleppung, die stören können werden sensible downstream-Anwendungen.
- Die restlichen Ethanol mit einer Pipette ziehen und dann an der Luft trocknen die RNA-Pellet bei Zimmertemperatur nicht länger als 5 bis 10 Minuten.
Hinweis: über getrockneten Pellets werden schwerlich wieder auszusetzen. - Fügen Sie 30-60 µL RNase-freies Wasser vorgewärmt auf 55-60 ° C, die RNA-Pellet solubilisieren hinzu.
- Legen Sie die RNA für sofortigen Einsatz oder Store bei-80 ° C für eine spätere Verwendung auf Eis.
(3) quantifizieren Sie RNA-Konzentration mit einem Micro-Volume Spektralphotometer
- Wechseln Sie die Einstellungen des Spektralphotometers zu RNA.
- Verwenden Sie 1-2 µL RNase-freies Wasser als Rohling.
- Verwenden Sie 1-2 µL jeder RNA-Probe, um die RNA-Menge zu beurteilen.
- Aufzeichnen der Messwerte bei 230 nm, 260 nm und 280 nm für jede Probe.
- Verdünnen Sie die RNA auf 200 ng/µL RNase-freies Wasser verwenden.
4. Synthese des komplementären DNA (cDNA) mit Gesamt-RNS
- Mischen Sie 1 µg Gesamt-RNS von Protokoll Nr. 2, 2 µM Oligo (dT), 0,5 mM dNTPs Mischung und bringen Sie das Endvolumen 10 µL mit Nuklease-freies Wasser. Hierzu bereiten Sie einen RT-Master-Mix nach der Anzahl der Proben und Kontrollen geprüft werden.
Hinweis: Die Steuerung ist ein Minus-Reverse-Transkriptase-Probe (-RT) wobei das RT-Enzym mit Nuklease-freies Wasser ersetzt wird (siehe Punkt 4.3) und einem keine Vorlage Kontrolle (NTC) wobei Nuklease-freies Wasser wird zu den Master-Mix statt RNA-Vorlage hinzugefügt.- Mischen Sie die Proben gut durch pipettieren, gefolgt von einer kurzen Zentrifugation.
- Inkubieren Sie die Proben bei 65 ° C für 5 min, gefolgt von einer 2 min Inkubation auf Eis.
- Zentrifugieren Sie kurz die Proben um die Flüssigkeit im unteren Teil der Rohre zu sammeln.
- 1 X Reverse Transkriptase Puffer, 100 U-Reverse-Transkriptase und bringen Sie das Endvolumen von jeder Probe zu 20 µL mit Nuklease-freies Wasser.
- Mischen Sie die Proben gut durch pipettieren, gefolgt von einer kurzen Zentrifugation.
- Inkubieren Sie die Proben bei 42 ° C für 60 min, gefolgt von 85 ° C für 5 min.
- Die synthetisierte cDNA zu einer gewünschten Konzentration durch Zugabe von einem entsprechenden Volumen der Nuklease-freies Wasser zu verdünnen und die cDNA auf Eis für den sofortigen Einsatz oder bei-20 ° C für die spätere Verwendung zu speichern.
(5) konventionelle PCR TiLV
- Verwenden Sie die cDNA, Proben und Kontrollen, im Protokoll Abschnitt 4 als Vorlagen für einen PCR-Reaktion mit einer etablierten Grundierung Paare gemäß Tabelle 1, zusammen mit einer DNA-Polymerase Wahl generiert.
Hinweis: Eine zusätzliche nicht-Template-Kontrolle (NTC) sollte hier aufgenommen werden, mit der Substitution von cDNA für Nuklease-freies Wasser in der PCR-Reaktion. Eine positive Kontrolle, falls vorhanden, sollte auch enthalten, bestehend aus zuvor verifizierte TiLV positiven Proben oder das entsprechende TiLV cDNA Fragment in auf einem Plasmid geklont. - Bereiten Sie einen PCR-Master-Mix nach den Richtlinien der DNA-Polymerase-System im Einsatz und die Anzahl der Proben und Kontrollen geprüft werden. Diese Mischung sollte der forward Primer, rückwärts-Primer, dNTPs, MgCl2 und die ausgewählten DNA-Polymerase, zusammen mit seinen Puffer enthalten.
- Kombinieren Sie nach den Richtlinien des ausgewählten DNA-Polymerase die benannten Volumen des Master-Mix mit vorgeschlagenen Betrag von cDNA-Proben und Kontrollproben.
Hinweis: Vorbereitung einer 0,5 x Reaktion Überschuss ist oft vorteilhaft da einige von den Master-Mix geht verloren beim Pipettieren. - Führen Sie PCR Radfahren Bedingungen nach den Richtlinien des Systems der verwendeten DNA-Polymerase und unter Verwendung eines geeigneten Anlasstemperatur für die Primer verwendet (Tabelle 1). Ein solches Programm wird gewöhnlich eine anfängliche Denaturierung bei 95 ° C für 2-5 min, gefolgt von 30-40 Zyklen der Denaturierung bei 95 ° C für 30 s, Glühen bei der empfohlenen Temperatur für 30 s und Dehnung bei 72 ° C für 30 s, gefolgt von einer endgültigen Dehnung bei 72 ° C für 5-10 min.
- 5-15 µL jeder PCR-Reaktion und eine entsprechende DNA-Leiter in in Vertiefungen von 1-2 % Agarosegel, abhängig von der erwarteten PCR-Produkt-Größe zu laden. Trennen Sie die amplifizierte DNA durch Gelelektrophorese und Beflecken Sie das Gel durch Interkalation Bromid (EthBr) zur Visualisierung von DNA-Bänder der erwarteten Größe (Tabelle 1) Erleichterung in einer Gel-Dokumentation-Maschine mit UV-Licht.
Achtung: EtBr ist giftig; Es sollte mit Vorsicht behandelt werden, durch das Tragen der richtigen Schutzkleidung und Schutzhandschuhe.
| TiLV Genom Zielsegment | Forward primer 5' - 3' | Rückwärts-primer 5' - 3' | PCR-Produkt-Größe (bp) | Tm ° C | Original-Referenz | ||||
| 1 | CCAAACGTTATCTCTTAATTACGCAC | GCAAATATTTCTCTCATTCGCCT | 1641 | 50 | Surachetpong Et Al., 2017 | ||||
| 1 | CCTCATTCCTCGTTGTGTAAGT | AGGAGTTGCTGTTGGGTTATAG | 1000 | 62 | Mugimba Et Al., 2018 | ||||
| 2 | ACTCTCTATTACCAAATACATTTACT | TTACCATATATATAGTGAAGGC | 1445 | 45 | Surachetpong Et Al., 2017 | ||||
| 2 | GTCCAGGGCGGTATGTATTG | CTTACGGCTGACAAGTCTCTAAG | 834 | 62 | Mugimba Et Al., 2018 | ||||
| 3 | GTTGGGCACAAGGCATCCTA | TATCACGTGCGTACTCGTTCAGT | 250 | 56 | Eyngor Et Al., 2014 | ||||
| 3 | TATGCAGTACTTTCCCTGCC | TTGCTCTGAGCAAGAGTACC | 491 | 57 | Eyngor Et Al., 2014 | ||||
| 3 | ACCCCTTAATCCTTAATAGACCGTTA | CCCATAATCCTCTATTAGAACGTCGT | 1352 | 50 | Surachetpong Et Al., 2017 | ||||
| 3 | GTCGAGGCATTCCAGAAGTAAG | GAGCTAAGGGAACGGCTATTG | 834 | 62 | Mugimba Et Al., 2018 | ||||
| 4 | AGCAGCAGCAGGAGAAAGAG | ACCGTCCTGTTTCTGAATGG | 358 | 60 | Nicholson Et Al., 2017 | ||||
| 4 | CCAAAGTTTACTCCTATTACCCAGA | GCAAATCTTTCTCCAATTACCGTCT | 1250 | 50 | Surachetpong Et Al., 2017 | ||||
| 4 | GCCCAATGGTTCCCATATCT | GCCCAATGGTTCCCATATCT | 524 | 62 | Mugimba Et Al., 2018 | ||||
| 5 | CCAAATGTTTCTCTTATCTCAGACTC | CTTTTTCTCAGTTTACCACTTTATG | 1087 | 57 | Surachetpong Et Al., 2017 | ||||
| 5 | CAACTCTTAGCCTCCGGAATAC | CGTTCTGCACTGGGTTACA | 696 | 62 | Mugimba Et Al., 2018 | ||||
| 6 | CCAAATTTTACCTCTCGCAT | TCAAGCACTTAAAACTGTACC | 1027 | 45 | Surachetpong Et Al., 2017 | ||||
| 6 | CCCACACGACAGGACATATAG | GAGTTGGCTTAGGGTGATAAGA | 948 | 62 | Mugimba Et Al., 2018 | ||||
| 7 | CTCTCTTTGCATTGCATACCGT | GACCAATTATCCCTGCTTTCA | 704 | 57 | Surachetpong Et Al., 2017 | ||||
| 7 | TCCTTTAGGGATTGGCACTAAC | TTCCATCGACTGCTCCTAGA | 486 | 62 | Mugimba Et Al., 2018 | ||||
| 8 | ACCTCATCTACACTAACATTTCCA | TCATCATTACACAAATGGAGTAGCT | 637 | 50 | Surachetpong Et Al., 2017 | ||||
| 8 | CTTAAGGGCCATCCTGTCATC | TGGCTCAAATCCCAACACTAA | 476 | 62 | Mugimba Et Al., 2018 | ||||
| 9 | TTGGTGATGTCACGATGGATA | AGTTCTATCGCCAGCCATGT | 351 | 60 | Nicholson Et Al., 2017 | ||||
| 9 | ACAAGTCCGATTACTTTTTCCGC | TCTTTCTCACGTCCTTAAAGTCA | 530 | 50 | Surachetpong Et Al., 2017 | ||||
| 9 | GATATCCTCCACATGACCCTTC | GTACGTCACTTTGTGCCATTAC | 261 | 62 | Mugimba Et Al., 2018 | ||||
| 10 | AACCCTACTAACACCAAATATAGCT | CTTTCCCTCTGACACCCTGT | 450 | 50 | Surachetpong Et Al., 2017 | ||||
| 10 | TCCTCTCTGTCCCTTCTGTT | CAGGATGAGTGTGGCAGATTAT | 276 | 62 | Mugimba Et Al., 2018 | ||||
Tabelle 1. Veröffentlichten Primer-Paaren für die Verstärkung der TiLV cDNA mit Endpunkt PCR. Die Grundierung set in fett dargestellt wurden verwendet, um die repräsentative Ergebnisse in Abbildung 3A und 3 b zu generieren.
6. TiLV Quantitative Polymerase-Kettenreaktion (qPCR)
- Ein Plasmid mit den entsprechenden TiLV mit genomischen Segment 3 cDNA als Standard, wie z. B. pTiLV32, bereiten eine doppelt oder dreifach 10-divisibel serielle Verdünnungsreihen.
- Bereiten Sie einen qPCR Master-Mix für alle Proben, Standards und Kontrollen, duplizieren oder dreifacher Nutzen 0.4 µL Nuklease-freies Wasser, 0,3 µL vorwärts Grundierung, 0,3 µL des rückwärts-Primer und 5 µL 2 x SYBR Green zu berücksichtigen, die die Reaktionen in durchgeführt werden sollte DNA-Polymerase Master-Mix pro Reaktion.
- Verwenden Sie die Primer bei einer Konzentration von 10 µM und die Primer-Informationen und der standard pTiLV wie folgt:
Grundierung zu übermitteln: TiLV-112F (5'-CTGAGCTAAAGAGGCAATATGGATT-3 ")
Reverse Primer: TiLV-112R (5'-CGTGCGTACTCGTTCAGTATAAGTTCT-3 ")
Standard pTiLV:10 Pg/µL
Hinweis: Wenn die Gesamtzahl der Proben und Kontrollen 10 ist und in Triplicates durchgeführt werden wird, das entspricht einem qPCR-Master-Mix bestehend aus 12 µL Nuklease-freies Wasser, 9 µL forward Primer, 9 µL-rückwärts-Primer und 150 µL SYBR Green DNA Polymerase Master-Mix. Kommerziell erworben 2 x universal SYBR Green DNA enthalten Polymerase Meister-Mischungen alle notwendigen Komponenten für die qPCR-Reaktion, nämlich die SYBR Green ich färben, hot Start Taq-DNA-Polymerase, dNTPs, MgCl2 und passive Referenz Farbstoffe. Schützen Sie die SYBR Green-master-Mix vor Licht.
- Verwenden Sie die Primer bei einer Konzentration von 10 µM und die Primer-Informationen und der standard pTiLV wie folgt:
- Verzichten Sie 6 µL qPCR Master-Mix in qPCR Streifen Röhren oder 96 well-Platte mit qPCR-Maschine im Einsatz.
- Fügen Sie 4 µL cDNA Vorlage, Steuerelemente oder seriell verdünnten TiLV Standards in den Rohren oder Vertiefungen der 96-well-Platte.
- Schließen Sie die qPCR-Rohre oder Versiegeln der 96-well-Platte mit einem kompatiblen Platte Deckel für die qPCR-Maschine im Einsatz
- Streichen Sie sanft qPCR Röhren um die Lösung und Spin-down qPCR Röhren oder 96 well-Platte mit einer Zentrifuge um zu sammeln alle die Flüssigkeit in den Boden der Gefäße zu mischen.
- Legen Sie die Rohre oder Platte in Echtzeit-Thermocycler.
- Programmieren der qPCR Thermocycler, führen Sie eine erste Denaturierung bei 95 ° C für 3 min, gefolgt von 40 Zyklen von 95 ° C für 10 s und 60 ° C für 30 s für Grundierung Glühen und Dehnung, endend mit einem schmelzenden Kurve Schritt von 65 ° C bis 95 ° C mit einem Inkrement von 0,5 ° C / 5 s.
- Wählen Sie SYBR als Fluorophor Farbstoff, dann wählen Sie unbekannte als eine Probe aus und legen Sie einen Namen in einer Probe Namensfeld.
- Öffnen Sie den Deckel der Maschine RT-qPCR und legen Sie die qPCR-Streifen in die zugeordneten Vertiefungen, dann schließen Sie den Deckel.
- Durchführen Sie die RT-qPCR-Assay mit den ausgewählten Bedingungen. Die Maschine beginnt zu laufen, nachdem der Deckel die gewünschte Temperatur erreicht hat. Sammeln Sie die Fluoreszenz jeder Probe nach jedem Schritt der Erweiterung zur Überwachung des Fortschritts der Reaktion.
Hinweis: Die qPCR-Maschine und die zugehörige Software automatisch berechnet alle Parameter des Assays und zeigt die Verstärkung Kurven in Echtzeit, während der Standardkurve und schmelzenden Kurve am Ende des Zyklus qPCR erzeugt werden. - Datenanalyse und Übernahme durch erste sicherzustellen, die das Schmelzen Kurven für jede Probe und Standard haben eine einheitliche Spitze auf die erwartete Temperatur für die Amplifikate.
- Bewerten die Verstärkung Kurven der Proben und Normen und setzen die Schwelle in einer Region waren, dass die Verstärkung der cDNAs in allen Proben gleich ist. Dies erfolgt normalerweise automatisch durch die Software aber sollte sorgfältig geprüft werden.
- Berechnen Sie die Anzahl von TiLV Exemplaren mit der Standardkurve.
Ergebnisse
Nach dem in Abschnitt 1 beschriebenen Protokoll sterbenden roten Hybrid Tilapia mit klinischen Anzeichen einer TiLV Infektion (Abbildung 1A) wurden durch das Baden in einer hohen Konzentration von Nelkenöl, fungiert als Narkosemittel eingeschläfert. Klinische Symptome sind variabel, aber die häufigsten Symptome zu Lethargie, Haut Erosion und Verfärbungen, Exophthalmia, freistehende Skalen, offene Wunden/Läsion und Verhaltensstörungen15,16,33zu sein scheinen, 35,36, einige davon ist deutlich in Figur 1A. Die Bauchdecke wurde entfernt, um innere Organe wie Leber, Milz oder Kopf Niere (Abbildung 1 b) zu sammeln. Schleim-Proben wurden auch in diesem Stadium von sanft kratzen die Haut vom vorderen zum hinteren des Fisches mit einem Deckglas oder chirurgische Messer37gesammelt.

Abbildung 1 . Tilapia Dissektion und Sample-Sammlung. A. TiLV-infizierten roten Hybrid Tilapia mit Haut Leisons, Rötung um den Mund und Operculum, Haut Erosion und Hornhauttrübung. B. Sectioned rot Hybrid Tilapia, um Gewebe-Sammlung aus der Leber (zum Zeitpunkt der blauer Pfeil), Milz oder Kopf Niere Organe zu ermöglichen. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
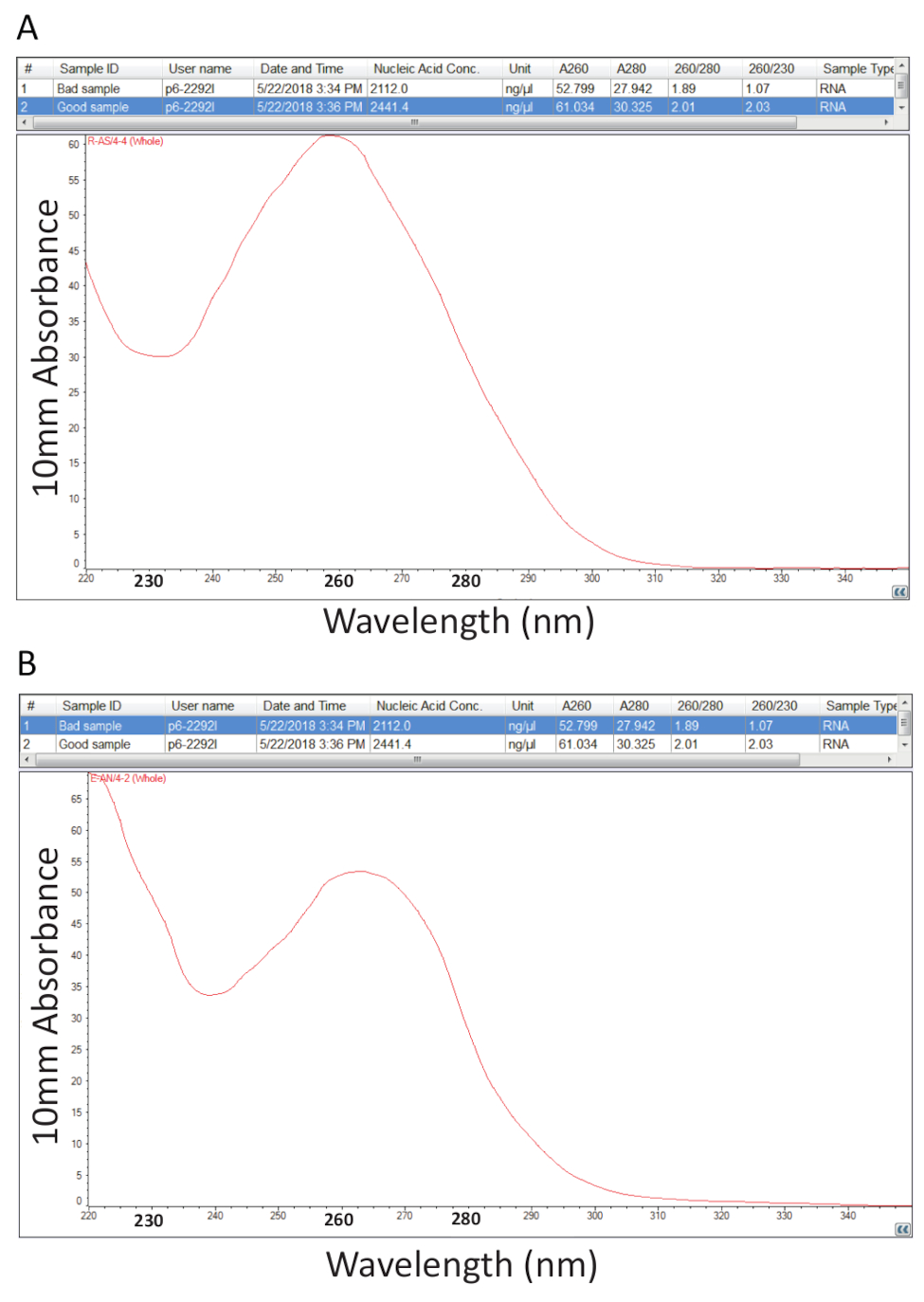
Danach folgte das Protokoll gemäß Abschnitt 2 für Guanidium Thiocyanat-Phenol-Chloroform Extraktion von Gesamt-RNS und RNA Quantifizierung wie in Abschnitt 3 dargelegt wurde durchgeführt, um Probe Reinheit beurteilen durch Berechnung des Verhältnisses von Reinheit und Untersuchung der spektralen Profile (Abbildung 2). Abbildung 2A zeigt ein repräsentatives Ergebnis aus ein erfolgreiches Gesamt RNA Extraktionsverfahren, während Abbildung 2 b eine schlechte Vorbereitung der RNA darstellt. Nukleinsäuren sind Absorption Maxima bei 260 während Proteine Ihnen bei 280 haben nm. Das Verhältnis der Messungen bei 260 nm und 280 nm zeigen die Reinheit jeder Probe und Verhältnisse von 1,9 bis 2,1 zeigen reine RNA, wie es der Fall für das Beispiel in Abbildung 2A. Niedrigere A260/280-Werte beobachtet in Abbildung 2 b zeigen mögliche Protein oder Phenol Belastung Überbleibsel aus der RNA-Extraktion-Verfahren. Extinktion bei 230 nm kann das Ergebnis einer Kontamination der Probe und das A260/230 nm Verhältnis wird auch aus diesem Grund berechnet. Dieses Verhältnis sollte im Bereich von 2.0-2.2 für reinen RNA-Präparaten wie dargestellt durch einen Wert von 2.03 für das Beispiel in Abbildung 2A, während Abbildung 2 b ein niedriges A260/230 Verhältnis von 1,07 hat und die spektrale Profil eine Verschiebung in den Trog mit 230 zeigt nm auf 240 nm, was bezeichnend für passives Guanidin oder Phenol in der Probe. Für das Beispiel in Abbildung 2 bgezeigt kann die Reinheit der Probe wieder Fällung der RNS um die Verunreinigung zu entfernen verbessert werden.

Abbildung 2 . Spektralfotometrische Quantifizierung von Gesamt-RNS erkrankten Tilapia Gewebe entnommen. A. Reinheit Verhältnisse und spektrale Profile aus einer erfolgreichen RNA-Vorbereitung. B. als A, außer Vertreter eines armen RNA-Extraktion-Verfahrens. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
Um TiLV durch RT-PCR zu erkennen, reine Proben wie man in Abbildung 2A vertreten waren reverse Transkription (Protokoll Nr. 4) in cDNA und verwendet, wie eine Vorlage für die PCR assay detailliert in Abschnitt 5 und repräsentative Ergebnisse sind in Abbildung 3Adargestellt. Primer gezeigt Fettdruck in Tabelle 1 wurden verwendet, um ein 491 bp Fragment TiLV genomische Segment 314zu verstärken. Die PCR-Produkte wurden durch Gelelektrophorese getrennt und zur Visualisierung mit EtBr befleckt. Abbildung 3A zeigt die Ergebnisse eines zweistufigen RT-PCR mit 4 cDNA Proben (S1-S4), abgeleitet aus der Leber von erkrankten Tilapia isoliert in Thailand und in jeder Probe, eine saubere single-Band von ca. 500 bp beobachtet werden und somit sind die Proben 1-4 TiLV positiv. Das gleiche PCR-Produkt wurde aus der Positivkontrolle Probe, bestehend aus cDNA TiLV Segment 3 in ein Plasmid32 kloniert, während der keine Vorlage-Kontrolle (NTC) PCR-Produkte nicht Ausbeute gewonnen. Die Probe in Abb. 3 b erfolgte mittels der gleichen Grundierungen wie in Abbildung 3A , aber in einem anderen Labor, mit einem One-Step RT-PCR-Ansatz und mit 5 RNA-Proben, abgeleitet aus dem Kopf Nieren-Gewebe von Tilapia mit Ursprung in der ägyptischen Aquakultur 15. es wurde festgestellt, dass mit dieser Erkennung-Assay, dass Proben 1, 3 und 5 TiLV positiv sind, während Proben 2 und 4 TiLV negativ sind, da keine PCR-Produkt in der richtigen Größe gefunden wurde. Den Negativkontrollen hat unter anderem zwei minus Reverse Transkriptase Kontrollen und zwei NTCs keiner PCR-Produkte generiert. Ein One-Step RT-PCR-Assay wurde ebenfalls durchgeführt targeting Tilapia ActinB gen. Die Größe der Amplifikate 217 bp wurde als erwarteten38in jeder Probe (S1-S5) generiert. Dieser Assay diente als ein Steuerelement für die Integrität der RNA-Proben sowie für eine semi-quantitative Untersuchung der TiLV positive Proben ermöglicht. Angesichts der Tatsache, dass die generierten Tilapia ActB Produkt relativ gleich ist, können Unterschiede in der Höhe von TiLV spezifischen PCR-Produkt erzeugt als ein getreues Spiegelbild der Höhe der TiLV in einem bestimmten Gewebeprobe interpretiert werden.

Abbildung 3 . TiLV RT-PCR. A. cDNA herstellbare Produkte von Leber Gewebe des Kranken Tilapia, gesammelt aus Thailand wurden Bildschirm für TiLV Infektion mit spezifischen Zündkapseln für Segment 3 (gezeigt Fett in Tabelle 1) des TiLV mit einem 2-Schritt-RT-PCR-Assay. M = Marker angezeigt in Basenpaaren; S1-S4 = Proben 1-4; C1 = Positivkontrolle mit pTiLV als Kopiervorlage PCR; und C2 = keine Vorlage-Kontrolle (NTC). B. One-Step RT-PCR unter Verwendung der gleichen Zündkapseln a und Proben vom Kopf Niere Gewebe des Kranken Tilapia aus Ägypten15gesammelt. M = Marker angezeigt in Basenpaaren; S1-S5 = Proben 1-5. Kontrollen C1-C2 sind abzüglich Reverse Transkriptase steuert und C3-C4 NTCs. Bodenplatte nutzt eine One-Step RT-PCR Primer gegen Tilapia ActinB38 (für Details siehe Text) produzieren ein PCR-Produkt von 217 Basenpaaren. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
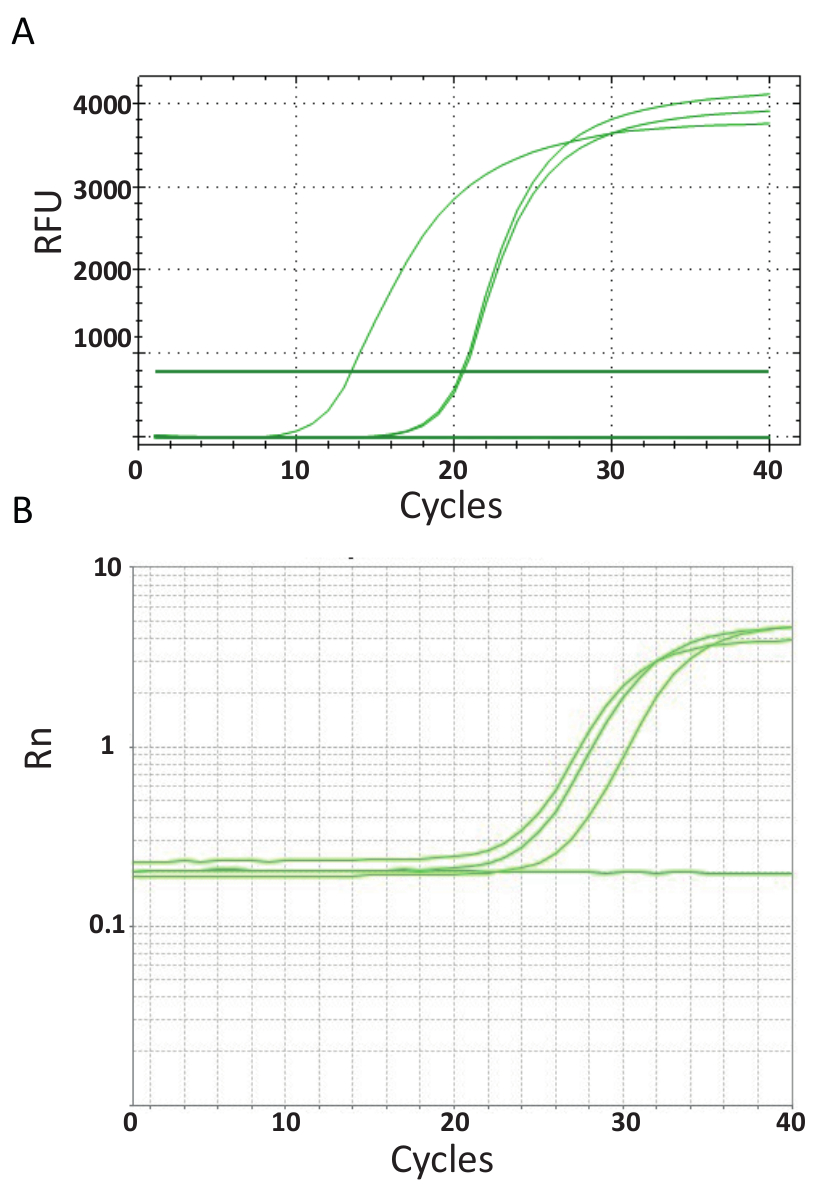
Im Gegensatz zu den Endpunkt PCRs dargestellt in Abbildung 3, qPCR-Assays, die im Protokoll 6, erläutert werden Messen Sie die Menge des PCR Produktes nach jedem PCR-Zyklus. Die Verstärkung der Ziel-DNA-Erkennung erfolgt anhand fluoreszierende Moleküle, die Interaktion mit DNA aus jeder Runde Reaktion erzeugt. Hier wurde SYBR Green ich färben genutzt, die Plasmamembrane mit doppelsträngige DNA. Das Fluoreszenzsignal folgt während der Reaktion und ihre Intensität bezieht sich auf die Menge des Produktes gebildet 39,40,41,42,43. TiLV qPCR Tests wurden durchgeführt, wie im Protokoll 6 in verschiedenen Laboratorien mit verschiedenen SYBR Green Reagenzien, qPCR Maschinen und Proben aus verschiedenen Ländern beschrieben. Die daraus resultierende Verstärkung Kurven sind in Abb. 4A und 4 bgezeigt. Es kann beobachtet werden, dass für jeden Test, der Kurs des Experiments vier Phasen hat: die Phase linear Boden, exponentielle Frühphase, späten exponentiellen Phase und die Plateau-Phase. Die lineare Boden Phase tritt während der frühen Zyklen wo kann DNA-Vervielfältigung noch durch DNA-Mengen produzieren nicht genügend Signal/Hintergrund-Verhältnis nicht identifiziert werden. Grundlinie Fluoreszenz wird während dieser Phase berechnet. Danach beginnt Ziel DNA doppelte Konzentration bei jedem Zyklus induzieren das Signal nachweisbar über Hintergrund und exponentiell. Die Verstärkung Effizienz (E) eine gut optimierte qPCR-Assay ist sehr hoch (in der Nähe von 100 %) am Anfang der Reaktion und bleibt stabil in dieser frühen exponentielle Phase von der Verstärkung und es ist an dieser Stelle, dass Quantifizierung, wenn durchgeführt wird Effizienz der Reaktion ist immer noch stabil. In späteren Zyklen beginnt das Signal Plateau und die Intensität der Fluoreszenz ist nicht mehr auf die Startnummer Vorlage Kopie korreliert, weil die Reaktionskomponenten erschöpft44sind. Sättigung kann auch aufgrund der Konkurrenz wieder Glühen Reaktionen, die veränderten Konzentrationsverhältnis der Komponenten oder die Menge des Enzyms Einheiten zu DNA Substrat Molekülen auftreten. Solche Parameter entfallen möglicherweise, die Unterschiede zwischen den Kurven Verstärkung für die Assays gezeigt in Abb. 4A und 4 b. Die enthaltenen Steuerelemente hat nicht diese charakteristischen Verstärkung Kurven generiert.

Abbildung 4 . Verstärkung-plots, um die Ansammlung von Produkt über die Dauer des Echtzeit-PCR-Tests zeigen. A. Verstärkung Kurven des TiLV-positiven Proben abgeleitet aus Thailand, NTCs und positive Plasmid-Steuerung über ein SYBR Green 2-stufige qPCR-Assay. Das Diagramm wurde durch Plotten relative Fluoreszenz (RFU) vs. Zykluszahl generiert. B. Verstärkung Kurven der TiLV positiven Proben aus Ägypten, wie in Abbildung 3 b und ein NTC abgeleitet. Die Verstärkung-Kurve ist die Fluoreszenz des Reporter-Signals auf die Fluoreszenz des passiven ROX Farbstoffs in die Assay (Rn) versus Zykluszahl enthalten normalisiert. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
Am Ende der qPCR-Thermocyclings auf die verschiedenen Maschinen in jedem Labor wurde die Daten erworben und analysiert. Abbildung 5A und 5 b zeigen repräsentative schmelzende Kurven aus der Assays in jedem Labor durchgeführt. Jede qPCR-Maschine wurde programmiert, eine schmelzende Kurvenanalyse am Ende durchführen. Dies wurde erreicht durch die schrittweise Erhöhung der Temperatur und Überwachung der Fluoreszenz als Funktion der Temperatur. Wenn die Temperatur hoch genug, um DsDNA denaturieren, ist ein großer Tropfen Fluoreszenz aufgezeichnet, weil das Fluorophor-Molekül freigesetzt wird. Die Software der Schuldtitel qPCR berechnet Anlasstemperatur (Tm) von den schmelzenden Kurvendaten durch Auftragen der ersten Derivative Vs Minustemperatur (Abbildung 5). Es ist ersichtlich, dass in Abbildung 5A und 5 b , dass die Produkte in den verschiedenen Sample-Sets gebildet einheitliche schmelzen Übergang auf die erwartete Temperatur ca. 80 ° c zum Test haben. Keine andere Spitzen bei niedrigeren Temperaturen wurden beobachtet. Aufgrund ihrer geringen Größe ist die T-m der Primer-Dimere in der Regel niedriger als die des Ziels DNA-Sequenz. Dieser Unterschied zwischen den T-merleichtert daher das potenzielle Grundierung-Dimere oder andere unspezifische Amplifikationsprodukte zu identifizieren. Die Steuerung hat Schmelze Kurven wie TiLV positiven Proben und Standards nicht generiert und können als eine fast horizontale Linie am unteren Rand die Diagramme in Abbildung 5A und 5 bgesehen werden.

Abbildung 5 . Schmelzen Sie Kurvenanalyse um sicherzustellen, dass die Spezifität des Assays und verschiedenen PCR-Produkte durch ihre schmelzenden Merkmale unterschieden werden können. Schmelzen Sie A. Kurvenanalyse des TiLV-positiven Proben aus Thailand, negativ-Kontrolle und positive Plasmid Kontrolle. B. Kurvenanalyse von TiLV positive Proben aus Ägypten, pTiLV Standards und einem NTC schmelzen. Die Charts in A und B beide die Änderung Fluoreszenz dividiert durch die Veränderung der Temperatur zeigen aufgetragen gegen Temperatur, ein klares Bild von den schmelzenden Dynamik zu produzieren. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
Die meisten qPCR-Maschinen verfügen über eine Software erleichtert die weitere Auswertung der qPCR laufen und werden die Proben durch die Erzeugung einer Standardkurve durch Plotten automatisch die Zyklus-Schwelle (Ct) gegen den Logarithmus der pTiLV Standards Vorlage zu quantifizieren Kopieren Sie die Nummer wie in Abbildung 6A und 6 b für die zwei unabhängigen Labors gezeigt. Ct ist die Maßeinheit für die Auswertung qPCR Ergebnisse. Der C-t -Wert bezeichnet die Anzahl der Zyklen erforderlich, um einen festgelegten Schwellenwert Fluoreszenz Signal zu erreichen. Je größer die Datenmenge ab Vorlage, desto weniger Zyklen es braucht, um einen nachweisbaren Fluoreszenz-Niveau zu erreichen. In der Tat werden Proben mit einer hohen Belastung der TiLV niedriger C-t -Werte als bei Proben mit einer geringen Belastung der TiLV wie z. B. in Fisch mit eine subklinische Infektion haben. Ermitteln, C-t -Werte, sind natürliche Fluoreszenz Grundbelastung Rohdaten zunächst abgezogen. Als nächstes wählt die Software dem qPCR Gerät zugeordnet automatisch eine Fluoreszenz-Schwelle bei der Suche die Datenkurven für jede Probe und unter Einbeziehung einer Ct vertreten, wo die Probe die Schwelle überschritten. Dies erfolgt getrennt für jede Probe und jede Schwelle sollten sorgfältig geprüft werden, um sicherzustellen, dass der Schwellenwert im logarithmischen Teil der Verstärkung Kurven und an einem Ort, wo alle Kurven parallel sind. So die spezifischen Ct erworben ist ein relativer Wert und es ist relativ ab Vorlage Kopie Nummer45, aber es ist auch speziell für die qPCR-Maschine und Reagenzien, die Effizienz der PCR-Amplifikation und die Nachweisempfindlichkeit. Diese Parameter dazu beitragen, die Unterschiede mit den gleichen Test in Abbildung 6.
Von den standard Kurven in Abbildung 6, Regressionsanalysen, einschließlich Berechnung der Standardkurve Pisten (m) und fängt, Verstärkung Wirkungsgrade (100 x (101/m -1))46 und Linearität der Reaktion wurden durchgeführt. Standardkurve Analysen wurden auch verwendet, um Empfindlichkeit (Begrenzung auf Erkennung), Wiederholbarkeit und Reproduzierbarkeit des Tests zu bestätigen. Theoretisch wird die Menge an DNA verdoppelt, mit jedem PCR-Zyklus, was bedeutet, dass die Effizienz (E) 100 % entspricht. Allerdings ist in der Praxis selten ein idealer Wirkungsgrad aufgrund der suboptimalen PCR-Bedingungen wie Hemmung der DNA-Polymerase, Verunreinigungen, zuviel cDNA und Pipettieren Fehler47erreicht. In der Regel Verstärkung E reichen von 90-110 % für gute Assays in Abbildung 6A ein Wirkungsgrad von 94,5 % mit 8 seriell verwässerten pTiLV Proben während der Assay-Effizienz in der Probe berechnet wurde, dargestellt in Abbildung 6 b mit 7 seriell verdünnten pTiLV Proben betrug 101,2 %. Ein Wirkungsgrad von über 100 % ist in der Regel aufgrund des Vorhandenseins von PCR-Inhibitoren in der Probe. Lineare Regressionsanalyse der standard Handlung ermöglicht auch für die Berechnung der Anzahl der TiLV Exemplare in jeder Probe41,42,45, wie für die drei TiLV Proben in rot in Abbildung 6 gezeigten beobachtet werden können ist im Einklang mit den Ergebnissen für Proben S1, S3 und S5 in Abbildung 3 bgezeigt.

Abbildung 6 . RT-qPCR Standardkurven. Real-Time PCR von 10-divisibel serielle Verdünnungen von pTiLV, der Standard in beiden Laboratorien verwendet. A. 8 seriell verwässerten pTiLV Proben wurden getestet, alle bekannter Konzentration und korreliert mit der Anzahl der TiLV Kopien / Reaktion. Die Standardkurve wurde durch Plotten Log Kopie Nummer vs. Zyklus Schwelle (Ct) generiert. Die Steigung =-3.462, R2 = 0.9992 und der Wirkungsgrad liegt bei 94.47 %. B. a, außer 7 seriell verwässerten pTiLV Proben (grün) wurden getestet und das Diagramm zeigt die Schwelle-Zyklus auf der y-Achse und die Kopienzahl des TiLV (Menge) auf der x-Achse. Der y-Achsenabschnitt = 32.327, Steigung =-3.292, R2 = 0,98 und der Wirkungsgrad liegt 101,2 %. Für beide Normen-Kurven werden in A und B, Steigung, y-Achsenabschnitt und Korrelationskoeffizient Werte (R2) genutzt, um die Leistung des Assays zu verstehen. Wichtig ist, sollte R2 Wert nahe bei 1 sein, denn es ist ein Maß für die Linearität der Standardkurve. Die Steigung wird verwendet, um PCR-Effizienz, wobei 100 % Wirkungsgrad einer Steigung von-3.32 entspricht, zu messen, siehe Haupttext für die Gleichung und weitere Details. Eine gute qPCR Reaktion hat in der Regel einen Wirkungsgrad zwischen 90-110 % Korrelation zu einer Neigung von zwischen-3.58 und-3.10. Die Standardkurve für absolute Quantifizierung von unbekannten TiLV positive Proben verwendet wird und bestimmt die genaue Anzahl der TiLV Kopien / Reaktion, wie ist der Fall für die drei TiLV positive Proben rot b.
Diskussion
TiLV wurde erstmals im Jahr 2014 in Israel14 gemeldet und seitdem wurde es in mehreren Ländern, darunter Ägypten, Kolumbien, Indien, Malaysia, Uganda, Tansania und Thailand15,16,18, identifiziert 35 , 48. globales Bewusstsein, insbesondere in Tilapia produzierenden Ländern mehr Aufmerksamkeit auf das Virus und verschiedene Einschränkungen und Kontrollmaßnahmen von Regierungsbehörden haben versucht, die Ausbreitung von TiLV umgesetzt worden. Hier wurde ein detailliertes Protokoll zur TiLV Erkennung im Tilapia Gewebe, für Musterkollektion, RNA-Isolierung, cDNA Synthese, PCR und qPCR Assays erläutert. Es gibt verschiedene Aspekte dieser Methoden, die eine spezifische Diskussion zu rechtfertigen. TiLV wurde in Fisch überspannt eine Vielzahl Größen9,12,14,15,49 und Arten von Tilapia bisher einschließlich gezüchteten Hybriden Tilapia (O. identifiziert Niloticus x O. Aureus)11,14, Nil Tilapia (O. Niloticus)9,10,14,15,16, 33 , 35 , 36 , 49 , 50 und roter Tilapia (Oreochromis SP.()16,33,48,51, wie auch in wilden Nile Tilapia9,12, schwarz Tilapia51, T. Zilli14,15, S. Galilaeus, O. Aureus und T. Simonis Intermedia14 und vor kurzem TiLV wurde in Wildkarpfen (Barbonymus identifiziert Schwanenfeldii)52. Gewebeproben von inneren Organen (Kieme, Milz, Leber, Herz, Kopf Niere) oder Schleim37 können von gesunden sowie sterbenden Tilapia unabhängig von Alter, Größe oder Art gesammelt und zur RNA-Isolierung verarbeitet werden. Das Gesamt RNA Extraktion Protokoll beschriebenen verwendet hier eine monophasische Lösung von Phenol und Guanidinium-Thiocyanat, die ein chaotropen denaturierenden Agent ist. Die Gewebe werden direkt in dieser Lösung gefolgt von dem Zusatz von Chloroform und Zentrifugation Phasentrennung zu erreichen, wobei eine klare RNA mit oberen wässrigen Phase, eine Interphase und eine geringere organische Phase erzeugt wird, homogenisiert. Die RNA ist isoliert von der wässrigen Phase durch Isopropanol-Fällung, gefolgt von der wiederhergestellten RNA get rid of Verschmutzungen waschen. Isolierung der RNA durch diese Methodik wurde Pionierarbeit von Piotr Chomczynski und Nicoletta Sacchi und wurde als Guanidinium-Thiocyanat-Phenol-Chloroform Extraktion53,54bezeichnet. Diese Art von Reagenz für die RNA-Extraktion verwendet im Handel gekauft werden oder im Labor gemacht (siehe die Tabelle der Materialien für weitere Informationen). Dieses Protokoll dauert etwas länger als das Spalte-basierte Methoden wie die Silica-basierten Reinigung, aber im Allgemeinen es ist kostengünstiger und ergibt mehr RNA.
In diesem Protokoll Quantifizierung der RNA mit A260 Werten wurde skizziert, wobei Spektralphotometrie Werte RNS-Qualität zeigen können (A260/A280 = 1,9-2,1). Während diese Methode einen guten Indikator für Probe Reinheit geben wird, kann nicht es absolut über die Qualität der extrahierten RNA informieren. Um richtig festzustellen, ob die RNA intakt oder teilweise abgebaut ist, können Proben Trennung durch Agarose-Gelelektrophorese wobei Verschmieren der die EtBr 18 gebeizt und 28 s rRNA zeigen RNA-Abbau. Weitere Überprüfung der RNS-Qualität zählen mit einem Lab-on-a-Chip-Instrument. Darüber hinaus ist es auch wichtig, die gereinigte RNA mit DNase verdauen ich verunreinigen entfernen host genomischen DNA, die abhängig von den nachgelagerten Anwendungen zu falschen Ergebnissen führen kann. Wenn Host gDNA RNS-Probe zu einem großen Teil noch verunreinigt ist, kann auch eine zusätzliche Behandlung der DNaseI am Ende des Verfahrens RNA-Extraktion durchgeführt werden (siehe Tabelle der Materialien).
Komplementären DNA-Synthese kann großen Einfluss auf das Gesamtergebnis qPCR und ist ein Aspekt der Methode, die Variation einführen kann. Die cDNA-Protokoll befürwortet hier besteht aus einer einzelnen Komponente-Aufstellung mit Oligo (dT) und somit nur transkribiert mRNAs mit PolyA Schwänzen. Es ermöglicht die Benutzer die Kontrolle der genau welche, die Komponenten zur Verwendung in der reversen Transkription Reaktion und diese Art der cDNA Synthese für TiLV Erkennung32bewährt hat. Eine Alternative zu diesem Set-up ist ein kommerziell gekauften Master-Mix enthält alle Komponenten für die reverse Transkription Reaktion und ist sehr schnell und einfach ohne die üblichen mehrstufigen, pipettieren und Multi-Temperatur-Protokoll. Dies ist vorteilhaft, weil es minimiert die Handhabung und fördert die Einheitlichkeit in allen Proben. Solche Meister-Mischungen enthalten häufig oligo(dT) und zufällige Primer, so dass es für verschiedene RNA-Vorlagen und repräsentative cDNA Kopien der Sequenzen aus der gesamten Länge des RNAs in einer Population zu generieren (virale und Tilapia mRNA hosten) und in der Theorie, jede gewünschte RNA-Spezies kann dann durch konventionelle PCR oder qPCR von solch einer Probe gemessen werden. Diese Vielseitigkeit ist der Hauptvorteil eines 2-Stufen-RT-PCR-Ansatzes; Freuen Sie sich auf einen langfristigen Pool, der für viele verschiedene Experimente verwendet werden kann. In den Ergebnissen ist ein One-Step RT-PCR-Ansatz vertreten wobei Sequenz spezifische Primer (Tabelle 1) dienten und die RT und PCR wurden in eine Röhre durchgeführt (siehe Materialliste). In der Regel spezifische Primer Sequenz ermöglichen eine höhere Effizienz der RT der zielgruppenspezifischen RNA als zufällige Grundierung zu verwenden, aber das bestimmte Ziel-RNA ist die einzige, die in solch einer cDNA-Probe quantifiziert werden kann, die das einzige Ziel von bestimmten Laboratorien sein können (siehe Tabelle der Materialien für Produktvorschläge cDNA Synthese).
Während konventionelle RT-PCR erscheint häufig verwendet werden bisher in der TiLV Diagnose9,13,14,15,16,17,18, 33 , 35 , 48 , 55. RT-qPCR hat gezeigt, dass ein leistungsfähigeres Tool für die Erkennung und Quantifizierung von geringen Mengen an TiLV in Fisch Geweben oder Schleim32,37sein. Im Allgemeinen ist qPCR in klinische Virologie Diagnostiklabors aufgrund seiner hohen Empfindlichkeit, Spezifität, gute Reproduzierbarkeit, Dynamikbereich und Geschwindigkeit21verbreitet. Während qPCR zunächst teurer als bei herkömmlichen RT-PCR kann, bietet es viele wichtige Vorteile gegenüber konventionellen PCR; Es hat eine schnellere Turnaround-Zeit von Probe zu Ergebnissen und Post-PCR-Schritte ist nicht erforderlich. Dieser letzte Punkt bedeutet, dass es minimales Risiko für Labor-Kontamination gibt und es sich leichter anpassen Hochdurchsatz-Situationen wie z. B. bei Ausbrüchen lässt. Darüber hinaus ist es von Natur aus empfindlicher als herkömmliche RT-PCR, die von entscheidender Bedeutung für niedrige Viruslast in subklinische Infektionen21zu erkennen ist. Dies würde einen verschachtelten PCR-Ansatz erfordert reverse Transkription, zwei weitere PCR-Reaktionen und Analyse von Agarose-gel-Elektrophorese. Diese viele Schritte nehmen viel Zeit und erhöhen die Chancen auf Fehler oder Kontamination. Dennoch aufgrund seiner hohen Empfindlichkeit verlangt RT-qPCR sorgfältige experimentelles Design und ein gründliches Verständnis der Quantifizierung Techniken zur Erzeugung von präzise Ergebnisse56,57.
Die DNA-Bindung Fluorophor, SYBR Green nachgewiesen in diesem Protokoll. Es ist ein DsDNA unspezifischen DNA bindenden Farbstoff und die Spezifität des Assays liegt somit vollständig in das Set der Zündkapseln, die Fehlalarme58erzeugen kann. Daher, während die DsDNA schmelzen Kurvenanalyse am Ende von jedem PCR durchgeführt ein besonders wichtiger Bestandteil der PCR-Reaktion ist, denn es, dass nur eine PCR Amplifikate der richtige T bestätigtm entsteht (sollte auch dies durch Gel Elektrophorese, wenn neue Assays umgesetzt werden). Tm eines DNA-Fragments ist abhängig von einer Vielzahl von Funktionen wie Länge, GC Zusammensetzung, Sequenz, Strang Komplementarität, Konzentration sowie auf Puffer-Komponenten und PCR-Enhancer. Die schmelzenden Kurve Analysen in die repräsentativen Ergebnisse von zwei Labors ergab das Vorhandensein von Primer-Dimere oder andere unerwünschte PCR-Produkte, aber wenn dies mit anderen Proben und/oder Versuchsanordnungen beobachtet wird, dann sollte der Test sein neu optimiert. Erweiterte qPCR Technologien erfordern keine solchen schmelzenden Kurve Schritt und in der Tat, da diese Methoden, wie Papier geschrieben wurde, eine TaqMan basierend TiLV RT-qPCR entwickelt wurde unter Verwendung zwei Zündkapseln und einer Sonde, so dass es sehr spezifische TiLV34.
Zweifellos die Primer entwickelt für RT-qPCR-Assays sind grundlegend für den Erfolg des Tests und die Primer hier wurden entwickelt, basiert auf öffentlich zugänglichen TiLV genomischen Daten zur Zeit32. Allerdings RNA-Viren sind bekannt für hohe Mutationsraten aufweisen und mögliche Belastungen werden die aktuellen diagnostischen Tests zu entkommen, da für ISAV59beobachtet wurde. Werden immer schwierig für solche Virustypen generieren eine universelle Pfanne-TiLV RT-qPCR-Assay und solche Tests nur kontinuierlich verbessert werden wenn mehr TiLV Genomdaten von weitreichenden Orte und Zeiträume zur Verfügung stehen.
Schließlich ist es wichtig, doppelte laufen oder wenn möglich, dreifacher Reaktionen in beide Intra und inter qPCR-Assays. Wenn die C-t -Werte sehr hoch sind, ist die Verwendung von Wiederholungen besonders wichtig festzustellen, dass die PCR-Reaktion zuverlässig und reproduzierbar ist. Im Allgemeinen wenn Daten replizieren Reaktionen variiert mehr als 0,5 Zyklen, die Reaktionen sollte wiederholt werden und wenn die C-t -Werte konsequent > 0,5 Zyklen variieren repliziert, die Probe erneut optimiert werden sollte. Die Verwendung einer integrierten qPCR Pipettierroboter hilft ungemein mit diesem Thema, aber es ist ein Luxus-Werkzeug. Wie bei jedem Experiment sind die Aufnahme geeignete und angemessene Kontrollen von größter Bedeutung für die Entwicklung von robusten molekularer Assays, vor allem in diagnostischen Labors, wo solche Tests müssen akkreditiert sein. Kontrollen sollten positiv (positive TiLV Probe, TiLV Plasmid standard) und Negativkontrollen (NTC und -RT) Proben sowie die Erkennung von endogenen Tilapia-Housekeeping-Gene enthalten. Solche Kontrollen darf nicht unterschätzt werden und sollte in jeder Probe, die Qualität der einzelnen Schritte des Tests richtig zu verstehen und richtig interpretieren die Ergebnisse aufgenommen werden.
Offenlegungen
Die Autoren haben nichts preisgeben.
Danksagungen
Wir sind dankbar, das Institut für Veterinär Bakteriologie, Vetsuisse-Fakultät, Universität Bern für ihre Unterstützung. Diese Arbeit wurde vom Komitee für akademische Förderung des wissenschaftlichen Nachwuchses und der Gleichstellung der Geschlechter an der Vetsuisse-Fakultät, Universität Bern von 120 % Finanzierung Modell finanziert an PN vergeben. WS und PR werden vom Center for Advanced Studies unterstützt für Landwirtschaft und Nahrungsmittel, Institute for Advanced Studies, Kasetsart University, Bangkok, Thailand unter der Hochschulbildung Forschungsförderung und nationale Forschung Universität Projekt von Thailand, Büro der Hochschulbildung Kommission, Bildungsministerium, Thailand. Wir möchten danken Dr. Kwanrawee Sirikanchana für ihre Erzählung und Piyawatchara Sikarin für die Videobearbeitung.
Materialien
| Name | Company | Catalog Number | Comments |
| Tissue collection | Step 1 | ||
| Tricaine methanesulfonate | Sigma-Aldrich | E10521 | An alternative to clove oil. Step 1.1 |
| RNAlater stabilization solution | Thermo Fisher Scientific | AM7020 | For storing tissues if they cannot be processed immediately Step 1.3 |
| RNA extraction | Step 2 | ||
| TRIreagent | Sigma-Aldrich | Step 2.1 | |
| TRIzol | Thermo Fisher Scientific (Invitrogen) | 15596026 | Step 2.1 |
| GENEzol | Geneaid | GZR100 | Step 2.1 |
| Trisure | Bioline | BIO-38032 | Step 2.1 |
| Homemade solution | - | - | 94.53 g/L (800 mM) guanidine thiocyanate 30.45 g/L (400 mM) ammonium thiocyanate 8.20 g/L (100 mM) sodium acetate 380 mL/L (38 % v/v) phenol 50 mL/L (5 % v/v) glycerol 1.0 g/L (0.1 % w/v) 8-quinolinol, pH 5.0 Store up to 2 years at 4oC Step 2.1 |
| MagNA Lyser Green Beads | Roche | 3358941001 | An alternative tissue homogenization method used in conjunction with tissue lysing machines detailed below Step 2.2 |
| Lysing Matrix D, 2 mL Tube | MP BIOMEDICALS | 116913050 | |
| Chloroform | Sigma-Aldrich | C2432 | Step 2.3 |
| Chloroform | RCI Labscan | AR1027E-G2.5L | Step 2.3 |
| 1-Bromo-3-chloropropane | Sigma-Aldrich | B9673 | A less toxic alternative to chloroform Step 2.3 |
| Isopropanol (GC) ≥ 99.8 % | Sigma-Aldrich | 59300 | Step 2.6 |
| Isopropanol (ACS, ISO Reag. Ph Eur) | Merck KGaA (EMSURE) | 1.09634.2500 | Step 2.6 |
| Glycogen, molecular biology grade (e.g., Sigma, cat. no. G1767) | Thermo Fisher Scientific (Thermo Scientific) | R0551 | Useful step if tissue starting material is small to maximise RNA precipitation optional |
| Ethanol (purity (GC) ≥ 99.9 % | Sigma-Aldrich (EMD Millipore) | 1.00983 | Step 2.9 |
| Ethanol (ACS, ISO Reag. Ph Eur) | Merck (EMSURE) | 1.00983.2500 | Step 2.9 |
| Nuclease-free water | Promega | P1193 | Step 2.13 |
| Nuclease-free water | Multicell | 809-115-CL | Step 2.13 |
| Ambion TURBO DNA-free kit | Thermo Fisher Scientific (Invitrogen) | AM1907 | Can be performed at the end of the RNA extraction protocol optional |
| cDNA synthesis | Step 4 | ||
| Viva cDNA Synthesis Kit | Vivantis | cDSK01 | Step 4.1 & 4.3 |
| ReverTra Ace qPCR RT MasterMix with gDNA remover | Toyobo | A1172K | An alternative option see discussion |
| ReverTra Ace qPCR RT Kit | Toyobo | FSQ-101 | An alternative option see discussion |
| AffinityScript Multiple Temperature Reverse Transcriptase | Agilent Technologies | 600107 | An alternative option |
| PCR | Step 5 | ||
| DNA polymerase systems: | Step 5.2 | ||
| - Platinum II Hot-Start Green PCR Master Mix (2X) | Thermo Fisher Scientific (Invitrogen) | 14001012a | Step 5.2 |
| - GoTaq Mastermix | Promega | M7122 | Step 5.2 |
| Separate PCR mixture components: | Step 5.2 | ||
| 10mM dNTP Mix | Vivantis | NP2409 | Step 5.2 |
| 25mM MgCl2 | Thermo Fisher Scientific | R0971 | Step 5.2 |
| 10X Taq Buffer with KCl | Thermo Fisher Scientific | 00348114 | Step 5.2 |
| Taq DNA polymerase | Vivantis | PL1202 | Step 5.2 |
| - Verso 1-step RT-PCR ReddyMix with ThermoPrime Taq | Thermo Fisher Scientific | AB1454 | One step RT-PCR exemplified in Figure 3B |
| Gel electrophoresis: | For visulation of PCR products from steps 5.1-5.4 | ||
| Ethidium Bromide solution (10 mg/mL) | Thermo Fisher Scientific | 17898 | Step 5.5 |
| Tris/Acetic/EDTA (TAE) buffer: | Step 5.5 | ||
| - Tris | Vivantis | PR0612-1KG | Step 5.5 |
| - Acetic acid (glacial) (ACS, ISO, Reag. Ph Eur) | Merck KGaA (EMSURE) | 1.00063.2500 | Step 5.5 |
| - Ethylenediaminetetraacetic acid (EDTA) | BIO-RAD | 161-0729 | Step 5.5 |
| Agarose | Vivantis | PC0701-100G | Step 5.5 |
| DNA ladders and markers | Vivantis | NL1405 | Step 5.5 |
| DNA gel loading dye (6X) | Thermo Fisher Scientific | R0611 | Step 5.5 |
| qPCR | Step 6 | ||
| PowerUP SYBR Green Master Mix | Thermo Fisher Scientific (Applied Biosystems) | A25779 | Exemplified in Figures 4-6B Step 6.2 |
| iTaq Universal SYBR Green Supermix | BIO-RAD | 1725120 | Exemplified in the video and in Figures 4-6A Step 6.2 |
| Equipment | |||
| Dounce tissue grinder pestle | Sigma-Aldrich | P1110 | Protocol 2 |
| MagNA Lyser Instrument | Roche | 3358976001 | An alternative tissue homogenizing option for protocol 2 which are used in conjunction with the lysing beads detailed above Step 2.2 |
| FastPrep-24 5G Homogenizer | MP BIOMEDICALS | 116005500 | |
| Refrigerated microcentrifuge | Eppendorf | Eppendorf 5427R | Protocol 2 Step 2.4, 2.7 & 2.10 |
| Refrigerated microcentrifuge | Eppendorf | Eppendorf 5418R | |
| Heat box | Labnet | AccuBlock Digital Dry Bath | Protocol 2 Step 2.13 |
| Microvolume spectrophotometer | Thermo Fisher Scientific (Applied Biosystems) | Nanodrop 2000 | Protocol 3 Step 3.1 - 3.4 |
| PCR machine | BIO-RAD | T100 Thermal Cycler | Protocol 5 Step 5.4 |
| Power supply | BIO-RAD | PowerPac HC | Protocol 5 Step 5.5 |
| Horizontal gel electrophoresis | BIO-RAD | Mini ReadySub-Cell GT Cell #1704487edu | Protocol 5 Step 5.5 |
| Mini microcentrifuge | Corning | LSE 6766 | Useful to quickly spin down PCR reaction tubes in protocols 4, 5 & 6 Step 6.5.1 |
| Microcentrifuge | LioFuge | LM-60 | Step 6.5.1 |
| qPCR machine and software | Thermo Fisher Scientific | 7500 Fast Real-Time PCR System with 7500 Software v2.0 | Protocol 6 Step 6.6-6.8 |
| qPCR machine and software | BIO-RAD | CFX96 Touch Real-Time PCR Detection System with CFX Manager software | |
| General Materials | |||
| Mayo scissors | Step 1.1-1.2 | ||
| Forceps | Step 1.1-1.2 | ||
| Pipette | Rainin | Pipette-Lite XLS | |
| Aerosol-barrier pipette tips | Sigma-Aldrich | Z333328, Z333336, Z333344 | |
| Nuclease-free 1.5-ml microcentrifuge tubes | Eppendorf |
Referenzen
- FAO. . The State of World Fisheries and Aquaculture, 2014. Opportunities and Challenges. , (2014).
- FAO. . The State of World Fisheries and Aquaculture, 2016. Contributing to Food Security and Nutrition for all. , (2016).
- WorldBank. . FISH TO 2030: Prospects for Fisheries and Aquaculture. Agriculture and Environmental Services Discussion Paper 03. , (2013).
- Wing-Keong, N., Nicholas, R. A review of the nutrition and feeding management of farmed tilapia throughout the culture cycle. Reviews in Aquaculture. 5 (4), 220-254 (2013).
- Cleasby, N., et al. The socio-economic context for improving food security through land based aquaculture in Solomon Islands: A peri-urban case study. Marine Policy. 45, 89-97 (2014).
- Ponzoni Raul, W., et al. Genetic improvement of Nile tilapia (Oreochromis niloticus) with special reference to the work conducted by the WorldFish Center with the GIFT strain. Reviews in Aquaculture. 3 (1), 27-41 (2011).
- Hounmanou, Y. M. G., et al. Tilapia lake virus threatens tilapiines farming and food security: Socio-economic challenges and preventive measures in Sub-Saharan Africa. Aquaculture (Amsterdam, Netherlands). 493, 123-129 (2018).
- OIE. . Tilapia Lake Virus (TiLV) - a novel orthomyxo-like virus. OIE technical disease cards. , (2018).
- Mugimba, K. K., et al. Detection of tilapia lake virus (TiLV) infection by PCR in farmed and wild Nile tilapia (Oreochromis niloticus) from Lake Victoria. Journal of Fish Diseases. , (2018).
- Koesharyani, I., Gardenia, L., Widowati, Z., Khumaira, D. D., Rustianti, Studi kasus infeksi tilapia lake virus (tilv) pada ikan nila (Oreochromis niloticus). Jurnal Riset Akuakultur. 13 (1), 85-92 (2018).
- OIE. . Tilapia lake virus disease (TiLV), Chinese Taipei. Immediate Notification. , (2017).
- OIE. . Tilapia Lake Virus Disease (TiLV), Peru. Immediate Notification. , (2018).
- Bacharach, E., et al. Characterization of a Novel Orthomyxo-like Virus Causing Mass Die-Offs of Tilapia. MBio. 7 (2), e00431-e00416 (2016).
- Eyngor, M., et al. Identification of a novel RNA virus lethal to tilapia. Journal of Clinical Microbiology. 52 (12), 4137-4146 (2014).
- Nicholson, P., et al. Detection of Tilapia Lake Virus in Egyptian fish farms experiencing high mortalities in 2015. Journal of Fish Diseases. 40 (12), 1925-1928 (2017).
- Surachetpong, W., et al. Outbreaks of Tilapia Lake Virus Infection, Thailand, 2015-2016. Emerging Infectious Diseases. 23 (6), 1031-1033 (2017).
- Tattiyapong, P., Dachavichitlead, W., Surachetpong, W. Experimental infection of Tilapia Lake Virus (TiLV) in Nile tilapia (Oreochromis niloticus) and red tilapia (Oreochromis spp.). Veterinary Microbiology. 207, 170-177 (2017).
- Kembou Tsofack, J. E., et al. Detection of Tilapia Lake Virus in Clinical Samples by Culturing and Nested Reverse Transcription-PCR. Journal of Clinical Microbiology. 55 (3), 759-767 (2017).
- Thangaraj, R. S., et al. Derivation of two tilapia (Oreochromis niloticus) cell lines for efficient propagation of Tilapia Lake Virus (TiLV). Aquaculture (Amsterdam, Netherlands). 492, 206-214 (2018).
- Hanson, L. A., Rudis, M. R., Vasquez-Lee, M., Montgomery, R. D. A broadly applicable method to characterize large DNA viruses and adenoviruses based on the DNA polymerase gene. Virology Journal. 3, 28-28 (2006).
- Josko, D. Molecular virology in the clinical laboratory. Clinical Laboratory Science. 23 (4), 231-236 (2010).
- Munir, K., Kibenge, F. S. Detection of infectious salmon anaemia virus by real-time RT-PCR. Journal of Virological Methods. 117 (1), 37-47 (2004).
- Snow, M., et al. Developement, application and validation of a Taqman real-time RT-PCR assay for the detection of infectious salmon anaemia virus (ISAV) in Atlantic salmon (Salmo salar). Developments in Biologicals. 126, 133-145 (2006).
- Matejusova, I., McKay, P., McBeath, A. J., Collet, B., Snow, M. Development of a sensitive and controlled real-time RT-PCR assay for viral haemorrhagic septicaemia virus (VHSV) in marine salmonid aquaculture. Diseases of Aquatic Organisms. 80 (2), 137-144 (2008).
- Garver, K. A., et al. Development and validation of a reverse transcription quantitative PCR for universal detection of viral hemorrhagic septicemia virus. Diseases of Aquatic Organisms. 95 (2), 97-112 (2011).
- Dalla Valle, L., et al. Development of a sensitive and quantitative diagnostic assay for fish nervous necrosis virus based on two-target real-time PCR. Veterinary Microbiology. 110 (3-4), 167-179 (2005).
- Hodneland, K., Garcia, R., Balbuena, J. A., Zarza, C., Fouz, B. Real-time RT-PCR detection of betanodavirus in naturally and experimentally infected fish from Spain. Journal of Fish Diseases. 34 (3), 189-202 (2011).
- Hodneland, K., Endresen, C. Sensitive and specific detection of Salmonid alphavirus using real-time PCR (TaqMan). Journal of Virological Methods. 131 (2), 184-192 (2006).
- Wang, X. W., Ao, J. Q., Li, Q. G., Chen, X. H. Quantitative detection of a marine fish iridovirus isolated from large yellow croaker, Pseudosciaena crocea, using a molecular beacon. Journal of Virological Methods. 133 (1), 76-81 (2006).
- van Beurden, S. J., et al. Development and validation of a real-time PCR assay for the detection of anguillid herpesvirus 1. Journal of Fish Diseases. 39 (1), 95-104 (2016).
- Ciulli, S., et al. Development and application of a real-time PCR assay for the detection and quantitation of lymphocystis disease virus. Journal of Virological Methods. 213, 164-173 (2015).
- Tattiyapong, P., Sirikanchana, K., Surachetpong, W. Development and validation of a reverse transcription quantitative polymerase chain reaction for tilapia lake virus detection in clinical samples and experimentally challenged fish. Journal of Fish Diseases. 41 (2), 255-261 (2018).
- Dong, H. T., et al. Emergence of tilapia lake virus in Thailand and an alternative semi-nested RT-PCR for detection. Aquaculture (Amsterdam, Netherlands). 476, 111-118 (2017).
- Waiyamitra, P., et al. A TaqMan RT-qPCR assay for tilapia lake virus (TiLV) detection in tilapia. Aquaculture (Amsterdam, Netherlands). 497, 184-188 (2018).
- Behera, B. K., et al. Emergence of Tilapia Lake Virus associated with mortalities of farmed Nile Tilapia Oreochromis niloticus (Linnaeus 1758) in India. Aquaculture (Amsterdam, Netherlands). 484, 168-174 (2018).
- Ferguson, H. W., et al. Syncytial hepatitis of farmed tilapia, Oreochromis niloticus (L.): a case report. Journal of Fish Diseases. 37 (6), 583-589 (2014).
- Liamnimitr, P., Thammatorn, W., U-thoomporn, S., Tattiyapong, P., Surachetpong, W. Non-lethal sampling for Tilapia Lake Virus detection by RT-qPCR and cell culture. Aquaculture (Amsterdam, Netherlands). 486, 75-80 (2018).
- Yang, C. G., et al. Evaluation of reference genes for quantitative real-time RT-PCR analysis of gene expression in Nile tilapia (Oreochromis niloticus). Gene. 527 (1), 183-192 (2013).
- Bustin, S. A. Real-time, fluorescence-based quantitative PCR: a snapshot of current procedures and preferences. Expert Review of Molecular Diagnostics. 5 (4), 493-498 (2005).
- Fleige, S., Pfaffl, M. W. RNA integrity and the effect on the real-time qRT-PCR performance. Molecular Aspects of Medicine. 27 (2-3), 126-139 (2006).
- Kubista, M., et al. The real-time polymerase chain reaction. Molecular Aspects of Medicine. 27 (2-3), 95-125 (2006).
- Mackay, I. M., Arden, K. E., Nitsche, A. Real-time PCR in virology. Nucleic Acids Research. 30 (6), 1292-1305 (2002).
- Wong, M. L., Medrano, J. F. Real-time PCR for mRNA quantitation. Biotechniques. 39 (1), 75-85 (2005).
- Bustin, S. A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. Journal of Molecular Endocrinology. 25 (2), 169-193 (2000).
- Heid, C. A., Stevens, J., Livak, K. J., Williams, P. M. Real time quantitative PCR. Genome Research. 6 (10), 986-994 (1996).
- Rutledge, R. G., Côté, C. Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Research. 31 (16), e93-e93 (2003).
- Svec, D., Tichopad, A., Novosadova, V., Pfaffl, M. W., Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomolecular Detection and Quantification. 3, 9-16 (2015).
- Amal, M. N. A., et al. A case of natural co-infection of Tilapia Lake Virus and Aeromonas veronii in a Malaysian red hybrid tilapia (Oreochromis niloticus × O. mossambicus) farm experiencing high mortality. Aquaculture (Amsterdam, Netherlands). 485, 12-16 (2018).
- Fathi, M., et al. Identification of Tilapia Lake Virus in Egypt in Nile tilapia affected by ‘summer mortality’ syndrome. Aquaculture (Amsterdam, Netherlands). 473, 430-432 (2017).
- OIE. . Tilapia Lake Virus disease (TiLV), Philippines. Immediate Notification. , (2017).
- OIE. . Tilapia lake virus disease (TiLV), Malaysia. Immediate Notification. , (2017).
- Abdullah, A., et al. First detection of tilapia lake virus (TiLV) in wild river carp (Barbonymus schwanenfeldii) at Timah Tasoh Lake, Malaysia. Journal of Fish Diseases. 41 (9), 1459-1462 (2018).
- Chomczynski, P., Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 162 (1), 156-159 (1987).
- Chomczynski, P., Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nature Protocols. 1 (2), 581-585 (2006).
- Del-Pozo, J., et al. Syncytial Hepatitis of Tilapia ( Oreochromis niloticus L.) is Associated With Orthomyxovirus-Like Virions in Hepatocytes. Veterinary Pathology. 54 (1), 164-170 (2017).
- Bustin, S. A., et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry. 55 (4), 611-622 (2009).
- Purcell, M. K., Getchell, R. G., McClure, C. A., Garver, K. A. Quantitative polymerase chain reaction (PCR) for detection of aquatic animal pathogens in a diagnostic laboratory setting. Journal of Aquatic Animal Health. 23 (3), 148-161 (2011).
- Simpson, D. A., Feeney, S., Boyle, C., Stitt, A. W. Retinal VEGF mRNA measured by SYBR green I fluorescence: A versatile approach to quantitative PCR. Molecular Vision. 6, 178-183 (2000).
- Kibenge, M. J., et al. Discovery of variant infectious salmon anaemia virus (ISAV) of European genotype in British Columbia, Canada. Virology Journal. 13, 3 (2016).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten