
Method Article
Séquençage de l’ultra longue lecture pour l’analyse de l’ADN génomique ensemble
Dans cet article
Résumé
Long-lire des séquences facilitent considérablement l’assemblage des génomes complexes et caractérisation de variation structurale. Les auteurs décrivent une méthode pour générer des séquences ultra longues de plateformes de séquençage nanopore-basé. L’approche adopte une extraction d’ADN optimisée suivie de préparations de bibliothèque modifiée pour générer des centaines de kilobases se lit avec une couverture modérée à partir des cellules humaines.
Résumé
Troisième génération de technologies de séquençage de l’ADN molécule unique offrent nettement plus longtemps lire longueur qui peut faciliter le montage des génomes complexes et l’analyse des variantes structurales complexes. Plates-formes Nanopore effectuent seule molécule séquençage en mesurant directement les changements actuels médiées par le passage de l’ADN à travers les pores et peuvent générer des centaines de lectures kilobases (kb) avec le coût en capital minime. Cette plate-forme a été adoptée par de nombreux chercheurs pour une variété d’applications. Réalisation de plus longues longueurs de lecture de séquençage est le facteur le plus critique de tirer parti de la valeur des plateformes de séquençage nanopore. Pour générer des lectures ultra longues, une attention particulière est nécessaire pour éviter les ruptures de l’ADN et de gagner en efficacité pour générer des modèles productifs de séquençage. Ici, nous fournissons le protocole détaillé de séquençage de l’ADN ultra long dont le poids moléculaire élevé (HMW) extraction de l’ADN des cellules fraîches ou congelées, construction de la bibliothèque de cisaillement mécanique ou une fragmentation de la transposase et le séquençage sur un périphérique nanopore. De 20 à 25 µg d’ADN HMW, la méthode peut atteindre N50 lire longueur de 50 à 70 KO avec le cisaillement mécanique et N50 de 90-100 kb lire la longueur avec la transposase médiée par fragmentation. Le protocole peut être appliqué à l’ADN extrait des cellules de mammifères pour effectuer le séquençage du génome entier pour la détection des variantes structurelles et assemblage de génome. Des améliorations supplémentaires sur l’extraction de l’ADN et les réactions enzymatiques vont encore augmenter la durée de lecture et élargir son utilité.
Introduction
Ces dix dernières années, massivement parallèle et technologies de haute précision deuxième génération haut débit séquençage ont conduit à une explosion de découverte biomédicale et de l’innovation technologique1,2,3. Malgré les progrès techniques, le court-lecture de données générées par les plateformes de deuxième génération est inefficaces à résoudre les régions génomiques complexes et est limités dans la détection des variants structurels génomiques (SVs), qui jouent un rôle important dans l’homme Evolution et maladies4,5. En outre, court-lecture de données est incapables de résoudre la répétition de variation et ne conviennent pas pour les hautes haplotype progressive des variants génétiques6.
Les progrès récents dans le séquençage de la molécule unique offre significativement plus longue lire la longueur, ce qui peut faciliter la détection de l’éventail complet des SVs7,8,9et offres Assemblée précise et complète du complexe génomes microbiens et mammifères6,10. La plate-forme nanopore effectue seule molécule séquençage en mesurant directement les changements actuels médiées par le passage de l’ADN dans les pores de12,11,13. Contrairement à toute chimie de séquençage ADN existante, nanopore séquençage peut générer long (des dizaines de milliers de kilobases) se lit comme suit en temps réel sans avoir à compter sur la cinétique de la polymérisation ou artificielle l’amplification de l’échantillon d’ADN. Par conséquent, nanopore long lire séquençage (NLR-seq) détient très prometteur pour la génération ultra grandes longueurs de lecture bien au-delà de 100 Ko, ce qui ferait progresser considérablement les analyses génomiques et biomédicales14, particulièrement dans la faible complexité ou riches en répétition régions des génomes15.
La caractéristique unique de séquençage nanopore est sa capacité à générer des longues lectures sans limitation de durée théorique. Par conséquent, la longueur de lecture dépend de la longueur physique de l’ADN qui est directement touchée par la qualité de modèle intégrité et séquençage de l’ADN. En outre, selon l’ampleur de la manipulation et le nombre d’étapes impliquées, comme forces de pipetage et les conditions d’extraction, la qualité de l’ADN est très variable. Il est donc difficile pour que l'on puisse céder lectures longues en appliquant simplement les protocoles d’extraction d’ADN standards et des méthodes de construction de la bibliothèque fournie du fabricant. À cette fin, nous avons développé une solide méthode permettant de générer ultra-longue lire (des centaines de kilobases) données de séquençage à partir de granulés de cellules récoltées. Nous avons adopté plusieurs améliorations dans les procédures de préparation ADN extraction et bibliothèque. Nous avons simplifié le protocole afin d’exclure des procédures inutiles qui causent des dommages-intérêts et la dégradation de l’ADN. Ce protocole est composé de poids moléculaire élevé (HMW) extraction d’ADN, ultra longue construction de bibliothèque d’ADN et le séquençage sur une plate-forme nanopore. Pour un biologiste moléculaire bien formé, cela prend généralement de 6 h de cellule récolte jusqu'à l’achèvement de HMW DNA extraction, 90 min ou 8 h pour la construction de la bibliothèque selon la méthode de cisaillement et jusqu'à une autre 48 h pour le séquençage de l’ADN. L’utilisation du protocole permet à la communauté de la génomique afin d’améliorer notre compréhension de la complexité du génome et avoir un nouvel aperçu variation génomique dans les maladies humaines.
Protocole
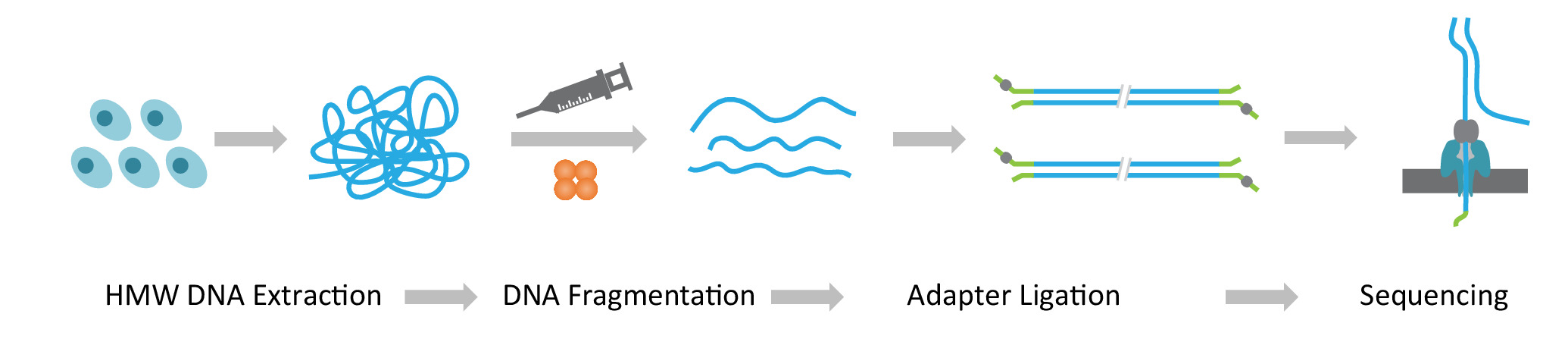
Remarque : Le protocole NLR-seq se compose de trois étapes successives : 1) extraction de grande molécularité poids ADN génomique (HMW) ; 2) ultra longue construction de bibliothèque d’ADN, qui comprend la fragmentation de l’ADN de HMW dans les tailles souhaitées et ligature des adaptateurs de séquençage de l’ADN se termine ; et 3) chargement de l’ADN adaptateur-ligaturé sur les baies de nanopores (Figure 1).
1. extraction d’ADN de HMW
- Configuration du réactif. Faire 1 x tampon de phosphate solution saline tamponnée (PBS) (1 000 mL) en ajoutant 100 mL de PBS (10 x) à 900 mL d’eau et bien mélanger. Faire le tampon de lyse (50 mL) en ajoutant 43,5 mL d’eau dans un tube de 50 mL. Ajouter 500 μl de Tris (1 M, pH 8,0), 1 mL de chlorure de sodium (NaCl) (5 M), 2,5 mL d’acide tétraacétique (EDTA) (0,5 M, pH 8,0) et 2,5 mL de sodium dodecyl sulfate (SDS) (10 %, wt/vol) dans le tube et bien mélanger.
Remarque : Ce tampon PBS peut être stocké à 4 ° C pendant 6 mois. Le tampon de lyse prédéfinis peut être stocké à ta jusqu'à 2 mois. - Vérifier le taux de mortalité cellulaire et compter les cellules non vides. Veiller à ce que le rapport direct est > 85 % et le nombre total de cellules est 30 x 106.
NOTE : Les cellules utilisées dans le présent protocole sont de la lignée cellulaire de HG00733, une lignée de cellules lymphoblastoïdes humaines d’origine portoricaine, largement utilisé dans le consortium de génomique de 1000 pour l’analyse de la variation structurale (voir le tableau des matériaux pour les informations à la commande), auquel appartient ressource d’échantillon International génome. - Recueillir les cellules par centrifugation à 200 x g pendant 5 min à RT. jeter le milieu et resuspendre le culot cellulaire (30 x 106 cellules) avec 5 mL de solution tampon PBS 1 x. Centrifuger à nouveau à 200 x g pendant 5 min à RT et éliminer le surnageant.
NOTE : 25-35 x 106 cellules sont acceptables pour cette approche. Plus la variation d’un montant de cellules utilisées devront encore optimisation. Le culot cellulaire peut être stocké à −80 ° C jusqu'à 6 mois. - Resuspendre le culot dans 200 μl de tampon de PBS 1 x. Si vous utilisez un culot congelés, laver avec 5 mL de tampon PBS 1 x. Centrifuger la solution à 200 x g pendant 5 min à RT, éliminer le surnageant et remettre en suspension les cellules dans 200 μl de 1 x tampon PBS.
- Préparer 10 mL de tampon de lyse dans un tube de 50 mL. Ajouter la suspension cellulaire de 200 μL au tampon de lyse et vortex à haute vitesse pour 3 s. incuber la solution à 37 ° C pendant 1 h.
- Ajouter 2 μL de RNase A (100 mg/mL) dans le lysate. Tourner doucement le tube de 50 mL pour mélanger l’échantillon. Incuber la solution à 37 ° C pendant 1 h.
- Ajouter 50 protéinase μL K (20 mg/mL) pour le lysate. Tourner doucement le tube de 50 mL pour mélanger l’échantillon. Incuber la solution à 50 ° C pendant 2 h. Durant l’incubation, mélanger doucement l’échantillon toutes les 30 minutes.
- Retirer le tube de 50 mL de 50 ° C et laisser reposer à RT pendant 5 min.
- Ajouter 10 mL de la couche de phénol de phénol : chloroforme : isoamylique alcool (25:24:1, vol/vol/vol) au lysat et tourner le tube sur une table de mixage rotator (voir Table des matières) à ta dans une hotte de laboratoire à 20 tr/min pendant 10 min. envelopper le capuchon du tube avec du parafilm pour éviter les fuites pendant la rotation.
- Préparer deux tubes de gel 50 mL (voir Table des matières) par centrifugation à 1 500 x g pendant 2 min à température ambiante.
Remarque : Le gel forme une barrière stable entre la phase aqueuse contenant de l’acide nucléique et de solvant organique. - Verser la solution de l’échantillon/phénol dans d'entre les prêt 50 mL tubes de gel de 1.10 étape. Centrifuger la solution à 3 000 x g pendant 10 min à température ambiante.
- Versez le liquide surnageant dans un nouveau tube de 50 mL. Ajouter 10 mL de la couche de phénol de phénol : chloroforme : isoamylique alcool (25:24:1, vol/vol/vol) et tourner le tube sur un mélangeur de rotateur à ta dans une hotte de laboratoire à 20 tr/min pendant 10 min.
- Répétez le tube gel étape 1.11 une fois avec le second établi.
- Versez le liquide surnageant dans un nouveau tube de 50 mL. Ajouter 25 mL d’éthanol glacé de 100 % et tourner doucement le tube à la main jusqu'à ce que l’ADN précipite (Figure 2).
Remarque : L’approche de précipitations contribue à stabiliser l’ADN HMW. - Plier un pourboire de 20 μL de faire un crochet. Soigneusement, retirez l’ADN HMW avec le crochet et laisser la goutte liquide.
- Placez l’ADN HMW dans un tube de 50 mL contenant 40 mL d’éthanol à 70 %. Laver l’ADN en retournant doucement le tube 3 fois.
- Répétez l’étape 1,15 fois pour recueillir l’ADN du tube de l’éthanol à 70 %.
- Placez l’ADN HMW dans un tube de 2 mL contenant 1,8 mL d’éthanol à 70 %.
- Centrifuger l’ADN HMW lavées à 10 000 x g pendant 3 s à RT. Enlevez autant de l’éthanol résiduel que possible en pipettant également.
Remarque : Ne pas déranger le culot d’ADN lors du pipetage l’éthanol résiduel. - Incuber le tube 2 mL à 37 ° C pendant 10 min avec le couvercle ouvert pour sécher l’échantillon.
- Si continuer avec l’étape 2.1 (avec cisaillement mécanique et 1 Kit de séquençage de la ligature D), ajouter 1 mL de TE (10 mM Tris et 1 mM EDTA, pH 8,0) dans le tube 2 mL.
- Si il continue avec l’étape 2.2 (avec axée sur la transposase fragmentation et Kit de séquençage rapide), ajouter 200 μl de 10 mM Tris (pH 8,0) avec 0,02 % X-100 Triton.
Remarque : Ne pas déranger le culot d’ADN. Laisser le tube s’élèvent à 4 ° C dans l’obscurité pendant 48 h aidera l’échantillon complètement remettre en suspension. L’ADN de HMW peuvent être stocké à 4 ° C pendant 2 semaines. Durée de stockage ou d’autres conditions de stockage peuvent introduire plus de courts fragments.
2. ultra longue construction de bibliothèque d’ADN
Remarque : Il y a deux façons pour construire les bibliothèques d’ADN ultra longues issus des deux méthodes différentes de cisaillement couplés avec nanopore kits de séquençage. Une bibliothèque de cisaillement mécanique génère des données avec un N50 de 50 à 70 Ko, prenant environ 8 h pour la construction de la bibliothèque. Une bibliothèque transposase fragmentation produit un N50 de 90-100 Ko de données, en prenant seulement 90 min pour la construction de la bibliothèque. Le protocole de cisaillement mécanique donne un rendement plus élevé de l’ADN même introduit au moyen de versions identiques de l’adaptateur de séquençage et de la qualité des cellules d’écoulement nanopore.
- Construction de la bibliothèque de base cisaillement mécanique
- Dégel et mélanger les réactifs de la ligature nécessaire (voir la Table des matières). Dégel FFPE ADN réparer le buffer et réparation/dA-tailing fin sur la glace, puis de vortex et filer vers le bas pour mélanger. Dégeler adaptateur mix (AMX) et tampon de liaison cordon adaptateur (ABB) sur la glace, puis la pipette et spin down pour mélanger. Dégel dévalant un tampon avec un mélange de carburant (RBF) et tampon d’élution (ELB) à RT, puis vortex et un essorage à mélanger. Décongelez bibliothèque perles (LLB) de chargement à RT et pipette à mélanger avant utilisation.
- Une fois décongelé, conserver tous les composants du kit sur la glace. Sortez les enzymes uniquement lorsque nécessaire. Mettre les billes magnétiques à RT pour utilisation.
Remarque : Pour obtenir des recommandations sur les billes magnétiques à utiliser, consultez le tableau des matériaux.
- Une fois décongelé, conserver tous les composants du kit sur la glace. Sortez les enzymes uniquement lorsque nécessaire. Mettre les billes magnétiques à RT pour utilisation.
- Vérifier la qualité et la quantité d’ADN de l’étape 1.21.1 HMW. Pipette à 20 μL d’ADN dans nouveaux tubes de 1,5 mL de trois endroits différents dans le tube de HMW ADN utilisant P200 large alésage conseils. Prendre 1 μL des trois aliquotes pour détecter la concentration à l’aide d’un fluorimètre et la qualité à l’aide d’un UV de lecture. Vérifier plusieurs fois pour confirmer les résultats.
NOTE : Les résultats attendus sont montrées dans la Figure 3 a. La valeur de260/280 OD est environ 1,9 et la valeur de260/230 OD est environ 2,3. - Transfert le μL 940 restants de HMW ADN dans un bouchon de tube de 50 mL avec un large P1000 alésage pointe.
- Aspirez tous les ADN dans une seringue de 1 mL sans l’aiguille.
- Mettez l’aiguille 27 G sur la seringue et éjecter tous les ADN dans la PAC doucement et lentement (environ 10 s). Décoller l’aiguille 27 G de la seringue.
- Répétez les étapes 2.1.4 et 2.1.5 pour 29 fois pour un total de 30 passages à travers l’aiguille.
Remarque : L’ADN HMW cisaillées peuvent être conservé à 4 ° C dans l’obscurité pendant 24 h. contrôle de la qualité (CQ) est hautement recommandée par électrophorèse sur gel en champ pulsé, mais il est coûteux et prend du temps. Si vous effectuez QC sur une machine automatisée impulsion champ gel électrophorèse utilisent un protocole de 5-150 kb pour un 20 h exécuter. Les résultats attendus sont indiquées à la Figure 4. - Préparer la réaction de réparation de l’ADN dans un tube de 0,2 mL en ajoutant 100 μL de coulissage HMW ADN (20 μg), 15 μL de tampon de réparation de l’ADN FFIP, 12 μL du mélange de réparation FFPE ADN et 16 μL d’eau exempte de nucléase. Mélanger la réaction en effleurant doucement 6 fois et filer vers le bas pour supprimer bulles.
- Incubez la réaction à 20 ° C pendant 60 min. transfert de l’échantillon dans un nouveau tube de 1,5 mL avec une pointe d’alésage large P200.
- Remettre en suspension les billes magnétiques en pipette ou l’agitation. Ajouter 143 perles μL (1 x) à la réaction de réparation de l’ADN et mélangez doucement en effleurant le tube 6 fois. Tourner le tube sur un mélangeur de rotateur à RT à 20 tr/min pendant 30 min.
- Tournez en bas de l’échantillon à 1 000 x g pendant 2 s à la Place le tube sur un support magnétique pour 10 min. de la RT. maintenir le tube sur le support magnétique et éliminer le surnageant.
- Gardez le tube sur le support magnétique, ajouter 400 ml d’éthanol à 70 % fraîchement préparés sans déranger le culot. Supprimer l’éthanol à 70 % après 30 s.
- Répétez une fois l’étape 2.1.11.
- Tournez en bas de l’échantillon à 1 000 x g pendant 2 s à RT. Replacer le tube sur le support magnétique. Retirer tout l’éthanol résiduel et laisser sécher pendant 30 s. Ne pas sécher le culot.
- Retirer le tube de la grille magnétique et ajouter 103 μL de TE (10 mM Tris et 1 mM EDTA, pH 8,0). Doucement, flick le tube pour que les perles sont couverts dans la mémoire tampon et incuber doucement sur un mélangeur de rotateur à RT pendant 30 min. balayer le tube toutes les 5 min pour remise en suspension de la pastille de l’aide.
- Les perles sur le support magnétique de granule pour au moins 10 min. transférer 100 μL d’éluat avec un P200 large trou de pointe dans un tube de 0,2 mL.
- Préparer la réparation de fin et la réaction de dA-tailing dans un tube de 0,2 mL en ajoutant 100 μL de réparé HMW ADN, 14 μL de tampon de réparation/dA-tailing fin et 7 μL du mélange de réparation/dA-tailing fin. Mélanger la réaction en effleurant doucement 6 fois et actionner jusqu'à supprimer bulles.
- Incubez la réaction à 20 ° C pendant 60 minutes suivie par 65 ° C pendant 20 min et puis maintenez à 22° C. Transfert de l’échantillon dans un nouveau tube de 1,5 mL à l’aide d’une large P200 alésage tip.
- Remettre en suspension les billes magnétiques en pipette ou l’agitation. Ajouter 48 μL de perles (0,4 x) pour la réaction de réparation/dA-tailing fin et mélangez doucement en effleurant le tube 6 fois. Tourner le tube sur un mélangeur de rotateur à RT à 20 tr/min pendant 30 min.
- Répétez les étapes 2.1.10-2.1.13 une fois.
- Retirer le tube de la grille magnétique et ajouter 33 μL de TE (10 mM Tris et 1 mM EDTA, pH 8,0). Doucement, flick le tube pour que les perles sont couverts dans la mémoire tampon et incuber doucement sur un mélangeur de rotateur à RT pendant 30 min. balayer le tube toutes les 5 min pour remise en suspension de la pastille de l’aide.
- Les perles sur le support magnétique de granule pour au moins 10 min. transférer 30 μL d’éluat avec un P200 large trou de pointe dans un nouveau tube de 1,5 mL. Prendre le μL de 1-2 supplémentaires pour détecter la concentration à l’aide d’un fluorimètre.
NOTE : Reprise de 5-6 μg à cette étape est attendue. - Préparer la réaction de ligature dans le tube d’échantillon de 1,5 mL en ajoutant 30 μL d’ADN de HMW réparé à la fin, 20 μL de mélange de l’adaptateur (AMX 1D) et 50 μL de mélange principal de ligature blunt/TA. Mélanger la réaction en effleurant doucement 6 fois entre chaque addition séquentielle et essorage jusqu'à supprimer bulles.
- Incubez la réaction à RT pendant 60 min.
- Remettre en suspension les billes magnétiques en pipette ou l’agitation. Ajouter 40 perles μL (0,4 x) pour la réaction de ligature et mélangez doucement en effleurant le tube 6 fois. Tourner le tube sur un mélangeur de rotateur à RT à 20 tr/min pendant 30 min.
- Répétez l’étape 2.1.10 une fois.
- Ajouter 400 μL de tampon de liaison (EBA) pour le cordon adaptateur dans le tube. Mettez le tube doucement 6 fois pour remettre en suspension les perles. Placer le tube sur la grille magnétique pour séparer les billes de la mémoire tampon et jeter le surnageant.
- Répétez l’étape 2.1.26 une fois.
- Tournez en bas de l’échantillon à 1 000 x g pendant 2 s à RT. Replacer le tube sur le support magnétique. Supprimer n’importe quel tampon résiduelle et laisser sécher pendant 30 s. Ne pas sécher le culot.
- Retirer le tube de la grille magnétique et resuspendre le culot dans 43 μL de tampon d’élution. Doucement flick le tube pour que les perles sont couverts dans la mémoire tampon et incuber doucement sur un mélangeur de rotateur à RT pendant 30 min. balayer le tube toutes les 5 min pour remise en suspension de la pastille de l’aide.
- Les perles sur le support magnétique de granule pour au moins 10 min. transférer 40 μL d’éluat avec un P200 large trou de pointe dans un nouveau tube de 1,5 mL. Prendre le μL de 1-2 supplémentaires pour détecter la concentration à l’aide d’un fluorimètre.
NOTE : Reprise de 1-2 μg à cette étape est attendue. La bibliothèque de cisaillement mécanique est prête pour le chargement. La bibliothèque peut être stockée sur la glace jusqu'à 2 h jusqu’au chargement pour le séquençage si nécessaire.
- Dégel et mélanger les réactifs de la ligature nécessaire (voir la Table des matières). Dégel FFPE ADN réparer le buffer et réparation/dA-tailing fin sur la glace, puis de vortex et filer vers le bas pour mélanger. Dégeler adaptateur mix (AMX) et tampon de liaison cordon adaptateur (ABB) sur la glace, puis la pipette et spin down pour mélanger. Dégel dévalant un tampon avec un mélange de carburant (RBF) et tampon d’élution (ELB) à RT, puis vortex et un essorage à mélanger. Décongelez bibliothèque perles (LLB) de chargement à RT et pipette à mélanger avant utilisation.
- Construction de bibliothèque axée sur la fragmentation de transposase
- Décongeler les réactifs de la transposase nécessaire (voir la Table des matières). Décongeler la fragmentation mix (FRA) et adaptateur rapide (RAP) sur la glace et la pipette pour mélanger. Décongeler le tampon de séquençage (CBRS), chargement de billes (LB), tampon (FLB) et rincer d’attache (FLT) à ta et pipeter pour mélanger. Décongeler le chargement de billes (LB) à RT et pipette à mélanger avant utilisation. Une fois décongelé, conserver tous les composants du kit sur la glace. Sortez les enzymes uniquement lorsque nécessaire.
- Vérifier la qualité et la quantité d’ADN de l’étape 1.21.2 HMW. Pipette à 20 μL d’ADN dans nouveaux tubes de 1,5 mL de trois endroits différents dans le tube de HMW ADN utilisant P200 large alésage conseils. Prendre 1 μL des trois aliquotes pour détecter la concentration à l’aide d’un fluorimètre et la qualité à l’aide d’un UV de lecture. Vérifier plusieurs fois pour confirmer les résultats.
NOTE : Les résultats attendus sont montrées dans la Figure 3 b. La valeur de260/280 OD est environ 1,9 et la valeur de260/230 OD est environ 2,3. - Préparer la réaction de tagmentation d’ADN dans un tube de 0,2 mL en ajoutant 22 μL de HMW ADN, 1 μL de 10 mM Tris (pH 8,0) avec 0,02 % Triton X-100 et 1 μL de fragmentation mélanger (FRA). Mix en pipettant également, avec une large P200 alésage de pointe aussi lentement que possible 6 fois, prenant soin de ne pas pour introduire des bulles.
- Incubez la réaction à 30 ° C pendant 1 min, suivie de 80 ° C pendant 1 min et puis maintenez à 4 ° C. Transfert le mélange dans un nouveau tube de 1,5 mL avec une large P200 alésage astuce et passez à l’étape suivante immédiatement.
- Ajouter 1 μL d’adaptateur rapide (RAP) dans le tube d’échantillon de 1,5 mL. Mix en pipettant également, avec une large P200 alésage de pointe aussi lentement que possible 6 fois, prenant soin de ne pas pour introduire des bulles.
- Incubez la réaction à RT pendant 60 min.
NOTE : La bibliothèque transposase fragmentation est prête pour le chargement. La bibliothèque peut être stockée sur la glace jusqu'à 2 h jusqu’au chargement pour le séquençage si nécessaire.
3. séquençage sur le périphérique nanopore
- Vérifier l’appareil de séquençage nanopore (voir Table des matières). Assurez-vous que les logiciels et matériels travaillent tant il y a assez de rangement.
- Vérifier les cellules de la circulation. Ouvrez une nouvelle cellule de flux et insérez la cellule d’écoulement dans l’appareil nanopore. Vérifier que la boîte de l’emplacement de la cellule d’écoulement a été insérée dans (X1-X5). Sélectionnez le type de cellule un flux correct. Cliquez sur le flux de travail de Vérifier les cellules de couler . Cliquez sur le bouton Démarrer le Test pour démarrer la cellule de flux analyse QC.
Remarque : Si le nombre total rapporté de pore actif est inférieure à 800, utilisez une nouvelle cellule de flux différents pour le séquençage. - Préparer le tampon d’amorçage. Pour une bibliothèque basée sur cisaillement mécanique, ajouter 576 μL de tampon avec un mélange de carburant (RBF) et 624 μL d’eau exempte de nucléase dans un tube de 1,5 mL. Vortex et un essorage jusqu'à mélange le tampon d’amorçage. Pour une bibliothèque d’axée sur la fragmentation de la transposase, ajouter 30 μL d’affleurement d’attache (FLT) dans le tube de tampon affleurant (FLB). Vortex et un essorage jusqu'à mélange le tampon d’amorçage.
- Sur la cellule de flux, le couvercle du port d’amorçage déplacer vers la droite pour exposer le port d’amorçage.
- Affectez une pipette P1000 100 μL et introduire l’embout dans le port d’amorçage. Faire reculer un petit volume de tampon (moins de 30 μL) pour supprimer toutes les bulles de la cellule de flux. Arrêter le pipetage une fois une petite quantité de liquide jaune pénètre dans la pointe.
- Utiliser une pipette P1000 pour charger 800 μL du mélange d’amorçage dans la cellule de flux via le port d’amorçage. Pour éviter d’introduire des bulles, ajoutez 30 μL du mélange d’amorçage pour couvrir le haut du port d’amorçage tout d’abord, puis introduire l’embout dans le port d’amorçage et ajouter lentement le reste du mélange d’amorçage. Sortir la pointe quand il y a environ 50 μL de gauche. Ajouter le reste du mélange d’amorçage sur le dessus de l’orifice d’amorçage. Le fluide ira à l’intérieur de lui-même.
- Quitter le programme d’installation à incuber pendant 5 min. Pendant ce temps, préparer le mélange de bibliothèque dans le tube de 1,5 mL contenant la bibliothèque.
Remarque : Pour une bibliothèque de cisaillement mécanique ajouter 35 µL de l’exécution de tampon avec un mélange de carburant (RBF) à 40 µL de la bibliothèque de l’ADN. Pour une bibliothèque de base fragmentation transposase ajouter 34 µL de tampon de séquençage (CBRS) et 16 µL d’eau exempte de nucléase à 25 µL de la bibliothèque de l’ADN. - Ouvrez le couvercle de port flux cellulaire échantillon doucement afin d’exposer le port de l’échantillon. Utiliser une pipette P1000 pour ajouter 200 μL du mélange d’amorçage via le port d’amorçage dans la cellule de flux, comme indiqué au point 3.5. Assurez-vous que le mélange d’amorçage n’est pas chargé dans la cellule de flux via le port de l’échantillon.
- Affectez une pipette P200 80 μL. Mélanger doucement la bibliothèque avec une alésage large pointe de pipetage et descendre 6 fois juste avant le chargement.
- Charger le mélange bibliothèque goutte à goutte par le port de l’échantillon dans la cellule de flux. Ajouter chaque goutte qu’après que la chute de la précédente est complètement chargée dans le port.
- Remettre en place le couvercle du port échantillon doucement et assurez-vous que le port de l’échantillon est entièrement couvert. Déplacer le cache-port d’amorçage dans le sens anti-horaire pour couvrir l’orifice d’amorçage. Fermer le couvercle de l’appareil.
- Cliquez sur le flux de travail de Nouvelle expérience . Tapez le nom de la bibliothèque, sélectionnez le kit correct selon les procédures utilisées et vérifiez que les paramètres sont correct (48 h exécuter, en temps réel ON base d’appel).
- Cliquez sur Démarrer Exécuter. Après 10 min, consigner l’identification de cellule de flux et les numéros de nanopore actif (nombre total et les numéros de chacun des quatre groupes) d’après les informations d’exécution.
- Analyse des données. Copier les données vers un ordinateur local ou sur un cluster à tout moment du séquençage ou lorsque la série est terminée. Minimap216 (https://github.com/lh3/minimap2) permet d’aligner les données de séquence dans le génome de référence. Résumer les performances de séquençage de séquences brutes et les alignements de NanoPlot17 (https://github.com/wdecoster/NanoPlot).
Résultats
HMW ADN s’applique le protocole de séquençage ADN ultra long pour la construction de la bibliothèque. Par conséquent, il est essentiel de choisir les cellules bien cultivées avec un rapport direct > 85 % à la cellule à l’étape de récolte. La quantité de cellules utilisée pour l’extraction d’ADN aura une incidence sur la qualité et la quantité de l’ADN de HMW. La lyse cellulaire ne fonctionne pas bien si commençant par trop de cellules. En utilisant des cellules trop peu ne génère pas suffisamment d’ADN pour la construction de la bibliothèque, car la précipitation de l’ADN HMW est effectuée à l’aide de rotation douce à la main au lieu de centrifugation à haute vitesse. Un exemple de l’ADN de HMW après que ajout glacé 100 % d’éthanol et la rotation sont montré comme le précipité blanc de coton-comme à la Figure 2.
Il est important de vérifier la qualité de l’ADN d’entrée avant de commencer la construction de la bibliothèque. Dégradation, quantification incorrecte, contamination (par exemple, protéines, ARN, détergents, tensioactifs et phénol résiduel ou éthanol) et faible poids moléculaire ADN peuvent avoir un effet significatif sur les procédures ultérieures et sur la finale lire la longueur. Nous recommandons de procéder de l’analyse QC à l’aide de l’ADN de trois endroits différents dans le tube contenant de l’ADN de HMW. De UV lecture des résultats de l’ADN de HMW, la valeur do260/OD280 est environ 1,9 et la valeur do260/OD230 est environ 2.3 (Figure 3 aB). Ces valeurs de ratio sont compatibles entre les trois tests pour un échantillon d’ADN de HMW bon. Différentes méthodes de cisaillement nécessite des volumes différents de l’ADN d’entrée. La concentration d’ADN de HMW doit être > 200 ng/µL de cisaillement mécanique alors qu’il doit être > 1 µg/µL de fragmentation de la transposase. La concentration détectée par un fluorimètre est un peu inférieure à UV lecture. Toutefois, le coefficient de variation de la concentration de l’échantillon d’ADN de HMW même devra être inférieur à 15 % avec le fluorimètre et le UV, lecture d’épreuves. Tonte mécanique s’applique une seringue avec une aiguille pour casser l’ADN HMW afin que le nombre de passes par l’intermédiaire de l’aiguille aura une incidence la taille de l’ADN cisaillé et lire de la finale de la longueur. Il est recommandé d’effectuer la taille QC après que aiguille tonte afin d’assurer la majorité de l’ADN de HMW est supérieure à 50 Ko comme illustré à la Figure 4. Dans la méthode de cisaillement mécanique, 30 passes a généré les meilleurs résultats de séquençage, compte tenu de la longueur et la sortie.
La N50 d’une bibliothèque de base cisaillement mécanique est 50-70 kb tandis qu’une transposase axée sur la fragmentation bibliothèque est de 90-100 Ko. Les résultats de quatre essais en utilisant la lignée cellulaire de HG00733 sont indiqués dans le tableau 1. Tous les quatre descentes ont plus 2 300 lectures avec une longueur de plus de 100 Ko. La longueur maximale est plus longue dans les transposase axée sur la fragmentation bibliothèques (455 Ko et 489ko) comparés avec les bibliothèques de base cisaillement mécaniques (348ko et 387 kb) alors que ce dernier produit plus de lectures totales, ce qui indique un rendement plus élevé. La construction de bibliothèque fragmentation transposase a moins de marches et de temps de préparation plus courte afin qu’elle introduira moins courts fragments. Les deux pistes à l’aide de la transposase ont une longueur moyenne supérieure (> 30 Ko) et la durée médiane (> 10 Ko). En outre, les données montrent une qualité élevée constante dans toutes les séries (score de qualité moyenne est environ 10,0, précision de base d’origine ~ 90 %). Plus de 97 % des bases totales étaient alignés au génome humain de référence (hg19) à l’aide de Minimap216 avec les paramètres par défaut. Les distributions de la taille attendue des lectures brutes sont indiquées à la Figure 5. Toutes les séries ont une grande partie des données dépassant les 50 Ko tandis que transposase axée sur la fragmentation des bibliothèques ont un ratio plus élevé de lectures ultra longues (p. ex. > 100 Ko). Ce protocole a été appliqué avec succès dans plusieurs lignées de cellules humaines (complémentaires tableau 1).

Figure 1 : vue d’ensemble schématique du workflow nanopore séquençage long-read (NLR-seq). Orange, la transposase complexe. Jaune-vert, l’adaptateur nanopore. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 2 : précipitations d’ADN représentant de méthode d’extraction de phénol-chloroforme. La flèche blanche indique l’ADN HMW. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 3 : résultats de QC de l’exemple de l’ADN de HMW de lecture UV. ADN de HMW (A) de l’étape 1.21.1 prêt pour la construction de la bibliothèque de base cisaillement mécanique. ADN de HMW (B) de l’étape 1.21.2 pour construction de bibliothèque axée sur la fragmentation de transposase. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 4 : résultats QC de l’aiguille cisaillement HMW ADN par électrophorèse sur gel en champ pulsé. L1 : Quick charge 1Ko échelle d’ADN ; L2 : Quick charge 1Ko étendre échelle d’ADN. 1-8 : ADN avec des temps de passage différents par le biais de la tonte de l’aiguille. 1-3, aucune tonte ; 4, 10 fois ; 5, 20 fois ; 6, 30 fois ; 7, 40 fois ; 8, 50 fois. Cette étape QC est facultative. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 5 : prévu des distributions granulométriques des bibliothèques nanopore ADN ultra-longue. MS, bibliothèques de base cisaillement mécaniques. TF, transposase axée sur la fragmentation des bibliothèques. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
| Shearing_rep1 mécanique | Shearing_rep2 mécanique | Transposase fragmentation_rep1 | Transposase fragmentation_rep2 | |
| Lignée cellulaire | HG00733 | HG00733 | HG00733 | HG00733 |
| N50 les lectures | 55 180 | 63 007 | 98 237 | 95 629 |
| Nombre de lectures plus de 100ko | 2 500 | 3 082 | 2 386 | 2 355 |
| Nombre de lit total | 97 859 | 80 465 | 24 166 | 21 032 |
| Longueur maximale (bp) | 348 482 | 387 113 | 454 660 | 489 426 |
| Longueur moyenne (bp) | 17 861 | 20 395 | 33 528 | 38 175 |
| Durée médiane (bp) | 5 335 | 5 894 | 10 249 | 15 656 |
| Qualité moyenne des lectures | 10.0 | 10.1 | 9,9 | 10.0 |
| Totales bases de lectures brutes | 1,747,849,822 | 1,641,058,932 | 810,229,733 | 802,886,304 |
| Totales bases alignées lectures | 1,693,300,832 | 1,607,975,925 | 791,422,077 | 778,417,627 |
| Mappés ratio du totales bases (hg19, Minimap2) | 96,9 % | 98,0 % | 97,7 % | 97,0 % |
| Nombre de pores actives | 1225 : 480, 402, 254, 89 | 1058 : 480, 356, 176, 46 | 958 : 452, 328, 148, 30 | 1092 : 487, 367, 195, 43 |
Tableau 1 : Sommaire des indicateurs de Performance fonctionne avec différents protocoles de cisaillement.
| Bibliothèque 1 | Bibliothèque 2 | |
| Lignée cellulaire | K562 | GM19240 |
| Cellule Information de commande | ATCC, cat. Lol CCL-243 | Coriell Institute, cat. Lol GM19240 |
| Protocole | tonte mécanique | tonte mécanique |
| N50 les lectures | 60 063 | 55 295 |
| Nombre de lit total | 193 783 | 120 807 |
| Durée médiane (bp) | 1 843 | 4 688 |
| Longueur moyenne (bp) | 9 825 | 17 408 |
| Longueur maximale (bp) | 548 780 | 212 338 |
| Totales bases de lectures brutes | 1,903,989,686 | 2,103,015,331 |
| Totales bases alignées lectures | 1,837,350,047 | 1,997,419,761 |
| Mappés ratio du totales bases (hg19, Minimap2) | 96,6 % | 95,0 % |
| Nombre de pores actives | 1111 : 482, 371, 203, 55 | 1032 : 447, 333, 196, 56 |
Supplémentaire tableau 1 : Résumé de deux courses de NLR-seq en utilisant d’autres lignées cellulaires avec le protocole de cisaillement mécanique.
Discussion
En principe, nanopore séquençage est capable de produire 100 Ko de megabase lectures en longueur11,12,13. Quatre principaux facteurs affectera les performances de la qualité de course et les données de séquençage : 1) nombre de pores actifs et l’activité des pores ; 2) protéine moteur, qui contrôle la vitesse de l’ADN en passant par le nanopore ; 3) modèle ADN (longueur, pureté, qualité, masse) ; 4) séquençage adaptateur ligature efficacité, ce qui détermine l’ADN utilisable de l’échantillon d’entrée. Les deux premiers facteurs dépendent de la version de la cellule d’écoulement et le kit de séquençage fournies par le fabricant. Les deux facteurs sont des étapes cruciales dans le présent protocole (extraction d’ADN de HMW, cisaillement et ligature).
Ce protocole requiert patience et pratique. La qualité de l’ADN de HMW est importante pour les bibliothèques6de l’ADN ultra longue. Le protocole commence par cellules recueillies avec viabilité élevée (> 85 % des cellules viables préféré), limitant l’ADN dégradé des cellules mortes. Tout processus rigoureux qui peuvent introduire des dommages à l’ADN (par exemple, fort dérangeant, secouant, vortex, multiples pipetage, répétées gel et dégel) doit être évitée. Dans la conception du protocole, nous omettons pipetage dans l’ensemble du processus d’extraction de l’ADN. Conseils d’alésage large doivent être utilisées lorsque le pipetage est nécessaire après le cisaillement mécanique pendant la construction de la bibliothèque et le séquençage. Comme les nanopores sont sensibles aux chimies dans le tampon de chambre12, il doit être aussi peu contaminants résiduels (par exemple, les détergents, agents tensio-actifs, phénol, éthanol, protéines, ARN, etc.) que possible dans l’ADN. Compte tenu de la longueur et le rendement, la méthode d’extraction de phénol montre des résultats meilleurs et plus reproductibles comparés avec plusieurs différentes méthodes d’extraction testées jusqu'à présent.
Malgré la capacité du présent protocole pour produire des séquences de lecture longue, plusieurs limitations subsistent. Tout d’abord, ce protocole a été optimisé basé sur l’appareil de séquençage nanopore disponible au moment de la publication ; ainsi, il est limité à la chimie sélective axée sur les nanopore séquençage et pourrait être sous-optimal lorsque effectuées dans d’autres types d’appareils de séquençage long à lire. En second lieu, le résultat est fortement tributaire de la qualité de l’ADN extrait de la matière (tissus ou cellules). Longueur de lecture sera compromise si l’ADN de départ est déjà dégradé ou endommagé. Troisièmement, bien que plusieurs étapes QC sont incorporés dans le protocole pour vérifier la qualité de l’ADN, le rendement final et la longueur de la lecture peuvent être affectées par la cellule de flux et activité, ce qui pourrait être variable à ce stade précoce de la plate-forme de séquençage nanopore des pores développement.
Le protocole décrit ici utilise des échantillons de ligne de cellules humaines de suspension pour l’extraction de l’ADN. Nous avons optimisé les temps de passage en aiguille de cisaillement, le ratio d’ADN HMW transposase et le moment de ligature pour produire les résultats décrits. Le protocole peut être augmenté de quatre façons. Tout d’abord, les utilisateurs peuvent démarrer avec d’autres cellules mammifères cultivées et avec des montants différents de cellules, tissus, échantillons cliniques ou d’autres organismes. Optimisation sur les temps d’incubation de lyse, volume réactionnel et centrifugation est nécessaires. Deuxièmement, il est difficile de prédire la taille cible pour le séquençage ultra longue lecture. Si la longueur de lecture est plus courte que prévu, les utilisateurs peuvent ajuster les temps de passage dans la méthode cisaillement mécanique ou changer le ratio de l’ADN de HMW à transposase dans la méthode axée sur la fragmentation de transposase. Liaison et élution longtemps durant les étapes de nettoyage sont utiles parce que l’ADN HMW est très visqueux. Troisièmement, avec nanopore différents appareils de séquençage, on peut ajuster la quantité et le volume de l’ADN pour remplir les critères du séquenceur. Quatrièmement, seulement ces ADN ligaturé aux adaptateurs de séquençage va être séquencé. Afin d’améliorer l’efficacité de la ligature, on peut essayer de titrer les concentrations de l’adaptateur et la ligase. Ligature des mis à jour le temps et les agents encombrement moléculaires comme PEG18 peuvent être appliquées à l’avenir. Le protocole de séquençage de l’ADN ultra long combiné avec CRISPR19,20 peut offrir un outil efficace pour l’ordonnancement de l’enrichissement de cible.
Déclarations de divulgation
Les auteurs déclarent qu’ils n’ont aucun intérêt financier concurrentes.
Remerciements
Les auteurs remercient Y. Zhu pour ses commentaires sur le manuscrit. Recherche rapporté dans cette publication a été partiellement financée par le National Cancer Institute de la National Institutes of Health, sous attribution numéro P30CA034196. Le contenu est la seule responsabilité des auteurs et ne représente pas nécessairement l’opinion officielle de la National Institutes of Health.
matériels
| Name | Company | Catalog Number | Comments |
| Reagents | |||
| Absolute ethanol | Sigma-Aldrich | E7023 | |
| Agencourt AMPure XPbeads | Beckman | A63881 | magnetic beads for cleanup |
| BD conventional needles | Becton Dickinson | 305136 | 27G, for mechanical shearing |
| BD Luer-Lok syringe | Becton Dickinson | 309628 | for mechanical shearing |
| Blunt/TA Ligase Master Mix | NEB | M0367S | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | for cell counting |
| EDTA | Invitrogen | AM9261 | pH 8.0, 0.5 M, 500 mL |
| Flow Cell | Oxford Nanopore Technologies | FLO-MIN106 | R9.4.1 |
| HG00773 cells | Coriell Institute | HG00733 | cells used in this protocol |
| Ligation Sequencing Kit 1D | Oxford Nanopore Technologies | SQK-LSK108 | nanopore ligation kit |
| MaXtract High Density tubes | Qiagen | 129073 | gel tubes |
| NEBNext FFPE DNA Repair Mix | NEB | M6630S | |
| NEBNext Ultra II End Repair/dA-Tailing Module | NEB | M7546S | |
| Nuclease-free water | Invitrogen | AM9937 | |
| Phosphate-Buffered Saline, PBS | Gibco | 70011044 | 10X, pH 7.4 |
| Phenol:chloroform:IAA | Invitrogen | AM9730 | |
| Proteinase K | Qiagen | 19131 | 20 mg/mL |
| Qubit dsDNA BR Assay Kit | Invitrogen | Q32850 | fluorometer assays for DNA quantification |
| Rapid Sequencing Kit | Oxford Nanopore Technologies | SQK-RAD004 | nanopore transposase kit |
| RNase A | Qiagen | 19101 | 100 mg/mL |
| SDS | Invitrogen | AM9822 | 10% (wt/vol) |
| Sodium chloride solution | Invitrogen | AM9759 | 5.0 M |
| TE buffer | Invitrogen | AM9849 | pH 8.0 |
| Tris | Invitrogen | AM9856 | pH 8.0, 1 M |
| Triton X-100 solution | Sigma-Aldrich | 93443 | ~10% |
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| Bio-Rad C1000 Thermal Cycler | Bio-Rad | 1851196EDU | |
| Centrifuge 5810R | Eppendorf | 22628180 | |
| Countess II FL Automated Cell Counter | Life Technologies | AMQAF1000 | for cell counting |
| DynaMag-2 Magnet | Life Technologies | 12321D | magnetic rack |
| Eppendorf ThermoMixer | Eppendorf | 5382000023 | for incubation |
| Freezer | LabRepCo | LHP-5-UFMB | |
| GridION | Oxford Nanopore Technologies | GridION X5 | nanopore device used in this protocol |
| HulaMixer Sample Mixer | Thermo Fisher Scientific | 15920D | rotator mixer |
| MicroCentrifuge | Benchmark Scientific | C1012 | |
| NanoDrop ND-1000 Spectrophotometer | Thermo Fisher Scientific | ND-1000 | for UV reading |
| Pippin Pulse | Sage Science | PPI0200 | pulsed-field gel electrophoresis instrument |
| Qubit 3.0 Fluorometer | Invitrogen | Q33216 | fluorometer |
| Refrigerator | LabRepCo | LABHP-5-URBSS | |
| Vortex-Genie 2 | Scientific Industries | SI-A236 | |
| Water bath | VWR | 89501-464 |
Références
- Mardis, E. R. Next-generation sequencing platforms. Annual Review of Analytical Chemistry. 6, 287-303 (2013).
- Goodwin, S., McPherson, J. D., McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nature Reviews Genetics. 17 (6), 333-351 (2016).
- Shendure, J., et al. DNA sequencing at 40: past, present and future. Nature. 550 (7676), 345-353 (2017).
- Alkan, C., Coe, B. P., Eichler, E. E. Genome structural variation discovery and genotyping. Nature Reviews Genetics. 12 (5), 363-376 (2011).
- Weischenfeldt, J., Symmons, O., Spitz, F., Korbel, J. O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nature Reviews Genetics. 14 (2), 125-138 (2013).
- Pollard, M. O., Gurdasani, D., Mentzer, A. J., Porter, T., Sandhu, M. S. Long reads: their purpose and place. Human Molecular Genetics. 27 (R2), R234-R241 (2018).
- Cretu Stancu, M., et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nature Communications. 8 (1), 1326(2017).
- Gong, L., et al. Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nature Methods. 15 (6), 455-460 (2018).
- Sedlazeck, F. J., et al. Accurate detection of complex structural variations using single-molecule sequencing. Nature Methods. 15 (6), 461-468 (2018).
- Jain, M., et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology. 36 (4), 338-345 (2018).
- Jain, M., et al. Improved data analysis for the MinION nanopore sequencer. Nature Methods. 12 (4), 351-356 (2015).
- Deamer, D., Akeson, M., Branton, D. Three decades of nanopore sequencing. Nature Biotechnology. 34 (5), 518-524 (2016).
- Jain, M., Olsen, H. E., Paten, B., Akeson, M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biology. 17 (1), 239(2016).
- Editorial, The long view on sequencing. Nature Biotechnology. 36 (4), 287(2018).
- Jain, M., et al. Linear assembly of a human centromere on the Y chromosome. Nature Biotechnology. 36 (4), 321-323 (2018).
- Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094-3100 (2018).
- De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34, 2666-2669 (2018).
- Akabayov, B., Akabayov, S. R., Lee, S. J., Wagner, G., Richardson, C. C. Impact of macromolecular crowding on DNA replication. Nature Communications. 4, 1615(2013).
- Gabrieli, T., Sharim, H., Michaeli, Y., Ebenstein, Y. Cas9-Assisted Targeting of CHromosome segments (CATCH) for targeted nanopore sequencing and optical genome mapping. bioRxiv. , (2017).
- Gabrieli, T., et al. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Research. , (2018).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.