Method Article
Ultra-lange lesen Sequenzierung für ganze genomische DNA-Analysen
In diesem Artikel
Zusammenfassung
Long-Read Sequenzen erleichtern erheblich die Montage komplexer Genome und Charakterisierung von strukturellen Variation. Wir beschreiben eine Methode um ultra-lange Sequenzen durch Sequenzierung Nanopore-basierte Plattformen zu generieren. Das Konzept nimmt eine optimierte DNA-Extraktion, gefolgt von modifizierten Bibliothek Vorbereitungen, Hunderte von Kilobase liest mit moderaten Abdeckung aus menschlichen Zellen zu generieren.
Zusammenfassung
Dritter Generation bieten Einzelmolekül-DNA-Sequenzierung Technologien deutlich mehr lesen Länge, die die Montage von komplexen Genomen und Analyse von komplexen strukturellen Varianten erleichtern können. Nanopore Plattformen führen Einzelmolekül-Sequenzierung durch direkte Messung der aktuellen Veränderungen vermittelt durch DNA-Passage durch die Poren und können Hunderte von Kilobase (kb) liest mit minimalen Investitionskosten zu generieren. Diese Plattform wurde von vielen Forschern für eine Vielzahl von Anwendungen übernommen. Längere Sequenzierung lesen Sie Längen zu erreichen ist der kritischste Faktor, den Wert der Nanopore Sequenzierung Plattformen zu nutzen. Um ultralangen liest zu generieren, ist besondere Aufmerksamkeit erforderlich, um DNA-Brüche zu vermeiden und Effizienz um produktive Sequenzierung Vorlagen zu erzeugen. Hier bieten wir das ausführliche Protokoll der ultra-lange DNA-Sequenzierung einschließlich hohes Molekulargewicht (HMW) DNA-Extraktion aus frischen oder gefrorenen Zellen, Bibliotheksbau durch mechanische Scherung oder Transposase Fragmentierung und Sequenzierung auf einem Nanopore Gerät. Von 20-25 µg HMW DNA die Methode erreichen N50 lesen Länge von 50-70 kb mit mechanischen Scheren und N50 von 90-100 kb Länge mit Transposase lesen vermittelt Fragmentierung. Das Protokoll kann auf aus Säugetierzellen extrahierte DNA Sequenzierung des gesamten Genoms für den Nachweis von strukturellen Varianten und Genom Montage durchführen angewendet werden. Weitere Verbesserungen auf der DNA-Extraktion und enzymatischen Reaktionen weiter lesen Sie Länge zu erhöhen und ihre Nützlichkeit zu erweitern.
Einleitung
Im letzten Jahrzehnt haben massiv parallel und hochgenaue Hochdurchsatz-zweite Generation Sequenziertechnologien eine Explosion von biomedizinischen Entdeckung und technologische Innovation1,2,3. Trotz der technischen Fortschritte die kurze lesenden Daten generiert durch die zweite Generation-Plattformen sind unwirksam bei der Lösung von komplexen genomische Regionen und sind begrenzt bei der Erkennung von genomischen strukturelle Varianten (SVs), die beim Menschen eine wichtige Rolle spielen Evolution und Krankheiten4,5. Darüber hinaus kurze gelesene Daten nicht wiederholen Variation lösen und sind ungeeignet für anspruchsvolle Haplotyp Ausstieg von genetischen Varianten6.
Jüngste Fortschritte in Einzelmolekül-Sequenzierung bietet deutlich mehr lesen Länge, die die Aufdeckung der das gesamte Spektrum der SVs7,8,9und bietet genaue und vollständige Montage des Komplexes zu erleichtern, können mikrobielle und Säugetier-Genome6,10. Die Nanopore-Plattform führt Einzelmolekül-Sequenzierung durch direkte Messung der aktuellen Veränderungen vermittelt durch DNA-Passage durch die Poren11,12,13. Im Gegensatz zu bestehenden DNA-Sequenzierung Chemie, erzeugen Nanopore Sequenzierung langer (zig Tausende von Kilobases) liest in Echtzeit ohne auf Polymerase Kinetik oder künstliche Verstärkung des DNA-Probe. Daher lange lesende Nanopore Sequenzierung (NLR-Seq) hält große Verheißung für die Erzeugung von ultra-lange lesen Sie Längen weit über 100 kb, die stark genomische und biomedizinische Analysen14fördern würde, insbesondere bei geringer Komplexität oder Repeat-reich Regionen der Genome15.
Die Besonderheit der Nanopore Sequenzierung ist, sein Potenzial zu lange ohne eine theoretische Längenbegrenzung liest. Deshalb lesen Sie Länge abhängig von der physikalischen Länge der DNA durch die DNA-Integrität und Sequenzierung Vorlage Qualität direkt betroffen ist. Darüber hinaus ist je nach Ausmaß der Manipulation und die Anzahl der Schritte, wie Pipettieren Kräfte und Extraktionsbedingungen, die Qualität der DNA sehr variabel. Daher ist es schwierig für einen zu langen liest nur mittels Standardprotokollen der DNA-Extraktion und des Herstellers mitgelieferte Bibliothek Bauweisen. Zu diesem Zweck haben wir eine robuste Methode zur Erzeugung ultralangen lesen (Hunderte von Kilobases) Sequenzierungsdaten geernteten Zelle Pellets ab. Wir haben mehrere Verbesserungen bei den DNA-Extraktion und Bibliothek Vorbereitung angenommen. Wir optimiert das Protokoll um unnötige Verfahren auszuschließen, die DNA-Schädigung und Schäden verursachen. Dieses Protokoll besteht aus hochmolekularen (HMW) DNA-Extraktion, ultra-lange DNA Bibliotheksbau und Sequenzierung auf einer Nanopore Plattform. Für ein gut ausgebildeter Molekularbiologe dauert es in der Regel 6 h aus Zelle Ernte bis zur Fertigstellung der HMW DNA-Extraktion, 90 min. oder 8 h für Bibliotheksbau je nach der Schur Methode und bis zu weitere 48 h für die DNA-Sequenzierung. Die Verwendung des Protokolls wird die Genomik Gemeinschaft zu einem besseren Verständnis des Genoms Komplexität und neue Einblicke in Genom Variation im menschlichen Krankheiten stärken.
Protokoll
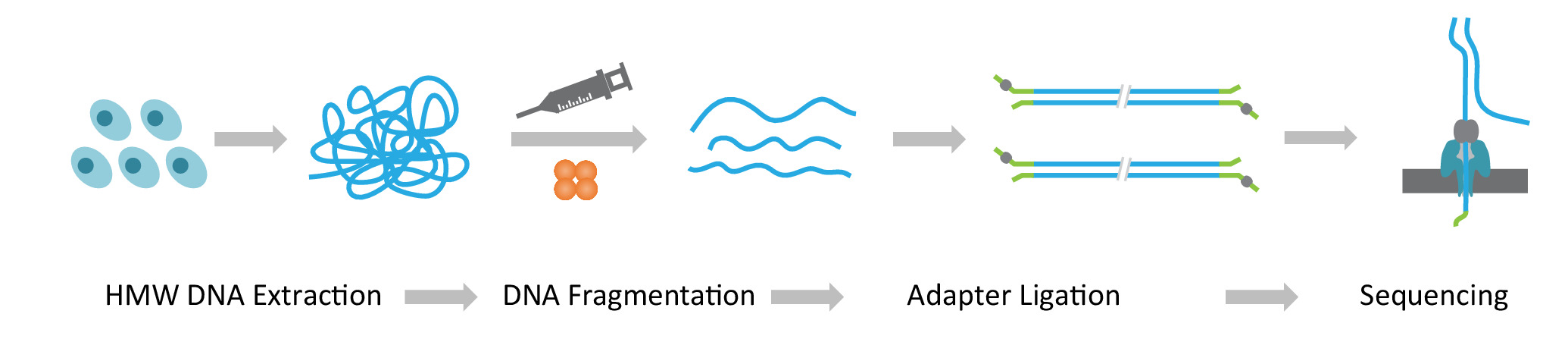
Hinweis: Das NLR-Seq-Protokoll besteht aus drei aufeinander folgenden Schritten: 1) Gewinnung von hochmolekularen Gewicht (HMW) genomischen DNA; (2) ultra-lange DNA Bibliotheksbau, die Fragmentierung der HMW DNA in die gewünschte Korngröße und Unterbindung der Sequenzierung Adapter an die DNA enthält endet; und 3) Laden der Adapter ligiert DNA auf die Arrays von Nanoporen (Abbildung 1).
(1) HMW DNA-Extraktion
- Reagenz-Setup. 1 x Phosphat-gepufferter Kochsalzlösung (PBS)-Puffer (1.000 mL) durch Zugabe von 100 mL PBS zu machen (10 X), 900 mL Wasser und gut mischen. Stellen Sie Lyse Puffer (50 mL) indem Sie eine 50 mL-Tube 43,5 mL Wasser hinzufügen. Fügen Sie 500 μl Tris (1 M, pH 8,0), Natriumchlorid (NaCl) (5 M), 1 mL 2,5 mL der Ethylenediaminetetraacetic Säure (EDTA) (0,5 M, pH 8,0) und 2,5 mL Sodium Dodecyl Sulfat (SDS) (10 %, wt/Vol) am Rohr und gut mischen.
Hinweis: Dieses PBS-Puffer kann bei 4 ° C für bis zu 6 Monate gespeichert werden. Die vorgefertigten Lyse-Puffer kann bei RT für bis zu 2 Monate gespeichert werden. - Überprüfen Sie die Zelle Sterblichkeit und zählen Sie die Zellen zu. Sicherstellen Sie, dass das live-Verhältnis liegt bei > 85 % der gesamten Zelle ist 30 x 106.
Hinweis: In diesem Protokoll verwendeten Zellen sind von der HG00733-Zell-Linie, eine menschliche Lymphoblastoid Zelllinie Puerto-Ricanischer Herkunft am meisten benutzt in den 1000 Genome Consortium für strukturelle Variation Analyse (siehe Tabelle der Materialien für Bestellinformationen), dem gehört auf internationalen Genom Probe Ressource. - Sammeln Sie die Zellen durch Zentrifugieren bei 200 X g für 5 min bei RT. verwerfen das Medium und Aufschwemmen der Zelle Pellet (30 x 106 Zellen) mit 5 mL 1 X PBS-Puffer. Zentrifugieren Sie wieder bei 200 X g für 5 min bei RT und verwerfen Sie den überstand.
Hinweis: 25-35 x 106 Zellen sind akzeptabel für diesen Ansatz. Variation in Höhe von Zellen verwendet wird weiter weitere Optimierung benötigen. Die Zelle-Pellets kann bei −80 ° C für bis zu 6 Monate gespeichert werden. - Aufschwemmen der Zelle Pellet in 200 μl 1 X PBS-Puffer. Wenn einen gefrorene Zelle Pellet, mit 5 mL 1 X PBS-Puffer waschen. Die Lösung bei 200 X g für 5 min bei RT Zentrifugieren, überstand verwerfen und Aufschwemmen der Zellen in 200 μl 1 x PBS-Puffer.
- Bereiten Sie 10 mL Lyse-Puffer in einem 50 mL-Tube. Hinzu kommen 200 μl Zellsuspension die Lyse Puffer und Wirbel mit der höchsten Geschwindigkeit für 3 S. Inkubation die Lösung bei 37 ° C für 1 h.
- Fügen Sie 2 μl RNase A (100 mg/mL) um die lysate. Drehen Sie behutsam die 50 mL-Tube um die Probe zu mischen. Inkubieren Sie die Lösung bei 37 ° C für 1 h.
- Fügen Sie 50 μL Proteinase K (20 mg/mL hinzu) der lysate. Drehen Sie behutsam die 50 mL-Tube um die Probe zu mischen. Inkubieren Sie die Lösung bei 50 ° C für 2 h. Während der Inkubation vorsichtig mischen der Probenmaterials alle 30 Minuten.
- Entfernen Sie die 50 mL-Tube von 50 ° C und bei RT 5 min stehen lassen.
- Die lysate 10 mL der Phenol-Schicht von Phenol: Chloroform: Isoamyl Alkohol (25:24:1, Vol/Vol/Vol) hinzu, und drehen Sie das Rohr auf einem Rotator-Mixer (siehe Tabelle der Materialien) bei RT in einer Dampfhaube bei 20 u/min für 10 min. wickeln die Rohr-Kappe mit Parafilm auslaufen zu verhindern während der Rotation.
- Bereiten Sie zwei 50 mL Gel Tuben (siehe Tabelle der Materialien) durch Zentrifugieren bei 1.500 X g für 2 min bei RT
Hinweis: Das Gel bildet eine stabile Barriere zwischen der Nukleinsäure-haltigen wässrigen Phase und dem organischen Lösungsmittel. - Gießen Sie die Probe/Phenol-Lösung eines vorbereiteten 50 mL Gel Tuben aus Schritt 1.10. Zentrifugieren Sie die Lösung bei 3.000 X g für 10 min bei RT
- Gießen Sie den Überstand in eine neue 50 mL Tube. Fügen Sie 10 mL der Phenol-Schicht von Phenol: Chloroform: Isoamyl Alkohol (25:24:1, Vol/Vol/Vol) und drehen Sie das Rohr auf einem Rotator-Mixer bei RT in einer Dampfhaube bei 20 u/min für 10 Minuten.
- Wiederholen Sie Schritt 1.11 einmal mit dem zweiten vorbereitet Gel Tube.
- Gießen Sie den Überstand in eine neue 50 mL Tube. Fügen Sie 25 mL eiskaltes 100 % Ethanol und drehen Sie sanft das Rohr von hand, bis die DNA (Abbildung 2) ausfällt.
Hinweis: Der Niederschlag Ansatz hilft, um die HMW-DNA zu stabilisieren. - Biegen Sie einen 20 μl-Tipp, um einen Haken machen. Sorgfältig die HMW-DNA mit den Haken herausnehmen und die Flüssigkeitstropfens ablassen.
- Legen Sie die HMW-DNA in ein 50 mL-Tube mit 40 mL 70 % igem Ethanol. Waschen Sie die DNA durch sanft das Rohr 3 mal umdrehen.
- Wiederholen Sie Schritt 1.15 einmal DNA aus der Tube 70 % Ethanol zu sammeln.
- Legen Sie die HMW-DNA in eine 2 mL-Röhrchen mit 1,8 mL 70 % igem Ethanol.
- Zentrifugieren Sie die gewaschenen HMW DNA bei 10.000 X g für 3 s bei RT. Entfernen Sie so viel von den restlichen Ethanol wie möglich durch pipettieren.
Hinweis: Stören Sie das DNA-Pellet nicht, wenn die verbleibende Ethanol pipettieren. - Inkubieren Sie der 2-mL-Tube bei 37 ° C für 10 min bei geschlossenem Deckel offen für die Probe trocknen.
- Wenn weiter mit Schritt 2.1 (mit mechanischen Scheren und 1 D Ligation Sequencing Kit), Hinzufügen der 2 mL Tube 1 mL TE (10 mM Tris und 1 mM EDTA, pH 8,0).
- Wenn weiter mit Schritt 2.2 (mit Transposase-basierte Fragmentierung und schnelle Sequencing Kit), fügen Sie 200 μl 10 mm Tris (pH 8,0) mit 0,02 % Triton x-100.
Hinweis: Bitte nicht stören Sie das DNA-Pellet. Das Rohr für 48 h bei 4 ° C im Dunkeln stehen zu lassen hilft die Probe vollständig aufzuwirbeln. Die HMW-DNA kann bis zu 2 Wochen bei 4 ° C aufbewahrt werden. Längere Lagerzeit oder anderen Lagerbedingungen können mehr kurze Fragmente einführen.
(2) ultra-lange DNA Bibliotheksbau
Hinweis: Es gibt zwei Möglichkeiten, die Ultra-langen DNA-Bibliotheken basierend auf zwei verschiedene Scheren Methoden gepaart mit Nanopore Sequenzierung Kits zu konstruieren. Eine mechanische Scheren-basierte Bibliothek produziert Daten mit einem N50 von 50-70 kb, Einnahme von ca. 8 h für die Bibliotheksbau. Eine Transposase Fragmentierung-basierte Bibliothek erzeugt ein N50 von 90-100 kb Daten, wobei nur 90 min. für den Bibliotheksbau. Das mechanische Scherkräfte Protokoll gibt höhere Ausbeute aus der gleichen DNA Eingang mit identischen Versionen der Sequenzierung Adapter und Qualität der Nanopore Durchflusszellen.
- Mechanische Scheren-basierte Bibliotheksbau
- Tauwetter und Mischen der Reagenzien aus der Ligatur kit (siehe Tabelle der Materialien). Tauwetter FFPE DNA Reparatur Puffer und Ende Reparatur/dA-Tailing Puffer auf Eis, dann Wirbel und Spinnen bis zu mischen. Tauen Sie Adapter-Mix (AMX) und Adapter Wulst Bindung Puffer (ABB) auf Eis, dann Pipette und Spin hinunter um zu mischen. Tauwetter liefen Puffer mit Kraftstoff-Gemisch (RBF) und Elution Buffer (ELB) bei RT, dann Wirbel und Spin um zu mischen. Tauen Sie Bibliothek laden Perlen (LLB auf) bei RT und Pipette mischen Sie vor dem Gebrauch.
- Einmal aufgetaut, halten Sie alle Kit-Komponenten auf dem Eis. Nehmen Sie die Enzyme nur im Bedarfsfall. RT bringen Sie die magnetische Beads für den Einsatz.
Hinweis: Für eine Empfehlung zur magnetischen Beads zu verwenden siehe Tabelle Materialien.
- Einmal aufgetaut, halten Sie alle Kit-Komponenten auf dem Eis. Nehmen Sie die Enzyme nur im Bedarfsfall. RT bringen Sie die magnetische Beads für den Einsatz.
- Überprüfen Sie die Qualität und Quantität der HMW DNA aus Schritt 1.21.1. Pipette, 20 μl DNA in neuen 1,5 mL Röhrchen aus drei verschiedenen Standorten in der HMW DNA-Röhre mit P200 breite Bohrung Tipps. Nehmen Sie 1 μL aus den drei Aliquote, die Konzentration, die mit einem Fluorometer und die Qualität mit einem UV lesen zu erkennen. Prüfen Sie mehrmals um die Ergebnisse zu bestätigen.
Hinweis: Die erwarteten Ergebnisse sind in Abbildung 3Aangezeigt. Der OD-260/280 -Wert ist ca. 1,9 und der OD260/230 Wert ist ungefähr 2,3. - Übertragung der verbleibenden 940 μL der HMW DNA in eine 50 mL Tube Kappe mit einem breiten P1000 trug Tipp.
- Aspirieren Sie alle DNA in eine 1 mL Spritze ohne Nadel.
- Die 27 G Nadel auf die Spritze setzen und alle DNA in die Kappe auswerfen, sanft und langsam (~ 10 s). Die 27 G Nadel von der Spritze abnehmen.
- Wiederholen Sie die Schritte 2.1.4 und 2.1.5 für 29 Mal für insgesamt 30 Pässe durch die Nadel.
Hinweis: Die sheared HMW-DNA können bei 4 ° C im Dunkeln gespeichert werden, für bis zu 24 Std. Qualitätskontrolle (QC) dringend, durch pulsieren-auffangen Gel-Elektrophorese empfohlen wird, aber es teuer und zeitaufwändig ist. Wenn ein 5-150 kb-Protokoll durchführen QC auf eine automatisierte Puls Feld Gel-Elektrophorese-Maschine für eine 20 h laufen verwendet werden. Die erwarteten Ergebnisse sind in Abbildung 4dargestellt. - Vorbereiten die DNA-Reparatur Reaktion in einem 0,2 mL Röhrchen durch Zugabe von 100 μl DNA-sheared HMW (20 μg), 15 μl der Proteinkinase DNA Reparatur Puffer, 12 μL FFPE DNA-Reparatur-Mix und 16 μL der Nuklease-freies Wasser. Mischen Sie die Reaktion durch Streichen sanft 6 Mal und drehen Sie auf Luftblasen entfernen.
- Inkubieren Sie der Reaktion bei 20 ° C für 60 min. Transfer der Probenmaterials in ein neues 1,5 mL Röhrchen mit P200 breite Bohrung Spitze.
- Die magnetische Beads durch Pipettieren oder vortexen aufzuwirbeln. Hinzu kommen 143 μL Perlen (1 X) der DNA Reparatur Reaktion und Mischung sanft durch Streichen der Röhre 6 x. Drehen Sie das Rohr auf einem Rotator-Mixer bei RT bei 20 u/min für 30 min.
- Spin-down der Probe bei 1.000 X g für 2 s bei rt das Rohr auf einer magnetischen Zahnstange für 10 min. halten Sie das Rohr auf die magnetische Rack und den Überstand verwerfen.
- Halten das Rohr auf die magnetische Rack, fügen Sie 400 μl von frisch zubereiteten 70 % Ethanol ohne zu stören das Pellet. Entfernen Sie das 70 % Ethanol nach 30 s.
- Wiederholen Sie Schritt 2.1.11 einmal.
- Spin-down der Probe bei 1.000 X g für 2 s bei RT der Schlauch wieder auf die magnetische Rack. Entfernen Sie alle verbleibenden Ethanol und an der Luft trocknen für 30 s. Trocknen Sie die Pellets nicht über.
- Entfernen Sie den Schlauch aus dem magnetischen Rack und fügen Sie 103 μL TE (10 mM Tris und 1 mM EDTA, pH 8,0 hinzu). Sanft flick Flick das Rohr um sicherzustellen, dass Perlen in den Puffer fallen und sanft auf einem Rotator-Mixer für 30 min. bei RT inkubieren das Rohr alle 5 min. zur Wiederfreisetzung des Pellet-Unterstützung.
- Pellet-die Perlen der magnetischen Zahnstange für mindestens 10 min. Transfer 100 μL Eluat mit einem breiten P200 Tipp in einem 0,2 mL-Tube Bohrung.
- Bereiten Sie die Ende-Reparatur und dA-Tailing Reaktion in einer 0,2 mL Tube indem 100 μL der reparierten HMW DNA, 14 μL Ende Reparatur/dA-Tailing Puffer und 7 μL Ende Reparatur/dA-Tailing-Mix. Mischen Sie die Reaktion durch Streichen sanft 6 Mal, und drehen Sie auf Luftblasen entfernen.
- Inkubieren Sie die Reaktion bei 20 ° C für 60 min, gefolgt von 65 ° C für 20 min, und halten Sie dann bei 22° C. Tipp: Transfer, die die Probe in eine neue 1,5 mL-Tube mit einem breiten P200 trug.
- Die magnetische Beads durch Pipettieren oder vortexen aufzuwirbeln. Fügen Sie 48 μL der Perlen (0,4 X) zum Ende Reparatur/dA-Tailing Reaktion und Mischung sanft durch Streichen der Röhre 6 Mal. Drehen Sie das Rohr auf einem Rotator-Mixer bei RT bei 20 u/min für 30 min.
- Wiederholen Sie Schritte 2.1.10-2.1.13 einmal.
- Entfernen Sie den Schlauch aus dem magnetischen Rack und fügen Sie 33 μL TE (10 mM Tris und 1 mM EDTA, pH 8,0 hinzu). Sanft flick Flick das Rohr um sicherzustellen, dass Perlen in den Puffer fallen und sanft auf einem Rotator-Mixer für 30 min. bei RT inkubieren das Rohr alle 5 min. zur Wiederfreisetzung des Pellet-Unterstützung.
- Pellet-die Perlen der magnetischen Zahnstange für mindestens 10 min. Transfer 30 μL Eluat mit einem breiten P200 Tipp in eine neue 1,5 mL Tube Bohrung. Nehmen Sie die zusätzliche 1-2 μl, die Konzentration, die mit einem Fluorometer zu erkennen.
Hinweis: Erholung der 5-6 μg bei diesem Schritt wird erwartet. - Bereiten Sie die Ligatur Reaktion in der 1,5 mL Probe Tube durch Hinzufügen von 30 μl Ende repariert HMW DNA, 20 μL des Adapter-Mix (AMX 1D) und 50 μL der Blunt/TA-Ligatur-master-Mix. Mischen Sie die Reaktion durch Streichen sanft 6 Mal zwischen jedes aufeinanderfolgende Zugabe und Spin auf Luftblasen entfernen.
- Inkubieren Sie die Reaktion bei RT 60 min.
- Die magnetische Beads durch Pipettieren oder vortexen aufzuwirbeln. 40 μl Perlen (0,4 X) die Ligatur Reaktions-und Mischung sanft durch Streichen der Röhre 6 Mal. Drehen Sie das Rohr auf einem Rotator-Mixer bei RT bei 20 u/min für 30 min.
- Wiederholen Sie Schritt 2.1.10 einmal.
- Fügen Sie 400 μl der Adapter Wulst Bindung (ABB) Puffer in die Röhre. Wechseln Sie das Rohr sanft 6 Mal um die Perlen aufzuwirbeln. Setzen Sie den Schlauch wieder auf die magnetischen Zahnstange, die Perlen aus dem Puffer zu trennen und entsorgen Sie überstand.
- Wiederholen Sie Schritt 2.1.26 einmal.
- Spin-down der Probe bei 1.000 X g für 2 s bei RT der Schlauch wieder auf die magnetische Rack. Entfernen Sie alle verbleibenden Puffer und an der Luft trocknen für 30 s. Trocknen Sie die Pellets nicht über.
- Entfernen Sie den Schlauch aus der magnetischen Halterung und aufzuwirbeln Sie das Pellet in 43 μL der Elution Buffer. Sanft flick Flick das Rohr um sicherzustellen, dass Perlen im Puffer fallen und sanft auf einem Rotator-Mixer für 30 min. bei RT inkubieren das Rohr alle 5 min. zur Wiederfreisetzung des Pellet-Unterstützung.
- Pellet-die Perlen der magnetischen Zahnstange für mindestens 10 min. Transfer 40 μL Eluat mit einem breiten P200 Tipp in eine neue 1,5 mL Tube Bohrung. Nehmen Sie die zusätzliche 1-2 μl, die Konzentration, die mit einem Fluorometer zu erkennen.
Hinweis: Erholung der 1-2 μg bei diesem Schritt wird erwartet. Die mechanische Scheren-basierte Bibliothek ist zum Laden bereit. Die Bibliothek kann auf Eis für bis zu 2 h bis Laden für die Sequenzierung gespeichert werden, bei Bedarf.
- Tauwetter und Mischen der Reagenzien aus der Ligatur kit (siehe Tabelle der Materialien). Tauwetter FFPE DNA Reparatur Puffer und Ende Reparatur/dA-Tailing Puffer auf Eis, dann Wirbel und Spinnen bis zu mischen. Tauen Sie Adapter-Mix (AMX) und Adapter Wulst Bindung Puffer (ABB) auf Eis, dann Pipette und Spin hinunter um zu mischen. Tauwetter liefen Puffer mit Kraftstoff-Gemisch (RBF) und Elution Buffer (ELB) bei RT, dann Wirbel und Spin um zu mischen. Tauen Sie Bibliothek laden Perlen (LLB auf) bei RT und Pipette mischen Sie vor dem Gebrauch.
- Transposase Fragmentierung-basierte Bibliotheksbau
- Tauwetter die Reagenzien aus der Transposase kit (siehe Tabelle der Materialien). Tauen Sie Fragmentierung Mix (FRA) und schnelle Adapter (RAP) auf Eis und Pipette zu mischen. Auftauen Sequenzierung Puffer (SQB), Perlen (LB) laden, Puffer (FLB) spülen und spülen Tether (FLT) bei RT und pipette um zu mischen. Tauen Sie laden Perlen (LB) bei RT und Pipette, vor Gebrauch zu mischen. Einmal aufgetaut, halten Sie alle Kit-Komponenten auf dem Eis. Nehmen Sie die Enzyme nur im Bedarfsfall.
- Überprüfen Sie die Qualität und Quantität der HMW DNA aus Schritt 1.21.2.. Pipette, 20 μl DNA in neuen 1,5 mL Röhrchen aus drei verschiedenen Standorten in der HMW DNA-Röhre mit P200 breite Bohrung Tipps. Nehmen Sie 1 μL aus den drei Aliquote, die Konzentration, die mit einem Fluorometer und die Qualität mit einem UV lesen zu erkennen. Prüfen Sie mehrmals um die Ergebnisse zu bestätigen.
Hinweis: Die erwarteten Ergebnisse sind in Abbildung 3 bangezeigt. Der OD-260/280 -Wert ist ca. 1,9 und der OD260/230 Wert ist ungefähr 2,3. - Bereiten Sie die DNA-Tagmentation Reaktion in einem 0,2 mL Röhrchen durch Zugabe von 22 μL der HMW DNA, 1 μl 10 mm Tris (pH 8,0) mit 0,02 % Triton X-100 und 1 μL der Fragmentierung (FRA) mischen. Mix von Pipettieren mit einem breiten P200 trug Tipp so langsam wie möglich 6 Mal, kümmert sich nicht um Luftblasen einzuführen.
- Inkubieren Sie die Reaktion bei 30 ° C für 1 min, gefolgt von 80 ° C für 1 min, und halten Sie dann bei 4 ° C. Transfer der Mischung in eine neue 1,5 mL Tube mit einem breiten P200 trug Tipp und gehen sofort weiter.
- Das Probenröhrchen 1,5 mL 1 μL der schnellen Adapter (RAP) hinzufügen. Mix von Pipettieren mit einem breiten P200 trug Tipp so langsam wie möglich 6 Mal, kümmert sich nicht um Luftblasen einzuführen.
- Inkubieren Sie die Reaktion bei RT 60 min.
Hinweis: Die Transposase Fragmentierung-basierte Bibliothek ist zum Laden bereit. Die Bibliothek kann auf Eis für bis zu 2 h bis Laden für die Sequenzierung gespeichert werden, bei Bedarf.
3. Sequenzierung auf dem Nanopore-Gerät
- Überprüfen Sie die Nanopore-Sequenzierung-Gerät (siehe Tabelle der Materialien). Stellen Sie sicher, die Software und Hardware arbeiten sowohl genügend Speicherplatz vorhanden ist.
- Überprüfen Sie die Messzelle. Öffnen Sie eine neue Messzelle und setzen Sie die Messzelle in die Nanopore Gerät. Überprüfen Sie das Feld des Standortes der Messzelle an (X1-X5) eingefügt wurde. Wählen Sie den korrekten Fluss Zelle. Klicken Sie auf Überprüfen Flow Zellen Workflow. Klicken Sie auf Test starten , um die Durchflusszelle QC Analyse starten.
Hinweis: Wenn die Anzahl der gemeldeten total aktiven Pore weniger als 800 ist, verwenden Sie eine andere neue Messzelle für die Sequenzierung. - Vorbereiten des Priming-Puffers. Für eine mechanische Scheren-basierte Bibliothek hinzufügen 576 μL des laufenden Puffer mit Kraftstoff-Gemisch (RBF) und 624 μL der Nuklease-freies Wasser in einem 1,5 mL-Tube. Wirbel und drehen bis die Mischung der Priming-Puffer. Fügen Sie für eine Transposase Fragmentierung-basierte Bibliothek 30 μl bündig Kräfte (FLT hinzu) das Rohr bündig Puffer (FLB). Wirbel und drehen bis die Mischung der Priming-Puffer.
- Bewegen Sie auf der Durchflusszelle Priming-Port-Abdeckung im Uhrzeigersinn, um den Priming-Port verfügbar zu machen.
- Legen Sie eine Pipette P1000 auf 100 μL und stecken Sie die Spitze in den Priming-Anschluss. Ziehen Sie zurück, ein kleines Volumen des Puffers (weniger als 30 μl), eventuell Luftblasen aus der Durchflusszelle zu entfernen. Pipettieren, sobald eine kleine Menge der gelben Flüssigkeit die Spitze tritt zu stoppen.
- Verwenden Sie eine P1000 Pipette 800 μl des Priming-Mix in der Messzelle über den Priming-Anschluss zu laden. Zur Vermeidung von Luftblasen Einführung fügen Sie 30 μl des Priming-Mix, decken Sie die Spitze des Hafens Grundierung zuerst, dann stecken die Spitze in der Grundierung Anschluss und fügen Sie langsam den Rest des Priming-Mix hinzu. Nehmen Sie die Spitze bei ca. 50 μL links. Fügen Sie den Rest des Priming-Mix auf der Oberseite der Priming-Port. Die Flüssigkeit wird im Inneren von selbst gehen.
- Verlassen Sie das Setup für 5 min inkubieren. In der Zwischenzeit bereiten Sie die Bibliothek-Mix in der 1,5 mL Tube mit der Bibliothek.
Hinweis: Für eine mechanische Scheren-basierte Bibliothek fügen Sie 35 µL Puffer mit Kraftstoff-Gemisch (RBF) ausführen, um 40 µL DNA-Bibliothek hinzu. Für eine Transposase Fragmentierung-basierte Bibliothek hinzufügen, 34 µL Sequenzierung Puffer (SQB) und 16 µL Nuklease-freies Wasser zu 25 µL DNA-Bibliothek. - Öffnen Sie die Strömung Zelle Probe Port Abdeckung vorsichtig, um die Probenöffnung verfügbar zu machen. Verwenden Sie eine Pipette P1000 200 μL des Priming-Mix über den Priming-Port in der Messzelle hinzufügen, wie in Schritt 3.5 beschrieben. Stellen Sie sicher, dass der Priming-Mix in der Messzelle durch die Probenöffnung nicht geladen wird.
- Legen Sie eine P200 Pipette auf 80 μl. Mischen Sie die Bibliothek mit einer großen Bohrung Spitze durch Pipettieren nach oben und unten 6 mal gerade vor der Verladung.
- Laden Sie die Bibliothek Mischung tropfenweise durch den Probenport in der Messzelle. Tropfen Sie jeder erst nach der letzte Tropfen in den Hafen vollständig geladen ist.
- Die Probe-Port-Abdeckung vorsichtig wieder und sicherstellen Sie, dass die Probenöffnung vollständig bedeckt ist. Verschieben Sie die Grundierung die Schutzkappe gegen den Uhrzeigersinn um die Grundierung zu schützen. Schließen Sie den Deckel Gerät.
- Klicken Sie auf den Neuen Test -Workflow. Geben Sie den Namen der Bibliothek, wählen Sie das richtige Zubehör gemäß den Verfahren verwendet, und überprüfen Sie, ob die Einstellungen korrekt (48 h laufen, Echtzeit-Basis-Calling ON) sind.
- Klicken Sie auf "Start" ausführen. Nach 10 min Aufzeichnen der Flow Cell-ID und die aktive Nanopore Zahlen (Gesamtzahl und jeweils vier Gruppen zahlen) aus der Informations.
- Analyse der Daten. Kopieren Sie die Daten auf einem lokalen Computer oder einem Cluster jederzeit die Sequenzierung oder wenn die Ausführung abgeschlossen ist. Verwenden Sie Minimap216 (https://github.com/lh3/minimap2), um die Sequenzdaten an die Referenz-Genom auszurichten. Die Sequenzierung Leistung von der rohen Sequenzdaten und die Achsen nach NanoPlot17 (https://github.com/wdecoster/NanoPlot) zusammenfassen.
Ergebnisse
Die ultra-lange DNA-Sequenzierung Protokoll gilt für Bibliotheksbau HMW DNA. Daher ist es wichtig, gut kultivierte Zellen mit dem live-Verhältnis zu wählen > 85 % bei der Ernte Schritt Zelle. Die Menge von Zellen für die DNA-Extraktion verwendet wird die Qualität und Quantität der HMW DNA beeinflussen. Die lyse der Zelle funktioniert nicht gut, wenn zu viele Zellen ab. Mit zu wenig Zellen erzeugt nicht genügend DNA für Bibliotheksbau, da die HMW DNA-Fällung erfolgt mit sanften Drehung von hand statt High-Speed-Zentrifugation. Ein Beispiel für die HMW-DNA nach Hinzufügen von eiskalten 100 % Ethanol und drehen als der weiße baumwollähnliche Niederschlag in Abbildung 2gezeigt wird.
Es ist wichtig, die Qualität der Eingabe DNA vor Beginn der Bibliotheksbau zu überprüfen. Abbau, falsche Quantifizierung, Kontamination (z. B. Proteine, RNAs, Waschmittel, Tenside, und passives Phenol oder Ethanol) und niedrigem Molekulargewicht DNA haben erhebliche Auswirkungen auf die nachfolgenden Verfahren und auf die endgültige Länge zu lesen. Es wird empfohlen, die QC-Analyse mit der DNA von drei verschiedenen Standorten in der Röhre mit HMW DNA durchzuführen. Von UV Leseergebnisse für HMW-DNA die OD260/OD280 Wert ist ca. 1,9 und der OD260/OD230 Wert ist ungefähr 2,3 (Abb. 3AB). Diese Verhältniswerte entsprechen den drei Tests für eine gute HMW DNA-Probe. Schere Methoden erfordert unterschiedliche Volumina der Eingabe DNA. Die Konzentration der HMW DNA muss > 200 ng/µL für mechanische Scheren, während es sein muss > 1 µg/µL für Transposase Fragmentierung. Die Konzentration von einem Fluorometer erkannt ist ein wenig niedriger als UV lesen. Der Variationskoeffizient der Konzentration der gleichen HMW DNA-Probe ist jedoch erforderlich, weniger als 15 % der Fluorometer mit UV-Assays zu lesen sein. Mechanische Scheren gilt eine Spritze mit einer Nadel, die HMW-DNA zu brechen, so dass die Anzahl von Durchläufen durch die Nadel auf die Größe der DNA geschoren Auswirkungen wird und das Finale Länge lesen. Es wird empfohlen, führen Sie Größe QC, nachdem Nadel Scheren um sicherzustellen die Mehrheit der HMW DNA größer als 50 kb wie in Abbildung 4dargestellt ist. In der mechanischen Scheren-Methode erzeugt 30 Pässe die besten Sequenzierung Ergebnisse unter Berücksichtigung der Dauer und Ausgang.
Die N50 einer mechanischen Scheren-basierte Bibliothek ist 50-70 kb während eine Transposase Fragmentierung-basierte Bibliothek 90-100 kb. Die Ergebnisse der vier Läufe unter Verwendung der HG00733-Zell-Linie sind in Tabelle 1dargestellt. Alle vier Läufe haben mehr als 2.300 liest mit mehr als 100 kb Länge. Die maximale Länge ist länger in die Transposase Fragmentierung-basierte Bibliotheken (455 kb und 489 kb) im Vergleich mit den mechanischen Scheren-basierte Bibliotheken (348 kb und 387 kb) während die letzteren mehr Gesamtanzahl der Lesezugriffe produziert, zeigt eine höhere Ausbeute. Die Transposase Fragmentierung-basierte Bibliotheksbau hat weniger Schritte und kürzere Zubereitungszeit, so dass es weniger kurze Fragmente einführen wird. Die zwei Läufe mit Transposase haben eine längere durchschnittliche Länge (> 30 kb) und mittlere Länge (> 10 kb). Darüber hinaus zeigt die Daten gleich bleibend hohen Qualität in allen Läufen (mittlere Qualitätsfaktor ist ca. 10,0, ~ 90 % Basis Genauigkeit). Mehr als 97 % der gesamten Grundlagen waren die menschlichen Bezug Genom (hg19) mit Minimap216 mit den Standardeinstellungen ausgerichtet. Die erwarteten Größenverteilung des rohen lautet sind in Abbildung 5dargestellt. Alle Ausführungen haben einen Großteil der Daten über 50 kb Transposase Fragmentierung-basierte Bibliotheken ein höheres Verhältnis von ultra-lange liest (z. B. > 100 kb). Dieses Protokoll wurde in mehreren humanen Zelllinien (ergänzende Tabelle1) erfolgreich angewendet.

Abbildung 1: Schematische Übersicht über die Nanopore lange lesende Sequenzierung (NLR-Seq) Workflow. Orange, die Transposase Komplex. Gelb-grün, die Nanopore-Adapter. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2: repräsentative DNA Niederschlag aus Phenol-Chloroform Extraktionsmethode. Der weiße Pfeil zeigt die HMW-DNA. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3: Beispiel QC Ergebnisse der HMW DNA aus UV lesen. (A) HMW DNA aus Schritt 1.21.1 mechanische Scheren-basierte Bibliothek baureif. (B) HMW DNA aus Schritt 1.21.2. für Transposase Fragmentierung-basierte Bibliotheksbau. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 4: QC-Ergebnisse der Nadel geschert HMW DNA durch pulsieren-auffangen Gel-Elektrophorese. L1: Schnell-Ladung 1 kb DNA-Leiter; L2: Quick-Load 1 kb DNA-Leiter zu verlängern. 1-8: DNA mit verschiedenen vorbeifahrenden Zeiten durch die Nadel Scheren. 1-3, keine Scheren; 4, 10 Mal; 5, 20 Mal; 6, 30-Mal; 7, 40-Mal; 8, 50-Mal. Dieser QC-Schritt ist optional. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 5: Größenverteilung der Nanopore ultra-lange DNA-Bibliotheken erwartet. MS, mechanische Scheren-basierte Bibliotheken. TF, Transposase Fragmentierung-basierte Bibliotheken. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
| Mechanische shearing_rep1 | Mechanische shearing_rep2 | Transposase fragmentation_rep1 | Transposase fragmentation_rep2 | |
| Zell-Linie | HG00733 | HG00733 | HG00733 | HG00733 |
| N50 des lautet | 55.180 | 63.007 | 98.237 | 95.629 |
| Anzahl der Lesevorgänge länger als 100 Kb | 2.500 | 3.082 | 2.386 | 2.355 |
| Anzahl der insgesamt mal gelesen | 97.859 | 80.465 | 24.166 | 21.032 |
| Maximale Länge (bp) | 348.482 | 387.113 | 454.660 | 489.426 |
| Mittlere Länge (bp) | 17.861 | 20.395 | 33.528 | 38.175 |
| Mittlere Länge (bp) | 5.335 | 5.894 | 10.249 | 15.656 |
| Mittlere Qualität des lautet | 10.0 | 10.1 | 9.9 | 10.0 |
| Gesamtunterseiten roh mal gelesen | 1,747,849,822 | 1,641,058,932 | 810,229,733 | 802,886,304 |
| Gesamtunterseiten ausgerichteten mal gelesen | 1,693,300,832 | 1,607,975,925 | 791,422,077 | 778,417,627 |
| Zugeordneten Verhältnis von Gesamtunterseiten (hg19, Minimap2) | 96,9 % | 98.0 % | 97,7 % | 97,0 % |
| Anzahl der aktiven Poren | 1225: 480, 402, 254, 89 | 1058: 480, 356, 176, 46 | 958: 452, 328, 148, 30 | 1092: 487, 367, 195, 43 |
Tabelle 1: Zusammenfassung von Leistungskennzahlen läuft mit verschiedenen Scheren Protokollen.
| Bibliothek 1 | Bibliothek 2 | |
| Zell-Linie | K562 | GM19240 |
| Zelle Bestellinformationen | ATCC, Kat. Nein. CCL-243 | Coriell Institut, Kat. Nein. GM19240 |
| Protokoll | mechanische Scheren | mechanische Scheren |
| N50 des lautet | 60.063 | 55.295 |
| Anzahl der insgesamt mal gelesen | 193.783 | 120.807 |
| Mittlere Länge (bp) | 1.843 | 4.688 |
| Mittlere Länge (bp) | 9.825 | 17.408 |
| Maximale Länge (bp) | 548.780 | 212.338 |
| Gesamtunterseiten roh mal gelesen | 1,903,989,686 | 2,103,015,331 |
| Gesamtunterseiten ausgerichteten mal gelesen | 1,837,350,047 | 1,997,419,761 |
| Zugeordneten Verhältnis von Gesamtunterseiten (hg19, Minimap2) | 96,6 % | 95,0 % |
| Anzahl der aktiven Poren | 1111: 482, 371, 203, 55 | 1032: 447, 333, 196, 56 |
Zusätzliche Tabelle 1: Zusammenfassung der zwei NLR-Seq-Läufe mit anderen Zelllinien mit dem mechanischen Scheren-Protokoll.
Diskussion
Im Prinzip ist Nanopore Sequenzierung 100 kb zu Megabase liest in Länge11,12,13zu generieren. Vier wichtige Faktoren wirkt sich die Leistung der Sequenzierung ausführen und Daten: 1) aktive Pore Zahlen und die Aktivität der Poren; (2) motor-Protein, das die Geschwindigkeit der DNA durch die Nanopore steuert; (3) DNA-Vorlage (Länge, Reinheit, Qualität, Masse); (4) Sequenzierung Adapter Ligatur Effizienz, die die nutzbare DNA aus der Eingabe Probe bestimmt. Die ersten beiden Faktoren hängen von der Version von der Messzelle und das Sequencing Kit des Herstellers. Die zweiten zwei Faktoren sind wichtige Schritte in diesem Protokoll (HMW DNA-Extraktion, Scheren und Ligatur).
Dieses Protokoll erfordert Geduld und Übung. Die Qualität der HMW DNA ist wichtig für ultra-lange DNA-Bibliotheken-6. Das Protokoll beginnt mit Zellen mit hoher Rentabilität (> 85 % lebensfähigen Zelle bevorzugt), Begrenzung der degradierten DNS aus abgestorbenen Zellen. Harten Prozess, der Schäden an der DNA (z. B. stark stören, schütteln, Wirbel, mehrere pipettieren, wiederholtes Einfrieren und Auftauen) vorstellen kann, sollte vermieden werden. Bei der Gestaltung des Protokolls lassen wir Pipettieren in den gesamten Prozess der DNA-Extraktion. Große Bohrung Tipps müssen verwendet werden, wenn Pipettieren nach dem mechanischen Scheren während Bibliotheksbau und Sequenzierung erforderlich ist. Da die Nanoporen empfindlich auf Chemikalien in die Kammer Puffer12sind, sollte es möglichst wenige verbleibende Verunreinigungen (z.B. Waschmittel, Tenside, Phenol, Ethanol, Proteine RNAs, etc.) wie möglich in der DNA. Angesichts der Länge und der Ertrag zeigt die Phenol-Extraktions-Verfahren die beste und am meisten reproduzierbare Ergebnisse im Vergleich mit mehreren verschiedenen Extraktionsmethoden bisher getestet.
Trotz der Fähigkeit dieses Protokolls, lange lesende Sequenzen produzieren bleiben noch mehrere Einschränkungen. Zunächst wurde dieses Protokoll optimiert, basierend auf dem Nanopore Sequenzierung Gerät zum Zeitpunkt der Veröffentlichung verfügbar; so, es beschränkt sich auf die selektive Nanopore-basierte Sequenzierung Chemie und suboptimal, wenn in anderen Gerätetypen lange lesende Sequenzierung durchgeführt werden konnte. Zweitens ist das Ergebnis stark abhängig von der Qualität der DNA extrahiert aus dem Ausgangsmaterial (Gewebe oder Zellen). Lesen Sie Länge wird gefährdet, wenn die Start DNA bereits beeinträchtigt oder beschädigt wird. Drittens, obwohl mehrere QC Schritte in das Protokoll um die DNA-Qualität aufgenommen werden, den endgültigen Ertrag und die Länge des lautet können durch die Flusszelle beeinflusst werden und pore-Aktivität, die Variable in diesem frühen Stadium der Nanopore Sequenzierung Plattform sein könnte Entwicklung.
Das Protokoll beschrieben verwendet hier menschliche Aussetzung Linie Zellproben für DNA-Extraktion. Wir haben die Weitergabe in Nadeln, Scheren, das Verhältnis von HMW DNA zu Transposase und die Ligatur-Zeit, um die beschriebenen Ergebnisse produzieren optimiert. Das Protokoll kann auf vier verschiedene Arten erweitert werden. Erstens können Benutzer mit anderen kultivierten Säugerzellen und unterschiedliche Anzahl von Zellen, Geweben, klinischen Proben oder andere Organismen beginnen. Eine weitere Optimierung auf Lyse Inkubationszeit, Reaktionsvolumen und Zentrifugation wird benötigt. Zweitens ist es schwer vorherzusagen, die Zielgröße für ultra-lange lesen Sie Sequenzierung. Wenn die lesen Sie Längen kürzer als erwartet sind, können die Benutzer stellen Sie die Zeiten vorbei in die mechanische Scheren-basierte Methode oder ändern das Verhältnis der HMW DNA in Transposase in die Transposase Fragmentierung-basierte Methode. Bindung und Elution länger während der Bereinigung Schritte sind hilfreich, weil die HMW DNA sehr zähflüssig ist. Drittens kann man mit verschiedenen Nanopore Sequenzierung Geräte, die Höhe und das Volumen der DNA die Kriterien des Sequenzers einstellen. Viertens wird nur die DNA Sequenzierung Adapter ligiert sequenziert werden. Zur Unterbindung Effizienz weiter zu steigern, kann man versuchen, die Adapter und Ligase Konzentrationen zu titrieren. Modifizierte Ligatur Zeit und molekulare crowding Mittel wie PEG18 können in Zukunft angewendet werden. Die ultra-lange DNA-Sequenzierung Protokoll kombiniert mit CRISPR19,20 kann ein effektives Ziel Bereicherung Sequenzierung anbieten.
Offenlegungen
Die Autoren erklären, dass sie keine finanziellen Interessenkonflikte.
Danksagungen
Die Autoren danken für ihre Bemerkungen auf die Handschrift Y. Zhu. Forschung, die in dieser Publikation berichtet wurde teilweise durch das National Cancer Institute der National Institutes of Health unter Preis Anzahl P30CA034196 unterstützt. Der Inhalt ist ausschließlich in der Verantwortung der Autoren und nicht unbedingt die offizielle Meinung der National Institutes of Health.
Materialien
| Name | Company | Catalog Number | Comments |
| Reagents | |||
| Absolute ethanol | Sigma-Aldrich | E7023 | |
| Agencourt AMPure XPbeads | Beckman | A63881 | magnetic beads for cleanup |
| BD conventional needles | Becton Dickinson | 305136 | 27G, for mechanical shearing |
| BD Luer-Lok syringe | Becton Dickinson | 309628 | for mechanical shearing |
| Blunt/TA Ligase Master Mix | NEB | M0367S | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | for cell counting |
| EDTA | Invitrogen | AM9261 | pH 8.0, 0.5 M, 500 mL |
| Flow Cell | Oxford Nanopore Technologies | FLO-MIN106 | R9.4.1 |
| HG00773 cells | Coriell Institute | HG00733 | cells used in this protocol |
| Ligation Sequencing Kit 1D | Oxford Nanopore Technologies | SQK-LSK108 | nanopore ligation kit |
| MaXtract High Density tubes | Qiagen | 129073 | gel tubes |
| NEBNext FFPE DNA Repair Mix | NEB | M6630S | |
| NEBNext Ultra II End Repair/dA-Tailing Module | NEB | M7546S | |
| Nuclease-free water | Invitrogen | AM9937 | |
| Phosphate-Buffered Saline, PBS | Gibco | 70011044 | 10X, pH 7.4 |
| Phenol:chloroform:IAA | Invitrogen | AM9730 | |
| Proteinase K | Qiagen | 19131 | 20 mg/mL |
| Qubit dsDNA BR Assay Kit | Invitrogen | Q32850 | fluorometer assays for DNA quantification |
| Rapid Sequencing Kit | Oxford Nanopore Technologies | SQK-RAD004 | nanopore transposase kit |
| RNase A | Qiagen | 19101 | 100 mg/mL |
| SDS | Invitrogen | AM9822 | 10% (wt/vol) |
| Sodium chloride solution | Invitrogen | AM9759 | 5.0 M |
| TE buffer | Invitrogen | AM9849 | pH 8.0 |
| Tris | Invitrogen | AM9856 | pH 8.0, 1 M |
| Triton X-100 solution | Sigma-Aldrich | 93443 | ~10% |
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| Bio-Rad C1000 Thermal Cycler | Bio-Rad | 1851196EDU | |
| Centrifuge 5810R | Eppendorf | 22628180 | |
| Countess II FL Automated Cell Counter | Life Technologies | AMQAF1000 | for cell counting |
| DynaMag-2 Magnet | Life Technologies | 12321D | magnetic rack |
| Eppendorf ThermoMixer | Eppendorf | 5382000023 | for incubation |
| Freezer | LabRepCo | LHP-5-UFMB | |
| GridION | Oxford Nanopore Technologies | GridION X5 | nanopore device used in this protocol |
| HulaMixer Sample Mixer | Thermo Fisher Scientific | 15920D | rotator mixer |
| MicroCentrifuge | Benchmark Scientific | C1012 | |
| NanoDrop ND-1000 Spectrophotometer | Thermo Fisher Scientific | ND-1000 | for UV reading |
| Pippin Pulse | Sage Science | PPI0200 | pulsed-field gel electrophoresis instrument |
| Qubit 3.0 Fluorometer | Invitrogen | Q33216 | fluorometer |
| Refrigerator | LabRepCo | LABHP-5-URBSS | |
| Vortex-Genie 2 | Scientific Industries | SI-A236 | |
| Water bath | VWR | 89501-464 |
Referenzen
- Mardis, E. R. Next-generation sequencing platforms. Annual Review of Analytical Chemistry. 6, 287-303 (2013).
- Goodwin, S., McPherson, J. D., McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nature Reviews Genetics. 17 (6), 333-351 (2016).
- Shendure, J., et al. DNA sequencing at 40: past, present and future. Nature. 550 (7676), 345-353 (2017).
- Alkan, C., Coe, B. P., Eichler, E. E. Genome structural variation discovery and genotyping. Nature Reviews Genetics. 12 (5), 363-376 (2011).
- Weischenfeldt, J., Symmons, O., Spitz, F., Korbel, J. O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nature Reviews Genetics. 14 (2), 125-138 (2013).
- Pollard, M. O., Gurdasani, D., Mentzer, A. J., Porter, T., Sandhu, M. S. Long reads: their purpose and place. Human Molecular Genetics. 27 (R2), R234-R241 (2018).
- Cretu Stancu, M., et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nature Communications. 8 (1), 1326 (2017).
- Gong, L., et al. Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nature Methods. 15 (6), 455-460 (2018).
- Sedlazeck, F. J., et al. Accurate detection of complex structural variations using single-molecule sequencing. Nature Methods. 15 (6), 461-468 (2018).
- Jain, M., et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology. 36 (4), 338-345 (2018).
- Jain, M., et al. Improved data analysis for the MinION nanopore sequencer. Nature Methods. 12 (4), 351-356 (2015).
- Deamer, D., Akeson, M., Branton, D. Three decades of nanopore sequencing. Nature Biotechnology. 34 (5), 518-524 (2016).
- Jain, M., Olsen, H. E., Paten, B., Akeson, M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biology. 17 (1), 239 (2016).
- Editorial, The long view on sequencing. Nature Biotechnology. 36 (4), 287 (2018).
- Jain, M., et al. Linear assembly of a human centromere on the Y chromosome. Nature Biotechnology. 36 (4), 321-323 (2018).
- Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094-3100 (2018).
- De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34, 2666-2669 (2018).
- Akabayov, B., Akabayov, S. R., Lee, S. J., Wagner, G., Richardson, C. C. Impact of macromolecular crowding on DNA replication. Nature Communications. 4, 1615 (2013).
- Gabrieli, T., Sharim, H., Michaeli, Y., Ebenstein, Y. Cas9-Assisted Targeting of CHromosome segments (CATCH) for targeted nanopore sequencing and optical genome mapping. bioRxiv. , (2017).
- Gabrieli, T., et al. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Research. , (2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten