Method Article
Muy largo leer la secuencia para el análisis de la DNA de Genomic entero
En este artículo
Resumen
Secuencias de largo leer facilitan enormemente el montaje de genomas complejos y la caracterización de la variación estructural. Se describe un método para generar secuencias muy largas por plataformas de secuenciación basada en nanopore. El enfoque adopta una extracción de ADN optimizada seguida por las preparaciones de la biblioteca modificada para generar cientos de kilobases Lee con una cobertura moderada de células humanas.
Resumen
Tercera generación de tecnologías de secuenciación de ADN de una sola molécula ofrecen significativamente más Lee longitud que puede facilitar el ensamblaje de complejos genomas y análisis de variantes estructurales complejas. Plataformas Nanopore realizan secuenciación de una sola molécula por medición directa de los cambios actuales mediados por el paso de ADN a través de los poros y pueden generar cientos de kilobases (kb) dice con mínimo costo de capital. Esta plataforma ha sido adoptada por muchos investigadores para una variedad de aplicaciones. Lograr más largos de lectura de la secuencia es el factor más crítico para aprovechar el valor de las plataformas de secuenciación nanopore. Para generar lecturas ultra-larga, consideración especial es necesaria para evitar roturas de DNA y ganar eficiencia para generar plantillas de secuencia productiva. Presentamos el protocolo detallado de ultra larga secuencia de la DNA incluyendo la extracción de ADN de alto peso molecular (APM) de las células frescas o congeladas, construcción de biblioteca por corte mecánico o fragmentación de la transposasa y la secuencia en un dispositivo nanopore. De 20-25 μg de ADN de HMW, el método puede alcanzar N50 leer longitud de 50-70 kb con el corte mecánico y N50 de 90-100 kb leer longitud con transposasa mediada fragmentación. El protocolo se puede aplicar al ADN extraído de células de mamíferos para realizar la secuenciación del genoma entero para la detección de variantes estructurales y ensamblaje del genoma. Mejoras adicionales sobre la extracción de ADN y las reacciones enzimáticas más aumentará la longitud leer y ampliar su utilidad.
Introducción
En la última década, masivamente paralelo y tecnologías de secuenciación de alto rendimiento segunda generación altamente exacto han llevado a una explosión de descubrimientos biomédicos e innovación tecnológica1,2,3. A pesar de los avances técnicos, el corto-leer datos generados por las plataformas de segunda generación son eficaces en la resolución de regiones genómicas complejas y están limitados en la detección de variantes estructurales genómicas (SVs), que desempeñan papeles importantes en humanos evolución y enfermedades4,5. Además, corto-leer datos no puede resolver variación repetición y son inadecuados para los más exigentes haplotipo eliminación de variantes genéticas6.
Últimos avances en secuenciación de una sola molécula ofrece mucho más leer longitud, que puede facilitar la detección de la gama completa de SVs7,8,9y ofrece montaje precisa y completa del complejo genomas microbianos y mamíferos6,10. La plataforma nanopore realiza la secuenciación de una sola molécula por medición directa de los cambios actuales mediados por el paso de ADN a través de los poros11,12,13. A diferencia de cualquier química existente de secuenciación de ADN, secuenciación nanopore puede generar largos (decenas o miles de kilobases) lecturas en tiempo real sin depender de la cinética de la polimerasa o artificial la amplificación de la muestra de ADN. Por lo tanto, nanopore larga lectura secuencia (NLR-seq) tiene gran promesa para la generación de ultra largos leerlas más allá de 100 kb, lo que adelantaría mucho análisis genómico y biomédica14, particularmente en la repetición-ricas o baja complejidad regiones del genoma15.
La característica única de la secuencia nanopore es su potencial para generar largas lee sin una limitación de la longitud teórica. Por lo tanto, la longitud leer depende de la longitud física de la DNA que es afectada directamente por la calidad de plantilla de integridad y secuenciación de ADN. Por otra parte, dependiendo del grado de manipulación y el número de pasos implicados, como el pipeteo de las fuerzas y condiciones de extracción, la calidad de la DNA es muy variable. Por lo tanto, es un reto para uno para lecturas largas aplicando sólo los protocolos de extracción de ADN estándar y métodos de construcción de biblioteca suministrado del fabricante. Con este fin, hemos desarrollado un sólido método para generar muy largo leer (cientos de kilobases) datos de la secuencia a partir de gránulos de células cosechadas. Adoptaron múltiples mejoras en los procedimientos de preparación de extracción y biblioteca de ADN. Optimizamos el protocolo para excluir procedimientos innecesarios que causan daños y degradación del ADN. Este protocolo está compuesto de alto peso molecular (HMW) extracción de ADN, ultra larga construcción de biblioteca de ADN y la secuencia en una plataforma nanopore. Para un biólogo molecular bien entrenado, normalmente tarda 6 h desde la cosecha hasta la finalización de la extracción de ADN de HMW, 90 minutos o 8 h para la construcción de la biblioteca según el método de corte y hasta un más 48 h para la secuencia de la DNA de la célula. El uso del Protocolo será empoderar a la comunidad genómica para mejorar nuestra comprensión de la complejidad del genoma y conocer nueva variación del genoma en enfermedades humanas.
Protocolo
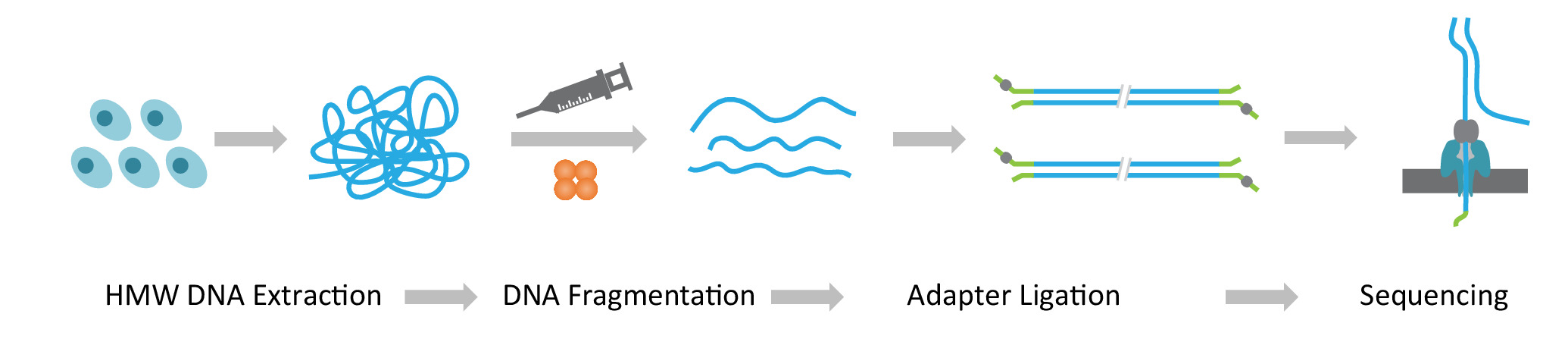
Nota: El protocolo de NLR-seq consta de tres pasos consecutivos: 1) extracción de peso molecular alto peso (HMW) ADN genómico; 2) ultra largo DNA biblioteca de la construcción, que incluye la fragmentación del ADN de HMW en los tamaños deseados y la ligadura de adaptadores de secuenciación del ADN termina; y 3) carga de la DNA adaptador de ligarse a los arreglos de discos de interconectivos (figura 1).
1. extracción de ADN de HMW
- Configuración de reactivos. Hacer 1 x de tampón de solución salina tamponada (PBS) fosfato (1.000 mL), añadir 100 mL de PBS (10 x) en 900 mL de agua y mezclar bien. Hacer el tampón de lisis (50 mL) añadiendo 43,5 mL de agua a un tubo de 50 mL. Añadir 500 μL de Tris (1 M, pH 8.0), 1 mL de cloruro de sodio (NaCl) (5 M), 2,5 mL de ácido etilendiaminotetracético (EDTA) (0,5 M, pH 8.0) y 2,5 mL de sodio dodecil sulfato (SDS) (10%, peso/vol) en el tubo y mezclar bien.
Nota: Este tampón PBS puede almacenarse a 4 ° C hasta por 6 meses. El tampón de lisis predefinidos puede almacenarse a temperatura ambiente para hasta 2 meses. - Verificar la mortalidad celular y contar las células. Asegúrese de que la relación viva es > 85% y el número total de células es de 30 x 106.
Nota: Las células utilizadas en este protocolo son de la línea celular de HG00733, una línea de células linfoblastoides humanas de origen puertorriqueño, ampliamente utilizado en el consorcio del genoma de 1000 para el análisis de la variación estructural (véase tabla de materiales para ordenar información), que pertenece a genoma internacional muestra recursos. - Recogen las células por centrifugación a 200 x g durante 5 min a TA. descartar el medio y resuspender el precipitado de células (30 x 106 células) con 5 mL de tampón de PBS 1 x. Centrifugar nuevamente a 200 x g durante 5 min a temperatura ambiente y descarte el sobrenadante.
Nota: 25-35 x 106 células son aceptables para este enfoque. Más variación en la cantidad de células tendrán mayor optimización. El precipitado de células puede almacenarse en el ° C −80 hasta por 6 meses. - Resuspender el precipitado de células en 200 μL de tampón de PBS 1 x. Si utiliza un precipitado de células congeladas, lavar con 5 mL de tampón de PBS 1 x. Centrifugue la solución a 200 x g durante 5 min a temperatura ambiente, desechar el sobrenadante y resuspender las células en 200 μL de 1 x de tampón PBS.
- Preparar 10 mL de tampón de lisis en un tubo de 50 mL. Añadir la suspensión de 200 μL de células al tampón de lisis y vortex a máxima velocidad para incubar s. 3 la solución a 37 ° C durante 1 hora.
- Añadir 2 μL de Rnasa A (100 mg/mL) para el lisado. Gire suavemente el tubo de 50 mL para mezclar la muestra. Incubar la solución a 37 ° C durante 1 hora.
- Añadir proteinasa μL 50 K (20 mg/mL) para el lisado. Gire suavemente el tubo de 50 mL para mezclar la muestra. Incubar la solución a 50 ° C por 2 h. Durante la incubación, mezclar suavemente la muestra cada 30 minutos.
- Quite el tubo de 50 mL de 50 ° C y dejar reposar a temperatura ambiente durante 5 minutos.
- Añadir 10 mL de la capa de fenol de alcohol fenol: cloroformo: isoamílico (25:24:1, vol/vol/vol) de lisado y gire el tubo en un mezclador de rotor (véase Tabla de materiales) a temperatura ambiente en una campana de humos a 20 rpm durante 10 minutos envolver la tapa del tubo con parafilm para evitar fugas durante la rotación.
- Preparar dos tubos de gel de 50 mL (véase Tabla de materiales) por centrifugación a 1.500 x g durante 2 min a TA.
Nota: El gel forma una barrera estable entre la fase acuosa que contiene ácido nucleico y el solvente orgánico. - Verter la solución de muestra/fenol en uno de los preparados 50 mL tubos de gel de paso 1.10. Centrifugue la solución a 3.000 x g durante 10 min a TA.
- Vierta el sobrenadante en un nuevo tubo de 50 mL. Añadir 10 mL de la capa de fenol de alcohol fenol: cloroformo: isoamílico (25:24:1, vol/vol/vol) y gire el tubo en un mezclador de rotor a RT en una campana de humos a 20 rpm durante 10 minutos.
- Repita el tubo del gel de paso 1.11 una vez con el segundo preparado.
- Vierta el sobrenadante en un nuevo tubo de 50 mL. Añadir 25 mL de etanol 100% helado y gire suavemente el tubo con la mano hasta que el ADN precipita (figura 2).
Nota: El método de precipitación ayuda a estabilizar el ADN de HMW. - Doblar una punta de 20 μL para hacer un gancho. Cuidadosamente saque el ADN de HMW con el gancho y deje la gota líquida.
- Coloque el ADN de HMW en un tubo de 50 mL que contiene 40 mL de etanol al 70%. Lavar el ADN suavemente invirtiendo el tubo 3 veces.
- Repita el paso 1.15 una vez para recoger el ADN del tubo de etanol al 70%.
- Coloque el ADN de HMW en un tubo de 2 mL que contiene 1,8 mL de etanol al 70%.
- Centrifugue el ADN de HMW lavado a 10.000 x g durante 3 s en RT. quitar tanto el etanol residual posible mediante pipeteo.
Nota: No molestar el pellet de ADN cuando el etanol residual el pipeteo. - Incubar el tubo de 2 mL a 37 ° C por 10 min con la tapa abierta para secar la muestra.
- Si continúa con el paso 2.1 (con esquila mecánica y 1 Kit de secuenciación de la ligadura de D), añadir 1 mL de TE (10 mM Tris y 1 mM EDTA, pH 8.0) para el tubo de 2 mL.
- Si continúa con el paso 2.2 (con fragmentación basada en la transposasa y Kit rápido de la secuencia), añadir 200 μL de 10 mM Tris (pH 8.0), 0.02% Tritón X-100.
Nota: No molestar el pellet de DNA. Dejar el tubo reposar a 4 ° C en oscuridad 48 h ayudará la muestra resuspender completamente. El ADN de HMW pueden almacenarse a 4 ° C hasta por 2 semanas. Tiempo de almacenamiento u otras condiciones de almacenamiento de información pueden introducir más pequeños fragmentos.
2. Ultra largo DNA biblioteca de la construcción
Nota: Hay dos formas de construir las bibliotecas de ADN muy largas basadas en dos métodos diferentes de corte juntados con kits de secuenciación nanopore. Una biblioteca de base de esquila mecánica produce datos con una N50 de 50-70 kb, teniendo alrededor de 8 h para la construcción de la biblioteca. Una biblioteca basada en la fragmentación de la transposasa produce una N50 de 90-100 kb de datos, tomando sólo 90 min para la construcción de la biblioteca. El protocolo de corte mecánico da mayor rendimiento desde el mismo ADN de entrada utilizar versiones idénticas de adaptador de la secuencia y calidad de las células de flujo nanopore.
- Construcción de biblioteca base de esquila mecánica
- Descongelar y mezclar los reactivos de la ligadura kit (véase Tabla de materiales). Deshielo FFPE ADN reparación final dA/reparación-tailing tampón en el hielo, entonces el vórtice y búfer y exprimido hasta la mezcla. Descongele la mezcla de adaptador (AMX) y adaptador grano enlace buffer (ABB) en hielo, después pipeta y vuelta abajo para mezclar. Deshielo bajando buffer de combustible (RBF) y tampón de elución (ELB) en RT, entonces vortex y vuelta a mezclar. Descongele la biblioteca carga granos (LLB) en RT y pipeta para mezclar antes de usar.
- Una vez descongelado, mantenga todos los componentes del kit en el hielo. Sacar las enzimas sólo cuando sea necesario. Llevar las bolas magnéticas a RT de uso.
Nota: Recomendaciones sobre las bolas magnéticas a utilizar consulte la tabla de materiales.
- Una vez descongelado, mantenga todos los componentes del kit en el hielo. Sacar las enzimas sólo cuando sea necesario. Llevar las bolas magnéticas a RT de uso.
- Compruebe la calidad y cantidad del ADN de HMW de paso 1.21.1. Pipeta de 20 μL de ADN en nuevos tubos de 1,5 mL de tres lugares diferentes en el tubo de ADN de HMW con P200 amplia dio consejos. Toma 1 μL de las tres alícuotas para detectar la concentración con un fluorómetro y la calidad, utilizando una lectura de UV. Revise varias veces para confirmar los resultados.
Nota: Los resultados esperados se muestran en la Figura 3A. El valor de260/280 de OD es aproximadamente 1,9 y el valor de OD260/230 es aproximadamente 2.3. - Transferencia el restante 940 μL de ADN de HMW en un tapón de tubo de 50 mL con una P1000 amplia alesaje de punta.
- Aspire todo ADN en una jeringa de 1 mL sin la aguja.
- Colocar la aguja de 27 G en la jeringa y expulse todo ADN en la tapa suavemente y lentamente (~ 10 s). Sacar la aguja de 27 G de la jeringa.
- Repita los pasos 2.1.4 y 2.1.5 para 29 veces para un total de 30 pasa a través de la aguja.
Nota: El ADN de HMW esquilado pueden almacenarse a 4 ° C en la oscuridad de hasta 24 h. control de calidad (QC) es altamente recomendado por electroforesis en gel de campo pulsado, pero es costoso y desperdiciador de tiempo. Si se realiza control de calidad en una máquina de electroforesis de pulso automático campo gel utilizar un protocolo de 5-150 kb para un 20 h ejecutar. Los resultados esperados se muestran en la figura 4. - Preparar la reacción de reparación de ADN en un tubo de 0.2 mL por adición de 100 μL de ADN de HMW esquilado (20 μg), 15 μL de tampón de reparación de ADN FFPE, 12 μL de mezcla de reparación de ADN FFPE y 16 μL de agua libre de nucleasa. La reacción de la mezcla por sacudiendo suavemente 6 veces y girar hacia abajo para quitar las burbujas.
- Incubar la reacción a 20 ° C por 60 min transferencia de la muestra en un tubo de 1,5 mL nueva con una punta de calibre ancho P200.
- Resuspender las bolas magnéticas por pipeteo o vórtex. Agregar granos μL 143 (1 x) a la reacción de reparación de ADN y mezcla suavemente agitando el tubo 6 veces. Gire el tubo en un mezclador de rotor a temperatura ambiente a 20 rpm por 30 min.
- Desactivación de la muestra a 1.000 x g durante 2 s en lugar de RT. el tubo en un estante magnético durante 10 minutos, mantenga el tubo en la rejilla magnética y descarte el sobrenadante.
- Manteniendo el tubo en la rejilla magnética, añadir 400 μL de etanol 70% recién preparada sin perturbar el pellet. Eliminar el etanol del 70% después de 30 s.
- Repita el paso 2.1.11 una vez.
- Desactivación de la muestra a 1.000 x g durante 2 s en RT. Coloque el tubo de vuelta sobre la parrilla magnética. Retire cualquier etanol residual y del aire seco de 30 s. No secar sobre el sedimento.
- Retire el tubo de la rejilla magnética y añadir 103 μL de TE (10 mM Tris y 1 mM EDTA, pH 8.0). Suavemente flick el tubo para asegurar que los granos están cubiertos en el buffer e incuban en un mezclador de rotor a temperatura ambiente durante 30 minutos suavemente flick el tubo cada 5 min para facilitar la resuspensión de la pelotilla.
- Los granos en la rejilla magnética de pellets para por lo menos 10 minutos transferir 100 μL de eluido con una P200 amplia punta en un tubo de 0.2 mL.
- Preparar la final reparación y dA seguimiento la reacción en un tubo de 0.2 mL mediante la adición de 100 μL de ADN de HMW reparado, 14 μL de tampón de reparación/dA-relaves final y 7 μL de la mezcla de reparación/dA-relave final. La reacción de la mezcla por sacudiendo suavemente 6 veces y girar hacia abajo para quitar las burbujas.
- Incubar la reacción a 20 ° C durante 60 min seguido de 65 ° C por 20 min y luego mantener a 22° C. Transferencia de la muestra en un tubo de 1,5 mL nuevo utilizando una P200 gran diámetro punta.
- Resuspender las bolas magnéticas por pipeteo o vórtex. Añadir 48 μL de granos (0.4 x) a la reacción de reparación/dA-relaves final y mezcla suavemente por sacudiendo el tubo 6 veces. Gire el tubo en un mezclador de rotor a temperatura ambiente a 20 rpm por 30 min.
- Repita los pasos 2.1.10-2.1.13 una vez.
- Retire el tubo de la rejilla magnética y añadir 33 μL de TE (10 mM Tris y 1 mM EDTA, pH 8.0). Suavemente flick el tubo para asegurar que los granos están cubiertos en el buffer e incuban en un mezclador de rotor a temperatura ambiente durante 30 minutos suavemente flick el tubo cada 5 min para facilitar la resuspensión de la pelotilla.
- Los granos en la rejilla magnética de pellets para al menos 10 min transferencia 30 μL de eluido con una P200 amplia punta en un nuevo tubo de 1,5 mL. Tomar el extra 1-2 μL para detectar la concentración con un fluorómetro.
Nota: Se espera una recuperación de 5-6 μg en este paso. - Preparar la reacción de la ligadura en el tubo de muestra de 1,5 mL por añadir 30 μL de ADN de HMW final reparado, 20 μL de mezcla de adaptador (AMX 1D) y 50 μL de la mezcla principal de ligadura blunt/TA. Mezcla de la reacción por sacudiendo suavemente 6 veces entre cada adición secuencial y vuelta hacia abajo para quitar las burbujas.
- Incubar la reacción a temperatura ambiente durante 60 minutos.
- Resuspender las bolas magnéticas por pipeteo o vórtex. Añadir 40 perlas μL (0.4 x) a la reacción de la ligadura y la mezcla suavemente por sacudiendo el tubo 6 veces. Gire el tubo en un mezclador de rotor a temperatura ambiente a 20 rpm por 30 min.
- Repita el paso 2.1.10 una vez.
- Añadir 400 μL de tampón de unión (ABB) de grano de adaptador en el tubo. Flick el tubo suavemente 6 veces a suspender las cuentas. Coloque el tubo en la rejilla magnética para separar los granos del buffer y descartar el sobrenadante.
- Repita el paso 2.1.26 una vez.
- Desactivación de la muestra a 1.000 x g durante 2 s en RT. Coloque el tubo de vuelta sobre la parrilla magnética. Elimine cualquier tampón residual y del aire seco de 30 s. No secar sobre el sedimento.
- Quite el tubo de la rejilla magnética y resuspender el precipitado en 43 μL de tampón de elución. Suavemente flick el tubo para asegurar que los granos están cubiertos en el buffer e incuban en un mezclador de rotor a temperatura ambiente durante 30 minutos suavemente flick el tubo cada 5 min para facilitar la resuspensión de la pelotilla.
- Los granos en la rejilla magnética de pellets para al menos 10 min transferencia 40 μL del eluido con una P200 amplia punta en un nuevo tubo de 1,5 mL. Tomar el extra 1-2 μL para detectar la concentración con un fluorómetro.
Nota: Se espera una recuperación de 1-2 μg en este paso. La biblioteca base de esquila mecánica está lista para la carga. La biblioteca se puede almacenar en hielo para un máximo de 2 h hasta la carga de la secuencia si es necesario.
- Descongelar y mezclar los reactivos de la ligadura kit (véase Tabla de materiales). Deshielo FFPE ADN reparación final dA/reparación-tailing tampón en el hielo, entonces el vórtice y búfer y exprimido hasta la mezcla. Descongele la mezcla de adaptador (AMX) y adaptador grano enlace buffer (ABB) en hielo, después pipeta y vuelta abajo para mezclar. Deshielo bajando buffer de combustible (RBF) y tampón de elución (ELB) en RT, entonces vortex y vuelta a mezclar. Descongele la biblioteca carga granos (LLB) en RT y pipeta para mezclar antes de usar.
- Construcción de biblioteca basada en la fragmentación transposasa
- Descongelar los reactivos de la transposasa kit (véase Tabla de materiales). Descongele la fragmentación mix (FRA) y adaptador rápido (RAP) en el hielo y pipeta para mezclar. Descongelar el búfer de la secuencia (SQB), granos (LB) de carga, descarga buffer (FLB) y flush tether (FLT) en RT y pipeta para mezclar. Descongelar los granos de carga (LB) en RT y pipeta para mezclar antes de usar. Una vez descongelado, mantenga todos los componentes del kit en el hielo. Sacar las enzimas sólo cuando sea necesario.
- Compruebe la calidad y cantidad del ADN de HMW de paso 1.21.2. Pipeta de 20 μL de ADN en nuevos tubos de 1,5 mL de tres lugares diferentes en el tubo de ADN de HMW con P200 amplia dio consejos. Toma 1 μL de las tres alícuotas para detectar la concentración con un fluorómetro y la calidad, utilizando una lectura de UV. Revise varias veces para confirmar los resultados.
Nota: Los resultados esperados se muestran en la figura 3B. El valor de260/280 de OD es aproximadamente 1,9 y el valor de OD260/230 es aproximadamente 2.3. - Preparar la reacción de tagmentation de ADN en un tubo de 0.2 mL agregando 22 μL de ADN de HMW, 1 μL de 10 mM Tris (pH 8.0) con 0,02% Tritón X-100 y 1 μL de fragmentación (FRA) de la mezcla. Mezclar mediante pipeteo con una P200 ancho diámetro punta tan lentamente como sea posible 6 veces, teniendo cuidado de no para introducir burbujas.
- Incubar la reacción a 30 ° C durante 1 min seguido por 80 ° C por 1 min y luego mantener a 4 ° C. Transferencia de la mezcla en un tubo nuevo de 1,5 mL con una P200 amplia diámetro punta e ir al siguiente paso inmediato.
- Añadir 1 μL de adaptador rápido (RAP) para el tubo de muestra de 1,5 mL. Mezclar mediante pipeteo con una P200 ancho diámetro punta tan lentamente como sea posible 6 veces, teniendo cuidado de no para introducir burbujas.
- Incubar la reacción a temperatura ambiente durante 60 minutos.
Nota: La biblioteca basada en la fragmentación de la transposasa está lista para la carga. La biblioteca se puede almacenar en hielo para un máximo de 2 h hasta la carga de la secuencia si es necesario.
3. la secuencia en el dispositivo nanopore
- Verifique el dispositivo de secuencia nanopore (véase Tabla de materiales). Asegúrese de que tanto el software y el hardware están trabajando y hay suficiente espacio de almacenamiento.
- Compruebe la célula de flujo. Abrir una nueva célula de flujo e insertar la celda de flujo en el dispositivo nanopore. Compruebe que el cuadro de la ubicación de la celda de flujo fue insertado en (X1-X5). Seleccione el tipo de la célula de flujo correcto. Haga clic en el flujo de trabajo de Controlar flujo de células . Haga clic en el botón Iniciar prueba para iniciar la célula de flujo análisis de CC.
Nota: Si el número de activo total divulgado del poro es menos de 800, utilice una nueva célula de flujo diferentes de secuenciación. - Preparar el buffer de cebado. Para una biblioteca de base de corte mecánica, añada 576 μL de funcionamiento amortiguador de combustible (RBF) y 624 μL de agua libre de nucleasas en un tubo de 1,5 mL. Vórtice y vuelta hasta mezcla el búfer de cebado. Para una biblioteca basada en la fragmentación transposasa, Añadir 30 μL de anclaje al ras (FLT) al tubo de tampón de lavado (FLB). Vórtice y vuelta hasta mezcla el búfer de cebado.
- En la celda de flujo, gire la tapa del puerto cargado hacia la derecha para exponer el puerto de cebado.
- Establezca una pipetas P1000 en 100 μL e inserte la punta en el orificio de cebado. Retroceder un pequeño volumen de tampón (menos de 30 μL) para eliminar las posibles burbujas de la célula de flujo. Deje de pipeteado una vez una pequeña cantidad de líquido amarillo entra en la punta.
- Utilice una pipeta P1000 carga 800 μL de la mezcla de cebado en la célula de flujo vía el puerto de cebado. Para evitar la introducción de burbujas, Añadir 30 μL de la mezcla de cebado a la tapa del puerto de cebado en primer lugar, y luego introduzca la punta en el orificio de cebado y agregue lentamente el resto de la mezcla de cebado. Sacar la punta cuando hay alrededor de 50 μL de izquierda. Agregar el resto de la mezcla de oscurecimiento en la parte superior del puerto de cebado. El fluido irá dentro de sí mismo.
- Dejar la configuración a incubar durante 5 minutos. Mientras tanto, preparar la mezcla de la biblioteca en el tubo de 1,5 mL que contiene la biblioteca.
Nota: Para una biblioteca de base de esquila mecánica añadir 35 μl de que buffer de combustible (RBF) 40 μl de la biblioteca de ADN. Para una biblioteca basada en la fragmentación de la transposasa añadir 34 μl de tampón de secuenciación (SQB) y 16 μl de agua libre de nucleasa para 25 μl de la biblioteca de ADN. - Abra la cubierta de puerto de flujo celular muestra con suavidad para exponer el puerto de la muestra. Utilice una pipeta P1000 para añadir 200 μL de la mezcla de cebado a través del puerto de cebado en la célula de flujo, como se describe en el paso 3.5. Asegúrese de que la mezcla de cebado no está cargada en la celda de flujo a través del puerto de la muestra.
- Ponga una pipeta P200 en 80 μL. Mezclar suavemente la biblioteca con una punta de gran diámetro mediante pipeteo arriba abajo 6 veces antes de cargar.
- Cargar la mezcla de biblioteca gota a gota a través del puerto de la muestra en la celda de flujo. Agregar cada gota sólo después de la caída anterior es completamente cargada en el puerto.
- Vuelva a colocar la tapa del puerto muestra suavemente y asegúrese de que el puerto de la muestra es totalmente cubierto. Mueva la cubierta del puerto de cebado en sentido antihorario hasta que cubra el orificio de cebado. Cierre la tapa del dispositivo.
- Haga clic en el flujo de trabajo Nuevo experimento . Escriba el nombre de la biblioteca, seleccionar el kit correcto según los procedimientos utilizados y compruebe que la configuración es correcta (48 h ejecutar, en tiempo real ON base-calling).
- Haga clic en Inicio ejecutar. Después de 10 minutos, registrar la ID de celda de flujo y los números de activos nanopore (número total y número de cada cuatro grupos) de la información de ejecución.
- Análisis de los datos. Copiar los datos en un equipo local o un grupo en cualquier momento de la secuenciación o cuando termine la carrera. Utilice Minimap216 (https://github.com/lh3/minimap2) para alinear los datos en secuencia el genoma de referencia. Resumir el funcionamiento de la secuencia de los datos sin procesar de la secuencia y las alineaciones por NanoPlot17 (https://github.com/wdecoster/NanoPlot).
Resultados
El protocolo de secuenciación de ADN ultra largo aplica APM ADN para la construcción de la biblioteca. Por lo tanto, es fundamental elegir bien cultivos de células con el cociente directo > 85% en la celda de recolección paso. La cantidad de células utilizadas para la extracción de ADN afecta la calidad y la cantidad de ADN de HMW. La lisis celular no funciona bien si a partir de muchas células. Con muy pocas células no genera suficiente ADN para la construcción de la biblioteca porque la precipitación de ADN de HMW se realiza mediante rotación suave a mano en lugar de centrifugación de alta velocidad. Un ejemplo del ADN de HMW después agregar helado 100% de etanol y de rotación se muestran como el precipitado blanco de algodón-como en la figura 2.
Es importante comprobar la calidad de la entrada de ADN antes de comenzar la construcción de la biblioteca. Degradación, cuantificación incorrecta, contaminación (p. ej., proteínas, ARN, detergentes, surfactante y fenol residual o etanol) y ADN de bajo peso molecular pueden tener un efecto significativo sobre los procedimientos posteriores y sobre el final leer longitud. Se recomienda realizar el análisis de control de calidad utilizando el ADN de tres lugares diferentes en el tubo que contiene el ADN de HMW. De lectura de los resultados de los ADN de HMW de Ultravioleta, el OD260/do280 valor es aproximadamente 1,9 y el OD260/do230 valor es aproximadamente 2.3 (Figura 3AB). Estos valores de relación son consistentes entre las tres pruebas de una buena muestra de ADN de HMW. Diferentes métodos de corte requiere diferentes volúmenes de DNA entrada. La concentración de ADN de HMW debe ser > 200 ng/μL para mecánico de corte mientras que debe ser > 1 μg/μl de fragmentación de la transposasa. La concentración detectada por un Fluorímetro es un poco menor que UV leyendo. Sin embargo, el coeficiente de variación de la concentración de la misma muestra de ADN de HMW debe ser inferior al 15% con el fluorómetro y la UV leer ensayos. Esquila mecánica, aplica una jeringa con una aguja para romper el ADN de HMW lo que el número de pasadas a través de la aguja tendrá un impacto del tamaño de ADN esquilado y la final Lee longitud. Se recomienda realizar tamaño del control de calidad después de aguja de corte para asegurar la mayoría de los ADN de HMW es mayor que 50 kb como se ilustra en la figura 4. En el método de esquila mecánico, 30 pasadas generan los mejores resultados de la secuencia teniendo en cuenta la longitud y la salida.
N50 de una biblioteca de base de esquila mecánica es de 50-70 kb mientras que una biblioteca basada en la fragmentación de la transposasa es 90-100 kb. Los resultados de cuatro carreras utilizando la línea celular de HG00733 se muestran en la tabla 1. Todas las ejecuciones de cuatro tienen más de 2.300 lecturas con longitud superior a 100 kb. La longitud máxima es mayor en la transposasa fragmentación basado en bibliotecas (455 kb y 489 kb) en comparación con las bibliotecas de base de corte mecánicas (348 kb y 387 kb) mientras que estos últimos producen más lecturas total, indicando un rendimiento superior. La construcción de biblioteca basada en la fragmentación de la transposasa tiene menos pasos y menor tiempo de preparación para que presentará menos fragmentos cortos. Las dos carreras con la transposasa tienen una mayor longitud media (> 30 kb) y de longitud mediana (> 10 kb). Además, los datos demuestran alta calidad constante en todas las ejecuciones (puntuación de calidad media es aproximadamente 10.0, ~ 90% de precisión base). Más del 97% de total de bases fueron alineado con el genoma humano de referencia (hg19) usando Minimap216 con la configuración predeterminada. Las distribuciones de tamaño esperado de la materia prima Lee se muestran en la figura 5. Todas las ejecuciones tienen una gran proporción de datos por encima de 50 kb mientras que transposasa basado en la fragmentación de las bibliotecas tienen un cociente más alto de Lee muy largo (por ejemplo de > 100 kb). Este protocolo se ha aplicado con éxito en múltiples líneas celulares humanas (complementario tabla 1).

Figura 1: Descripción esquemática del flujo de trabajo de la secuencia larga de leer (NLR-seq) nanopore. Naranja, la transposasa complejo. Amarillo-verde, el adaptador nanopore. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: precipitación de ADN representativas de método de extracción de fenol-cloroformo. La flecha blanca indica el ADN de HMW. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: resultados del ejemplo del control de calidad del ADN de HMW de lectura UV. ADN de HMW (A) de paso 1.21.1 listo para construcción de biblioteca basado en el corte mecánico. ADN de HMW (B) de paso 1.21.2 para construcción de biblioteca basada en la fragmentación transposasa. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: resultados del control de calidad de la aguja esquilan HMW ADN por electroforesis del gel del pulsar-campo. L1: Rápido-carga escalera de DNA de 1 kb; L2: Rápido-carga 1 kb ampliar la escalera de ADN. 1-8: ADN con tiempos de paso diferentes a través de la aguja de corte. 1-3, ninguna corte; 4, 10 veces; 5, 20 veces; 6, 30 veces; 7, 40 veces; 8, 50 veces. Este paso de control de calidad es opcional. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: espera que las distribuciones de tamaño de las bibliotecas de ADN ultra-larga nanopore. MS, bibliotecas de base de corte mecánicas. TF, transposasa bibliotecas basado en la fragmentación. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Shearing_rep1 mecánica | Shearing_rep2 mecánica | Transposasa fragmentation_rep1 | Transposasa fragmentation_rep2 | |
| Línea celular | HG00733 | HG00733 | HG00733 | HG00733 |
| N50 de Lee | 55.180 | 63.007 | 98.237 | 95.629 |
| Número de lecturas de más de 100 Kb | 2.500 | 3.082 | 2.386 | 2.355 |
| Número de lecturas total | 97.859 | 80.465 | 24.166 | 21.032 |
| Longitud máxima (bp) | 348.482 | 387.113 | 454.660 | 489.426 |
| Longitud media (bp) | 17.861 | 20.395 | 33.528 | 38.175 |
| Longitud media (bp) | 5.335 | 5.894 | 10.249 | 15.656 |
| Calidad media de las lecturas | 10.0 | 10.1 | 9.9 | 10.0 |
| Total de bases de la materia prima Lee | 1,747,849,822 | 1,641,058,932 | 810,229,733 | 802,886,304 |
| Total de bases de Lee alineada | 1,693,300,832 | 1,607,975,925 | 791,422,077 | 778,417,627 |
| Proporción asignada de total de bases (hg19, Minimap2) | 96.9% | 98.0% | 97.7% | 97.0% |
| Número de poros activos | 1225: 480, 402, 254, 89 | 1058: 480, 176, 356, 46 | 958: 452, 328, 148, 30 | 1092: 487, 367, 195, 43 |
Tabla 1: Resumen de indicadores de rendimiento funciona con diferentes protocolos de corte.
| Biblioteca 1 | Biblioteca 2 | |
| Línea celular | K562 | GM19240 |
| Información de la célula | ATCC, cat. lol CCL-243 | Instituto Coriell, cat. lol GM19240 |
| Protocolo | esquila mecánica | esquila mecánica |
| N50 de Lee | 60.063 | 55.295 |
| Número de lecturas total | 193.783 | 120.807 |
| Longitud media (bp) | 1.843 | 4.688 |
| Longitud media (bp) | 9.825 | 17.408 |
| Longitud máxima (bp) | 548.780 | 212.338 |
| Total de bases de la materia prima Lee | 1,903,989,686 | 2,103,015,331 |
| Total de bases de Lee alineada | 1,837,350,047 | 1,997,419,761 |
| Proporción asignada de total de bases (hg19, Minimap2) | 96,6% | 95.0% |
| Número de poros activos | 1111: 482, 371, 203, 55 | 1032: 447, 333, 196, 56 |
Complementario tabla 1: Resumen de dos carreras de NLR-seq con otras líneas celulares con el protocolo de corte mecánico.
Discusión
En principio, la secuencia nanopore es capaz de generar 100 kb a megabase lee en longitud11,12,13. Cuatro principales factores afectarán el desempeño de la calidad de ejecución y los datos de la secuencia: 1) activo poro números y la actividad de los poros; 2) proteína motor, que controla la velocidad de ADN pasando por nanopore; 3) plantilla ADN (longitud, pureza, calidad, total); 4) secuencia adaptador ligadura eficiencia, que determina el ADN utilizable de la muestra de entrada. Los dos primeros factores dependen de la versión de la célula de flujo y el kit de secuenciación proporcionado por el fabricante. El segundo dos factores son pasos críticos en este protocolo (extracción de ADN de HMW, corte y ligadura).
Este protocolo requiere paciencia y práctica. La calidad del ADN de HMW es importante ultra-larga de bibliotecas de ADN6. El protocolo comienza con las células con alta viabilidad (> 85% de células viables preferido), limitando el ADN degradado de las células muertas. Debe evitarse cualquier proceso áspero que puede presentar daños a la DNA (por ejemplo, fuerte inquietante, sacudiendo, vortex, múltiples pipeteo, repetida congelación y descongelación). En el diseño del Protocolo, omitimos el pipeteo en todo el proceso de extracción de ADN. Consejos de gran diámetro deben usarse cuando el pipeteo es necesario después de la esquila mecánica durante la construcción de la biblioteca y la secuencia. Como el interconectivos son sensibles a los químicos en la cámara buffer12, debe haber contaminantes residuales como pocos (p. ej., detergentes, tensioactivos, fenol, etanol, proteínas ARN, etc.) como sea posible en la DNA. Teniendo en cuenta la longitud y el rendimiento, el método de extracción de fenol muestra los resultados mejores y más reproducibles en comparación con varios diferentes métodos de extracción probados hasta ahora.
A pesar de la capacidad de este protocolo para producir secuencias de lectura larga, varias limitaciones siguen. En primer lugar, este protocolo fue optimizada basada en el dispositivo de secuencia nanopore disponible en el momento de su publicación; por lo tanto, se limita a la química de secuenciación selectiva basada en nanopore y podría ser subóptima cuando se realiza en otros tipos de dispositivos de lectura larga secuencia. En segundo lugar, el resultado es altamente dependiente de la calidad del ADN extraído de material de partida (tejidos o las células). Longitud de lectura podría deteriorarse si el ADN partido ya está degradado o dañado. En tercer lugar, aunque varios pasos de control de calidad están incorporados en el protocolo para comprobar la calidad del ADN, el rendimiento final y la longitud de las lecturas pueden verse afectadas por la celda de flujo y poro actividad, que puede ser variable en esta primera etapa de la plataforma de secuenciación nanopore desarrollo.
El protocolo descrito aquí utiliza muestras de línea celular humana suspensión para extracción de ADN. Hemos optimizado los tiempos de paso de aguja de corte, la relación del ADN de HMW transposasa y el momento de la ligadura para producir los resultados descritos. El protocolo puede ser ampliado en cuatro maneras. En primer lugar, los usuarios pueden iniciar con otras células mamíferos cultivadas y con diferente cantidad de células, tejidos, muestras clínicas u otros organismos. Se necesitará mayor optimización en el tiempo de lisis de incubación, volumen de reacción y centrifugación. En segundo lugar, es difícil predecir el tamaño de destino para la secuencia de lectura ultra largo. Si la lectura son más cortas de lo esperado, los usuarios pueden ajustar los tiempos de paso en el método basado en el corte mecánico o cambiar la relación entre el ADN de HMW a transposasa en el método basado en la fragmentación de la transposasa. Tiempo de encuadernación y elución durante los pasos de limpieza son útiles porque el ADN de HMW es altamente viscoso. En tercer lugar, con dispositivos de secuencia diferentes nanopore, uno puede ajustar la cantidad y el volumen de la DNA para cumplir los criterios del secuenciador. Cuarto, solamente ésos DNA ligada a adaptadores de secuencia va ser secuenciado. Para mejorar aún más la eficacia de la ligadura, uno puede intentar valorar las concentraciones de adaptador y ligasa. Tiempo de ligadura modificada y agentes crowding moleculares como PEG18 pueden aplicarse en el futuro. El protocolo de secuenciación de ADN ultra largo combinado con CRISPR19,20 puede ofrecer una herramienta eficaz para la secuencia de enriquecimiento objetivo.
Divulgaciones
Los autores declaran que no tienen intereses financieros que compiten.
Agradecimientos
Los autores agradecen a Zhu Y. por sus comentarios sobre el manuscrito. Investigación en esta publicación fue apoyada parcialmente por el Instituto Nacional del cáncer de los institutos nacionales de salud bajo la concesión número P30CA034196. El contenido es responsabilidad exclusiva de los autores y no representan necesariamente las opiniones oficiales de los institutos nacionales de salud.
Materiales
| Name | Company | Catalog Number | Comments |
| Reagents | |||
| Absolute ethanol | Sigma-Aldrich | E7023 | |
| Agencourt AMPure XPbeads | Beckman | A63881 | magnetic beads for cleanup |
| BD conventional needles | Becton Dickinson | 305136 | 27G, for mechanical shearing |
| BD Luer-Lok syringe | Becton Dickinson | 309628 | for mechanical shearing |
| Blunt/TA Ligase Master Mix | NEB | M0367S | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | for cell counting |
| EDTA | Invitrogen | AM9261 | pH 8.0, 0.5 M, 500 mL |
| Flow Cell | Oxford Nanopore Technologies | FLO-MIN106 | R9.4.1 |
| HG00773 cells | Coriell Institute | HG00733 | cells used in this protocol |
| Ligation Sequencing Kit 1D | Oxford Nanopore Technologies | SQK-LSK108 | nanopore ligation kit |
| MaXtract High Density tubes | Qiagen | 129073 | gel tubes |
| NEBNext FFPE DNA Repair Mix | NEB | M6630S | |
| NEBNext Ultra II End Repair/dA-Tailing Module | NEB | M7546S | |
| Nuclease-free water | Invitrogen | AM9937 | |
| Phosphate-Buffered Saline, PBS | Gibco | 70011044 | 10X, pH 7.4 |
| Phenol:chloroform:IAA | Invitrogen | AM9730 | |
| Proteinase K | Qiagen | 19131 | 20 mg/mL |
| Qubit dsDNA BR Assay Kit | Invitrogen | Q32850 | fluorometer assays for DNA quantification |
| Rapid Sequencing Kit | Oxford Nanopore Technologies | SQK-RAD004 | nanopore transposase kit |
| RNase A | Qiagen | 19101 | 100 mg/mL |
| SDS | Invitrogen | AM9822 | 10% (wt/vol) |
| Sodium chloride solution | Invitrogen | AM9759 | 5.0 M |
| TE buffer | Invitrogen | AM9849 | pH 8.0 |
| Tris | Invitrogen | AM9856 | pH 8.0, 1 M |
| Triton X-100 solution | Sigma-Aldrich | 93443 | ~10% |
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| Bio-Rad C1000 Thermal Cycler | Bio-Rad | 1851196EDU | |
| Centrifuge 5810R | Eppendorf | 22628180 | |
| Countess II FL Automated Cell Counter | Life Technologies | AMQAF1000 | for cell counting |
| DynaMag-2 Magnet | Life Technologies | 12321D | magnetic rack |
| Eppendorf ThermoMixer | Eppendorf | 5382000023 | for incubation |
| Freezer | LabRepCo | LHP-5-UFMB | |
| GridION | Oxford Nanopore Technologies | GridION X5 | nanopore device used in this protocol |
| HulaMixer Sample Mixer | Thermo Fisher Scientific | 15920D | rotator mixer |
| MicroCentrifuge | Benchmark Scientific | C1012 | |
| NanoDrop ND-1000 Spectrophotometer | Thermo Fisher Scientific | ND-1000 | for UV reading |
| Pippin Pulse | Sage Science | PPI0200 | pulsed-field gel electrophoresis instrument |
| Qubit 3.0 Fluorometer | Invitrogen | Q33216 | fluorometer |
| Refrigerator | LabRepCo | LABHP-5-URBSS | |
| Vortex-Genie 2 | Scientific Industries | SI-A236 | |
| Water bath | VWR | 89501-464 |
Referencias
- Mardis, E. R. Next-generation sequencing platforms. Annual Review of Analytical Chemistry. 6, 287-303 (2013).
- Goodwin, S., McPherson, J. D., McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nature Reviews Genetics. 17 (6), 333-351 (2016).
- Shendure, J., et al. DNA sequencing at 40: past, present and future. Nature. 550 (7676), 345-353 (2017).
- Alkan, C., Coe, B. P., Eichler, E. E. Genome structural variation discovery and genotyping. Nature Reviews Genetics. 12 (5), 363-376 (2011).
- Weischenfeldt, J., Symmons, O., Spitz, F., Korbel, J. O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nature Reviews Genetics. 14 (2), 125-138 (2013).
- Pollard, M. O., Gurdasani, D., Mentzer, A. J., Porter, T., Sandhu, M. S. Long reads: their purpose and place. Human Molecular Genetics. 27 (R2), R234-R241 (2018).
- Cretu Stancu, M., et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nature Communications. 8 (1), 1326 (2017).
- Gong, L., et al. Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nature Methods. 15 (6), 455-460 (2018).
- Sedlazeck, F. J., et al. Accurate detection of complex structural variations using single-molecule sequencing. Nature Methods. 15 (6), 461-468 (2018).
- Jain, M., et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology. 36 (4), 338-345 (2018).
- Jain, M., et al. Improved data analysis for the MinION nanopore sequencer. Nature Methods. 12 (4), 351-356 (2015).
- Deamer, D., Akeson, M., Branton, D. Three decades of nanopore sequencing. Nature Biotechnology. 34 (5), 518-524 (2016).
- Jain, M., Olsen, H. E., Paten, B., Akeson, M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biology. 17 (1), 239 (2016).
- Editorial, The long view on sequencing. Nature Biotechnology. 36 (4), 287 (2018).
- Jain, M., et al. Linear assembly of a human centromere on the Y chromosome. Nature Biotechnology. 36 (4), 321-323 (2018).
- Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094-3100 (2018).
- De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34, 2666-2669 (2018).
- Akabayov, B., Akabayov, S. R., Lee, S. J., Wagner, G., Richardson, C. C. Impact of macromolecular crowding on DNA replication. Nature Communications. 4, 1615 (2013).
- Gabrieli, T., Sharim, H., Michaeli, Y., Ebenstein, Y. Cas9-Assisted Targeting of CHromosome segments (CATCH) for targeted nanopore sequencing and optical genome mapping. bioRxiv. , (2017).
- Gabrieli, T., et al. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Research. , (2018).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados