
Method Article
Detection of Tilapia Lake Virus Using Conventional RT-PCR and SYBR Green RT-qPCR
In This Article
Summary
This protocol diagnoses Tilapia Lake Virus (TiLV) in tilapia tissues using RT-PCR methodologies. The entire method is described from tissue dissection to total RNA extraction, followed by cDNA synthesis and detection of TiLV by either conventional PCR or quantitative PCR using dsDNA binding a fluorescent binding dye.
Abstract
The aim of this method is to facilitate the fast, sensitive and specific detection of Tilapia Lake Virus (TiLV) in tilapia tissues. This protocol can be used as part of surveillance programs, biosecurity measures and in TiLV basic research laboratories. The gold standard of virus diagnostics typically involves virus isolation followed by complementary techniques such as reverse-transcription polymerase chain reaction (RT-PCR) for further verification. This can be cumbersome, time-consuming and typically requires tissue samples heavily infected with virus. The use of RT-quantitative (q)PCR in the detection of viruses is advantageous because of its quantitative nature, high sensitivity, specificity, scalability and its rapid time to result. Here, the entire method of PCR based approaches for TiLV detection is described, from tilapia organ sectioning, total ribonucleic acid (RNA) extraction using a guanidium thiocyanate-phenol-chloroform solution, RNA quantification, followed by a two-step PCR protocol entailing, complementary deoxyribonucleic acid (cDNA) synthesis and detection of TiLV by either conventional PCR or quantitative identification via qPCR using SYBR green I dye. Conventional PCR requires post-PCR steps and will simply inform about the presence of the virus. The latter approach will allow for absolute quantification of TiLV down to as little as 2 copies and thus is exceptionally useful for TiLV diagnosis in sub-clinical cases. A detailed description of the two PCR approaches, representative results from two laboratories and a thorough discussion of the critical parameters of both have been included to ensure that researchers and diagnosticians find their most suitable and applicable method of TiLV detection.
Introduction
The global capita fish supply reached a new record of 20 kg in 2014 and this was due to vigorous growth in aquaculture. Aquaculture remains one of the fastest growing animal food producing sectors worldwide and is the only animal food producing sector that is growing faster than the human population1. Tilapiine cichilds comprise the second most important fresh water fish farmed worldwide with a total global production of 6.4 million tons (MT) and an estimated value of 9.8 billion US dollars in 20152. The top ten producers of tilapia are China (1.78 MT), Indonesia (1.12 MT) and Egypt (0.88 MT), followed by Bangladesh, Vietnam, the Philippines, Brazil, Thailand, Colombia and Uganda2. It is expected that global tilapia production will be around 7.3 MT by 20303. Tilapia have become such an important global food source not only because they are an inexpensive source of protein4 but also because they are easy to breed in capacity under a wide range of water and climate conditions5,6. Just a few decades ago, it was believed that there were few commercially significant diseases threatening tilapia farming but this is no longer true. An emerging viral disease called tilapia lake virus disease (TiLVD) is the first ever critical disease epidemic found in tilapia and the entire industry is at risk. This disease has serious socio-economic consequences and is a direct threat on food security for millions of people in Africa7, Asia and South America. At the start of 2018, the World Organization for Animal Health (OIE) reported that the etiological agent of this disease, TiLV, had been officially detected on three continents covering eight countries8 and since this pathogen information card was updated there have been more reports of TiLV in Tanzania, Uganda9, Indonesia10, Taiwan11 and Peru12. TiLV is a novel single-stranded RNA virus described to be an orthomyxo-like virus because it contains a variety of characteristics reminiscent of other orthomyoxoviruses such as Influenza or Infectious salmon anaemia virus (ISAV)13. It was first identified in the aftermath of massive losses of wild and farmed tilapia in the Lake of Galilee, Israel14. Thereafter, similar disease outbreaks referred to as summer mortalities and the one month mortality syndrome associated with TiLV infection were reported in Nile tilapia (Oreochromis niloticus) in Egypt15 and Nile and red hybrid tilapia (Oreochromis spp.) in Thailand16, respectively. Detection of aquatic animal viruses is historically carried out by growth and isolation of viruses in cell culture. Various cell lines have been tested for the propagation and isolation of TiLV including, E-11 cells derived from snakehead fish (Ophiocephalus striatus)17,18, OmB and TmB originating from Oreochromis mossambicus18, and OnlB and OnlL originating from Nile tilapia (O. niloticus)19. While virus culture has the advantage that it provides material for further experiments, it has the disadvantage that it requires at least 4-7 days to observe the formation of cytopathic effects (CPE) and crucially, different piscine viruses, that are more fit to replicate may be propagated and produce similar CPE.
In the last few decades, there has been a move away from traditional, often time-consuming diagnostic methods such as cell culture, serology and antigen detection and replacement with faster and more sensitive nucleic acid based detection tests20,21. This is evident by the fact that many qPCR assays have been developed as important diagnostic methods for a multitude of viral diseases in aquatic animals, such as for ISAV22,23, viral haemorrhagic septicaemia virus (VHSV)24,25, betanodavirus26,27 salmonid alphavirus28, fish iridovirus29, Anguillid herpesvirus 1 (AngHV1)30, and Lymphocystis disease virus (LCDV)31. Robust methods for diagnosis and pathogen surveillance are urgently required to reduce the spread of TiLV. Such methods should allow early detection of infection before clinical signs develop and detection of low virus loads. To date, different PCR protocols including RT-PCR14,32, nested RT-PCR18, semi-nested RT-PCR33, and RT-qPCR32,34 have been developed for the detection of TiLV in fish tissues. A comparison of RT-qPCR and virus isolation in susceptible cell lines for TiLV detection revealed that RT-qPCR was 1,000 times more sensitive than the virus isolation32. Although each published PCR protocol has reported different sensitivities for the detection of TiLV, most assays are highly sensitive with the detection limits of viral copies at 7.5 copies33, 7 copies18 or 2 copies32 per reaction.
The aim of this methods article is to explain, in detail, how to perform TiLV detection assays, starting with tilapia tissue collection, to total RNA extraction, cDNA synthesis and then TiLV specific PCR based assays. Specifically, comprehensive protocols of both conventional RT-PCR and also SYBR green-based RT-qPCR have been described to appeal to a wide range of scientists aiming to detect TiLV. The former is less sensitive but is typically a cheaper detection option. The latter requires more elaborate infrastructure such as a quantitative PCR machine and more expensive reagents, but it has the advantages of being quantitative, fast and highly sensitive, meaning that it can be used for the detection of TiLV in sub-clinically infected fish. The RT-PCR and RT-qPCR protocols were performed in two different laboratories with distinct geographical isolates of TiLV and the included results highlight the sensitivity and reproducibility of the assays described here.
Protocol
The animal use protocol for this study was approved by the Kasetsart University Animal Ethics Committee under permit number ACKU 59-VET-016.
NOTE: Please refer to the Table of Materials for extended information concerning the reagents and equipment suggested for this protocol.
1. Tissue Sample Collection
- Euthanize the fish using an overdose of clove oil (the volume depends on the size of fish and concentration of products, usually more than 3 mL/L). Immerse one-fourth of the forceps and mayo scissors into 95% (v/v) ethanol followed by burning the equipment using an alcohol burner to sterilize the equipment.
NOTE: Tricaine methanesulfonate (MS-222) can be used instead of clove oil. - Find the liver and cut off a small piece (approximately 20-100 mg) or collect 200 µL of mucus using cover glass or surgical blade to remove mucus from anterior to posterior of the tilapia fish and place the samples into a 1.5 mL microcentrifuge tube.
- Process samples immediately, store in a RNA stabilizing solution or move them to -80 °C until further use.
NOTE: The biggest task in working with RNA is preparing intact RNA molecules and keeping them undamaged throughout any subsequent handlings. The RNA backbone is innately more sensitive to damage than DNA. Extraction and isolation of total RNA from tissue cells necessitates careful laboratory technique; take all provisions to prevent RNase contamination by wearing gloves, using RNase free water, reagents, equipment, plastic ware, glassware, working space and by using filter tips for pipetting.
2. Guanidium Thiocyanate - Phenol - Chloroform Extraction of RNA
- Add 1 mL of monophasic solution containing phenol and guanidine isothiocyanate into a tube containing tissue sample from section 1.
CAUTION: This solution is very toxic and must be handled with care in a laminar flow hood with protective equipment and by wearing the proper protective eyewear, clothing and safety gloves. - Grind the tissue sample using a tissue pestle homogenizer until homogenous.
NOTE: Samples can also be homogenized using a power homogenizer combined with ceramic beads. Ensure that the tissue sample is completely homogenized before proceeding to the next step or stop the protocol here and store the fully homogenized samples at -80 °C until further use. - Add 200 µL of chloroform for phase separation.
CAUTION Chloroform is a potential narcotic and is extremely hazardous. It should be handled with care in a laminar flow hood with protective equipment as well as by wearing the proper protective eyewear, clothing and safety gloves. As a less toxic alternative, 1-Bromo-3-chloropropane can also be used.
NOTE: Scale the volumes up or down where appropriate. For example, if only 500 µL of monophasic solution containing phenol and guanidine isothiocyanate was used, then only add 100 µL of chloroform at this step.- Mix samples well by inversion for 15 s.
- Incubate samples for 3 min at room temperature (RT).
- Centrifuge for 15 min at 12,000 × g and 4 °C.
NOTE: There should be a clear separation into a lower organic phase, a white interphase and an upper aqueous phase containing RNA. This top phase is normally colorless but depending on the type and amount of homogenized tissue, it can have a light pink appearance. - Transfer the upper aqueous layer (approximately 500 µL) to a fresh microcentrifuge tube without disturbing the interphase.
NOTE: do not try to transfer the entire aqueous phase, leave a small amount to prevent any potential contamination of the RNA containing aqueous phase with the organic or interphase. - Add 1 volume of 100% isopropanol to precipitate the RNA.
- Optionally, if very small amounts of tissue were used, then add 1 µL (5-10 µg) of RNase-free glycogen to each sample to promote efficient RNA precipitation. This will aid the identification of the RNA pellet in step 2.8.
NOTE: The glycogen acts as a carrier of the RNA and will prevent small amounts of RNA from sticking to the side of the tube. - Mix tubes well by inversion several times.
- Store the samples for 2 h to overnight at -20 °C.
- Optionally, if very small amounts of tissue were used, then add 1 µL (5-10 µg) of RNase-free glycogen to each sample to promote efficient RNA precipitation. This will aid the identification of the RNA pellet in step 2.8.
- Centrifuge samples for 15 min at 12,000 x g and 4 °C.
- Discard the supernatant, being careful not to dislodge the RNA pellet at the bottom of microcentrifuge tube.
- Add 1 mL of 75% ethanol (v/v) and mix RNA samples by inverting the tube several times.
- Centrifuge for 15 min at 10,000 x g and 4 °C.
NOTE: The protocol can be stopped here and the samples comprising the RNA pellet in 75% ethanol can be stored at -20 °C until further use. - Discard the supernatant, being watchful not to dislodge the RNA pellet at the bottom of microcentrifuge tube.
- Optionally, repeat steps 2.9 - 2.11 using 70% ethanol (v/v). Thoroughly washing the RNA pellet will minimize any salt or contaminant carry-over that may interfere will sensitive downstream applications.
- Draw out the remaining ethanol using a pipette and then air-dry the RNA pellet at room temperature for no longer than 5 to 10 min.
NOTE: over-dried pellets will be difficult to re-suspend. - Add 30-60 µL of RNase-free water, pre-warmed to 55-60 °C to solubilize the RNA pellet.
- Place the RNA on ice for immediate use or store at -80 °C for later use.
3. Quantify RNA Concentration using a Micro-Volume Spectrophotometer
- Switch the settings of the spectrophotometer to RNA.
- Use 1-2 µL of RNase-free water as a blank.
- Use 1-2 µL of each RNA sample to assess the RNA quantity.
- Record the readings at 230 nm, 260 nm and 280 nm for each sample.
- Dilute the RNA to 200 ng/µL using RNase-free water.
4. Synthesis of complementary DNA (cDNA) using total RNA
- Mix 1 µg of total RNA from protocol 2, 2 µM oligo (dT), 0.5 mM dNTPs mixture and bring the final volume to 10 µL with nuclease-free water. For this, prepare a RT-master-mix according to the number of samples and controls to be tested.
NOTE: The controls are a minus-reverse transcriptase sample (-RT) wherein the RT enzyme is replaced with nuclease-free water (see step 4.3) and a no template control (NTC) wherein nuclease-free water is added to the master-mix instead of RNA template.- Mix the samples well by pipetting followed by a brief centrifugation.
- Incubate the samples at 65 °C for 5 min followed by a 2 min incubation on ice.
- Briefly centrifuge the samples to collect all of the liquid in the bottom of the tubes.
- Add 1x reverse transcriptase buffer, 100 U reverse transcriptase and bring the final volume of each sample to 20 µL using nuclease-free water.
- Mix the samples well by pipetting followed by a brief centrifugation.
- Incubate the samples at 42 °C for 60 min followed by 85 °C for 5 min.
- Dilute the synthesized cDNA to a desired concentration by adding an appropriate volume of nuclease-free water and place the cDNA on ice for immediate use or store it at -20 °C for later use.
5. TiLV Conventional PCR
- Use the cDNA, samples and controls, generated in protocol section 4 as templates for a PCR reaction using any of the established primer pairs detailed in Table 1, along with a DNA polymerase of choice.
NOTE: An additional no-template control (NTC) should be included here by substituting cDNA for nuclease-free water in the PCR reaction. A positive control, if available, should also be included comprising previously verified TiLV positive samples or the appropriate TiLV cDNA fragment cloned in to a plasmid. - Prepare a PCR master-mix according to the guidelines of the DNA polymerase system in use and the number of samples and controls to be tested. This mix should include the forward primer, reverse primer, dNTPs, MgCl2 and the selected DNA polymerase, along with its buffer.
- According to the guidelines of the selected DNA polymerase, combine the designated volume of master-mix with suggested amount of cDNA samples and control samples.
NOTE: Preparing a 0.5x reaction excess is often beneficial since some of the master-mix is lost during pipetting. - Perform PCR cycling conditions according to the guidelines of the utilized DNA polymerase system and using an appropriate annealing temperature for the primers in use (Table 1). Usually, such a program will involve an initial denaturation at 95 °C for 2-5 min, followed by 30-40 cycles of denaturation at 95 °C for 30 s, annealing at the recommended temperature for 30 s and elongation at 72 °C for 30 s, followed by a final elongation at 72 °C for 5-10 min.
- Load 5-15 µL of each PCR reaction and an appropriate DNA ladder in to wells of a 1-2% agarose gel, depending on the expected PCR product size. Separate the amplified DNA by gel electrophoresis and stain the gel by ethidium bromide (EthBr) to facilitate visualisation of DNA bands of the expected size (Table 1) in a gel documentation machine using UV light.
CAUTION: EtBr is toxic; it should be handled with care by wearing the proper protective clothing and safety gloves.
| Target TiLV genome segment | Forward primer 5' - 3' | Reverse primer 5' - 3' | PCR product size (bp) | Tm °C | Original reference | ||||
| 1 | CCAAACGTTATCTCTTAATTACGCAC | GCAAATATTTCTCTCATTCGCCT | 1641 | 50 | Surachetpong et al., 2017 | ||||
| 1 | CCTCATTCCTCGTTGTGTAAGT | AGGAGTTGCTGTTGGGTTATAG | 1000 | 62 | Mugimba et al., 2018 | ||||
| 2 | ACTCTCTATTACCAAATACATTTACT | TTACCATATATATAGTGAAGGC | 1445 | 45 | Surachetpong et al., 2017 | ||||
| 2 | GTCCAGGGCGGTATGTATTG | CTTACGGCTGACAAGTCTCTAAG | 834 | 62 | Mugimba et al., 2018 | ||||
| 3 | GTTGGGCACAAGGCATCCTA | TATCACGTGCGTACTCGTTCAGT | 250 | 56 | Eyngor et al., 2014 | ||||
| 3 | TATGCAGTACTTTCCCTGCC | TTGCTCTGAGCAAGAGTACC | 491 | 57 | Eyngor et al., 2014 | ||||
| 3 | ACCCCTTAATCCTTAATAGACCGTTA | CCCATAATCCTCTATTAGAACGTCGT | 1352 | 50 | Surachetpong et al., 2017 | ||||
| 3 | GTCGAGGCATTCCAGAAGTAAG | GAGCTAAGGGAACGGCTATTG | 834 | 62 | Mugimba et al., 2018 | ||||
| 4 | AGCAGCAGCAGGAGAAAGAG | ACCGTCCTGTTTCTGAATGG | 358 | 60 | Nicholson et al., 2017 | ||||
| 4 | CCAAAGTTTACTCCTATTACCCAGA | GCAAATCTTTCTCCAATTACCGTCT | 1250 | 50 | Surachetpong et al., 2017 | ||||
| 4 | GCCCAATGGTTCCCATATCT | GCCCAATGGTTCCCATATCT | 524 | 62 | Mugimba et al., 2018 | ||||
| 5 | CCAAATGTTTCTCTTATCTCAGACTC | CTTTTTCTCAGTTTACCACTTTATG | 1087 | 57 | Surachetpong et al., 2017 | ||||
| 5 | CAACTCTTAGCCTCCGGAATAC | CGTTCTGCACTGGGTTACA | 696 | 62 | Mugimba et al., 2018 | ||||
| 6 | CCAAATTTTACCTCTCGCAT | TCAAGCACTTAAAACTGTACC | 1027 | 45 | Surachetpong et al., 2017 | ||||
| 6 | CCCACACGACAGGACATATAG | GAGTTGGCTTAGGGTGATAAGA | 948 | 62 | Mugimba et al., 2018 | ||||
| 7 | CTCTCTTTGCATTGCATACCGT | GACCAATTATCCCTGCTTTCA | 704 | 57 | Surachetpong et al., 2017 | ||||
| 7 | TCCTTTAGGGATTGGCACTAAC | TTCCATCGACTGCTCCTAGA | 486 | 62 | Mugimba et al., 2018 | ||||
| 8 | ACCTCATCTACACTAACATTTCCA | TCATCATTACACAAATGGAGTAGCT | 637 | 50 | Surachetpong et al., 2017 | ||||
| 8 | CTTAAGGGCCATCCTGTCATC | TGGCTCAAATCCCAACACTAA | 476 | 62 | Mugimba et al., 2018 | ||||
| 9 | TTGGTGATGTCACGATGGATA | AGTTCTATCGCCAGCCATGT | 351 | 60 | Nicholson et al., 2017 | ||||
| 9 | ACAAGTCCGATTACTTTTTCCGC | TCTTTCTCACGTCCTTAAAGTCA | 530 | 50 | Surachetpong et al., 2017 | ||||
| 9 | GATATCCTCCACATGACCCTTC | GTACGTCACTTTGTGCCATTAC | 261 | 62 | Mugimba et al., 2018 | ||||
| 10 | AACCCTACTAACACCAAATATAGCT | CTTTCCCTCTGACACCCTGT | 450 | 50 | Surachetpong et al., 2017 | ||||
| 10 | TCCTCTCTGTCCCTTCTGTT | CAGGATGAGTGTGGCAGATTAT | 276 | 62 | Mugimba et al., 2018 | ||||
Table 1. Published primer pairs for the amplification of TiLV cDNA using endpoint PCR. The primer set shown in bold were used to generate the representative results shown in Figure 3A and 3B.
6. TiLV Quantitative Polymerase Chain Reaction (qPCR)
- Using a plasmid containing the appropriate TiLV genomic segment 3 cDNA as a standard, such as pTiLV32, prepare a duplicated or triplicated 10-fold serial dilution series.
- Prepare a qPCR master-mix for all samples, standards and controls, taking into account that the reactions should be performed in duplicate or triplicate utilizing 0.4 µL of nuclease-free water, 0.3 µL of forward primer, 0.3 µL of reverse primer and 5 µL of 2x SYBR Green DNA polymerase master-mix per reaction.
- Use the primers at a concentration of 10 µM, and the primer information and the standard pTiLV as follows:
Forward primer: TiLV-112F (5'-CTGAGCTAAAGAGGCAATATGGATT-3')
Reverse primer:TiLV-112R (5'-CGTGCGTACTCGTTCAGTATAAGTTCT-3')
Standard pTiLV:10 pg/µL
NOTE: If the total number of samples and controls is 10 and will be performed in triplicates, this equates to a qPCR master-mix comprising 12 µL nuclease-free water, 9 µL forward primer, 9 µL reverse primer and 150 µL of SYBR Green DNA polymerase master-mix. Commercially purchased 2x universal SYBR Green DNA polymerase master-mixes contain all the necessary components for the qPCR reaction, namely, the SYBR Green I dye, hot-start Taq DNA polymerase, dNTPs, MgCl2 and passive reference dyes. Protect the SYBR Green master mix from light.
- Use the primers at a concentration of 10 µM, and the primer information and the standard pTiLV as follows:
- Dispense 6 µL of the qPCR master-mix into qPCR strip tubes or a 96 well plate compatible with the qPCR machine in use.
- Add 4 µL of cDNA template, controls or serially diluted TiLV standards into the tubes or wells of the 96 well plate.
- Close the qPCR tubes or seal the 96 well plate with a compatible plate cover for the qPCR machine in use
- Gently flick the qPCR tubes to mix the solution and spin down the qPCR tubes or 96 well plate using a centrifuge to collect all of the liquid in the bottom of the vessels.
- Place the tubes or plate into the real-time thermal cycler.
- Program the qPCR thermocycler to perform an initial denaturation at 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s for primer annealing and elongation, ending with a melting curve step of 65 °C to 95 °C with an increment of 0.5 °C/ 5 s.
- Select SYBR as a fluorophore dye, then select unknown as a sample type and insert a name into a sample name box.
- Open the lid of the RT-qPCR machine and place the qPCR strip into the assigned wells, then close the lid.
- Perform the RT-qPCR assay with the selected conditions. The machine will start to run after the lid has reached the desired temperature. Collect the fluorescence of each sample after each extension step to monitor the progress of the reaction.
NOTE: The qPCR machine and related software will automatically calculate all parameters of the assay and display the amplification curves in real-time, while the standard curve and melting curve will be generated at the end of qPCR cycle. - Perform data analysis and acquisition by first ensuring that the melting curves for each sample and standard have one uniform peak at the expected temperature for the amplicon.
- Evaluate the amplification curves of the samples and standards and set the threshold in a region were the amplification rate of the cDNAs is the same in all samples. This is normally performed automatically by the software but should be carefully checked.
- Calculate the number of TiLV copies using the standard curve.
Results
Following the protocol described in section 1, moribund red hybrid tilapia displaying clinical signs of TiLV infection (Figure 1A) were euthanized by bathing in a high concentration of clove oil, which acts as an anesthetic. Reported clinical symptoms are variable but the common symptoms appear to be lethargy, skin erosion and discoloration, exophthalmia, detached scales, open wounds/Lesion and abnormal behaviour15,16,33,35,36, some of these can be clearly seen in Figure 1A. The abdominal wall was removed to collect internal organs such as the liver, spleen or head kidney (Figure 1B). Mucus samples were also collected at this stage by gently scraping the skin from the anterior to posterior of the fish using a cover glass or surgical blade37.

Figure 1. Tilapia dissection and sample collection.A. TiLV-infected red hybrid tilapia with skin leisons, redness around the mouth and operculum, skin erosion and corneal opacity. B. Sectioned red hybrid tilapia to allow for tissue collection from the liver (at the point of blue arrow), spleen or head kidney organs. Please click here to view a larger version of this figure.
{kind=link}
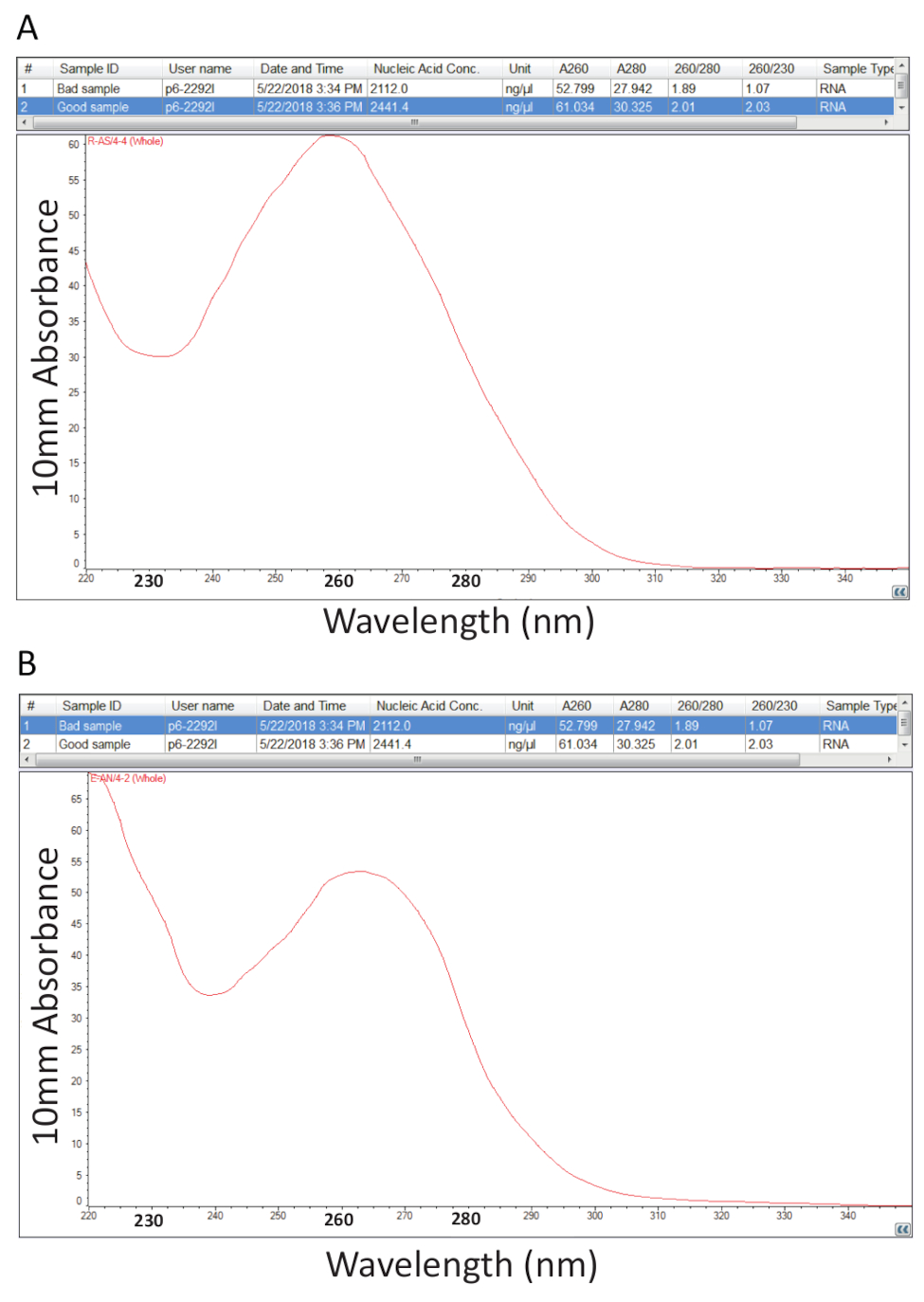
Thereafter, the protocol detailed in section 2 for Guanidium Thiocyanate-Phenol-Chloroform extraction of total RNA was followed and RNA quantification as outlined in section 3 was performed to assess sample purity by calculation of the purity ratios and examination of spectral profiles (Figure 2). Figure 2A shows a representative result from a successful total RNA extraction procedure, while Figure 2B represents a poor RNA preparation. Nucleic acids have absorbance maxima at 260 while proteins have theirs at 280 nm. The ratio of the measurements at 260 nm and 280 nm indicate the purity of each sample and ratios of 1.9 to 2.1 indicate pure RNA as is the case for the sample in Figure 2A. Lower A260/280 ratios observed in Figure 2B indicate possible protein or phenol contamination leftover from the RNA extraction procedure. Absorbance at 230 nm can be the result of sample contamination and the A260/230 nm ratio is also calculated for this reason. This ratio should be in the range of 2.0-2.2 for pure RNA preparations as illustrated by a value of 2.03 for the sample in Figure 2A, while Figure 2B has a low A260/230 ratio of 1.07 and the spectral profile shows a shift in the trough at 230 nm towards 240 nm which is indicative of residual guanidine or phenol in the sample. For the sample shown in Figure 2B, re-precipitating the RNA to remove the contamination may improve the purity of the sample.

Figure 2. Spectrophotometric quantification of total RNA extracted from diseased tilapia tissues. A. purity ratios and spectral profiles from a successful RNA preparation. B. As A, except representative of a poor RNA extraction procedure. Please click here to view a larger version of this figure.
{kind=link}
To detect TiLV by RT-PCR, pure samples such as the one represented in Figure 2A were reverse transcribed (protocol 4) into cDNA and used as a template for the PCR assay detailed in section 5 and representative results are shown in Figure 3A. Primers shown in bold in Table 1 were used to amplify a 491 bp fragment of TiLV genomic segment 314. The PCR products were separated by gel electrophoresis and stained with EtBr for visualization. Figure 3A shows the results of a two-step RT-PCR using 4 cDNA samples (S1-S4), derived from the liver of diseased tilapia isolated in Thailand, and in each sample, a clean single band of approximately 500 bp can be observed and thus, samples 1-4 are TiLV positive. The same PCR product was obtained from the positive control sample, comprising cDNA of TiLV segment 3 cloned into a plasmid32 while the no template control (NTC) did not yield PCR products. The assay in Figure 3B was performed using the same primers as in Figure 3A but in a different laboratory, using a one-step RT-PCR approach and with 5 RNA samples derived from the head kidney tissues of tilapia originating in Egyptian aquaculture15. It was determined using this detection assay that samples 1, 3 and 5 are TiLV positive while samples 2 and 4 are TiLV negative since no PCR product was found at the correct size. The negative controls, including two minus reverse transcriptase controls and two NTCs did not generate any PCR products. A one-step RT-PCR assay was also performed targeting tilapia ActinB gene. The amplicon size of 217 bp was generated in every sample (S1-S5) as expected38. This assay served as a control for the integrity of the RNA samples as well as allowing for a semi-quantitative examination of the TiLV positive samples. Given that the generated Tilapia ActB product is relatively equal, then differences in the amount of TiLV specific PCR product generated can be interpreted as a true reflection of the amount of TiLV in a given tissue sample.

Figure 3. TiLV RT-PCR. A. cDNA samples produced from liver tissues of diseased tilapia, collected from Thailand were screen for TiLV infection using specific primers for segment 3 (shown in bold in Table 1) of TiLV using a 2-step RT-PCR assay. M = marker shown in base-pairs; S1-S4 = samples 1-4; C1 = positive control using pTiLV as a PCR template; and C2 = no template control (NTC). B. One-step RT-PCR using the same primers as in A and samples from head kidney tissues of diseased tilapia collected from Egypt15. M = marker shown in base-pairs; S1 -S5 = samples 1-5. Controls C1-C2 are minus reverse transcriptase controls and C3-C4 are NTCs. Bottom panel is a one-step RT-PCR using primers directed against tilapia ActinB38 (see text for details) producing a PCR product of 217 base-pairs. Please click here to view a larger version of this figure.
{kind=link}
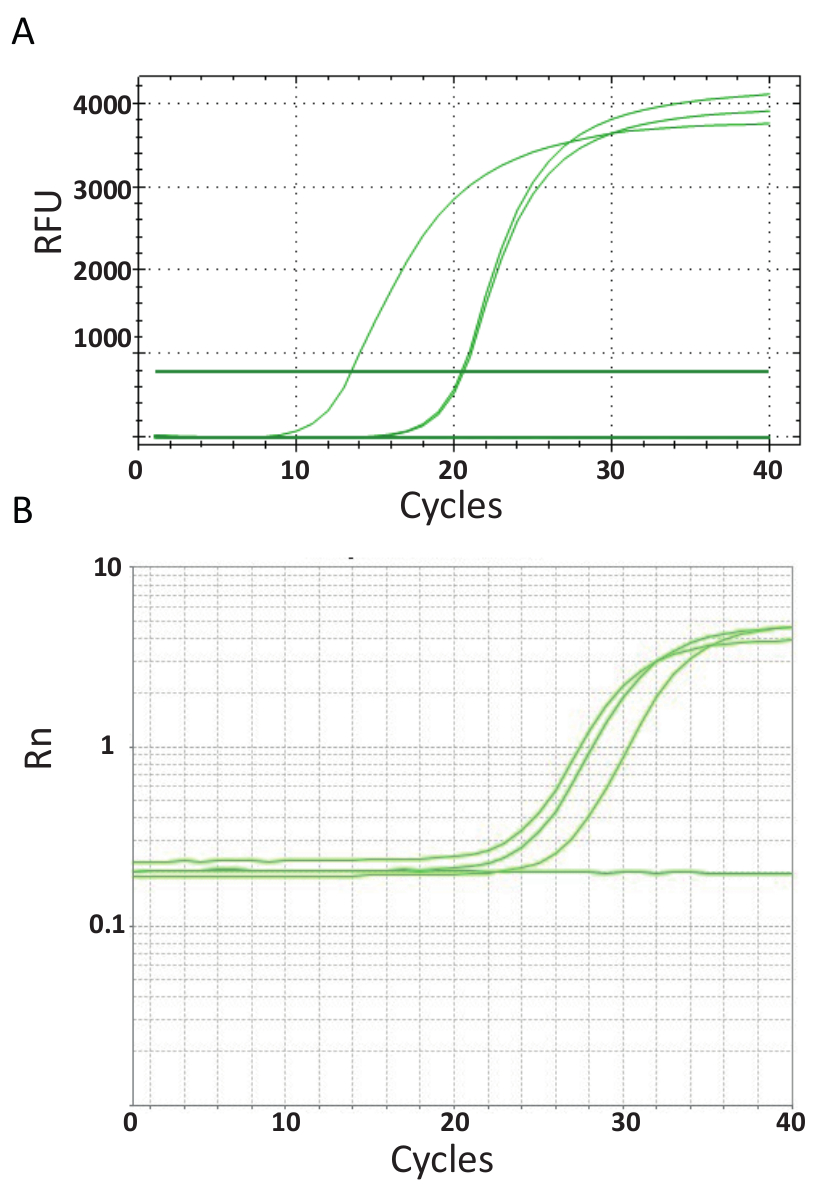
Unlike the endpoint PCRs represented in Figure 3, qPCR assays which are explained in protocol 6, measure the amount of PCR product after each PCR cycle. The amplification of target DNA is detected using fluorescent molecules that interact with DNA generated from each round of reaction. Here, SYBR Green I dye was utilized, which intercalates with double-stranded DNA. The fluorescent signal is followed during the reaction and its intensity relates to the amount of product formed 39,40,41,42,43. TiLV qPCR assays were carried out as described in protocol 6 in different laboratories using different SYBR Green reagents, qPCR machines and samples from different countries. The resulting amplification curves are shown in Figure 4A and 4B. It can be observed that for each assay, the course of the experiment has four phases: the linear ground phase, early exponential phase, late exponential phase and the plateau phase. The linear ground phase occurs during the early cycles where DNA duplication cannot yet be identified due to DNA quantities producing insufficient signal/background ratio. Baseline fluorescence is calculated during this phase. Thereafter, target DNA starts to double in concentration with each cycle inducing the signal to become detectable above background and increase exponentially. The amplification efficiency (E) of a well-optimized qPCR assay is very high (near 100%) in the beginning of the reaction and remains stable during this early exponential phase of the amplification and it is at this point that quantification is performed, when reaction efficiency is still steady. In later cycles the signal starts to plateau, and the intensity of the fluorescence is no longer correlated to the starting template copy number because the reaction components are exhausted44. Saturation may also occur due to competition from re-annealing reactions, the changing concentration ratios of the components, or the amount of enzyme units to DNA substrate molecules. Possibly, such parameters account for the differences between the amplification curves for the assays shown in Figure 4A and 4B. The included controls did not generate these characteristic amplification curves.

Figure 4. Amplification plots to show the accumulation of product over the duration of the real-time PCR assay. A. Amplification curves of TiLV-positive samples derived from Thailand, NTCs, and positive plasmid control using a SYBR-Green I 2-step qPCR assay. The chart was generated by plotting relative fluorescence (RFU) vs. cycle number. B. Amplification curves of TiLV positive samples derived from Egypt, as in Figure 3B and a NTC. The amplification curve is the fluorescence of the reporter signal normalized to the fluorescence of the passive ROX dye included in the assay (Rn) versus cycle number. Please click here to view a larger version of this figure.
{kind=link}
At the end of the qPCR thermocycling on the different machines in each laboratory, the data was acquired and analyzed. Figure 5A and 5B show representative melting curves from the assays performed in each laboratory. Each qPCR machine was programmed to perform a melting curve analysis at the end. This was achieved by incrementally increasing the temperature and monitoring the fluorescence as a function of the temperature. When the temperature is high enough to denature dsDNA, a large drop in fluorescence is recorded because the fluorophore molecule is released. The software of each qPCR instrument calculated the annealing temperature (Tm) from the melting curve data by plotting the negative first derivative vs temperature (Figure 5). It can be seen that in Figure 5A and 5B that the products formed in the different sample sets have uniform melting transition at the expected temperature of approximately 80 °C for the assay. No other peaks at lower temperatures were observed. Due to their small size, the Tm of primer-dimers is typically lower than that of the target DNA sequence. Therefore, this difference between the Tm's makes it easy to identify potential primer-dimers or other non-specific amplification products. The controls did not generate melt curves like TiLV positive samples and standards and can be seen as an almost horizontal line at the bottom of the charts in Figure 5A and 5B.

Figure 5. Melt curve analysis to ensure assay specificity and different PCR products can be differentiated by their melting features. A. Melt curve analysis of TiLV-positive samples originating from Thailand, negative control, and positive plasmid control. B. Melt curve analysis of TiLV positive samples derived from Egypt, pTiLV standards and a NTC. The charts in A and B both show the change in fluorescence divided by the change in temperature plotted against temperature to produce a clear picture of the melting dynamics. Please click here to view a larger version of this figure.
{kind=link}
Most qPCR machines come with a software facilitating further evaluation the qPCR run and will quantify the samples by generating a standard curve by automatically plotting the cycle threshold (Ct) against the logarithm of the pTiLV standards' template copy number as shown in Figure 6A and 6B for the two independent laboratories. Briefly, the Ct is the unit used for evaluating qPCR results. The Ct value denotes the number of cycles required to reach a set threshold fluorescence signal level. The greater the amount of starting template, the fewer cycles it takes to attain a detectable fluorescence level. Indeed, samples with a high load of TiLV will have lower Ct values than samples with a low load of TiLV such as in fish with a sub-clinical infection. To determine Ct values, background fluorescence levels are first deducted from raw data. Next, the software associated with the qPCR device will automatically select a fluorescence threshold by searching the data curves for each sample and incorporating a Ct representing where the sample crossed the threshold.This is done separately for each assay and each threshold should be carefully evaluated, ensuring that the threshold has been set in the logarithmic part of the amplification curves and at a place where all curves are parallel. Thus, the specific Ct acquired is a relative value and it is relative to the starting template copy number45, but it is also specific for the qPCR machine and reagents used, the efficiency of the PCR amplification and the sensitivity of detection. These parameters contribute to the differences observed using the same assay in Figure 6.
From the standard curves in Figure 6, regression analyses, including calculation of standard curve slopes (m) and intercepts, amplification efficiencies (100 x (101/m -1))46 and linearity of the reaction were performed. Standard curve analyses were also used to confirm sensitivity (limit of detection), repeatability and reproducibility of the assay. Theoretically, the amount of DNA is doubled with every PCR cycle, meaning that the efficiency (E) is equal to 100%. However, in practice such an ideal efficiency is seldom reached due to sub-optimal PCR conditions such as, DNA polymerase inhibition, contaminants, too much cDNA and pipetting errors47. Typically, amplification E range from 90-110% for good assays, in Figure 6A an efficiency of 94.5% was calculated using 8 serially diluted pTiLV samples, while, the assay efficiency in the assay shown in Figure 6B using 7 serially diluted pTiLV samples was 101.2%. An efficiency of over 100% is usually due to the presence of PCR inhibitors in the assay. Linear regression analysis of the standard plot also allows for the calculation of the number of TiLV copies in each sample41,42,45, as can be observed for the three TiLV samples shown in red in Figure 6B which is in line with the results for samples S1, S3 and S5 shown in Figure 3B.

Figure 6. RT-qPCR standard curves. Real-time PCR of 10-fold serial dilutions of pTiLV, the standard used in both laboratories. A. 8 serially diluted pTiLV samples were tested, all of known concentration and correlated to the number of TiLV copies/ reaction. The standard curve was generated by plotting log copy number vs. cycle threshold (Ct). The slope = -3.462, R2 = 0.9992 and the efficiency is 94.47%. B. As in A, except 7 serially diluted pTiLV samples (green) were tested and the chart displays the threshold cycle on the y-axis and the copy number of TiLV (Quantity) on the x-axis. The y-intercept = 32.327, slope = -3.292, R2 = 0.98 and the efficiency is 101.2%. For both standards curves in A and B, the slope, y-intercept and correlation coefficient values (R2) are utilized to understand the performance of the assay. Importantly, R2 value should be close to 1 since it is a measure of the linearity of the standard curve. The slope is used to measure PCR efficiency wherein 100% efficiency corresponds to a slope of -3.32, see main text for the equation and further details. A good qPCR reaction generally has an efficiency between 90-110% correlating to a slope of between -3.58 and -3.10. The standard curve is used for absolute quantification of unknown TiLV positive samples and determines the exact number of TiLV copies / reaction, as is the case for the three TiLV positive samples colored red in B.
Discussion
TiLV was first reported in 2014 in Israel14 and since then, it has been identified in multiple countries including Egypt, Colombia, India, Malaysia, Uganda, Tanzania and Thailand15,16,18,35,48. Global awareness, particularly, in tilapia producing countries has placed more attention on the virus and various restrictions and control measures from government authorities have been implemented attempting to prevent the spread of TiLV. Here, a detailed protocol for TiLV detection in tilapia tissue, covering sample collection, RNA isolation, cDNA synthesis, PCR and qPCR assays has been explained. There are various aspects to these methods that warrant specific discussion. TiLV has been identified in fish spanning a variety sizes9,12,14,15,49 and species of tilapia so far, including farmed hybrid tilapia (O. niloticus x O. aureus)11,14, Nile tilapia (O. niloticus)9,10,14,15,16,33,35,36,49,50 and red tilapia (Oreochromis sp.)16,33,48,51, as well as in wild Nile tilapia9,12, black tilapia51, T. zilli14,15, S. galilaeus, O. aureus and T. simonis intermedia14 and very recently TiLV was identified in wild carp (Barbonymus schwanenfeldii)52. Tissue samples from internal organs (gill, spleen, liver, heart, head kidney) or mucus37 can be collected from healthy as well as moribund tilapia regardless of age, size or species and processed for RNA isolation. The total RNA extraction protocol outlined here uses a monophasic solution of phenol and guanidinium thiocyanate, which is a chaotropic denaturing agent. The tissues are directly homogenized in this solution followed by the addition of chloroform and centrifugation to achieve phase separation wherein a clear RNA containing upper aqueous phase, an interphase and a lower organic phase is generated. The RNA is isolated from the aqueous phase by isopropanol precipitation, followed by washing of the recovered RNA to get rid of contaminants. Isolation of RNA by this methodology was pioneered by Piotr Chomczynski and Nicoletta Sacchi and was referred to as guanidinium thiocyanate-phenol-chloroform extraction53,54. This type of reagent used for RNA extraction can be commercially purchased or made in the laboratory (see the Table of Materials for further information). This protocol takes slightly longer than column-based methods such as the silica-based purification, but in general, it is more cost effective and yields more RNA.
In this protocol, quantifying RNA using A260 values was outlined whereby spectrophotometry values can indicate RNA quality (A260/A280 = 1.9 - 2.1). While this method will give a good indication of sample purity, it cannot absolutely inform about the quality of the extracted RNA. To properly determine whether the RNA is intact or partially degraded, samples can be separation by agarose gel electrophoresis wherein smearing of the EtBr stained 18S and 28S rRNA bands indicate RNA degradation. Further verification of RNA quality may include using a lab-on-a-chip instrument. Furthermore, it is also important to digest the purified RNA with DNase I to remove contaminating host genomic DNA, which depending on the downstream applications may lead to false results. If host gDNA is still contaminating the RNA sample to a large extent, an additional DNaseI treatment can also be performed at the end of the RNA extraction procedure (see Table of Materials).
Complementary DNA synthesis can greatly affect the overall qPCR results and is an aspect of the method that can introduce variation. The cDNA protocol advocated here comprises of a single component set-up using oligo (dT) and thus only transcribes mRNAs containing polyA tails. It allows the user control of exactly which components to use in the reverse transcription reaction and this mode of cDNA synthesis has proved successful for TiLV detection32. An alternative to this set-up is a commercially bought master-mix containing all components required for the reverse transcription reaction and is very fast and simple without the usual multi-step, pipetting and multi-temperature protocol. This is advantageous because it minimizes handling and promotes uniformity across all samples. Such master-mixes often include both oligo(dT) and random primers making it applicable to different RNA templates and generating representative cDNA copies of sequences from the entire length of the RNAs in a population (viral and tilapia host mRNA) and in theory, every desired RNA species can then be measured by conventional PCR or qPCR from such a sample. This versatility is the main advantage of a 2-step RT-PCR approach; it provides a long-term pool that can be used for many different experiments. In the results, a one-step RT-PCR approach has been represented wherein sequence specific primers (Table 1) were used and the RT and PCR were performed in one tube (see material list). In general, sequence specific primers allow for a higher RT efficiency of the specific target RNA than using random priming, but the specific target RNA is the only one that can be quantified in such a cDNA sample which may be the only aim of certain laboratories (see Table of Materials for cDNA synthesis product suggestions).
While conventional RT-PCR appears to be commonly used so far in the TiLV diagnosis9,13,14,15,16,17,18,33,35,48,55. RT-qPCR has been shown to be a more powerful tool for the detection and quantification of small amounts of TiLV in fish tissues or mucus32,37. In general, qPCR is widely used in clinical virology diagnostic labs due to its high sensitivity, specificity, good reproducibility, wide dynamic range and speed21. While qPCR may be initially more expensive to implement than conventional RT-PCR, it does offer many important advantages over conventional PCR; it has a faster turn-around time from sample to results and it does not require any post-PCR steps. This latter point means that there is minimal risk for laboratory contamination and it can be more easily adapted to high-throughput situations such as in the event of outbreaks. Furthermore, it is inherently more sensitive than conventional RT-PCR, which is of vital importance to detect low viral loads in sub-clinical infections21. This would require a nested PCR approach requiring reverse transcription, two further PCR reactions and then analysis by agarose gel electrophoresis. These many steps take a lot of time and increase the chances of errors or contamination. Nonetheless, due to its high sensitivity, RT-qPCR demands meticulous experimental design and a thorough understanding of quantification techniques for generating precise results56,57.
The DNA binding fluorophore, SYBR Green I has been demonstrated in this protocol. It is a dsDNA unspecific DNA binding dye and thus the specificity of the assay lies entirely in the set of primers, which may generate false positives58. Therefore, while the dsDNA melting curve analysis performed at the end of each PCR is an especially important part of the PCR reaction because it confirms that only one PCR amplicon of the correct Tm is produced (this should also be achieved by gel electrophoresis when new assays are being implemented). The Tm of a DNA fragment is dependent on a variety of features such as its length, GC composition, sequence, strand complementarity, concentration as well as on buffer components and PCR enhancers. The melting curve analyses in the representative results shown from two laboratories did not reveal the presence of primer-dimers or other unwanted PCR products but if this is observed with other samples and/or experimental set-ups, then the assay should be re-optimized. More advanced qPCR technologies do not require such a melting curve step and indeed, since this methods paper was written, a TaqMan based TiLV RT-qPCR has been developed utilizing two primers and a probe making it highly TiLV specific34.
Undoubtedly, the primers designed for RT-qPCR assays are fundamental to the success of assay and the primers here were designed based upon the publicly available TiLV genomic data at the time32. However, RNA viruses are well-known to exhibit high mutation rates and possible strains will escape the current diagnostic tests, as was observed for ISAV59. It will always be difficult for such virus types to generate a universal pan-TiLV RT-qPCR assay and such assays will only be continually improved if more TiLV genomic data from far-reaching locations and time periods become available.
Finally, it is essential to run duplicate or if possible, triplicate reactions in both intra and inter qPCR assays. If the Ct values are very high, then the use of replicates is especially important to ascertain that the PCR reaction is reliable and reproducible. In general, if data from replicate reactions varies more than 0.5 cycles, the reactions should be repeated and if the Ct values consistently vary > 0.5 cycles in replicates, the assay should be re-optimized. The use of an integrated qPCR pipetting robot helps immensely with this issue, but it is a luxury tool. As with any experiment, the inclusion adequate and appropriate controls are of utmost importance to the development of robust molecular assays, especially in diagnostic laboratories where such assays have to be accredited. Controls should include positive (positive TiLV sample, TiLV plasmid standard) and negative controls (NTC and -RT) samples as well as the detection of endogenous tilapia housekeeping genes. Such controls cannot be underestimated and should be included in each assay to properly understand the quality of each step of the assay and to properly interpret the results.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We are grateful to the Institute of Veterinary Bacteriology, Vetsuisse Faculty, University of Bern for their support. This work was funded by the Committee for Academic Promotion of Early Career Researchers and gender equality at the Vetsuisse Faculty, University of Bern by 120% model funding awarded to PN. WS and PR are supported by Center for Advanced Studies for Agriculture and Food, Institute for Advanced Studies, Kasetsart University, Bangkok, Thailand under the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission, Ministry of Education, Thailand. We would like to thank Dr. Kwanrawee Sirikanchana for her narration and Piyawatchara Sikarin for editing the video.
Materials
| Name | Company | Catalog Number | Comments |
| Tissue collection | Step 1 | ||
| Tricaine methanesulfonate | Sigma-Aldrich | E10521 | An alternative to clove oil. Step 1.1 |
| RNAlater stabilization solution | Thermo Fisher Scientific | AM7020 | For storing tissues if they cannot be processed immediately Step 1.3 |
| RNA extraction | Step 2 | ||
| TRIreagent | Sigma-Aldrich | Step 2.1 | |
| TRIzol | Thermo Fisher Scientific (Invitrogen) | 15596026 | Step 2.1 |
| GENEzol | Geneaid | GZR100 | Step 2.1 |
| Trisure | Bioline | BIO-38032 | Step 2.1 |
| Homemade solution | - | - | 94.53 g/L (800 mM) guanidine thiocyanate 30.45 g/L (400 mM) ammonium thiocyanate 8.20 g/L (100 mM) sodium acetate 380 mL/L (38 % v/v) phenol 50 mL/L (5 % v/v) glycerol 1.0 g/L (0.1 % w/v) 8-quinolinol, pH 5.0 Store up to 2 years at 4oC Step 2.1 |
| MagNA Lyser Green Beads | Roche | 3358941001 | An alternative tissue homogenization method used in conjunction with tissue lysing machines detailed below Step 2.2 |
| Lysing Matrix D, 2 mL Tube | MP BIOMEDICALS | 116913050 | |
| Chloroform | Sigma-Aldrich | C2432 | Step 2.3 |
| Chloroform | RCI Labscan | AR1027E-G2.5L | Step 2.3 |
| 1-Bromo-3-chloropropane | Sigma-Aldrich | B9673 | A less toxic alternative to chloroform Step 2.3 |
| Isopropanol (GC) ≥ 99.8 % | Sigma-Aldrich | 59300 | Step 2.6 |
| Isopropanol (ACS, ISO Reag. Ph Eur) | Merck KGaA (EMSURE) | 1.09634.2500 | Step 2.6 |
| Glycogen, molecular biology grade (e.g., Sigma, cat. no. G1767) | Thermo Fisher Scientific (Thermo Scientific) | R0551 | Useful step if tissue starting material is small to maximise RNA precipitation optional |
| Ethanol (purity (GC) ≥ 99.9 % | Sigma-Aldrich (EMD Millipore) | 1.00983 | Step 2.9 |
| Ethanol (ACS, ISO Reag. Ph Eur) | Merck (EMSURE) | 1.00983.2500 | Step 2.9 |
| Nuclease-free water | Promega | P1193 | Step 2.13 |
| Nuclease-free water | Multicell | 809-115-CL | Step 2.13 |
| Ambion TURBO DNA-free kit | Thermo Fisher Scientific (Invitrogen) | AM1907 | Can be performed at the end of the RNA extraction protocol optional |
| cDNA synthesis | Step 4 | ||
| Viva cDNA Synthesis Kit | Vivantis | cDSK01 | Step 4.1 & 4.3 |
| ReverTra Ace qPCR RT MasterMix with gDNA remover | Toyobo | A1172K | An alternative option see discussion |
| ReverTra Ace qPCR RT Kit | Toyobo | FSQ-101 | An alternative option see discussion |
| AffinityScript Multiple Temperature Reverse Transcriptase | Agilent Technologies | 600107 | An alternative option |
| PCR | Step 5 | ||
| DNA polymerase systems: | Step 5.2 | ||
| - Platinum II Hot-Start Green PCR Master Mix (2X) | Thermo Fisher Scientific (Invitrogen) | 14001012a | Step 5.2 |
| - GoTaq Mastermix | Promega | M7122 | Step 5.2 |
| Separate PCR mixture components: | Step 5.2 | ||
| 10mM dNTP Mix | Vivantis | NP2409 | Step 5.2 |
| 25mM MgCl2 | Thermo Fisher Scientific | R0971 | Step 5.2 |
| 10X Taq Buffer with KCl | Thermo Fisher Scientific | 00348114 | Step 5.2 |
| Taq DNA polymerase | Vivantis | PL1202 | Step 5.2 |
| - Verso 1-step RT-PCR ReddyMix with ThermoPrime Taq | Thermo Fisher Scientific | AB1454 | One step RT-PCR exemplified in Figure 3B |
| Gel electrophoresis: | For visulation of PCR products from steps 5.1-5.4 | ||
| Ethidium Bromide solution (10 mg/mL) | Thermo Fisher Scientific | 17898 | Step 5.5 |
| Tris/Acetic/EDTA (TAE) buffer: | Step 5.5 | ||
| - Tris | Vivantis | PR0612-1KG | Step 5.5 |
| - Acetic acid (glacial) (ACS, ISO, Reag. Ph Eur) | Merck KGaA (EMSURE) | 1.00063.2500 | Step 5.5 |
| - Ethylenediaminetetraacetic acid (EDTA) | BIO-RAD | 161-0729 | Step 5.5 |
| Agarose | Vivantis | PC0701-100G | Step 5.5 |
| DNA ladders and markers | Vivantis | NL1405 | Step 5.5 |
| DNA gel loading dye (6X) | Thermo Fisher Scientific | R0611 | Step 5.5 |
| qPCR | Step 6 | ||
| PowerUP SYBR Green Master Mix | Thermo Fisher Scientific (Applied Biosystems) | A25779 | Exemplified in Figures 4-6B Step 6.2 |
| iTaq Universal SYBR Green Supermix | BIO-RAD | 1725120 | Exemplified in the video and in Figures 4-6A Step 6.2 |
| Equipment | |||
| Dounce tissue grinder pestle | Sigma-Aldrich | P1110 | Protocol 2 |
| MagNA Lyser Instrument | Roche | 3358976001 | An alternative tissue homogenizing option for protocol 2 which are used in conjunction with the lysing beads detailed above Step 2.2 |
| FastPrep-24 5G Homogenizer | MP BIOMEDICALS | 116005500 | |
| Refrigerated microcentrifuge | Eppendorf | Eppendorf 5427R | Protocol 2 Step 2.4, 2.7 & 2.10 |
| Refrigerated microcentrifuge | Eppendorf | Eppendorf 5418R | |
| Heat box | Labnet | AccuBlock Digital Dry Bath | Protocol 2 Step 2.13 |
| Microvolume spectrophotometer | Thermo Fisher Scientific (Applied Biosystems) | Nanodrop 2000 | Protocol 3 Step 3.1 - 3.4 |
| PCR machine | BIO-RAD | T100 Thermal Cycler | Protocol 5 Step 5.4 |
| Power supply | BIO-RAD | PowerPac HC | Protocol 5 Step 5.5 |
| Horizontal gel electrophoresis | BIO-RAD | Mini ReadySub-Cell GT Cell #1704487edu | Protocol 5 Step 5.5 |
| Mini microcentrifuge | Corning | LSE 6766 | Useful to quickly spin down PCR reaction tubes in protocols 4, 5 & 6 Step 6.5.1 |
| Microcentrifuge | LioFuge | LM-60 | Step 6.5.1 |
| qPCR machine and software | Thermo Fisher Scientific | 7500 Fast Real-Time PCR System with 7500 Software v2.0 | Protocol 6 Step 6.6-6.8 |
| qPCR machine and software | BIO-RAD | CFX96 Touch Real-Time PCR Detection System with CFX Manager software | |
| General Materials | |||
| Mayo scissors | Step 1.1-1.2 | ||
| Forceps | Step 1.1-1.2 | ||
| Pipette | Rainin | Pipette-Lite XLS | |
| Aerosol-barrier pipette tips | Sigma-Aldrich | Z333328, Z333336, Z333344 | |
| Nuclease-free 1.5-ml microcentrifuge tubes | Eppendorf |
References
- FAO. . The State of World Fisheries and Aquaculture, 2014. Opportunities and Challenges. , (2014).
- FAO. . The State of World Fisheries and Aquaculture, 2016. Contributing to Food Security and Nutrition for all. , (2016).
- WorldBank. . FISH TO 2030: Prospects for Fisheries and Aquaculture. Agriculture and Environmental Services Discussion Paper 03. , (2013).
- Wing-Keong, N., Nicholas, R. A review of the nutrition and feeding management of farmed tilapia throughout the culture cycle. Reviews in Aquaculture. 5 (4), 220-254 (2013).
- Cleasby, N., et al. The socio-economic context for improving food security through land based aquaculture in Solomon Islands: A peri-urban case study. Marine Policy. 45, 89-97 (2014).
- Ponzoni Raul, W., et al. Genetic improvement of Nile tilapia (Oreochromis niloticus) with special reference to the work conducted by the WorldFish Center with the GIFT strain. Reviews in Aquaculture. 3 (1), 27-41 (2011).
- Hounmanou, Y. M. G., et al. Tilapia lake virus threatens tilapiines farming and food security: Socio-economic challenges and preventive measures in Sub-Saharan Africa. Aquaculture (Amsterdam, Netherlands). 493, 123-129 (2018).
- OIE. . Tilapia Lake Virus (TiLV) - a novel orthomyxo-like virus. OIE technical disease cards. , (2018).
- Mugimba, K. K., et al. Detection of tilapia lake virus (TiLV) infection by PCR in farmed and wild Nile tilapia (Oreochromis niloticus) from Lake Victoria. Journal of Fish Diseases. , (2018).
- Koesharyani, I., Gardenia, L., Widowati, Z., Khumaira, D. D., Rustianti, Studi kasus infeksi tilapia lake virus (tilv) pada ikan nila (Oreochromis niloticus). Jurnal Riset Akuakultur. 13 (1), 85-92 (2018).
- OIE. . Tilapia lake virus disease (TiLV), Chinese Taipei. Immediate Notification. , (2017).
- OIE. . Tilapia Lake Virus Disease (TiLV), Peru. Immediate Notification. , (2018).
- Bacharach, E., et al. Characterization of a Novel Orthomyxo-like Virus Causing Mass Die-Offs of Tilapia. MBio. 7 (2), e00431-e00416 (2016).
- Eyngor, M., et al. Identification of a novel RNA virus lethal to tilapia. Journal of Clinical Microbiology. 52 (12), 4137-4146 (2014).
- Nicholson, P., et al. Detection of Tilapia Lake Virus in Egyptian fish farms experiencing high mortalities in 2015. Journal of Fish Diseases. 40 (12), 1925-1928 (2017).
- Surachetpong, W., et al. Outbreaks of Tilapia Lake Virus Infection, Thailand, 2015-2016. Emerging Infectious Diseases. 23 (6), 1031-1033 (2017).
- Tattiyapong, P., Dachavichitlead, W., Surachetpong, W. Experimental infection of Tilapia Lake Virus (TiLV) in Nile tilapia (Oreochromis niloticus) and red tilapia (Oreochromis spp.). Veterinary Microbiology. 207, 170-177 (2017).
- Kembou Tsofack, J. E., et al. Detection of Tilapia Lake Virus in Clinical Samples by Culturing and Nested Reverse Transcription-PCR. Journal of Clinical Microbiology. 55 (3), 759-767 (2017).
- Thangaraj, R. S., et al. Derivation of two tilapia (Oreochromis niloticus) cell lines for efficient propagation of Tilapia Lake Virus (TiLV). Aquaculture (Amsterdam, Netherlands). 492, 206-214 (2018).
- Hanson, L. A., Rudis, M. R., Vasquez-Lee, M., Montgomery, R. D. A broadly applicable method to characterize large DNA viruses and adenoviruses based on the DNA polymerase gene. Virology Journal. 3, 28-28 (2006).
- Josko, D. Molecular virology in the clinical laboratory. Clinical Laboratory Science. 23 (4), 231-236 (2010).
- Munir, K., Kibenge, F. S. Detection of infectious salmon anaemia virus by real-time RT-PCR. Journal of Virological Methods. 117 (1), 37-47 (2004).
- Snow, M., et al. Developement, application and validation of a Taqman real-time RT-PCR assay for the detection of infectious salmon anaemia virus (ISAV) in Atlantic salmon (Salmo salar). Developments in Biologicals. 126, 133-145 (2006).
- Matejusova, I., McKay, P., McBeath, A. J., Collet, B., Snow, M. Development of a sensitive and controlled real-time RT-PCR assay for viral haemorrhagic septicaemia virus (VHSV) in marine salmonid aquaculture. Diseases of Aquatic Organisms. 80 (2), 137-144 (2008).
- Garver, K. A., et al. Development and validation of a reverse transcription quantitative PCR for universal detection of viral hemorrhagic septicemia virus. Diseases of Aquatic Organisms. 95 (2), 97-112 (2011).
- Dalla Valle, L., et al. Development of a sensitive and quantitative diagnostic assay for fish nervous necrosis virus based on two-target real-time PCR. Veterinary Microbiology. 110 (3-4), 167-179 (2005).
- Hodneland, K., Garcia, R., Balbuena, J. A., Zarza, C., Fouz, B. Real-time RT-PCR detection of betanodavirus in naturally and experimentally infected fish from Spain. Journal of Fish Diseases. 34 (3), 189-202 (2011).
- Hodneland, K., Endresen, C. Sensitive and specific detection of Salmonid alphavirus using real-time PCR (TaqMan). Journal of Virological Methods. 131 (2), 184-192 (2006).
- Wang, X. W., Ao, J. Q., Li, Q. G., Chen, X. H. Quantitative detection of a marine fish iridovirus isolated from large yellow croaker, Pseudosciaena crocea, using a molecular beacon. Journal of Virological Methods. 133 (1), 76-81 (2006).
- van Beurden, S. J., et al. Development and validation of a real-time PCR assay for the detection of anguillid herpesvirus 1. Journal of Fish Diseases. 39 (1), 95-104 (2016).
- Ciulli, S., et al. Development and application of a real-time PCR assay for the detection and quantitation of lymphocystis disease virus. Journal of Virological Methods. 213, 164-173 (2015).
- Tattiyapong, P., Sirikanchana, K., Surachetpong, W. Development and validation of a reverse transcription quantitative polymerase chain reaction for tilapia lake virus detection in clinical samples and experimentally challenged fish. Journal of Fish Diseases. 41 (2), 255-261 (2018).
- Dong, H. T., et al. Emergence of tilapia lake virus in Thailand and an alternative semi-nested RT-PCR for detection. Aquaculture (Amsterdam, Netherlands). 476, 111-118 (2017).
- Waiyamitra, P., et al. A TaqMan RT-qPCR assay for tilapia lake virus (TiLV) detection in tilapia. Aquaculture (Amsterdam, Netherlands). 497, 184-188 (2018).
- Behera, B. K., et al. Emergence of Tilapia Lake Virus associated with mortalities of farmed Nile Tilapia Oreochromis niloticus (Linnaeus 1758) in India. Aquaculture (Amsterdam, Netherlands). 484, 168-174 (2018).
- Ferguson, H. W., et al. Syncytial hepatitis of farmed tilapia, Oreochromis niloticus (L.): a case report. Journal of Fish Diseases. 37 (6), 583-589 (2014).
- Liamnimitr, P., Thammatorn, W., U-thoomporn, S., Tattiyapong, P., Surachetpong, W. Non-lethal sampling for Tilapia Lake Virus detection by RT-qPCR and cell culture. Aquaculture (Amsterdam, Netherlands). 486, 75-80 (2018).
- Yang, C. G., et al. Evaluation of reference genes for quantitative real-time RT-PCR analysis of gene expression in Nile tilapia (Oreochromis niloticus). Gene. 527 (1), 183-192 (2013).
- Bustin, S. A. Real-time, fluorescence-based quantitative PCR: a snapshot of current procedures and preferences. Expert Review of Molecular Diagnostics. 5 (4), 493-498 (2005).
- Fleige, S., Pfaffl, M. W. RNA integrity and the effect on the real-time qRT-PCR performance. Molecular Aspects of Medicine. 27 (2-3), 126-139 (2006).
- Kubista, M., et al. The real-time polymerase chain reaction. Molecular Aspects of Medicine. 27 (2-3), 95-125 (2006).
- Mackay, I. M., Arden, K. E., Nitsche, A. Real-time PCR in virology. Nucleic Acids Research. 30 (6), 1292-1305 (2002).
- Wong, M. L., Medrano, J. F. Real-time PCR for mRNA quantitation. Biotechniques. 39 (1), 75-85 (2005).
- Bustin, S. A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. Journal of Molecular Endocrinology. 25 (2), 169-193 (2000).
- Heid, C. A., Stevens, J., Livak, K. J., Williams, P. M. Real time quantitative PCR. Genome Research. 6 (10), 986-994 (1996).
- Rutledge, R. G., Côté, C. Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Research. 31 (16), e93-e93 (2003).
- Svec, D., Tichopad, A., Novosadova, V., Pfaffl, M. W., Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomolecular Detection and Quantification. 3, 9-16 (2015).
- Amal, M. N. A., et al. A case of natural co-infection of Tilapia Lake Virus and Aeromonas veronii in a Malaysian red hybrid tilapia (Oreochromis niloticus × O. mossambicus) farm experiencing high mortality. Aquaculture (Amsterdam, Netherlands). 485, 12-16 (2018).
- Fathi, M., et al. Identification of Tilapia Lake Virus in Egypt in Nile tilapia affected by ‘summer mortality’ syndrome. Aquaculture (Amsterdam, Netherlands). 473, 430-432 (2017).
- OIE. . Tilapia Lake Virus disease (TiLV), Philippines. Immediate Notification. , (2017).
- OIE. . Tilapia lake virus disease (TiLV), Malaysia. Immediate Notification. , (2017).
- Abdullah, A., et al. First detection of tilapia lake virus (TiLV) in wild river carp (Barbonymus schwanenfeldii) at Timah Tasoh Lake, Malaysia. Journal of Fish Diseases. 41 (9), 1459-1462 (2018).
- Chomczynski, P., Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 162 (1), 156-159 (1987).
- Chomczynski, P., Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nature Protocols. 1 (2), 581-585 (2006).
- Del-Pozo, J., et al. Syncytial Hepatitis of Tilapia ( Oreochromis niloticus L.) is Associated With Orthomyxovirus-Like Virions in Hepatocytes. Veterinary Pathology. 54 (1), 164-170 (2017).
- Bustin, S. A., et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry. 55 (4), 611-622 (2009).
- Purcell, M. K., Getchell, R. G., McClure, C. A., Garver, K. A. Quantitative polymerase chain reaction (PCR) for detection of aquatic animal pathogens in a diagnostic laboratory setting. Journal of Aquatic Animal Health. 23 (3), 148-161 (2011).
- Simpson, D. A., Feeney, S., Boyle, C., Stitt, A. W. Retinal VEGF mRNA measured by SYBR green I fluorescence: A versatile approach to quantitative PCR. Molecular Vision. 6, 178-183 (2000).
- Kibenge, M. J., et al. Discovery of variant infectious salmon anaemia virus (ISAV) of European genotype in British Columbia, Canada. Virology Journal. 13, 3 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved