
Method Article
전체 게놈 DNA 분석을 위한 매우 긴 읽기 시퀀싱
요약
긴 읽기 시퀀스는 크게 복잡 한 유전자 및 구조 변화의 특성의 어셈블리를 촉진 한다. 연속 nanopore 기반 플랫폼으로 매우 긴 시퀀스를 생성 하는 방법을 설명 합니다. 접근은 최적화 된 DNA 추출이 수정된 라이브러리 준비 인간 세포에서 kilobase 읽기 적당 한 보도 수백을 생성 하 여 다음을 채택 한다.
초록
제 3 세대 단일 분자 DNA 시퀀싱 기술 제공 훨씬 더 이상 읽을 길이 복잡 한 유전자 및 복잡 한 구조 이체의 분석의 어셈블리를 촉진할 수 있다. Nanopore 플랫폼 직접 숨 구멍을 통해 DNA 통행에 의해 중재 현재 변화를 측정 하 여 단일 분자 시퀀싱을 수행 하 고 최소한의 자본 비용으로 kilobase (kb) 읽기의 수백을 생성할 수 있습니다. 이 플랫폼은 다양 한 응용 프로그램에 대 한 많은 연구자에 의해 채택 되었습니다. 긴 연속 읽기 길이 달성 nanopore 시퀀싱 플랫폼의 가치를 활용 하는 가장 중요 한 요소입니다. 매우 긴 읽기를 생성 하려면 특별 한 고려 DNA 파손 방지 및 생산성 시퀀싱 서식 파일을 생성 하는 효율을 얻이 필요 합니다. 여기, 우리는 매우 긴 DNA 시퀀싱 신선 또는 냉동 셀, 기계적 전단 또는 transposase 조각화, 도서관 건축에서에서 고 분자량 (HMW) DNA 추출 및 nanopore 장치에서 시퀀싱의 상세한 프로토콜을 제공 합니다. HMW DNA의 20-25 µ g에서 메서드 N50 읽기 50-70 kb 기계적 전단의 길이 달성할 수 있다 고 90-100의 transposase 길이 읽기 N50 중재 조각화 합니다. 프로토콜 구조 변형 및 게놈 어셈블리의 검색을 위한 전체 게놈 시퀀싱을 수행 하기 위해 포유류 세포에서 추출한 DNA를 적용할 수 있습니다. DNA 추출 및 효소 반응에 대 한 추가 개선 또한 읽기 길이 증가 하 고 그것의 유틸리티를 확장 합니다.
서문
지난 10 년 동안 대규모 병렬 하 고 매우 정확 하 게 2 세대 높은 처리량 시퀀싱 기술 주도 생물 발견 및 기술 혁신1,2,3의 폭발. 기술 진보에도 불구 하 고 2 세대 플랫폼에 의해 생성 된 짧은 읽기 데이터 복잡 한 게놈 영역 해결에 효과가 및 게놈 구조 변형 (SVs), 인간에서 중요 한 역할을의 검출에 제한 됩니다. 진화와 질병4,5. 또한, 짧은 읽기 데이터 반복 변화를 확인할 수 없습니다 하 고 분별 haplotype 유전 이체6의 단계적 적합 하지 않습니다.
단일 분자 시퀀싱 제공 훨씬 더 최근 진행 길이, SVs7,,89, 전체 스펙트럼 및 제공 정확 하 고 완벽 한 복잡 한 검색을 용이 하 게 수 읽기 미생물 및 포유류 게놈6,10. Nanopore 플랫폼 직접 모11,,1213DNA 통행에 의해 중재 현재 변화를 측정 하 여 단일 분자 시퀀싱을 수행 합니다. 모든 기존 DNA 시퀀싱 화학, 달리 nanopore 시퀀싱을 생성할 수 있습니다 오래 (kilobases 수천 수만) 읽기 실시간 중 합 효소 활동에 의존 하지 않고 또는 인공 DNA 샘플의 증폭. 따라서, nanopore 긴-읽기 (NLR-seq) 낮은 복잡성 또는 반복 풍부한에서 특히 크게 게놈 및 생물 의학 분석14사전 것, 100kb 이상의 매우 긴 읽기 길이 생성 하기 위한 위대한 약속을 보유 하 고 시퀀싱 게놈15의 영역입니다.
Nanopore 시퀀싱의 독특한 기능은 잠재적인 긴 생성을 이론적인 길이 제한 없이 읽습니다. 따라서, 읽기 길이 DNA 무결성 및 시퀀싱 템플릿 품질에 의해 영향을 받는 직접 DNA의 물리적 길이에 따라 달라 집니다. 또한, 조작 및 단계, pipetting 세력 및 추출 조건 등의 수의 정도 따라 DNA의 품질이 매우 다양 합니다. 따라서, 그것은 단지 표준 DNA 추출 프로토콜 및 제조업체의 제공 된 라이브러리 공법을 적용 하 여 긴 읽기를 한 도전 이다. 이 끝으로, 우리는 강력한 개발 했습니다 매우 긴 생성 하는 방법 읽기 (kilobases 수백) 시퀀싱 데이터 수확된 셀 펠 릿에서 시작. 우리는 DNA 추출 및 라이브러리 준비 절차에서 여러 개선 채택. 우리는 DNA 저하 및 손상 시키는 불필요 한 절차를 제외 하려면 프로토콜을 간소화. 이 프로토콜은 고 분자량 (HMW)의 구성 된 DNA 추출, 매우 긴 DNA 도서관 건축 및 연속 nanopore 플랫폼에. 잘 훈련 된 분자 생물학에 대 한 일반적으로 걸리는 6 h HMW DNA 추출, 90 분 또는 도서관 건설 깎는 방법, 그리고 DNA 연속을 위한 추가 48 h 8 h의 완료에 수확 하는 세포에서. 프로토콜을 사용 하는 게놈 복잡성에 대 한 우리의 이해를 개선 하 여 인간의 질병에 게놈 변이에 새로운 통찰력을 얻을 유전체학 커뮤니티 권한을 부여 합니다.
프로토콜
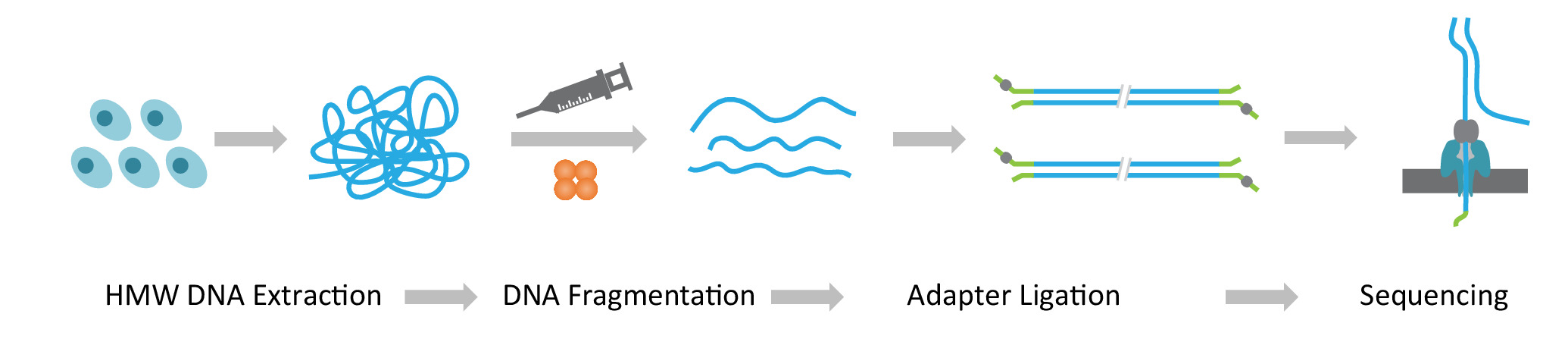
참고: NLR seq 프로토콜 3 연속 단계로 구성: 1) 추출 높은 분자의 무게 (HMW) 게놈 DNA; 2) 매우 긴 DNA 도서관 건설, 원하는 크기로 HMW DNA의 분열 그리고 DNA 시퀀싱 어댑터의 결 찰 포함 종료; 그리고 3) nanopores (그림 1)의 배열에 어댑터 출혈 DNA의 로드.
1. HMW DNA 추출
- 시 약 설치 합니다. PBS의 100 mL를 추가 하 여 인산 염 버퍼 식 염 수 (PBS) 버퍼 (1000 mL) x 1를 확인 (10 배) 900 ml의 물과 잘 혼합. 세포의 용 해 버퍼 (50 mL) 물 43.5 mL 50 mL 튜브에 추가 하 여 확인 합니다. 트리 스 (1 M, pH 8.0), 염화 나트륨 (NaCl) (5 M)의 1 mL의 500 μ ethylenediaminetetraacetic 산 (EDTA) (0.5 M, pH 8.0)의 2.5 mL을 추가 하 고 나트륨 라우릴의 2.5 mL 튜브 (SDS) (10%, wt/vol)을 황산과 잘 섞어.
참고:이 PBS 버퍼 저장할 수 있습니다 4 ° C에서 최대 6 개월까지. Premade 세포의 용 해 버퍼 최대 2 개월까지 실시간에 저장할 수 있습니다. - 세포 사망을 확인 하 고는 셀. 라이브 비율 > 85% 이며 전체 휴대폰 번호는 30 x 106확인 합니다.
참고: 셀이이 프로토콜에 사용 되는 HG00733 셀 라인, 구조 변화 분석을 위한 1000 게놈 컨소시엄에서 널리 이용 되는 푸에 토 리코 근원의 인간 lymphoblastoid 셀 라인에서 (주문 정보에 대 한 자료의 표 참조)에 속한 하 국제 게놈 샘플 리소스. - 실시간 삭제 매체에서 5 분 동안 200 x g 에서 centrifuging 여 세포를 수집 하 고 1 x PBS 버퍼의 5 mL와 함께 셀 펠 릿 (30 x 106 셀) resuspend. 200 x g RT 5 분에 다시 원심 및 삭제는 상쾌한.
참고: 25-35 x 106 세포의이 접근에 대 한 사용할 수 있습니다. 더 셀 사용 금액 변동 추가 최적화를 합니다. 셀 펠 릿 −80 ° C에서 최대 6 개월까지 저장할 수 있습니다. - 1 x PBS 버퍼의 200 μ에 셀 펠 릿을 resuspend. 고정된 셀 펠 릿을 사용 하 여, 1 x PBS 버퍼의 5 mL로 세척 한다. RT에 5 분 동안 200 x g 에서 솔루션 원심, 삭제는 상쾌한 고 PBS 버퍼 x 1의 200 μ 셀 resuspend.
- 50 mL 튜브에 세포의 용 해 버퍼의 10 mL를 준비 합니다. 3 미 품 1 h 37 ° C에서 솔루션에 대 한 세포의 용 해 버퍼 및 최고 속도로 소용돌이를 200 μ 세포 현 탁 액을 추가 합니다.
- lysate를 2 μ의 RNase A (100 mg/mL)를 추가 합니다. 부드럽게 혼합 샘플 50 mL 튜브를 회전 합니다. 1 시간 동안 37 ° C에서 솔루션을 품 어.
- lysate를 50 μ 가수분해 K (20 mg/mL)를 추가 합니다. 부드럽게 혼합 샘플 50 mL 튜브를 회전 합니다. 2 h 50 ° C에서 솔루션을 품 어. 부 화, 동안 부드럽게 혼합 샘플 마다 30 분.
- 50 ° C에서 50 mL 튜브를 제거 하 고 RT에 5 분 동안 서 보자.
- lysate를 페 놀: 클로 프롬: isoamyl 알콜 (25:24:1, vol/vol/vol)의 페 놀 층의 10 mL를 추가 하 고 회전 믹서에 튜브를 회전 ( 재료의 표참조) 10 분에 대 한 20 rpm에서 증기 두건에서 RT에서 누설을 방지 하기 위해 parafilm으로 튜브 캡 포장 동안 회전입니다.
- 실시간에서 2 분 동안 1500 x g 에서 centrifuging 여 두 50 mL 젤 튜브 ( 재료의 표참조)를 준비
참고: 젤 수성 단계 포함 하는 핵 산 및 유기 용 매 사이의 안정적인 장벽을 형성 한다. - 단계 1.10에서 젤 튜브 준비 50 mL 중에 샘플/페 놀 솔루션을 붓으십시오. 실시간에서 10 분 동안 3000 x g 에서 솔루션을 원심
- 새로운 50 mL 튜브에는 상쾌한 하 거 라. (25:24:1, vol/vol/vol) 페 놀: 클로 프롬: isoamyl 알콜 페 놀 층의 10 mL을 추가 하 고 10 분 20 rpm에서 증기 두건에서 RT에서 회전 믹서에 튜브를 회전.
- 준비와 한번 단계 1.11 젤 튜브를 반복 합니다.
- 새로운 50 mL 튜브에는 상쾌한 하 거 라. 얼음 처럼 차가운 100% 에탄올의 25 mL를 추가 하 고 부드럽게 돌려 튜브 손으로 DNA 침전 (그림 2).
참고: 강수량 접근 HMW DNA를 안정 시킬 수 있습니다. - 후크를 만들 20 μ 팁 벤드. 조심 스럽게 꺼내는 후크와 HMW DNA와 액체 방울을 풀어.
- HMW DNA를 포함 하는 70% 에탄올 40 mL 50 mL 튜브에 놓습니다. 부드럽게 반전 튜브 3 번 하 여 DNA를 씻어.
- 반복 단계 1.15 번 70% 에탄올 튜브에서 DNA를 수집 합니다.
- 70% 에탄올의 1.8 mL를 포함 하는 2 mL 튜브에 HMW DNA를 놓습니다.
- 10000 x g 3에서 씻어 HMW DNA를 원심 pipetting으로 가능한 잔여 에탄올의 실시간 제거에서 s.
참고: 때 잔여 에탄올 pipetting DNA 펠 렛을 방해 하지 마십시오. - 뚜껑을 샘플 건조 오픈 10 분 동안 37 ° C에서 2 mL 튜브를 품 어.
- 와 계속 2.1 (와 기계적 전단 1 D 결 찰 시퀀싱 키트) 단계, 2 mL 튜브에 테 (10 mM Tris 및 1 m m EDTA, pH 8.0)의 1 mL를 추가 합니다.
- 10 mm Tris (pH 8.0) 0.02 %200 μ 추가 계속 단계 2.2 (transposase 기반 조각화 및 급속 한 시퀀싱 키트), 트라이 톤 X-100.
참고: DNA 펠 렛을 방해 하지 마십시오. 어둠 속에서 4 ° C에서 48 h에 대 한 서 관 시키는 완전히 resuspend 샘플을 도움이 됩니다. HMW DNA는 4 ° C에서 최대 2 주 동안 저장할 수 있습니다. 긴 저장 시간 또는 다른 저장 조건 더 짧은 파편을 발생할 수 있습니다.
2. 매우 긴 DNA 도서관 건축
참고: nanopore 시퀀싱 키트와 결합 하 여 두 개의 다른 기울이기 방법에 따라 매우 긴 DNA 라이브러리를 구성 하는 두 가지 방법이 있다. 기계적 전단 기반 라이브러리 라이브러리 건설에 8 h 약을 복용 하는 50-70 킬로바이트의 N50 사용 하 여 데이터를 생성 합니다. Transposase 조각화 기반 라이브러리 라이브러리 건설에 대 일 분만 복용의 90-100 kb 데이터 N50 생성 합니다. 기계적 전단 프로토콜 같은 DNA 시퀀싱 어댑터와 nanopore 흐름 셀의 품질의 동일한 버전을 사용 하 여 입력에서 높은 수익률을 제공 합니다.
- 기계적 전단 기반 도서관 건축
- 해 동 하 고 믹스는 결 찰에서 시 약 키트 ( 재료의 표참조). Thaw FFPE DNA 버퍼와 얼음, 그 소용돌이에 최종 수리/다-미행 버퍼를 복구 및 혼합 아래로 회전. 섞어 녹여 내려 어댑터 믹스 (AMX)와 얼음, 그리고 피 펫 스핀에 어댑터 비드 바인딩 버퍼 (ABB). 연료 혼합 (RBF) 실행 버퍼 및 차입 버퍼 (ELB) RT, 그리고 소용돌이 스핀에 내려 섞어 녹여. RT와 피 펫을 사용 하기 전에 혼합 라이브러리 로드 구슬 (LLB)를 녹여.
- 일단 해 동, 얼음에 모든 키트 구성 요소를 계속. 필요할 때만 효소를 꺼내. 사용 하기 위해 RT 자석 구슬가지고.
참고: 사용 하는 자석 구슬에 추천 자료의 테이블 참조.
- 일단 해 동, 얼음에 모든 키트 구성 요소를 계속. 필요할 때만 효소를 꺼내. 사용 하기 위해 RT 자석 구슬가지고.
- 단계의 1.21.1 HMW DNA의 양과 품질을 확인 합니다. HMW DNA 튜브 넓은 p 200를 사용 하 여 세 가지 다른 위치에서 새로운 1.5 mL 튜브에 DNA의 20 μ 밖으로 피 펫 팁을 낳았다. 읽고 UV를 사용 하 여 품질과 fluorometer를 사용 하 여 농도 검출 하기 위하여 3 개의 aliquots에서 1 μ를 가져가 라. 여러 번 확인 결과를 확인 합니다.
참고: 그림 3A에서 예상 된 결과가 표시 됩니다. OD260/280 값은 약 1.9 하 고 OD260/230 값은 약 2.3. - 전송 P1000 넓은 50 mL 튜브 캡으로 HMW DNA의 나머지 940 μ 팁을 낳았다.
- 바늘 없는 주사기를 1 mL에 모든 DNA를 발음.
- 27 G 니 들을 주사기에 넣고 부드럽게 그리고 천천히 뚜껑으로 모든 DNA를 추출 (~ 10 s). 27 G 바늘 주사기에서 벗어.
- 바늘을 통해 30 패스의 총 29 시간 2.1.4-2.1.5 단계를 반복 합니다.
참고: 전단된 HMW DNA 24 h. 품질 관리 (QC) 최대 펄스 분야 젤 전기 이동 법으로 좋습니다 하지만 비용과 시간이 소요 그것은 어둠 속에서 4 ° C에서 저장할 수 있습니다. 자동된 펄스 분야 젤 전기 이동 법 기계에 QC를 수행 하는 경우 실행 20 h 5-150 kb 프로토콜을 사용 합니다. 예상된 결과 그림 4에 나와 있습니다. - 깎인된 HMW DNA의 100 μ를 추가 하 여 0.2 mL 튜브에 DNA 수리 반응 준비 (20 μ g), FFPE DNA 수리 버퍼의 15 μ, FFPE DNA 수리 믹스의 12 μ 및 16 μ nuclease 무료 물. 6 번 부드럽게 터치 하 여 반응 혼합 하 고 제거 거품 아래로 회전.
- P200 넓은 구멍 팁 새로운 1.5 mL 튜브에 60 분 전송 20 ° C에서 반응 샘플을 품 어.
- 마그네틱 구슬 pipetting 또는 vortexing resuspend. DNA 수리 반응과 믹스를 143 μ 구슬 (1 x) 추가 부드럽게 터치 튜브 6 번 하 여. 30 분 20 rpm에서 RT에서 회전 믹서에 튜브를 회전 합니다.
- 스핀에서 1000 x g 2에 대 한 샘플 다운 실시간 장소 10 분에 대 한 자석 선반에 튜브에서 s 자석 선반에 튜브를 유지 하 고는 상쾌한 삭제.
- 마그네틱 선반에 튜브를 유지, 펠 릿을 방해 하지 않고 갓된 70% 에탄올의 400 μ를 추가 합니다. 30 후 70% 에탄올을 제거 s.
- 2.1.11 단계를 한 번 반복 합니다.
- 스핀에서 1000 x g 2에 대 한 샘플 다운 튜브 자석 선반에 다시 실시간 장소 s. 모든 잔여 에탄올을 제거 하 고 공기 건조 30 s. 펠 릿 이상 건조 하지 마십시오.
- 마그네틱 선반에서 튜브를 제거 하 고 테 (10 mM Tris 및 1 m m EDTA, pH 8.0)의 103 μ를 추가. 부드럽게 톡 구슬, 버퍼에 적용 되 고 30 분에 대 한 RT에서 회전 믹서에 부드럽게 품 어 튜브 튜브 마다 5 분 터치 물의 resuspension 펠 릿의 원조 하.
- 작은 자석 선반에 구슬 와이드 P200와 eluate의 적어도 10 분 전송 100 μ 0.2 mL 관으로 팁을 지루하게 하는 대.
- 수리 HMW DNA의 100 μ, 최종 수리/다-미행 버퍼의 14 μ 및 최종 수리/다-미행 믹스의 7 μ를 추가 하 여 최종 수리 및 0.2 mL 튜브에 다 미행 반응 준비. 6 번 부드럽게 터치 하 여 반응 혼합 하 고 제거 거품 아래로 회전.
- 품 어 20 분, 65 ° c 60 분 동안 20 ° C에서 반응 하 고 22 ° c.에서 개최 넓은 p 200를 사용 하 여 새로운 1.5 mL 튜브에 샘플 보어 전송 팁.
- 마그네틱 구슬 pipetting 또는 vortexing resuspend. 구슬의 48 μ 추가 (0.4 x) 최종 수리/다-미행 반응을 부드럽게 터치 튜브 6 번 하 여 혼합. 30 분 20 rpm에서 RT에서 회전 믹서에 튜브를 회전 합니다.
- 한 번 2.1.10-2.1.13 단계를 반복 합니다.
- 마그네틱 선반에서 튜브를 제거 하 고 33 μ 테 (10 mM Tris 및 1 m m EDTA, pH 8.0)의 추가. 부드럽게 톡 구슬, 버퍼에 적용 되 고 30 분에 대 한 RT에서 회전 믹서에 부드럽게 품 어 튜브 튜브 마다 5 분 터치 물의 resuspension 펠 릿의 원조 하.
- 작은 자석 선반에 구슬 와이드 P200와 eluate의 적어도 10 분 전송 30 μ 구멍 팁 새로운 1.5 mL 튜브에 대 한. fluorometer을 사용 하 여 농도 검출 하 여분 1-2 μ를 가져가 라.
참고:이 단계에서 5-6 μ g의 복구 예정입니다. - 준비 끝 수리 HMW DNA의 추가 30 μ에 의해 1.5 mL 샘플 튜브에 결 찰 반응, 어댑터 믹스 (1 D AMX)의 20 μ, 블런트/TA 결 찰 마스터 믹스의 50 μ. 각 순차적 추가 및 제거 거품 아래로 회전 사이 6 번 부드럽게 터치 하 여 반응 혼합.
- 60 분에 대 한 RT에 반응을 품 어.
- 마그네틱 구슬 pipetting 또는 vortexing resuspend. 추가 40 μ 구슬 (0.4 x) 결 찰 반응을 부드럽게 터치 튜브 6 번 하 여 혼합. 30 분 20 rpm에서 RT에서 회전 믹서에 튜브를 회전 합니다.
- 2.1.10 단계를 한 번 반복 합니다.
- 튜브에 어댑터 비드 바인딩 (ABB) 버퍼의 400 μ를 추가 합니다. 6 번 부드럽게 구슬 resuspend으로 튜브를 터치 합니다. 버퍼에서 구슬을 분리 하 고 상쾌한 삭제 하 자석 선반에 다시 튜브를 놓습니다.
- 한 번 2.1.26 단계를 반복 합니다.
- 스핀에서 1000 x g 2에 대 한 샘플 다운 튜브 자석 선반에 다시 실시간 장소 s. 모든 잔여 버퍼를 제거 하 고 공기 건조 30 s. 펠 릿 이상 건조 하지 마십시오.
- 마그네틱 선반에서 튜브를 제거 하 고 차입 버퍼의 43 μ에 펠 릿을 resuspend. 부드럽게 톡 구슬 버퍼에 적용 되 고 30 분에 대 한 RT에서 회전 믹서에 부드럽게 품 어 튜브 튜브 마다 5 분 터치 물의 resuspension 펠 릿의 원조 하.
- 작은 자석 선반에 구슬 와이드 P200와 eluate의 적어도 10 분 전송 40 μ 구멍 팁 새로운 1.5 mL 튜브에 대 한. fluorometer을 사용 하 여 농도 검출 하 여분 1-2 μ를 가져가 라.
참고:이 단계에서 1-2 μ g의 복구 예정입니다. 기계적 전단 기반 라이브러리 로딩에 대 한 준비가 되어있습니다. 도서관 저장할 수 있습니다 최대 2 h ice에 시퀀싱에 대 한 로드까지 필요한 경우.
- 해 동 하 고 믹스는 결 찰에서 시 약 키트 ( 재료의 표참조). Thaw FFPE DNA 버퍼와 얼음, 그 소용돌이에 최종 수리/다-미행 버퍼를 복구 및 혼합 아래로 회전. 섞어 녹여 내려 어댑터 믹스 (AMX)와 얼음, 그리고 피 펫 스핀에 어댑터 비드 바인딩 버퍼 (ABB). 연료 혼합 (RBF) 실행 버퍼 및 차입 버퍼 (ELB) RT, 그리고 소용돌이 스핀에 내려 섞어 녹여. RT와 피 펫을 사용 하기 전에 혼합 라이브러리 로드 구슬 (LLB)를 녹여.
- Transposase 조각화 기반 도서관 건축
- 재개는 transposase에서 시 약 키트 ( 재료의 표참조). 녹여 조각화 믹스 (FRA)와 섞어 얼음과 피 펫에 빠른 어댑터 (랩). Thaw 시퀀싱 버퍼 (SQB), 구슬 (파운드) 로드, 플러시 버퍼 (FLB)와 RT에 밧줄 (FLT) 플러시 혼합 플라스틱. 로드 구슬 (파운드) RT와 피 펫을 사용 하기 전에 혼합에 녹여 일단 해 동, 얼음에 모든 키트 구성 요소를 계속. 필요할 때만 효소를 꺼내.
- 단계의 1.21.2 HMW DNA의 양과 품질을 확인 합니다. HMW DNA 튜브 넓은 p 200를 사용 하 여 세 가지 다른 위치에서 새로운 1.5 mL 튜브에 DNA의 20 μ 밖으로 피 펫 팁을 낳았다. 읽고 UV를 사용 하 여 품질과 fluorometer를 사용 하 여 농도 검출 하기 위하여 3 개의 aliquots에서 1 μ를 가져가 라. 여러 번 확인 결과를 확인 합니다.
참고: 그림 3B에서 예상 된 결과가 표시 됩니다. OD260/280 값은 약 1.9 하 고 OD260/230 값은 약 2.3. - HMW DNA, 10 mM Tris (pH 8.0) 조각화의 0.02% 트라이 톤 X-100 그리고 1 μ와 혼합 (FRA)의 1 μ의 22 μ를 추가 하 여 DNA tagmentation 반응 0.2 mL 튜브에서를 준비 합니다. 믹스와 와이드 P200 pipetting으로 낳았다 팁 가능한 천천히 6 번 거품을 소개 하도록 하지.
- 품 어 1 분 뒤에 1 분 동안 80 ° C에 30 ° C에서 반응 하 고 4 ° c.에서 개최 P200 광범위 새로운 1.5 mL 튜브에 혼합 전송 팁 및 즉시 다음 단계로 이동을 낳았다.
- 1.5 mL 샘플 튜브를 1 μ 빠른 어댑터 (랩)를 추가 합니다. 믹스와 와이드 P200 pipetting으로 낳았다 팁 가능한 천천히 6 번 거품을 소개 하도록 하지.
- 60 분에 대 한 RT에 반응을 품 어.
참고: transposase 조각화 기반 라이브러리는 로드에 대 한 준비. 도서관 저장할 수 있습니다 최대 2 h ice에 시퀀싱에 대 한 로드까지 필요한 경우.
3입니다. 연속 nanopore 장치에
- Nanopore 시퀀싱 장치 검사 ( 재료의 표참조). 소프트웨어와 하드웨어와 충분 한 저장 공간이 있는지 확인 합니다.
- 흐름 셀을 확인 합니다. 새로운 흐름 셀 열고 nanopore 장치로 흐름 셀을 삽입 합니다. 위치 흐름 셀의 상자 (X1-X5)에 삽입 된 확인 하십시오. 정확한 흐름 셀 유형을 선택 합니다. 흐름 세포 확인 워크플로 클릭 합니다. 테스트 시작 버튼 흐름 셀 QC 분석 시작을 클릭 합니다.
참고: 보고 된 총 활성 공 수 800 미만 이면 시퀀싱에 대 한 다른 새로운 흐름 셀을 사용 합니다. - 못쓰게 버퍼를 준비 합니다. 기계적 전단 기반 라이브러리에 대 한 추가 연료 혼합 (RBF) 버퍼와 624 576 μ μ nuclease 무료 물 1.5 mL 튜브에의. 와 동 그리고 믹스 못쓰게 버퍼 아래로 회전. Transposase 조각화 기반 라이브러리에 대 한 플러시 버퍼 (FLB)의 튜브를 플러시 밧줄 (편 명)의 30 μ를 추가 합니다. 와 동 그리고 믹스 못쓰게 버퍼 아래로 회전.
- 흐름 세포에서 못쓰게 포트 커버를 못쓰게 포트 노출 시계 방향으로 이동 합니다.
- P1000 피펫으로 100 μ를 설정 하 고 팁 못쓰게 포트에 삽입. 흐름 세포에서 모든 거품을 제거 하려면 버퍼 (30 μ)의 작은 볼륨을 다시 그립니다. 일단 노란색 액체의 작은 양의 입력 팁 pipetting 중지 합니다.
- P1000 피 펫을 사용 하 여 못쓰게 포트를 통해 흐름 셀으로 못쓰게 믹스의 800 μ를 로드. 소개 하는 거품을 피하기 위해, 먼저, 못쓰게 포트의 상단을 커버 다음 팁 못쓰게 포트에 넣고 천천히 못쓰게 혼합의 나머지를 추가 못쓰게 혼합의 30 μ를 추가 합니다. 약 50 μ 왼쪽 때 팁을 가져가 라. 못쓰게 포트 상단 못쓰게 혼합의 나머지를 추가 합니다. 액체는 자체적으로 안에 갈 것입니다.
- 설치 5 분 동안 품 어 둡니다. 한편, 라이브러리를 포함 하는 1.5 mL 튜브에 라이브러리 믹스를 준비 합니다.
참고: 기계적 전단 기반 라이브러리에 대 한 추가 35 µ L의 DNA 도서관의 40 µ L를 연료 혼합 (RBF)와 버퍼를 실행 합니다. Transposase 조각화 기반 라이브러리에 대 한 추가 시퀀싱 버퍼 (SQB)의 34 µ L 16 µ L 25 µ L의 DNA 도서관을 nuclease 무료 물. - 샘플 포트를 노출 하는 부드럽게 흐름 셀 샘플 포트 덮개를 엽니다. P1000 피 펫을 사용 하 여 3.5 단계에서 설명한 대로 못쓰게 포트를 통해 못쓰게 믹스 200 μ 흐름 셀에 추가. 못쓰게 혼합 샘플 포트를 통해 흐름 셀에 로드 되지 않은 다는 것을 확인 하십시오.
- 80 μ P200 피 펫을 설정 합니다. 믹스 넓은와 부드럽게 라이브러리 로드 직전 6 번 위아래로 pipetting으로 팁을 낳았다.
- 로드 라이브러리 믹스 dropwise 샘플 포트를 통해 흐름 셀에. 이전 드롭 포트에 완전히 로드 된 후에 각 방울을 추가 합니다.
- 다시 샘플 포트 커버 부드럽게 넣고 샘플 포트 완전히 덮여 있는지 확인 하십시오. 못쓰게 포트 커버 못쓰게 포트 커버 반시계 이동 합니다. 장치 뚜껑을 닫습니다.
- 새로운 실험 워크플로 클릭 합니다. 라이브러리 이름을 입력 하 고 사용, 절차에 따라 정확한 키트 선택한 설정이 올바른 (48 개의 h 실행, 실시간 자료 전화에) 확인.
- 시작 실행을클릭 합니다. 10 분 후 흐름 셀 ID와 실행된 정보에서 활성 nanopore 숫자 (총 수 및 각 4 개 그룹의 숫자)를 기록 합니다.
- 데이터 분석입니다. 언제 든 지 시퀀싱의 실행이 완료 되 면 로컬 컴퓨터 또는 클러스터에 데이터를 복사 합니다. Minimap216 (https://github.com/lh3/minimap2)를 사용 하 여 참조 게놈 시퀀스 데이터 정렬. NanoPlot17 (https://github.com/wdecoster/NanoPlot)에 의해 원시 시퀀스 데이터는 정렬에서 시퀀싱 성능 요약.
결과
매우 긴 DNA 시퀀싱 프로토콜 라이브러리 건설 HMW DNA를 적용합니다. 따라서, 그것은 라이브 비율 잘 경작된 한 세포를 선택 하는 중요 한 > 셀 수확 단계에서 85%. HMW DNA의 양과 질, DNA 추출에 사용 되는 셀의 크기에 영향을 미칩니다. 세포 세포의 용 해는 너무 많은 세포로 시작 하는 경우에 잘 작동 하지 않습니다. HMW DNA 강수량 고속 원심 분리 대신 손으로 부드러운 회전을 사용 하 여 수행 되기 때문에 너무 몇 가지 셀을 사용 하 여 라이브러리 건설에 대 한 충분 한 DNA을 생성 하지 않습니다. 얼음 처럼 차가운 100% 에탄올을 추가 하 고 회전 그림2에서 흰 목화 같은 침전으로 표시 됩니다 후 HMW DNA의 예입니다.
그것은 도서관 건축을 시작 하기 전에 입력 DNA의 품질을 확인 해야 합니다. 저하, 잘못 된 정량화, 오염 (예: 단백질, RNAs, 세제, 계면 활성 제, 그리고 잔여 석탄 산 또는 에탄올) 및 낮은 분자량 DNA 길이 읽고 마지막에 후속 절차에 상당한 효과 가질 수 있습니다. HMW DNA를 포함 하는 관에서 세 가지 다른 위치에서 DNA를 사용 하 여 품질 분석을 수행 하는 것이 좋습니다. UV HMW DNA에 대 한 결과 읽어에서 OD260/OD280 값 약 1.9 이며 OD260/OD230 값은 약 2.3 (그림 3AB). 이러한 비율 값은 좋은 HMW DNA 샘플에 대 한 세 가지 테스트 간에 일관. 다른 깎는 방법 입력된 DNA의 다른 볼륨을 필요 합니다. HMW DNA의 농도 필요 > 기계적 전단 필요 동안에 µ 200 ng/L > transposase 조각화에 대 한 µ 1 µ g/L. fluorometer에 의해 감지 농도 UV 읽기 보다 조금 낮습니다. 그러나, 동일한 HMW DNA 샘플의 농도의 변이 계수는 fluorometer와 UV 분석 실험을 읽고 미만 15% 필요 합니다. 기계적 전단 주사기 바늘을 통해 전달의 수 전단된 DNA의 크기에 영향을 줍니다 마지막 읽을 길이 있도록 HMW DNA를 바늘으로 적용 됩니다. 후 바늘 HMW DNA의 대부분을 위해 전단은 그림 4에서 볼 수 있듯이 50 kb 보다 큰 크기 품질 관리를 수행 하는 것이 좋습니다. 기계적 전단 방식에서 30 패스 길이 출력을 고려 최고의 시퀀싱 결과 생성.
기계적 전단 기반 라이브러리의 N50 transposase 조각화 기반 라이브러리는 90-100 킬로바이트 50-70 킬로바이트입니다. HG00733 셀 라인을 사용 하 여 4 개의 실행 결과 표 1에 표시 됩니다. 모든 4 개의 실행 100 kb 보다 긴 길이와 이상의 2300 읽기 있다. 최대 길이 transposase 조각화 기반 라이브러리에서 (455 kb 및 489 kb) 기계적 전단 기반 라이브러리 (348 kb 및 387 kb)와 비교 된 더 긴 동안 후자 더 총 읽습니다, 높은 수익률을 나타내는. 그것은 몇몇 짧은 조각을 소개 합니다 있도록 transposase 조각화 기반 라이브러리 건설 적은 단계와 짧은 준비 시간 있다. Transposase를 사용 하 여 두 개의 실행 긴 평균 길이 (> 30 kb)와 중간 길이 (> 10 kb). 또한, 데이터에서 모든 실행을 (평균 품질 평가 점수는 약 10.0, ~ 90% 기본 정확도) 일관 된 높은 품질을 보여줍니다. 총 기초의 97% 이상 인간의 참조 게놈 (hg19) Minimap216 을 사용 하 여 기본 설정으로 정렬 했다. 원시 읽기의 예상된 크기 분포는 그림 5에 나와 있습니다. 모든 실행 동안 transposase 조각화 기반 라이브러리는 매우 긴 읽기 (예: > 100 kb)의 높은 비율 50 kb 이상의 데이터의 큰 비율이 있다. 이 프로토콜은 여러 개의 인간 세포 라인 (보충 표 1)에 성공적으로 적용 되었다.

그림 1: nanopore 긴 읽기 시퀀싱 (NLR-seq) 워크플로의 도식 개요. 오렌지, 복잡 한 transposase입니다. 노란색-녹색, nanopore 어댑터입니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}

그림 2: 페 놀-클로 프롬 적 출 방법에서 대표 DNA 강수량. 흰색 화살표 HMW DNA를 나타냅니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}

그림 3: UV 독서에서 HMW DNA의 예제 QC 결과. 1.21.1 기계적 전단 기반 도서관 건축을 위한 준비 단계에서의 (A) HMW DNA 단계 1.21.2 transposase 라이브러리 조각화 기반 건설에서에서의 (B) HMW DNA 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}

그림 4: 바늘의 QC 결과 펄스 분야 젤 전기 이동 법에 의해 HMW DNA 전단. L1: 빠른 부하 1 kb DNA 사다리; L2: 빠른 로드 1 kb DNA 사다리 확장. 1-8: DNA 전단 하는 바늘을 통해 다른 지나가는 시간. 1-3, 전단; 4, 10 번; 5, 20 번; 6, 30 시간; 7, 40 번; 8, 50 번입니다. 이 품질 관리 단계는 선택 사항입니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}

그림 5: nanopore 매우 긴 DNA 라이브러리의 크기 분포를 예상. MS, 기계적 전단 기반 라이브러리입니다. TF, transposase 조각화 기반 라이브러리 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}
| 기계 shearing_rep1 | 기계 shearing_rep2 | Transposase fragmentation_rep1 | Transposase fragmentation_rep2 | |
| 셀 라인 | HG00733 | HG00733 | HG00733 | HG00733 |
| 읽기의 N50 | 55,180 | 63,007 | 98,237 | 95,629 |
| 100kb 이상 읽기 수 | 2500 | 3,082 | 2,386 | 2,355 |
| 총 읽기 수 | 97,859 | 80,465 | 24,166 | 21,032 |
| 최대 길이 (bp) | 348,482 | 387,113 | 454,660 | 489,426 |
| 평균 길이 (bp) | 17,861 | 20,395 | 33,528 | 38,175 |
| 중간 길이 (bp) | 5,335 | 5,894 | 10,249 | 15,656 |
| 읽기의 품질을 의미 | 10.0 | 10.1 | 9.9 | 10.0 |
| 원시 읽기의 총 기초 | 1,747,849,822 | 1,641,058,932 | 810,229,733 | 802,886,304 |
| 정렬 된 읽기의 총 기초 | 1,693,300,832 | 1,607,975,925 | 791,422,077 | 778,417,627 |
| 총 기초 (hg19, Minimap2)의 매핑된 비율 | 96.9% | 98.0% | 97.7% | 97.0% |
| 활성 숨 구멍의 수 | 1225: 480, 402, 254, 89 | 1058: 480, 356, 176, 46 | 958: 452, 328, 148, 30 | 1092: 487, 367, 195, 43 |
표 1: 다른 전단 프로토콜 성능 통계 요약에서 실행 됩니다.
| 도서관 1 | 도서관 2 | |
| 셀 라인 | K562 | GM19240 |
| 세포 주문 정보 | ATCC, 고양이입니다. 아니요. CCL-243 | Coriell 연구소, 고양이입니다. 아니요. GM19240 |
| 프로토콜 | 기계적 전단 | 기계적 전단 |
| 읽기의 N50 | 60,063 | 55,295 |
| 총 읽기 수 | 193,783 | 120,807 |
| 중간 길이 (bp) | 1,843 | 4,688 |
| 평균 길이 (bp) | 9,825 | 17,408 |
| 최대 길이 (bp) | 548,780 | 212,338 |
| 원시 읽기의 총 기초 | 1,903,989,686 | 2,103,015,331 |
| 정렬 된 읽기의 총 기초 | 1,837,350,047 | 1,997,419,761 |
| 총 기초 (hg19, Minimap2)의 매핑된 비율 | 96.6% | 95.0% |
| 활성 숨 구멍의 수 | 1111: 482, 371, 203, 55 | 1032: 447, 333, 196, 56 |
기계적 전단 프로토콜 다른 셀 라인을 사용 하 여 두 NLR seq의 보충 표 1: 요약.
토론
원칙적으로, nanopore 시퀀싱 길이11,,1213megabase 읽기 100 킬로바이트를 생성할 수 있다. 4 주요 요인 시퀀싱 실행 및 데이터 품질의 성능에 영향을 미칠 것입니다: 1) 활성 공 숫자와 모 공;의 활동 nanopore를 통과 하는 DNA의 속도 제어 하는 2) 모터 단백질 3) DNA 템플렛 (길이, 순도, 품질, 질량); 4) 시퀀싱 어댑터 결 찰 효율성, 입력된 샘플에서 유용한 DNA를 결정 합니다. 처음 두 가지 요소는 흐름 세포와 제조업체에서 제공 하는 시퀀싱 키트의 버전에 따라 달라 집니다. 두 번째 두 가지 요소 (HMW DNA 추출, 전단 및 결 찰)이이 프로토콜에 중요 한 단계가 있습니다.
이 프로토콜에는 인 내와 연습이 필요합니다. HMW DNA의 품질은 매우 긴 DNA 라이브러리6중요 합니다. 높은 생존 된 셀으로 시작 하는 프로토콜 (> 85% 가능한 셀 선호), 죽은 세포에서 저하 DNA를 제한. (예를 들어, 강력한 방해, 떨고, 소용돌이, 여러 pipetting, 반복 중지 및 재개) DNA에 손해를 도입할 수 있는 어떤 가혹한 과정은 피해 야 한다. 프로토콜의 디자인에 우리는 전체 DNA 추출 과정에서 pipetting 생략 합니다. 넓은 구멍 팁 pipetting 필요한 경우 도서관 건설 중 기계 전단 및 시퀀싱 후 사용할 필요가 있다. nanopores 챔버 버퍼12에서 화학 물질에 민감한 때문에, 이어야 한다 적은 잔여 오염 물질 (예를 들어, 세제, 계면 활성 제, 페 놀, 에탄올, RNAs 단백질, 등) 가능한 DNA에서. 길이 및 수율을 고려 하면 페 놀 추출 방법 지금까지 테스트 하는 여러 다른 추출 방법에 비해 최고의 그리고 가장 재현성 결과 보여 줍니다.
이 프로토콜의 생산 능력을 오래 읽기 시퀀스에 불구 하 고 몇 가지 제한이 여전히 남아 있다. 첫째,이 프로토콜 nanopore 시퀀싱 장치 간행물;의 시간에 사용할 수에 따라 최적화 되었다 따라서, 그것은 선택적 nanopore 기반 시퀀싱 화학 제한 되며 다른 유형의 긴 읽기 시퀀싱 장치에서 수행 하는 경우 최적이 될 수 있습니다. 둘째, 결과 매우 시작 물자 (조직 또는 세포)에서 추출한 DNA의 품질에 의존 합니다. 읽기 길이 시작 DNA 이미 저하 또는 손상 된 경우 손상 될 것 이다. 셋째, 여러 품질 관리 단계는 DNA 품질을 확인 하려면 프로토콜에 통합 됩니다, 비록 최종 수율과 읽기의 길이 흐름 세포에 의해 영향을 받을 수 및 활동, nanopore 시퀀싱 플랫폼의 초기 단계에서 변수를 수 있는 기 공 개발입니다.
설명 하는 프로토콜은 여기 DNA 추출에 대 한 인간의 현 탁 액 셀 라인 샘플을 사용 합니다. 우리는 바늘 전단, transposase와 설명된 결과를 내 고 시간을 HMW DNA의 비율에에서 지나가는 시간을 최적화 했습니다. 프로토콜 4 가지 방법으로 확장 될 수 있다. 첫째, 사용자가 다른 경작된 한 포유류 세포와 세포, 조직, 임상 샘플, 또는 다른 생물의 다른 금액으로 시작할 수 있습니다. 세포 배양 시간, 반응 볼륨 및 원심 분리에 더 최적화가 필요 합니다. 둘째, 그것은 매우 긴 읽기 시퀀싱에 대 한 대상 크기를 예측 하기 어렵다. 읽기 길이 예상 보다 짧은 경우에, 사용자가 기계적인 전단 기반 방법에 지나가는 시간을 조정 하거나 transposase 조각화 기반 메서드에서 transposase을 HMW DNA의 비율을 변경할 수 있습니다. 바인딩 및 차입 장시간 정리 단계 동안 HMW DNA는 높은 점성 때문에 도움이 됩니다. 셋째, 다른 nanopore 시퀀싱 장치와 하나 양과 DNA 시퀀서의 기준을 충족의 볼륨을 조정할 수 있습니다. 넷째, 시퀀싱 어댑터에 출혈 하는 DNA에만 시퀀싱 합니다. 추가로 결 찰 효율성을 개선 하기 위해, 하나의 어댑터와 리가 농도 적정 시도할 수 있습니다. 수정된 결 찰 시간 및 분자 크롤 링 에이전트 못18 등 미래에 적용할 수 있습니다. CRISPR19,20 와 함께 매우 긴 DNA 시퀀싱 프로토콜 대상 농축 시퀀싱을 위한 효과적인 도구를 제공할 수 있습니다.
공개
저자 들은 아무 경쟁 금융 관심사 선언 합니다.
감사의 말
저자는 원고에 대 한 그녀의 의견에 대 한 당 주를 감사합니다. 이 간행물에 보고 된 연구 보너스 번호 P30CA034196에서 국립 보건원의 국립 암 연구소에 의해 부분적으로 지원 되었다. 내용은 전적으로 저자의 책임 이며 반드시 국립 보건원의 공식 의견을 대표 하지 않는다.
자료
| Name | Company | Catalog Number | Comments |
| Reagents | |||
| Absolute ethanol | Sigma-Aldrich | E7023 | |
| Agencourt AMPure XPbeads | Beckman | A63881 | magnetic beads for cleanup |
| BD conventional needles | Becton Dickinson | 305136 | 27G, for mechanical shearing |
| BD Luer-Lok syringe | Becton Dickinson | 309628 | for mechanical shearing |
| Blunt/TA Ligase Master Mix | NEB | M0367S | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | for cell counting |
| EDTA | Invitrogen | AM9261 | pH 8.0, 0.5 M, 500 mL |
| Flow Cell | Oxford Nanopore Technologies | FLO-MIN106 | R9.4.1 |
| HG00773 cells | Coriell Institute | HG00733 | cells used in this protocol |
| Ligation Sequencing Kit 1D | Oxford Nanopore Technologies | SQK-LSK108 | nanopore ligation kit |
| MaXtract High Density tubes | Qiagen | 129073 | gel tubes |
| NEBNext FFPE DNA Repair Mix | NEB | M6630S | |
| NEBNext Ultra II End Repair/dA-Tailing Module | NEB | M7546S | |
| Nuclease-free water | Invitrogen | AM9937 | |
| Phosphate-Buffered Saline, PBS | Gibco | 70011044 | 10X, pH 7.4 |
| Phenol:chloroform:IAA | Invitrogen | AM9730 | |
| Proteinase K | Qiagen | 19131 | 20 mg/mL |
| Qubit dsDNA BR Assay Kit | Invitrogen | Q32850 | fluorometer assays for DNA quantification |
| Rapid Sequencing Kit | Oxford Nanopore Technologies | SQK-RAD004 | nanopore transposase kit |
| RNase A | Qiagen | 19101 | 100 mg/mL |
| SDS | Invitrogen | AM9822 | 10% (wt/vol) |
| Sodium chloride solution | Invitrogen | AM9759 | 5.0 M |
| TE buffer | Invitrogen | AM9849 | pH 8.0 |
| Tris | Invitrogen | AM9856 | pH 8.0, 1 M |
| Triton X-100 solution | Sigma-Aldrich | 93443 | ~10% |
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| Bio-Rad C1000 Thermal Cycler | Bio-Rad | 1851196EDU | |
| Centrifuge 5810R | Eppendorf | 22628180 | |
| Countess II FL Automated Cell Counter | Life Technologies | AMQAF1000 | for cell counting |
| DynaMag-2 Magnet | Life Technologies | 12321D | magnetic rack |
| Eppendorf ThermoMixer | Eppendorf | 5382000023 | for incubation |
| Freezer | LabRepCo | LHP-5-UFMB | |
| GridION | Oxford Nanopore Technologies | GridION X5 | nanopore device used in this protocol |
| HulaMixer Sample Mixer | Thermo Fisher Scientific | 15920D | rotator mixer |
| MicroCentrifuge | Benchmark Scientific | C1012 | |
| NanoDrop ND-1000 Spectrophotometer | Thermo Fisher Scientific | ND-1000 | for UV reading |
| Pippin Pulse | Sage Science | PPI0200 | pulsed-field gel electrophoresis instrument |
| Qubit 3.0 Fluorometer | Invitrogen | Q33216 | fluorometer |
| Refrigerator | LabRepCo | LABHP-5-URBSS | |
| Vortex-Genie 2 | Scientific Industries | SI-A236 | |
| Water bath | VWR | 89501-464 |
참고문헌
- Mardis, E. R. Next-generation sequencing platforms. Annual Review of Analytical Chemistry. 6, 287-303 (2013).
- Goodwin, S., McPherson, J. D., McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nature Reviews Genetics. 17 (6), 333-351 (2016).
- Shendure, J., et al. DNA sequencing at 40: past, present and future. Nature. 550 (7676), 345-353 (2017).
- Alkan, C., Coe, B. P., Eichler, E. E. Genome structural variation discovery and genotyping. Nature Reviews Genetics. 12 (5), 363-376 (2011).
- Weischenfeldt, J., Symmons, O., Spitz, F., Korbel, J. O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nature Reviews Genetics. 14 (2), 125-138 (2013).
- Pollard, M. O., Gurdasani, D., Mentzer, A. J., Porter, T., Sandhu, M. S. Long reads: their purpose and place. Human Molecular Genetics. 27 (R2), R234-R241 (2018).
- Cretu Stancu, M., et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nature Communications. 8 (1), 1326(2017).
- Gong, L., et al. Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nature Methods. 15 (6), 455-460 (2018).
- Sedlazeck, F. J., et al. Accurate detection of complex structural variations using single-molecule sequencing. Nature Methods. 15 (6), 461-468 (2018).
- Jain, M., et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology. 36 (4), 338-345 (2018).
- Jain, M., et al. Improved data analysis for the MinION nanopore sequencer. Nature Methods. 12 (4), 351-356 (2015).
- Deamer, D., Akeson, M., Branton, D. Three decades of nanopore sequencing. Nature Biotechnology. 34 (5), 518-524 (2016).
- Jain, M., Olsen, H. E., Paten, B., Akeson, M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biology. 17 (1), 239(2016).
- Editorial, The long view on sequencing. Nature Biotechnology. 36 (4), 287(2018).
- Jain, M., et al. Linear assembly of a human centromere on the Y chromosome. Nature Biotechnology. 36 (4), 321-323 (2018).
- Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094-3100 (2018).
- De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34, 2666-2669 (2018).
- Akabayov, B., Akabayov, S. R., Lee, S. J., Wagner, G., Richardson, C. C. Impact of macromolecular crowding on DNA replication. Nature Communications. 4, 1615(2013).
- Gabrieli, T., Sharim, H., Michaeli, Y., Ebenstein, Y. Cas9-Assisted Targeting of CHromosome segments (CATCH) for targeted nanopore sequencing and optical genome mapping. bioRxiv. , (2017).
- Gabrieli, T., et al. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Research. , (2018).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기더 많은 기사 탐색
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유